
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceKinetically controlled Ag+-coordinated chiral supramolecular polymerization accompanying a helical inversion†
Heekyoung
Choi
a,
Sojeong
Heo
a,
Seonae
Lee
a,
Ka Young
Kim
a,
Jong Hyeon
Lim
b,
Sung Ho
Jung
c,
Shim Sung
Lee
 a,
Hiroyuki
Miyake
d,
Jin Yong
Lee
*b and
Jong Hwa
Jung
*a
a,
Hiroyuki
Miyake
d,
Jin Yong
Lee
*b and
Jong Hwa
Jung
*a
aDepartment of Chemistry and Research Institute of Natural Sciences, Gyeongsang National University, Jinju 660-701, Republic of Korea. E-mail: jonghwa@gnu.ac.kr
bDepartment of Chemistry, Sungkyunkwan University, Suwon 16419, Republic of Korea. E-mail: jinylee@skku.edu
cDepartment of Liberal Arts, Gyeongnam National University of Science and Technology (GNTECH), Jinju 52725, Republic of Korea
dDepartment of Chemistry, Graduate School of Science, Osaka City University, Osaka 558-8585, Japan
First published on 14th November 2019
Abstract
We report kinetically controlled chiral supramolecular polymerization based on ligand–metal complex with a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (L:Ag+) stoichiometry accompanying a helical inversion in water. A new family of bipyridine-based ligands (D-L1, L-L1, D-L2, and D-L3) possessing hydrazine and D- or L-alanine moieties at the alkyl chain groups has been designed and synthesized. Interestingly, upon addition of AgNO3 (0.5–1.3 equiv.) to the D-L1 solution, it generated the aggregate I composed of the D-L1AgNO3 complex (D-L1:Ag+ = 1:1) as the kinetic product with a spherical structure. Then, aggregate I (nanoparticle) was transformed into the aggregate II (supramolecular polymer) based on the (D-L1)3Ag2(NO3)2 complex as the thermodynamic product with a fiber structure, which led to the helical inversion from the left-handed (M-type) to the right-handed (P-type) helicity accompanying CD amplification. In contrast, the spherical aggregate I (nanoparticle) composed of the D-L1AgNO3 complex with the left-handed (M-type) helicity formed in the presence of 2.0 equiv. of AgNO3 and was not additionally changed, which indicated that it was the thermodynamic product. The chiral supramolecular polymer based on (D-L1)3Ag2(NO3)2 was produced via a nucleation–elongation mechanism with a cooperative pathway. In thermodynamic study, the standard ΔG° and ΔHe values for the aggregates I and II were calculated using the van't Hoff plot. The enhanced ΔG° value of the aggregate II compared to that of the formation of aggregate I confirms that aggregate II was thermodynamically more stable. In the kinetic study, the influence of concentration of AgNO3 confirmed the initial formation of the aggregate I (nanoparticle), which then evolved to the aggregate II (supramolecular polymer). Thus, the concentration of the (D-L1)3Ag2(NO3)2 complex in the initial state plays a critical role in generating aggregate II (supramolecular polymer). In particular, NO3− acts as a critical linker and accelerator in the transformation from the aggregate I to the aggregate II. This is the first example of a system for a kinetically controlled chiral supramolecular polymer that is formed via multiple steps with coordination structural change.
:2 (L:Ag+) stoichiometry accompanying a helical inversion in water. A new family of bipyridine-based ligands (D-L1, L-L1, D-L2, and D-L3) possessing hydrazine and D- or L-alanine moieties at the alkyl chain groups has been designed and synthesized. Interestingly, upon addition of AgNO3 (0.5–1.3 equiv.) to the D-L1 solution, it generated the aggregate I composed of the D-L1AgNO3 complex (D-L1:Ag+ = 1:1) as the kinetic product with a spherical structure. Then, aggregate I (nanoparticle) was transformed into the aggregate II (supramolecular polymer) based on the (D-L1)3Ag2(NO3)2 complex as the thermodynamic product with a fiber structure, which led to the helical inversion from the left-handed (M-type) to the right-handed (P-type) helicity accompanying CD amplification. In contrast, the spherical aggregate I (nanoparticle) composed of the D-L1AgNO3 complex with the left-handed (M-type) helicity formed in the presence of 2.0 equiv. of AgNO3 and was not additionally changed, which indicated that it was the thermodynamic product. The chiral supramolecular polymer based on (D-L1)3Ag2(NO3)2 was produced via a nucleation–elongation mechanism with a cooperative pathway. In thermodynamic study, the standard ΔG° and ΔHe values for the aggregates I and II were calculated using the van't Hoff plot. The enhanced ΔG° value of the aggregate II compared to that of the formation of aggregate I confirms that aggregate II was thermodynamically more stable. In the kinetic study, the influence of concentration of AgNO3 confirmed the initial formation of the aggregate I (nanoparticle), which then evolved to the aggregate II (supramolecular polymer). Thus, the concentration of the (D-L1)3Ag2(NO3)2 complex in the initial state plays a critical role in generating aggregate II (supramolecular polymer). In particular, NO3− acts as a critical linker and accelerator in the transformation from the aggregate I to the aggregate II. This is the first example of a system for a kinetically controlled chiral supramolecular polymer that is formed via multiple steps with coordination structural change.
Introduction
Control of helicity in dynamic supramolecular assemblies based on metal–ligand coordinated architectures such as capsules, cages, and helices has rapidly developed into one of the most successful and versatile strategies in supramolecular chemistry,1–5 largely due to its essential role in biomolecules such as DNA and proteins. In addition, numerous artificial helical systems, ranging from oligonucleotides and peptide-mimetics to oligomers and polymers, have been developed.Metallosupramolecular polymers based on metal–ligand coordination have attracted much attention as a significant class of organic/inorganic hybrid supramolecular materials.6–30 Because of the incorporation of metal complexes in metallosupramolecular polymers, these metal-containing systems exhibit redox, optical, electrochromic, catalytic, and magnetic properties as well as the usual properties of organic polymers.31–34 Chiral metallosupramolecular polymers are particularly attractive because they are adaptable to chiral recognition and sensing, and asymmetric catalysis.35,36
In particular, recent advances in kinetic control over the dynamic structural and functional state of the metastable state (i.e., the out-of-equilibrium state) in supramolecular systems has allowed the development of supramolecular polymers with unprecedented structural complexity and sophisticated functionality.15–17,37–43 Such metastable states, in which spontaneous self-assembly is kinetically suppressed owing to competing aggregation and intramolecular hydrogen bonding, have been utilized to control the self-assembly behavior of supramolecular polymers.16,40,41 Furthermore, understanding artificial supramolecular systems from kinetic perspectives has improved in terms of pathway complexity.39,41,43–45 Moreover, the growth mechanisms of supramolecular polymerization can now be interpreted by the thermodynamic (cooperative or isodesmic) models proposed by Meijer et al.42,43,46 Such studies have demonstrated that self-assembly pathways play a significant role in determining thermodynamic or kinetic products. Thus, these previous works demonstrate that achieving kinetically programmable out-of-equilibrium states in supramolecular systems is a significant challenge related to achieving desired characteristics for dynamic self-assembly processes in supramolecular polymerization. For example, Besenius and co-workers demonstrated the kinetically controlled supramolecular polymerization of an Au(I)-metallopeptide in water.37 This metal complex kinetically assembles into metastable nanorods at room temperature but is converted into a thermodynamic equilibrium fibril state upon heating. Similarly, Fernández and co-workers recently demonstrated packing polymorphism in a supramolecular polymer achieved from an amphiphilic Pt(II) complex through the kinetically controlled pathway.15 However, kinetically controlled chiral supramolecular polymerization accompanying helical inversion, based on metal-coordinated building blocks by controlling the concentration of the unique complex structure has not been successful in water. Thus, kinetically controlled metal-coordinated chiral supramolecular polymerization that exhibits pathway complexity with unique metal–ligand building blocks remains highly challenging.
Based on these considerations, we designed a new family of chiral bipyridine-based ligands (D-L1, L-L1, D-L2, and D-L3) that have hydrazine and/or D- or L-alanine moieties at the alkyl chain groups as a chiral segment and a binding site for intermolecular hydrogen-bonding interactions in supramolecular polymerization (Fig. 1). The Ag+ ion was selected as a guest ion because it forms a variety of coordination geometries with bipyridine-based ligands, such as linear, triangular, square planar, and tetrahedral structures with different coordination numbers.47–49 In particular, there is a rarely observed Ag–Ag interaction in complexes obtained from Ag+ ions when three ligand molecules are coordinated with two Ag+ ions. Furthermore, the Ag–Ag interaction also acts as an additional driving force and a metal-coordinated chiral seed in chiral supramolecular polymerization.
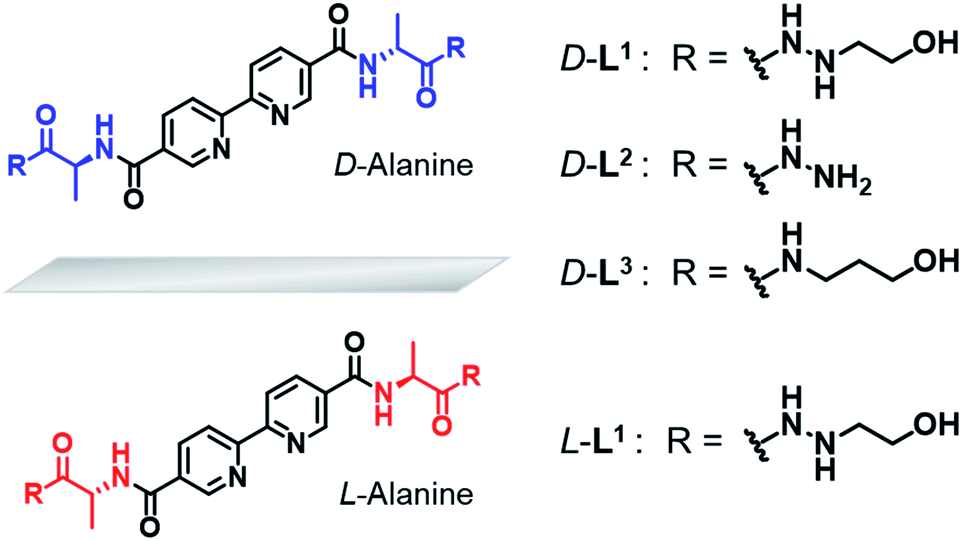 | ||
| Fig. 1 Chemical structures of chiral bipyridine derivatives with hydrazinoethanol (D-L1 and L-L1), hydrazine (D-L2) and 3-aminopropanol (D-L3). | ||
Surprisingly, we found that the left-handed (M-type) chiral bipyridine complexes (D-L1)2AgNO3 and D-L1AgNO3 are formed at less than 0.5 equiv. and at over 1.4 equiv. of AgNO3, respectively. More interestingly, the addition of 0.5–1.3 equiv. of AgNO3 to a D-L1 solution generates the D-L1AgNO3 complex, which forms aggregate I with a spherical structure. However, this eventually forms aggregate II with a fiber structure based on the (D-L1)3Ag2NO3 complex, involving left-handed (M-type) to right-handed (P-type) helicity conversion. The supramolecular polymerization that forms the fiber structure occurs via a cooperative pathway by a nucleation–elongation mechanism that is kinetically controlled by the concentrations of Ag+ and NO3−. Thus, we herein report kinetically controlled chiral supramolecular polymerization in water based on a unique complex building block.
Results and discussion
Stoichiometric ratios of self-assembled complexes with Ag+ by ESI-MS
Because bipyridine-based ligands form a variety of complexes with Ag+, we first investigated the complexation behavior of ligand D-L1, using high-resolution electrospray ionization mass spectrometry (HR ESI-MS) (Fig. S1–S4†). We confirmed that D-L1 formed mainly (D-L1)2AgNO3, (D-L1)3Ag2(NO3)2, and D-L1AgNO3 complexes with 0.4, 1.0, and 2.0 equiv. of Ag+, respectively (see ESI: explained in detail†).Helicity inversion in the formation of supramolecular polymers in the presence of Ag+ ion
To investigate the helicity of self-assembled complexes comprising D-L1 (with D-alanine moieties) and L-L1 (with L-alanine moieties) with Ag+, time-dependent circular dichroism (CD) and UV-Vis analysis of D-L1 (8 mM) was performed for three different concentrations (0.4, 1.0, and 2.0 equiv.) of AgNO3 at 25 °C (Fig. 2 and S5†). The negative CD signals immediately increase upon addition of 0.4 and 2.0 equiv. of AgNO3 but were not inverted to a positive CD signal (Fig. 2C: (a and c)). Conversely, the negative CD signal at 334 nm was inverted to a positive CD signal with 1.0 equiv. of AgNO3 after 70 min and was enhanced drastically (Fig. 2A and C: (b)). The transparent solution prepared with D-L1 (8 mM) and AgNO3 (1.0 equiv.) also changed to a colloidal suspension during the CD measurement (as observed by ESI/TOF-MS measurement). The CD inversion was due to the formation of the (D-L1)3Ag2(NO3)2 complex, which has right-handed (P-type) helicity. Furthermore, the weak CD signal intensity of D-L1 with 1.0 equiv. of AgNO3 is due to the presence of three complexes with different helicities in the initial state (Fig. 2C). The plot of the CD intensity and UV-Vis spectral changes showed a non-linear sigmoidal transition characterized by a nucleation–elongation mechanism (Fig. 2C and D). The long lag time in the CD and UV-Vis observations implies the existence of the kinetic aggregate.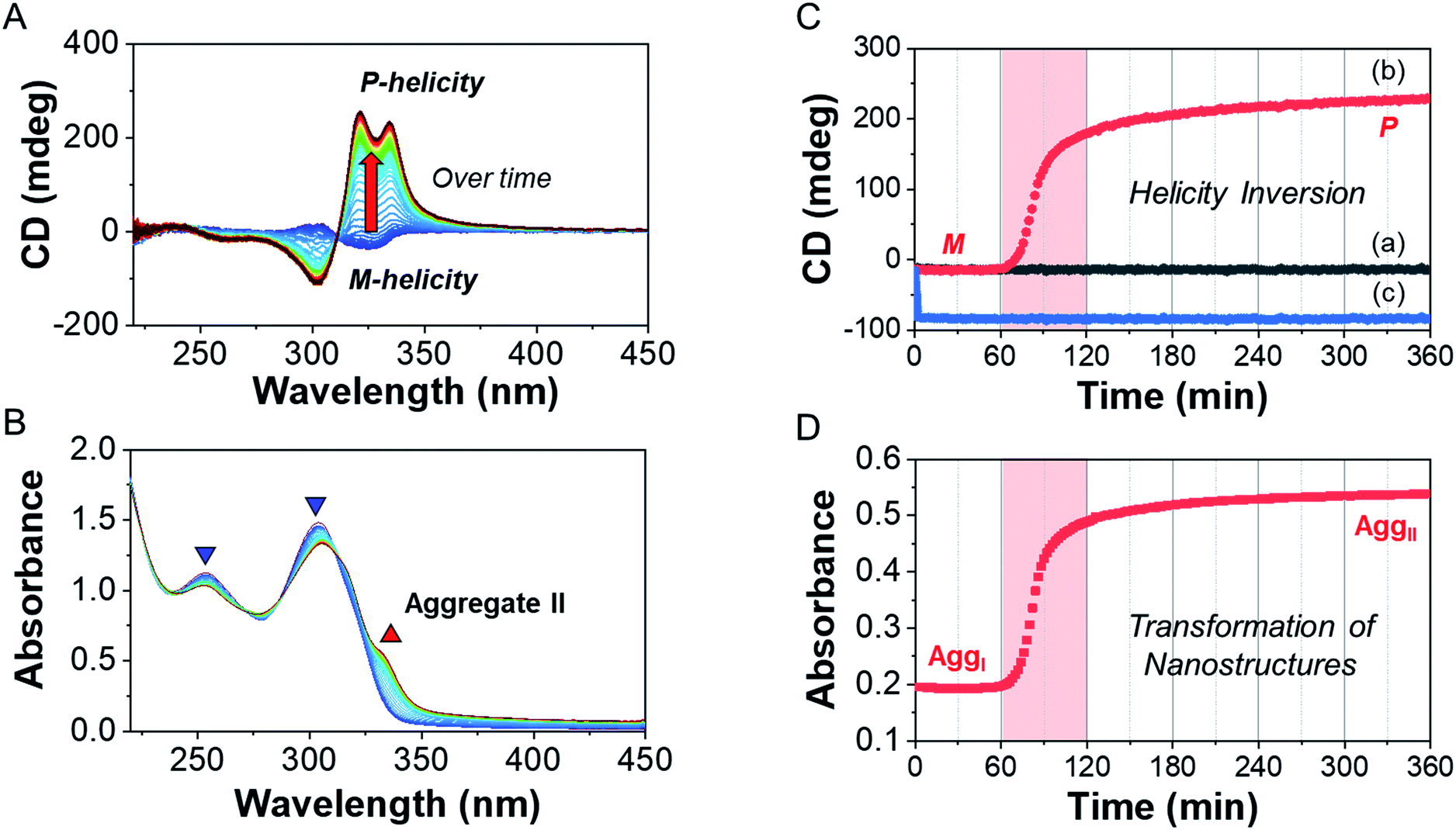 | ||
| Fig. 2 Time-dependent (A) CD and (B) UV spectra changes of D-L1 (8 mM) with AgNO3 (1.0 equiv.) in water. (C) Plot for CD spectra changes of D-L1vs. time in the presence of AgNO3 (a: 0.4, b: 1.0, and c: 2.0 equiv.) at 334 nm in water. (D) Plot for absorbance changes of D-L1vs. time in the presence of AgNO3 (1.0 equiv.) at 334 nm in water. | ||
We also observed the CD spectra of the supramolecular polymeric D-L1 complexes at various concentrations (0–2.0 equiv.) of Ag+ after aging for 3 h to eliminate the time effect (Fig. 3). The CD signal of complex D-L1 (8 mM) was observed at a mole ratio below 0.5 as a negative signal at 315 nm, which we ascribe to the bipyridine derivative D-L1 chromophores. These negative signals in the CD analysis indicate that complex (D-L1)2AgNO3 with four coordination was orientated to the left-handed direction (Schemes 1 and S1A†). Interestingly, the positive CD signal at 334 nm for supramolecular polymer D-L1 in cases where the Ag+-to-D-L1 mole ratio was greater than 0.4 was gradually enhanced (Fig. 3A and C), reaching a maximum at a Ag+-to-D-L1 mole ratio of 1.0 (Fig. 3C). This CD inversion was due to the transformation of (D-L1)2AgNO3 (anisotropic factor, |g| = 1.0 × 10−3 at 334 nm) and D-L1AgNO3 complexes into the (D-L1)3Ag2(NO3)2 complex, as shown in Scheme S1B.† Thus, the addition of 0.5–1.3 equiv. of AgNO3 generates aggregate I composed of the D-L1AgNO3 complex (|g| = 8.0 × 10−3 at 334 nm) with left-handed helicity as the kinetic product, and then finally aggregate II is formed based on the (D-L1)3Ag2(NO3)2 complex (|g| = 1.45 × 10−2 at 334 nm), which has right-handed (P-type) helicity (Schemes 1 and S1B†) as the thermodynamic product.
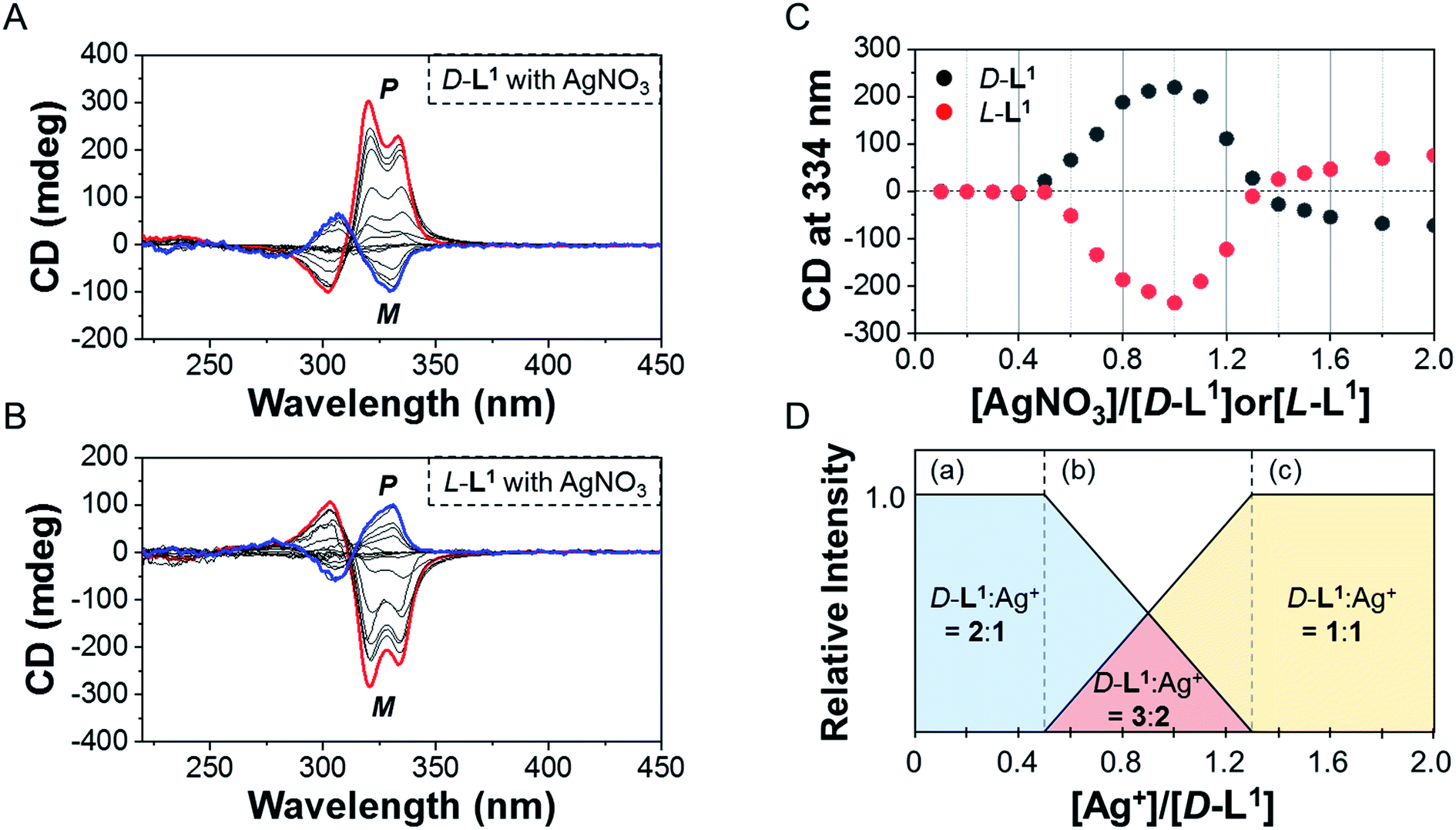 | ||
| Fig. 3 CD spectra of (A) D-L1 or (B) L-L1 upon addition of AgNO3 (0–2.0 equiv.) after aging for 3 h in water. (C) Plot for CD spectral changes of D-L1 and L-L1vs. equivalent of AgNO3 at 334 nm. (D) Complex structures existed in various concentration of AgNO3; (a) (D-L1)2AgNO3, (b) (D-L1)2AgNO3, (D-L1)3Ag2(NO3)2, and D-L1AgNO3, and (c) D-L1AgNO3. | ||
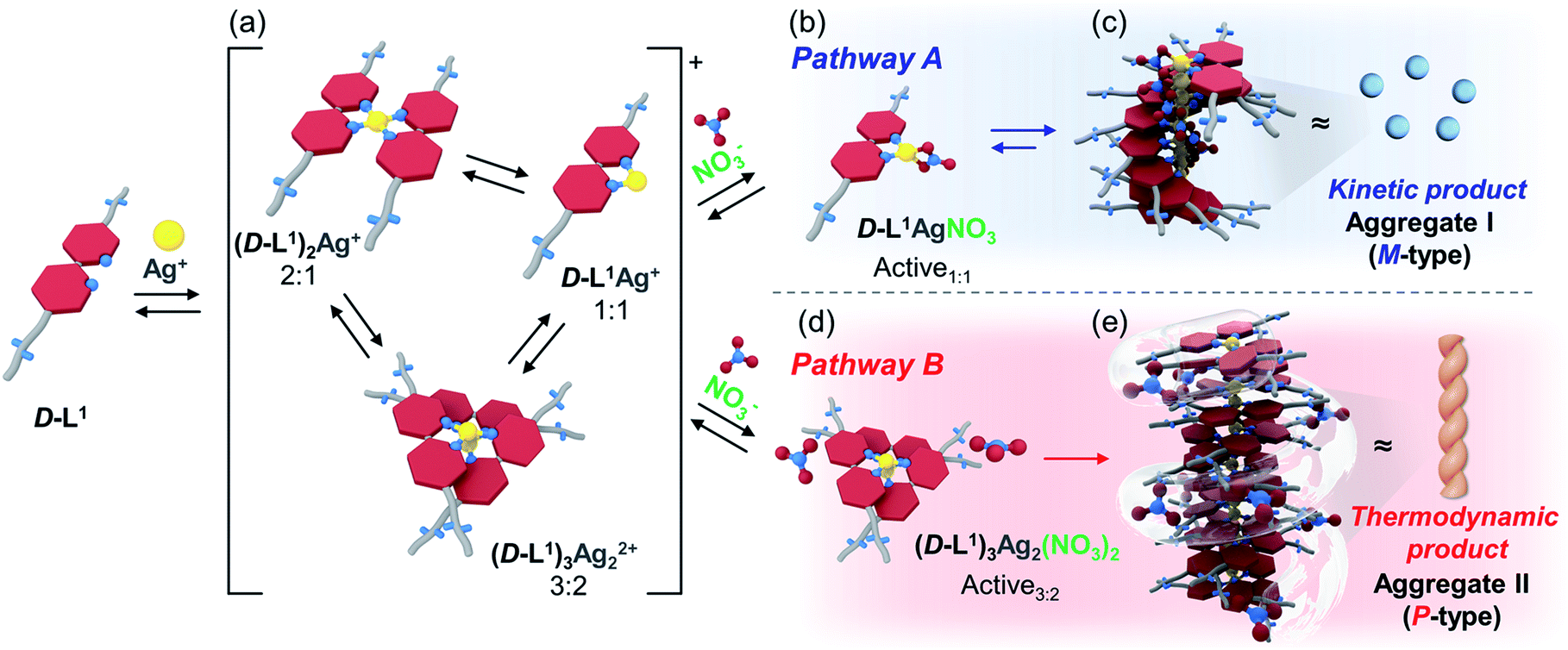 | ||
| Scheme 1 Proposed chiral supramolecular polymerization for D-L1 with AgNO3 (0.5–1.3 equiv.) showing the assembly by nucleation–elongation mechanism; (a) complex formation (D-L1:Ag+ = 2:1, 1:1 and 3:2), (b) 1:1 complex formation, (c) the formation of aggregate I based on D-L1AgNO3 complex with M-helicity, (d) 3:2 complex formation, and (e) formation of the aggregate II based on (D-L1)3Ag2(NO3)2 complex with P-helicity. | ||
Furthermore, the transparent solution became a colloidal suspension at 0.5–1.3 equiv. of Ag+. After separation, we observed the CD spectra of both the colloidal particles and the filtered solution. The colloidal particles dispersed in water exhibited an intense positive CD signal (Fig. S6a†), which was almost the same as that for the solution before filtration. However, the filtrate exhibited an extremely weak negative CD signal (Fig. S6b†). These findings indicate that complexes (D-L1)2AgNO3 and D-L1AgNO3 present before CD inversion change to (D-L1)3Ag2(NO3)2 as the colloidal aggregate II upon achieving chemical equilibrium, as shown in Schemes 1 and S1B.† These results provide clear evidence that the (D-L1)3Ag2(NO3)2 complex plays an essential role as a nucleus (seed) in the formation of aggregate II. In addition, it is noteworthy that AgNO3 acted as a key component in forming aggregates I and II. More importantly, the positive CD signal for the supramolecular polymer obtained with 1.0 equiv. of Ag+ did not change upon further addition of AgNO3 (Fig. S7†), indicating that aggregate II is the thermodynamic product. When the Ag+-to-D-L1 mole ratio increases beyond 1.0, the intensity of the CD signal for the supramolecular polymer gradually decreases again (Fig. 3A and C). This decrease in the positive CD signal was due to the coexistence of aggregate II and the monomeric D-L1AgNO3 complex species. At 1.3 equiv. of AgNO3, time-dependent CD spectra of D-L1 (8 mM) did not change after 180 min, indicating that the kinetic product was not existed (Fig. S8†). However, a small amount of monomeric 1:1 complex species, which does not form the spherical structure, exists with the thermodynamic product. Finally, the negative CD signal for D-L1 at over 1.4 equiv. of AgNO3 is attributed to the formation of the D-L1AgNO3 complex as the main product (Scheme S1C†), which indicates that the D-L1AgNO3 complex has M-helicity (left-handed helicity). In addition, the negative CD signal obtained at 1.5 equiv. of Ag+ was inverted to the positive signal when 0.5 equiv. of free D-L1 was added to a mixture of D-L1 and Ag+ (1.5 equiv.) (Fig. S9†). This CD inversion was due to the (D-L1)3Ag2(NO3)2 complex, which was generated from the D-L1AgNO3 complex. We conclude that the aggregate II as the thermodynamic product was only produced for 0.5–1.3 equiv. of Ag+ (Fig. 3D and Scheme 1).
In contrast, the CD spectra of complex L-L1 (8 mM) upon addition of AgNO3 (0–0.4 equiv.) exhibited a positive signal at 315 nm (Fig. 3B and C), which is exactly opposite to the behavior of complex D-L1 with Ag+. This positive CD signal indicates that the bipyridine moiety of L-L1 is orientated in the right-handed (P-type) direction. The positive CD signal of L-L1 was inverted into an intensely negative CD signal when 0.5–1.3 equiv. of Ag+ was added to L-L1. This chiral inversion is also due to the transformation from (L-L1)2AgNO3 and L-L1AgNO3 complexes into complex (L-L1)3Ag2(NO3)2, which led to the formation of the left-handed aggregate II. However, the positive CD signal of L-L1 was also observed when 1.4–2.0 equiv. of AgNO3 was added to L-L1, which was attributed to the formation of aggregate I (nanoparticles) based on the L-L1AgNO3 complex by path A in Scheme 1.
In order to investigate their linear dichroism (LD) effects, the LD spectra of D-L1 (8 mM) and L-L1 (8 mM) with AgNO3 (1.0 and 1.4 equiv.) were observed under the same conditions as those used for CD observations (Fig. S10†). Extremely weak LD signals were obtained for D-L1 and L-L1 in the presence of AgNO3 (1.0 and 1.4 equiv.). These results imply that the LD effect is only marginally affected by the CD response of supramolecular polymers.
To determine how anions effect the production of supramolecular polymers, we further observed the handedness of the supramolecular polymer upon addition of AgBF4, AgClO4, AgPF6, or AgOTf instead of NO3−. As shown in Fig. S11,† the negative CD signals for complex D-L1 formed with 0–2.0 equiv. of AgBF4, AgClO4, AgPF6, or AgOTf showed a slightly enhanced intensity, but they did not invert to the positive signal even though 3:2 complex (D-L1:Ag+) formation was confirmed by HR ESI-MS (Fig. S12–S15†). These findings imply that the NO3− anion plays a critical role in the formation of aggregate II, as shown in Schemes 1 and S1B.†
Role of NO3− anion in the supramolecular polymerization
To demonstrate the essential role of NO3− in the formation of the supramolecular polymer, we also observed the CD spectral changes for D-L1 in the presence of 1.0 equiv. of AgOTf, AgClO4, AgBF4, or AgPF6 upon the addition of 2.0 equiv. of NaNO3 (Fig. S16†). The weak negative CD signal became a positive CD signal when NaNO3 (0.4 equiv.) was added to a mixed D-L1 and AgOTf (1:1 equiv.) solution. The continuous addition of NaNO3 gradually enhanced the positive CD signals, and the signal reached a maximum at 2.0 equiv. of NaNO3 (Fig. 4), which was due to the generation of the aggregate II based on (D-L1)3Ag2(NO3)2. The peak for the [(D-L1)3Ag2NO3]+ fragmented from the (D-L1)3Ag2(NO3)2 complex was clearly observed in HR ESI/TOF-MS measurement (Fig. S17†). This led not only to the helical inversion but also to the formation of supramolecular polymers. These results indicate that NO3− plays a key role in not only the formation of the supramolecular polymer via the bridge effect but also helical inversion by path B, as shown in Scheme 1.
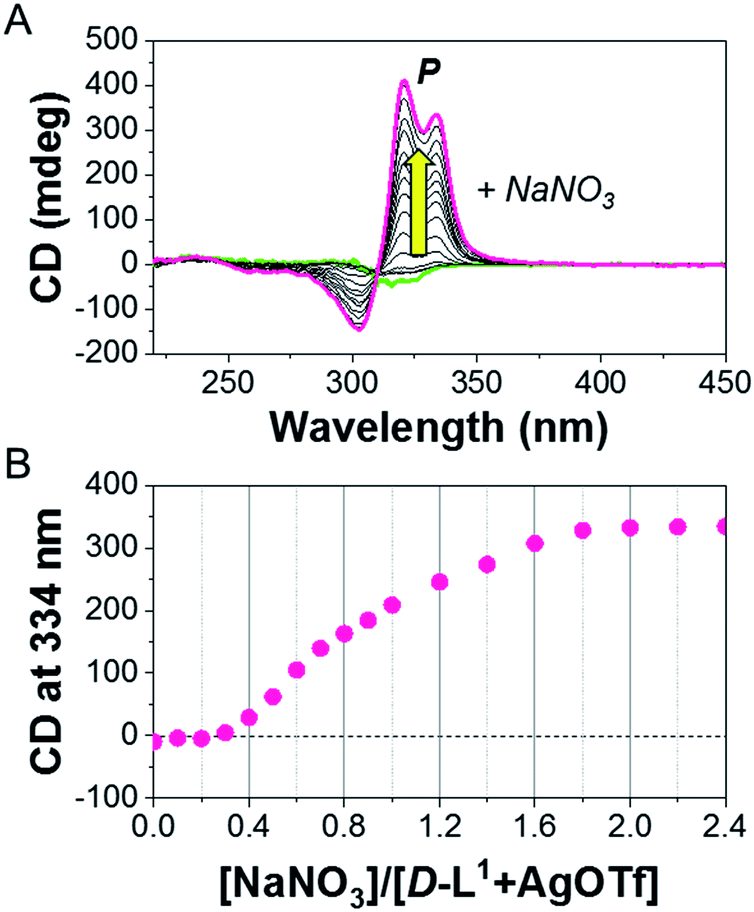 | ||
| Fig. 4 (A) CD spectra of D-L1 upon addition of NaNO3 in the presence of AgOTf (1.0 equiv.) in water. (B) Plot for CD intensity of D-L1 + AgOTf against added NaNO3 concentration. | ||
As a reference experiment, HR ESI-MS and CD spectra of D-L2, and D-L3 without ethyl alcohol or the hydrazine moiety at both ends were observed upon addition of AgNO3. The formation of the (D-L2)2AgNO3, (D-L2)3Ag2(NO3)2, or D-L2AgNO3 complexes obtained at different concentrations of Ag+ (0.4, 1.0, and 2.0 equiv. of Ag+) was confirmed via HR ESI-MS (Fig. S18–S20†). As expected, the D-L2 complex structures with different concentrations of Ag+ showed a similar tendency as that D-L1. The CD inversion of D-L2 (8 mM) with 0.8–1.1 equiv. of AgNO3 occurred due to the formation of aggregate II based on the (D-L2)3Ag2(NO3)2 complex (Fig. S21†). However, the CD intensity for the supramolecular polymer D-L2 was ca. 4.3-fold lower than that of supramolecular polymer D-L1 under a same condition, which was attributed to the weak complex formation between D-L2 and Ag+. However, the negative CD signal did not invert to a positive CD signal when 1.0 equiv. of AgNO3 was added to D-L3 (8 mM), which does not have the hydrazine moiety (Fig. S22†), because of no formation of 3:2 complex (D-L3:Ag+) as shown in Fig. S23.† These findings indicate that the amine in the hydrazine group plays a critical role in 3:2 complex formation (D-L3:Ag+).
Interestingly, upon addition of NaCl (0.9 equiv.) to aggregate I, a positive CD signal resulted, and the maximum positive CD signal was reached with 1.3 equiv. of NaCl (Fig. S24†). The (D-L1)2AgNO3 complex and free D-L1 were generated by decomplexation of D-L1AgNO3 in the presence of NaCl, and this eventually produced (D-L1)3Ag2(NO3)2, which enables the formation of aggregate II, as shown in Scheme S1C.† The largest positive CD signal induced via 1.3 equiv. of NaCl was ascribed to the formation of the largest amount of the (D-L1)3Ag2(NO3)2 complex. Thus, this CD inversion was due to transformation from the D-L1Ag to the (D-L1)3Ag2(NO3)2 complex, which led to further formation of aggregate II.
NMR and IR studies of aggregates I and II
To determine the exact binding site of D-L1 for NO3− in the formation of the supramolecular polymer, we obtained the 1H NMR spectrum of D-L1 in the presence and the absence of AgNO3 in D2O and H2O (1:1 v/v) (Fig. S25–S27†). These results suggest that the supramolecular polymer constructed from π–π stacking interactions, and the intermolecular hydrogen-bonding interactions between D-L1 and D-L1 were weakened due to the linkage with NO3− between the hydrazine moiety of D-L1 and D-L1 (see ESI: explained in detail†). The FT-IR results provide evidence for the intermolecular hydrogen bonds between the hydrazine NH and the NO3− in the chiral supramolecular polymers (Fig. S28 and S29, see ESI: explained in detail†). In addition, a sharp powder X-ray diffraction (XRD) peak for aggregate II was observed at 2θ = 32 (Fig. S30†), which corresponds to an Ag–Ag interaction with a distance of 2.8 Å.48 This suggests that NO3− did not coordinate with Ag+. The Ag–Ag interaction observed likely plays an important role in the chiral supramolecular polymerization. Thus, based on the FT-IR, powder XRD, and 1H NMR results, we determined that NO3− acted as a bridge to produce the helical supramolecular polymeric structure. This is in agreement with results previously reported by the Miyake group.50
Morphology observation of aggregates I and II by AFM
To confirm the morphology changes of the supramolecular polymers formed at different concentrations of AgNO3, we performed time-dependent atomic force microscopy (AFM) experiments (Fig. 5). For this purpose, freshly prepared solutions of D-L1 with and without AgNO3 (0.4, 1.0, 1.3, and 2.0 equiv.) were quickly dropped onto silicon wafers at 298 K after a constant aging time. This fixed the sample on the silicon wafer surface because it acts like a mode and prevents structural changes in the material during drying. | ||
| Fig. 5 AFM images of complex D-L1 with 1.0 equiv. of AgNO3 in water after aging of (A) 10 min, (B) 90 min, (C) 120 min and (D) 240 min. | ||
In the absence of AgNO3, the AFM image of pristine D-L1 showed fiber structures with widths of ca. 30 nm and lengths of several-hundred micrometers (Fig. S31†), indicating nanostructural formation of D-L1 by π–π stacking. This fiber structure did not change with time, which indicative that the fiber structure was the thermodynamic product. In contrast, no distinct morphology for the D-L1 solution with 0.4 equiv. of AgNO3 was observed regardless of aging time, indicating that the (D-L1)2AgNO3 complex did not elongate to form aggregates I or II. However, time-dependent AFM images of the D-L1 solution containing AgNO3 (2.0 equiv.), which formed complex D-L1AgNO3, were observed at different aging times. After aging for 10 min, the AFM image showed spherical structures with a diameter of ca. 50 nm (Fig. S32†). The spherical structures with dimensions close to 5 nm, as previously reported by Meijer's group,51 were similar to that of the micelle type containing a chiral molecular arrangement. This sphere would consist of the bilayer structure, because the full extended molecule length of D-L1 is ca. 3 nm.
No significant morphological changes were observed after aging for two weeks, indicating that the spherical structure was the thermodynamic product. The morphology changes were further supported by dynamic light scattering (DLS) study (Fig. S33b†). Note that a growth pattern in the scattering intensity was not observed, which means that the morphology did not change. This finding suggests that the nanoparticles formed in the presence of 2.0 equiv. of AgNO3 were a thermodynamic aggregate, as shown in Scheme S1C.†
Interestingly, the D-L1 solution containing AgNO3 (1.0 equiv.) had only small spherical structures with ca. 50 nm diameters at an aging time of 10 min (Fig. 5A), which originated from aggregate I (Scheme 1). After 90 min, the AFM image showed one-dimensional nanorods with 200 to 300 nm length and nanoparticles with ca. 50 nm diameter (Fig. 5B), which were due to aggregate II and the aggregate I, respectively. Upon allowing the solution to age for 120 min at 298 K, the nanorods grew into fibrous structures (Fig. 5C and D). This was clear evidence that the (D-L1)3Ag2(NO3)2 complex grew into one-dimensional fibers with intermolecular interactions as shown in Scheme 1. These fibers had an average thickness of ∼3 nm by AFM observation (Fig. S34†), which is consistent with the fully extended molecular length of ligand D-L1. Based on density functional theory (DFT) calculations, one fiber should consist of ca. 20 units of (D-L1)3Ag2(NO3)2 complex in the width direction (Fig. S34†). As shown by the DLS study (Fig. S33a†), the growth pattern in scattering intensity was similar to the time-dependent change in the CD signal at 334 nm. This clearly indicates that the spherical structure as the kinetic product was transformed into the fiber structure as the thermodynamic product. Furthermore, only fibrous structures were observed in the presence of 1.3 equiv. of AgNO3 after aging for 180 min (Fig. S35†), indicating that the thermodynamic product but not the kinetic product existed.
To confirm the anion effect in the formation of the supramolecular fibers, we obtained AFM images of the D-L1 solution containing AgOTf (1.0 equiv.). No significant morphological changes were observed, regardless of aging time. However, a narrow size distribution for the nanoparticles was observed during the lag time when NaNO3 (1.0 equiv.) was added to a D-L1 solution containing AgOTf (1.0 equiv.) (Fig. S36A†). These nanoparticles were transformed into fibrous structures several micrometers in length upon 90 min of aging (Fig. S36B and C†). This is indirect evidence that NO3− acted as a bridging ligand between the hydrazine groups of complex D-L1 to generate the chiral supramolecular polymer.
For D-L2 with AgNO3 (1.0 equiv.), the AFM image showed nanoparticles with ca. 50 nm diameter during the initial stage (Fig. S37A†). Then, helical fibrous structures with 100 μm length and 50 nm diameter was observed after 90 min of aging (Fig. S37B†). These results suggest that the supramolecular polymerization mechanism for D-L2 was the same as that for D-L1 and L-L1. We also confirmed the morphology of D-L3 (8 mM) with and without AgNO3 (1.0 equiv.) by AFM. The AFM image did not show any distinct morphologies for solutions of D-L3 in the presence and the absence of AgNO3 (1.0 equiv.) (Fig. S38†), because D-L3 could not form intermolecular hydrogen-bonding interactions between the –NH of the hydrazine moiety and NO3−.
DFT calculation for complexes formation with Ag+ ion
To gain insight into the influence of NO3− on formation of the D-L1 and Ag+ complex, we used DFT to optimize the structures of the complexes as confirmed by HR ESI-MS. During optimization, we considered the solvation effect of water by employing a conductor-like polarizable continuum model.52 Here, we optimized three complexes, i.e., D-L1AgNO3, (D-L1),2AgNO3, and (D-L1)3Ag2(NO3)2, depending on the number of NO3− ions and D-L1. As shown in Fig. 6, S39 and S40† (see ESI: explained in detail†), we found that the oxygen atoms of the NO3− moiety bonded to the hydrogen of the hydrazine group at the alkyl chain in the (D-L1)3Ag2(NO3)2 complex. The distances between the NO3− oxygens and the –NH hydrogen were in the range 1.93–1.95 Å, which is typical of hydrogen bonds (1.5–2.5 Å) between an oxygen acceptor and a nitrogen or oxygen donor.53,54 These results indicate that the NO3− group plays an essential role as an oxygen acceptor for hydrogen bonding in the complex formation of D-L1 and Ag+.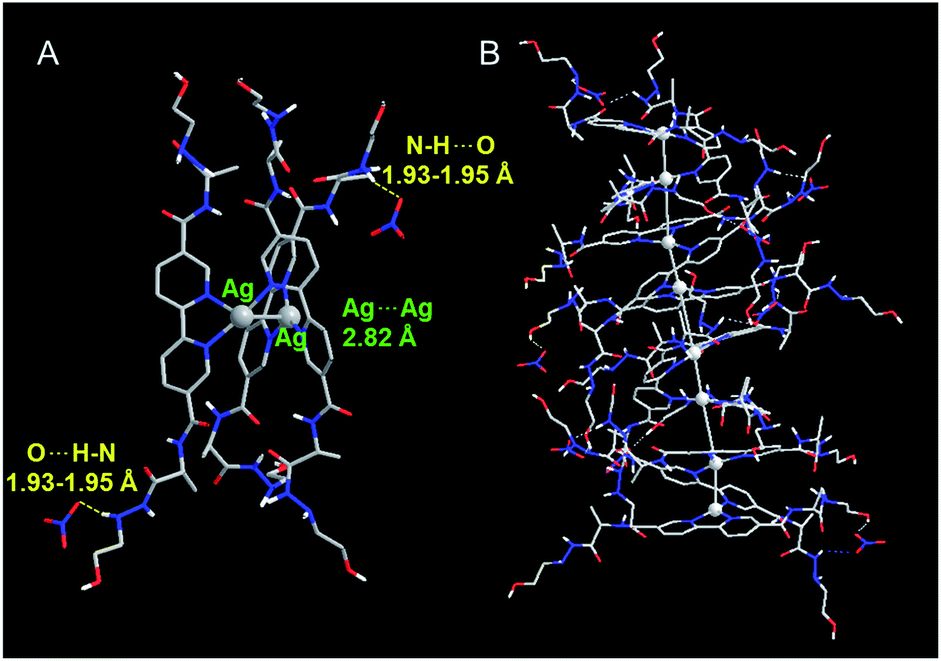 | ||
| Fig. 6 Optimized structure of (A) (D-L1)3Ag2(NO3)2 with P-helicity. (B) Proposed structure of the aggregate II consisting of four building block units of (D-L1)3Ag2(NO3)2. | ||
Thermodynamic study of aggregates I and II
To determine the thermodynamic parameters for supramolecular polymerization, temperature dependent spectra were observed at different concentrations (5–8 mM) of the supramolecular polymer formed with 1.0 equiv. of AgNO3 were observed from 293 to 373 K at a heating rate of 1 K min−1 (Fig. 7 and S41A†) because a long time is required for complex formation at room temperature. Further, the supramolecular polymerization (nucleation–elongation process) was observed only at concentrations >5 mM, due to the weak 3:2 complex formation. To determine the thermodynamic parameters, we calculated the results by referring to a previous study,55 which obtained thermodynamic parameters at high concentrations. The plot of CD intensity showed a non-sigmoidal curve that was characterized by the critical elongation temperature (Te) (Fig. 7 and S41B†). The onset temperatures for the elongation process were in the range 334.2–339.7 K, representing the critical temperatures between nucleation and elongation. The CD intensities of the supramolecular polymers enhanced with increasing concentration. The heating curves could be fitted using a nucleation–elongation model that has the characteristics of a cooperative assembly.56–58 We calculated the thermodynamic parameters using the van't Hoff plot.59
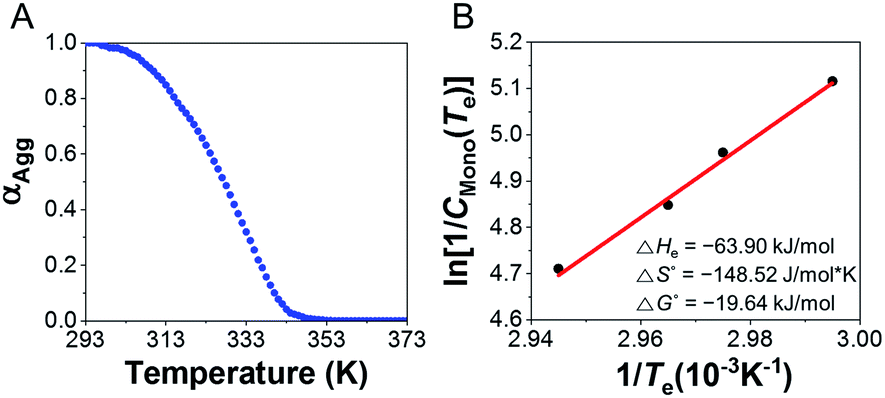 | ||
| Fig. 7 (A) Degree of aggregation of different concentrations of D-L1 (8 mM) complex with 1.0 equiv. of AgNO3 as a function of temperature at 334 nm in water. (B) Natural logarithm of the reciprocal CT as a function of the reciprocal Te showing the linear relationship. | ||
As shown in Fig. 7B, a linear correlation was evident between the concentration and Te in the van't Hoff plot. The standard ΔG° and ΔHe values for the supramolecular polymer (aggregate II) were calculated to be −19.64 kJ mol−1 and −63.90 kJ mol−1, respectively. The thermodynamic parameters of aggregate I at 2.0 equiv. of AgNO3 were also determined (Fig. S42†), even though the formation conditions of aggregate I differ slightly from those in Scheme S1B.† The standard ΔG° and ΔHe values for aggregate I were −15.82 kJ mol−1 and −33.08 kJ mol−1, respectively. The reduced ΔG° value compared to that for the formation of aggregate II (ΔG° = −19.64 kJ mol−1) confirms that aggregate II was thermodynamic product, as shown in Scheme S1B.† Accordingly, aggregate I was the kinetically favored state at 0.5–1.3 equiv. of AgNO3, i.e., aggregate II was the thermodynamically favored state in the energy landscape as related to pathway complexity (Scheme 1 and Fig. S43†). These results indicate that the pathway complexity in the self-assembly of aggregates I and II can be rationally modulated by controlling the conformation of the metal–ligand complex depending on the AgNO3 concentration. On the other hand, the standard ΔG° and ΔHe values for pristine D-L1 were calculated to be −15.23 and −25.26 kJ mol−1, respectively (Fig. S44†), which are lower than those for aggregates I and II.
Kinetic study of aggregate II at various concentrations of Ag+ and NO3−
To investigate kinetic control of the supramolecular polymer formation, we performed time-dependent CD experiments (Fig. 8). The kinetic profiles of supramolecular polymer D-L1 (8 mM) that formed with different concentrations of AgNO3 (0.6–1.0 equiv.) in aqueous solution at 25 °C showed non-sigmoidal curves with different lag times (Fig. 8). The t50 value, at which the transformation is 50% complete, became short as the AgNO3 concentration increased (Fig. 8B). The influence of AgNO3 concentration on the kinetics confirmed the initial formation of aggregate I consisting of D-L1AgNO3, which developed into aggregate II consisting of (D-L1)3Ag2(NO3)2. The largest amount of the (D-L1)3Ag2(NO3)2 complex produced during the initial stage in the presence of 1.0 equiv. of AgNO3, kinetically accelerated the transformation of aggregate I into the aggregate II. Thus, the concentration of the (D-L1)3Ag2(NO3)2 complex during the initial state played a critical role in producing aggregate II, which exhibited a helical fiber structure.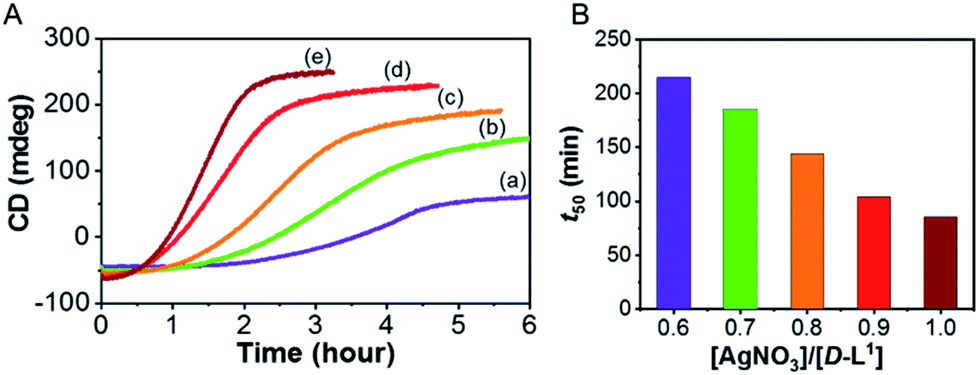 | ||
| Fig. 8 (A) Time course evolution of the aggregate I of D-L1 into the kinetically controlled the aggregate II with different concentrations of AgNO3; (a) 0.6, (b) 0.7, (c) 0.8, (d) 0.9 and (e) 1.0 equiv. at 334 nm in water. (B) Representation for t50 values vs. AgNO3 concentration. | ||
Furthermore, the kinetic profiles of the supramolecular polymer were also observed upon addition of NaNO3 (1.0, 1.4, 1.7, and 2.0 equiv.) in the presence of AgOTf (1.0 equiv.) (Fig. S45†). The highest concentration of NaNO3 showed the largest positive CD intensity (Fig. S45d†) owing to formation of the largest amount of the (D-L1)3Ag2(NO3)2 complex under these conditions. These results imply that the kinetics of aggregate II strongly influenced by NO3− anions. Furthermore, the lag time decreased with increasing NaNO3 concentration, indicating that the NO3− anion plays an essential role in the helical inversion from aggregate I to aggregate II. To our knowledge, this is the first example of a system in which the NO3− anion acts as an accelerator and linker in the transformation of aggregates. In addition, the kinetic profile of the supramolecular polymer D-L2 that does not have an ethyl alcohol moiety showed the same tendency as that of compound D-L1 (Fig. S46†). However, the supramolecular polymerization of D-L2 was observed in a narrower Ag+ concentration range compared to that for D-L1 because D-L2 forms the (D-L2)2AgNO3 complex as a main product, as shown by HR ESI-MS measurements.
Conclusions
We have demonstrated that supramolecular polymerization involving a distinct metal–ligand complex was controlled kinetically via the concentration of Ag+ and anions in water. This also enabled the controlled, reversible transformation between M- and P-type helices, with a change in complex structure as a function of Ag+ concentration. A change in the complex structure also accompanied the helical inversion, where the M-type helices of (D-L1)2AgNO3 and D-L1AgNO3 were inverted to the P-type helix of (D-L1)3Ag2(NO3)2. In the kinetic study, the (D-L1)3Ag2(NO3)2 complex was found to play an essential role in chiral inversion. The distinct (D-L1)3Ag2(NO3)2 complex produced the chiral supramolecular polymer (aggregate II) via a nucleation–elongation mechanism with a cooperative pathway in the range of 0.5–1.3 equiv. of AgNO3, and this process showed an unusually long lag time. In addition, D-L1, and L-L1 in the presence of 0.5–1.3 equiv. of AgNO3 formed intermediate aggregate I (nanoparticles) as the kinetic product that evolved to afford a helical supramolecular polymer (aggregate II). This transformation can be accelerated according to the Ag+ and NO3− concentration. Also, AgNO3 acted as a key component in forming aggregates I and II. In particular, NO3− bridged between hydrazine–NH groups of the complex via intermolecular hydrogen bonds in the supramolecular polymer. In contrast, aggregate I formed at over 1.4 equiv. of AgNO3 was the thermodynamic product.This system is an extremely rare example of chiral supramolecular polymerization based on metal–organic building blocks via control of coordination number. We believe that this concept will greatly contribute to the development of metal-coordinated supramolecular systems with optimized functional properties and may provide new insights into the understanding of the chiral assembly process in complex biological systems.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NRF (2017R1A4A1014595 and 2018R1A2B2003637) from the Ministry of Science, ICT and Future Planning, Korea and the Korea Basic Science Institute (KBSI) National Research Facilities & Equipment Center (NFEC) grant funded by the Korea government (Ministry of Education) (No. 2019R1A6C1010042). In addition, this work was partially supported by a grant from the Next-Generation BioGreen 21 Program (SSAC, grant#: PJ013186052019), Rural development Administration, Korea.Notes and references
- N. Ousaka, Y. Takeyama, H. Iida and E. Yashima, Nat. Chem., 2011, 3, 856–861 CrossRef CAS.
- J. Rebek, Jr, Angew. Chem., Int. Ed., 2005, 44, 2068–2078 CrossRef.
- P. J. Stang, B. Olenyuk, D. C. Muddiman, D. S. Wunschel and R. D. Smith, Organometallics, 1997, 16, 3094–3096 CrossRef CAS.
- E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752–13990 CrossRef CAS.
- A. R. A. Palmans and E. W. Meijer, Angew. Chem., Int. Ed., 2007, 46, 8948–8968 CrossRef CAS.
- M. Albrecht, Chem. Rev., 2001, 101, 3457–3498 CrossRef CAS.
- Q. Wan, X.-S. Xiao, W.-P. To, W. Lu, Y. Chen, K.-H. Low and C.-M. Che, Angew. Chem., Int. Ed., 2018, 57, 17189–17193 CrossRef CAS.
- Q. Wan, W.-P. To, C. Yang and C.-M. Che, Angew. Chem., Int. Ed., 2018, 57, 3089–3093 CrossRef CAS.
- J. L.-L. Tsai, T. Zou, J. Liu, T. Chen, A. O.-Y. Chan, C. Yang, C.-N. Lok and C.-M. Che, Chem. Sci., 2015, 6, 3823–3830 RSC.
- A. Mishra, D. B. Korlepara, M. Kumar, A. Jain, N. Jonnalagadda, K. K. Bejagam, S. Balasubramanian and S. J. George, Nat. Commun., 2018, 9, 1295 CrossRef.
- K. K. Kartha, N. K. Allampally, A. T. Politi, D. D. Prabhu, H. Ouchi, R. Q. Albuquerque, S. Yagai and G. Fernández, Chem. Sci., 2019, 10, 752–760 RSC.
- F. Mamiya, N. Ousaka and E. Yashima, Angew. Chem., Int. Ed., 2015, 54, 14442–14446 CrossRef CAS.
- N. Ousaka, K. Shimizu, Y. Suzuki, T. Iwata, M. Itakura, D. Taura, H. Iida, Y. Furusho, T. Mori and E. Yashima, J. Am. Chem. Soc., 2018, 140, 17027–17039 CrossRef CAS.
- M. E. Robinson, A. Nazemi, D. J. Lunn, D. W. Hayward, C. E. Boott, M.-S. Hsiao, R. L. Harniman, S. A. Davis, G. R. Whittell, R. M. Richardson, L. De Cola and I. Manners, ACS Nano, 2017, 11, 9162–9175 CrossRef CAS.
- A. Langenstroer, K. K. Kartha, Y. Dorca, J. Droste, V. Stepanenko, R. Q. Albuquerque, M. R. Hansen, L. Sánchez and G. Fernández, J. Am. Chem. Soc., 2019, 141, 5192–5200 CrossRef CAS PubMed.
- S. Ogi, C. Grzeszkiewicz and F. Würthner, Chem. Sci., 2018, 9, 2768–2773 RSC.
- L. Herkert, J. Droste, K. K. Kartha, P. A. Korevaar, T. F. A. de Greef, M. R. Hansen and G. Fernández, Angew. Chem., Int. Ed., 2019, 58, 11344–11349 CrossRef CAS.
- A. Langenstroer, Y. Dorca, K. K. Kartha, M. J. Mayoral, V. Stepanenko, G. Fernández and L. Sánchez, Macromol. Rapid Commun., 2018, 39, 1800191 CrossRef.
- N. A. M. S. Caturello, Z. Csók, G. Fernández and R. Q. Albuquerque, Chem.–Eur. J., 2016, 22, 17681–17689 CrossRef CAS.
- K. Y. Kim, J. Kim, C. J. Moon, J. Liu, S. S. Lee, M. Y. Choi, C. Feng and J. H. Jung, Angew. Chem., Int. Ed., 2019, 58, 11709–11714 CrossRef CAS.
- H. Park, K. Y. Kim, S. H. Jung, Y. Choi, H. Sato and J. H. Jung, Chem. Mater., 2018, 30, 2074–2083 CrossRef CAS.
- K. Y. Kim, J. Kim, H. Park, Y. Choi, K.-Y. Kwon and J. H. Jung, Bull. Korean Chem. Soc., 2018, 39, 988–994 CrossRef CAS.
- V. C.-H. Wong, C. Po, S. Y.-L. Leung, A. K.-W. Chan, S. Yang, B. Zhu, X. Cui and V. W.-W. Yam, J. Am. Chem. Soc., 2018, 140, 657–666 CrossRef CAS.
- S. Y.-L. Leung, K. M.-C. Wong and V. W.-W. Yam, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2845–2850 CrossRef CAS.
- M. H.-Y. Chan, S. Y.-L. Leung and V. W.-W. Yam, J. Am. Chem. Soc., 2018, 140, 7637–7646 CrossRef CAS.
- Y. Sakata, S. Chiba, M. Miyashita, T. Nabeshima and S. Akine, Chem.–Eur. J., 2019, 25, 2962–2966 CrossRef CAS.
- T. Nakamura, H. Kimura, T. Okuhara, M. Yamamura and T. Nabeshima, J. Am. Chem. Soc., 2016, 138, 794–797 CrossRef CAS.
- R. Matsuoka and T. Nabeshima, Front. Chem., 2018, 6, 349 CrossRef.
- B. Kemper, M. von Gröning, V. Lewe, D. Spitzer, T. Otremba, N. Stergiou, D. Schollmeyer, E. Schmitt, B. J. Ravoo and P. Besenius, Chem.–Eur. J., 2017, 23, 6048–6055 CrossRef CAS.
- B. Kemper, Y. R. Hristova, S. Tacke, L. Stegemann, L. S. van Bezouwen, M. C. A. Stuart, J. Klingauf, C. A. Strassert and P. Besenius, Chem. Commun., 2015, 51, 5253–5256 RSC.
- A. Winter and U. S. Schubert, Chem. Soc. Rev., 2016, 45, 5311–5357 RSC.
- L. Yu, Z. Wang, J. Wu, S. Tu and K. Ding, Angew. Chem., Int. Ed., 2010, 49, 3627–3630 CrossRef CAS.
- G. R. Whittell, M. D. Hager, U. S. Schubert and I. Manners, Nat. Mater., 2011, 10, 176–188 CrossRef CAS.
- M. Higuchi, J. Mater. Chem. C, 2014, 2, 9331–9341 RSC.
- D. Taura, S. Hioki, J. Tanabe, N. Ousaka and E. Yashima, ACS Catal., 2016, 6, 4685–4689 CrossRef CAS.
- L.-J. Chen, H.-B. Yang and M. Shionoya, Chem. Soc. Rev., 2017, 46, 2555–2576 RSC.
- B. Kemper, L. Zengerling, D. Spitzer, R. Otter, T. Bauer and P. Besenius, J. Am. Chem. Soc., 2018, 140, 534–537 CrossRef CAS.
- A. Sorrenti, J. Leira-Iglesias, A. J. Markvoort, T. F. A. de Greef and T. M. Hermans, Chem. Soc. Rev., 2017, 46, 5476–5490 RSC.
- T. Fukui, S. Kawai, S. Fujinuma, Y. Matsushita, T. Yasuda, T. Sakurai, S. Seki, M. Takeuchi and K. Sugiyasu, Nat. Chem., 2017, 9, 493–499 CrossRef CAS.
- W. Wagner, M. Wehner, V. Stepanenko, S. Ogi and F. Würthner, Angew. Chem., Int. Ed., 2017, 56, 16008–16012 CrossRef CAS.
- S. Ogi, V. Stepanenko, J. Thein and F. Würthner, J. Am. Chem. Soc., 2016, 138, 670–678 CrossRef CAS.
- M. F. J. Mabesoone, A. J. Markvoort, M. Banno, T. Yamaguchi, F. Helmich, Y. Naito, E. Yashima, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2018, 140, 7810–7819 CrossRef CAS.
- P. A. Korevaar, S. J. George, A. J. Markvoort, M. M. J. Smulders, P. A. J. Hilbers, A. P. H. J. Schenning, T. F. A. De Greef and E. W. Meijer, Nature, 2012, 481, 492–496 CrossRef CAS.
- S. Ogi, K. Sugiyasu, S. Manna, S. Samitsu and M. Takeuchi, Nat. Chem., 2014, 6, 188–195 CrossRef CAS.
- J. S. Valera, R. Gomez and L. Sánchez, Small, 2018, 14, 1702437 CrossRef PubMed.
- M. A. J. Gillissen, M. M. E. Koenigs, J. J. H. Spiering, J. A. J. M. Vekemans, A. R. A. Palmans, I. K. Voets and E. W. Meijer, J. Am. Chem. Soc., 2014, 136, 336–343 CrossRef CAS PubMed.
- T. Nakamura, H. Ube and M. Shionoya, Angew. Chem., Int. Ed., 2013, 52, 12096–12100 CrossRef CAS PubMed.
- A. Bellusci, M. Ghedini, L. Giorgini, F. Gozzo, E. I. Szerb, A. Crispini and D. Pucci, Dalton Trans., 2009, 7381–7389 RSC.
- A. Bellusci, A. Crispini, D. Pucci, E. I. Szerb and M. Ghedini, Cryst. Growth Des., 2008, 8, 3114–3122 CrossRef CAS.
- H. Miyake, K. Yoshida, H. Sugimoto and H. Tsukube, J. Am. Chem. Soc., 2004, 126, 6524–6525 CrossRef CAS PubMed.
- P. Besenius, G. Portale, P. H. H. Bomans, H. M. Janssen, A. R. A. Palmans and E. W. Meijer, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17888–17893 CrossRef CAS PubMed.
- J. Tomasi and M. Persico, Chem. Rev., 1994, 94, 2027–2094 CrossRef CAS.
- M. Shen, Y. Xie, H. F. Schaefer, III and C. A. Deakyne, J. Chem. Phys., 1990, 93, 3379–3388 CrossRef CAS.
- T. Vijayakumar, I. H. Joe, C. P. R. Nair and V. S. Jayakumar, J. Mol. Struct., 2008, 877, 20–35 CrossRef CAS.
- M. H.-Y. Chan, M. Ng, S. Y.-L. Leung, W. H. Lam and V. W.-W. Yam, J. Am. Chem. Soc., 2017, 139, 8639–8645 CrossRef CAS PubMed.
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS PubMed.
- M. Wehner, M. I. S. Röhr, M. Bühler, V. Stepanenko, W. Wagner and F. Würthner, J. Am. Chem. Soc., 2019, 141, 6092–6107 CrossRef CAS PubMed.
- S. Dhiman, A. Sarkar and S. J. George, RSC Adv., 2018, 8, 18913–18925 RSC.
- M. M. J. Smulders, M. M. L. Nieuwenhuizen, T. F. A. de Greef, P. van der Schoot, A. P. H. J. Schenning and E. W. Meijer, Chem.–Eur. J., 2010, 16, 362–367 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc04958d |
| This journal is © The Royal Society of Chemistry 2020 |