
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of copper-catalyzed enantioselective decarboxylative aldolization for the preparation of perfluorinated 1,3,5-triols featuring supramolecular recognition properties†
Céline
Sperandio
,
Jean
Rodriguez
and
Adrien
Quintard
 *
*
Aix Marseille Univ, CNRS, Centrale Marseille, iSm2, Marseille, France. E-mail: adrien.quintard@univ-amu.fr; Web: http://ism2.univ-amu.fr/fr/annuaire/stereo/quintardadrien
First published on 27th December 2019
Abstract
Fluorine is able to confer unique properties to organic molecules but the scarcity of natural organofluorine sources renders the development of new synthetic methods highly desirable. Using a chiral BOX/Cu combination, enantioselective decarboxylative aldolization of perfluorinated aldehydes has been developed. Most notably, the reaction occurring under mild conditions and with high enantiocontrol can create ketodiols in one single synthetic operation, which are precursors of crucial perfluorinated 1,3,5-triols. In addition, the reaction performed with chloral, validates the proposed transition state model based on steric interactions and provides the first enantioselective synthesis of hexachlorinated ketodiol of great synthetic utility. The ability of perfluorinated 1,3,5-triols to form a central hydrogen-bonding framework allows strong coordination of anions and the chirality obtained through the catalyst-controlled synthetic sequence demonstrates the selective chiral anion recognition ability of polyols.
Introduction
Organic molecules efficient in selective supramolecular recognition are central to numerous drugs, nano-devices, catalysts and materials.1 As a result, chemist's ability to design and prepare host molecules with improved binding affinities and selectivities is crucial. However, the preparation of original molecular objects has for long remained limited by the availability of starting materials or current existing synthetic methods. This is especially true regarding fluorine containing molecules, notably the ones possessing strong electron-withdrawing perfluorinated chains ((CF2)n-CF3), able to confer unique properties to numerous drugs, materials and catalysts.2 While fluorine insertion can impressively improve these supramolecular interactions, the preparation of these molecules depends exclusively on the synthetic chemist's ability to accommodate this particular atom through a reliable synthetic route, rendering the discovery of original synthetic methods of great value.3Given the progress made in the last few decades towards more efficient synthesis thanks to catalysis,4 it should now be possible to conceive on a routine basis the de novo design and synthesis of improved molecular objects through the parallel on demand development of original catalytic methods.
Among potential motifs for host molecule design, acyclic alcohols are particular due to the relatively weak interactions provided by a single alcohol hydrogen bond. This is why despite their ubiquity in numerous highly bioactive natural products and drugs, synthetic flexible acyclic polyols have only been identified recently for their coordination and catalytic properties.5 It was shown notably by the group of Kass that multiplication of alcohol functionalities5e–g or insertion of adjacent CF3 groups6 could increase the H-bonding strength of the core alcohol H-bonding frameworks. This role of the CF3 substituents in alcohol properties is further highlighted by the unique reactivity promoted by the use of hexafluoroisopropanol (HFIP) as the solvent.7 However, the enantioselective elaboration of perfluorinated alcohols still constitutes a major synthetic challenge. This considerably limits the construction of new scaffolds featuring enhanced supramolecular chiral recognition properties. As a result, designing new types of chiral perfluorinated alcohols and discovering new approaches for their enantioselective preparation is highly desirable.
In this context, we hypothesized that combining a central chiral 1,3,5-triol motif with adjacent electron-withdrawing perfluorinated chains would provide innovative molecules for application in selective supramolecular recognition by anion binding (Scheme 1a). These molecules would bridge the gap between known achiral or racemic flexible acyclic alcohols5,6 and more classical chiral receptors8 with strong implications in anion coordination or catalysis.
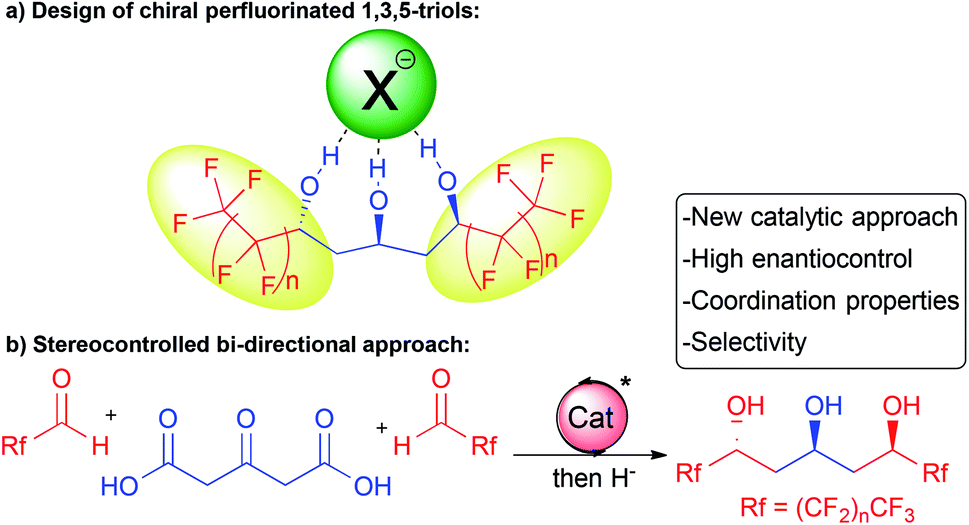 | ||
| Scheme 1 Proposed enantioselective approach towards perfluorinated 1,3,5-triols and their supramolecular coordination properties. | ||
In addition, in order to readily prepare these chiral complex molecules, we desired to perform parallel development of an original and modular enantioselective catalyst-controlled method also amenable to target other scaffolds of interest (Scheme 1b).
Herein, we report our success at fulfilling this double goal of constructing rapidly and with excellent stereocontrol the desired perfluorinated 1,3,5-triols with enhanced supramolecular properties starting from bio-sourced 1,3-acetonedicarboxylic acid through stereocontrolled bi-directional decarboxylative aldolization (Scheme 1b).
Results and discussion
Development of catalytic enantioselective perfluorinated 1,3,5-triol synthesis
Among the limited number of methods able to create enantioenriched perfluorinated secondary alcohols,9,10 the aldol reaction seems the most promising given its ability to potentially rapidly assemble elaborated fragments while controlling the hydroxy functionalized stereocenters.11 However, the reported aldolization reactions are unfortunately limited to the generation of simple trifluoromethylated ketols.12 In addition, the most efficient ones are incompatible with perfluorinated aldehyde hydrates thus requiring the application of more elaborated hemiacetals, while suffering from excessive reaction times (4 to 7 days). These slow kinetics constitute a severe limitation for their desired implementation to the proposed more challenging cascade preparation of 1,3,5-triol cores.12d,f,13To successfully create rapidly perfluorinated 1,3,5-triols, we proposed to take advantage of the high reactivity of 1,3-acetonedicarboxylic acid (2) towards multiple additions.14 Previous aldolizations using this precursor are based on substrate-controlled diastereoselective approaches15 and we set out to identify a suitable catalytic activation mode for the more challenging enantioselective addition to perfluorinated aldehyde hydrate 1a (Table 1).16
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Yielda | dr | ee |
| a Isolated yield. b Determined by 19F NMR and gas chromatography. c Determined by chiral gas chromatography. d Full conversion, yield not determined. e yield, dr and ee within parenthesis are given after purification by recrystallization. f Opposite enantiomer obtained. g Reaction run over 12 hours. The same result is observed using toluene as the solvent. | ||||
| 1 | (S,S)-TUC (10 mol%) | 38% | 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
2% |
| 2 | Quinine (10 mol%) | ndd | 1:1 |
17% |
| 3 | (DHQD)2PHAL (10 mol%) | ndd | 1:1 |
10% |
| 4 | Cu(OTf)2 (10 mol%)/L1-4 (13 mol%) | 34–64% | 1:1 |
0% |
| 5 | Cu(OTf) 2 (10 mol%)/L5 (13 mol%) | 74% (54%) |
4:1 (32:1)e |
88 (97%) |
| 6 | Cu(OTf)2 (10 mol%)/L6 (13 mol%) | 55% | 4:1 |
−62%f |
| 7 | Cu(OTf)2 (10 mol%) | 73% | 1:1 |
— |
| 8g | L5 (10 mol%) | 58% | 1:1 |
0% |
| 9 | Cu(OTf)2 (10 mol%)/L5 (13 mol%) + 4 Å MS | 33% | 4:1 |
86% |
| 10 | Cu(OTf)2 (10 mol%)/L5 (22 mol%) | 59% | 4:1 |
88% |
| 11 | Cu(OTf)2 (13 mol%)/L5 (10 mol%) | 56% | 4:1 |
82% |
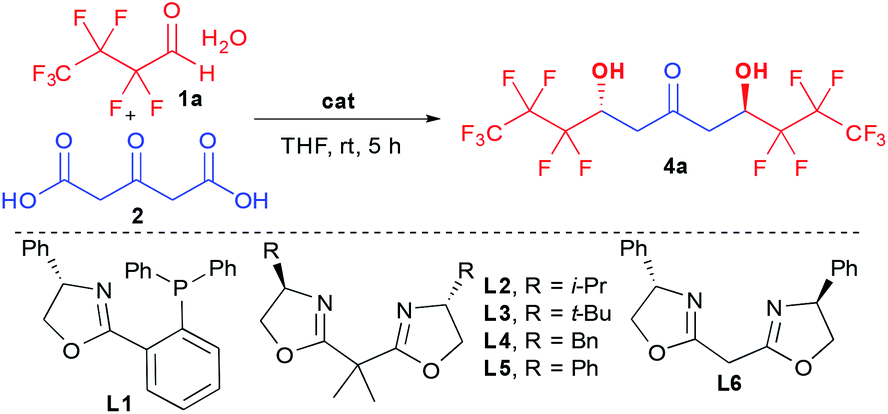
Given the behavior of a broad range of organocatalysts in various decarboxylative processes,14b–d we first checked the efficiency of such catalyst structures in our transformation. While it was previously shown that bi-directional aldolization was poorly efficient using organocatalytic activation modes on aliphatic aldehydes,15b the increased electrophilic character of perfluorinated aldehydes facilitated the formation of the ketodiol 4a albeit with poor stereocontrol (entries 1–3). Among all catalysts tested, 10 mol% quinine provided the best 17% ee with a total lack of diastereocontrol (1:1 dr) (entry 2). This absence of diastereocontrol indicates that the first generated stereocenter does not control the formation of the second.
The apparent lack of selectivity of most organocatalysts leads us turn our attention to the alternative use of Lewis acids such as chiral copper complexes, initially introduced in decarboxylative aldolizations by the group of Shair.17 Disappointingly, the phosphine containing ligand L1 or different bis-oxazoline (BOX) ligands containing isopropyl, tert-butyl or benzyl substituents (L1–4) were totally unable to control the stereoselectivity of the aldolization forming 4a as a racemate and a 1:1 anti-meso mixture (entry 4). In sharp contrast, phenyl substitution in L5 dramatically changed the transition state of the aldolization providing 4a in 4:1 dr and 88% ee (entry 5). Of interest, the stereopurity of the ketodiol could be easily improved by purification through simple recrystallization (32:1 dr and 97% ee). Finally, increasing the flexibility by removing the gem-dimethyl linker in L6 slightly decreased the selectivity, providing 4a in 62% ee of the opposite enantiomer (entry 6).
This unique enantiocontrol using the simple phenyl substituted BOX ligand L5 is in contrast to previous studies on aldolization17c,d,18 and suggests interactions between the aromatics and the perfluorinated chains during the enantiodetermining step.
In order to further understand this mechanism, several control experiments were also performed. Using Cu(OTf)2 alone, 4a was formed in 1:1 dr, indicating that the creation of the second stereocenter does not arise from substrate control but from control by the chiral ligand (entry 7). Using L5 alone as reported by Ma,12f4a could be formed after prolonged reaction time albeit without any diastereo- or enantiocontrol, confirming the role of the copper complex (entry 8).
Adding 4 Å molecular sieves to the reaction did not change the enantioselectivity (86% ee) while considerably decreasing the yield (33%, Table 1, entry 9). This suggests that the water present does not interfere in the transition state but might considerably help catalyst turn-over possibly through copper complex protonation.
In sharp contrast to the aldolization of aliphatic aldehydes reported by Shair, where excess BOX was responsible for the deprotonation of the pro-nucleophile,17c,d the use of a large excess of the ligand is not necessary to obtain a good efficiency during this transformation (entries 10–11). This is in agreement with the reported reactivity of ketodiacid 2, not requiring any base for its activation by simple Lewis acids.15 In addition, in our process, good levels of enantiocontrol are obtained regardless of the Cu/L* ratio indicating that the kinetics of aldolization by the generated chiral copper complex is much faster than that of the basic BOX ligand or Cu(OTf)2 alone (entries 10–11).19
More surprisingly, determination of the absolute configuration of the ketodiol (vide infra), indicated that the same Cu/L5 combination was inducing an opposite sense of enantioinduction compared to previously disclosed results by Shair on the aldolization using aliphatic aldehydes.17c,d A control experiment performing the reaction on aliphatic aldehyde 5 indicated that the aldol product 6 was formed without any stereocontrol using the same conditions as for perfluorinated aldehyde 1a (Scheme 2(a)).
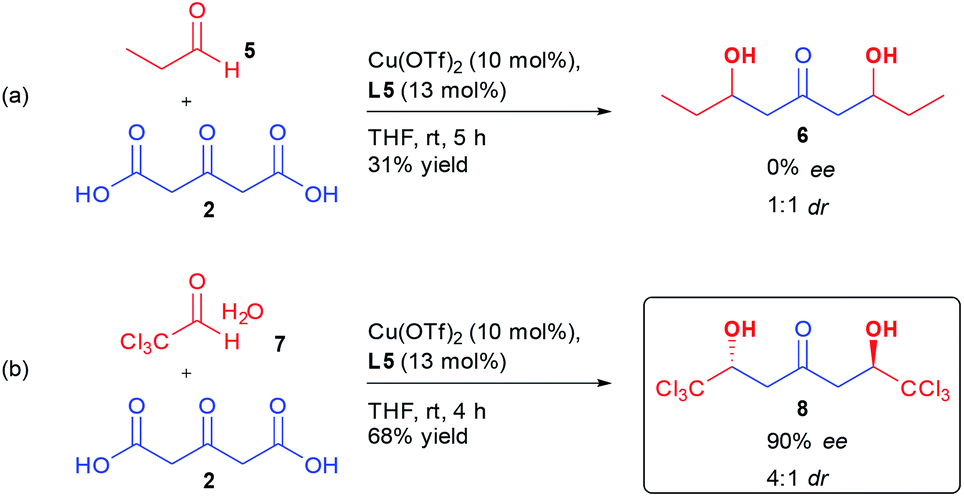 | ||
| Scheme 2 Bi-directional aldolization with other aldehydes. | ||
In order to better understand the interactions ongoing in our bi-directional aldolization, we subjected chloral hydrate 7 to the same conditions. Through the use of a Cu(OTf)2/L5 combination, ketodiol 8 was formed in 68% yield, 4:1 dr and 90% ee (Scheme 2(b)). Of importance, this hexachlorinated ketodiol 8 has the structure of a natural product that might explain the observed toxicity of oxytropis common feed plants.20 The corresponding racemic molecule has already been prepared and further derivatized through desymmetrization, highlighting the synthetic potential of this enantioselective preparation of 8.21
Altogether, these results suggest that the present process is mechanistically distinct from previous Cu/BOX-catalyzed decarboxylative aldolizations with non-fluorinated aliphatic aldehydes, pointing out a crucial role of the aromatic substituent of the ligand.
A catalytic cycle for the bi-directional aldolization taking into account all these observations is depicted in Scheme 3. From the carboxylic acid coordinated chiral complex A, deprotonation provides the reactive enolate B. As demonstrated previously, the aldolization event creating the new stereocenter precedes the decarboxylation.15c,17d With this experimental evidence, a potential preferred transition state accounting for the unique stereocontrol is presented in Scheme 3b(1). The crucial interaction governing the enantiodiscrimination might arise from F–π aromatic repulsion disfavoring transition state (2). This is further confirmed by the excellent result obtained using chloral 7 and eliminates a discrimination through less conventional F–π aromatic attractive interactions.22 In addition to additional interactions, the better results obtained with halogenated aldehydes vs. aliphatic ones might also originate from their higher electrophilicity. This might favor a faster nucleophilic addition prior to competing ligand exchange between the ketodiacid and the chiral ligand.15b From the resulting enolate C, ligand exchange followed by the second catalyst controlled aldolization-decarboxylation provides the new enolate E. The control of the second aldolization event by the chiral ligand is responsible for an overall increase in the enantiomeric excess on the product at the expense of diastereoselectivity through an amplification of chirality.23 At this stage, enolate protonation potentially assisted by the water coming from the aldehyde hydrate source provides ketoalcoholate F. Final ligand exchange with a new molecule of ketodiacid 2 liberates the enantioenriched product and regenerates the active catalytic species A.
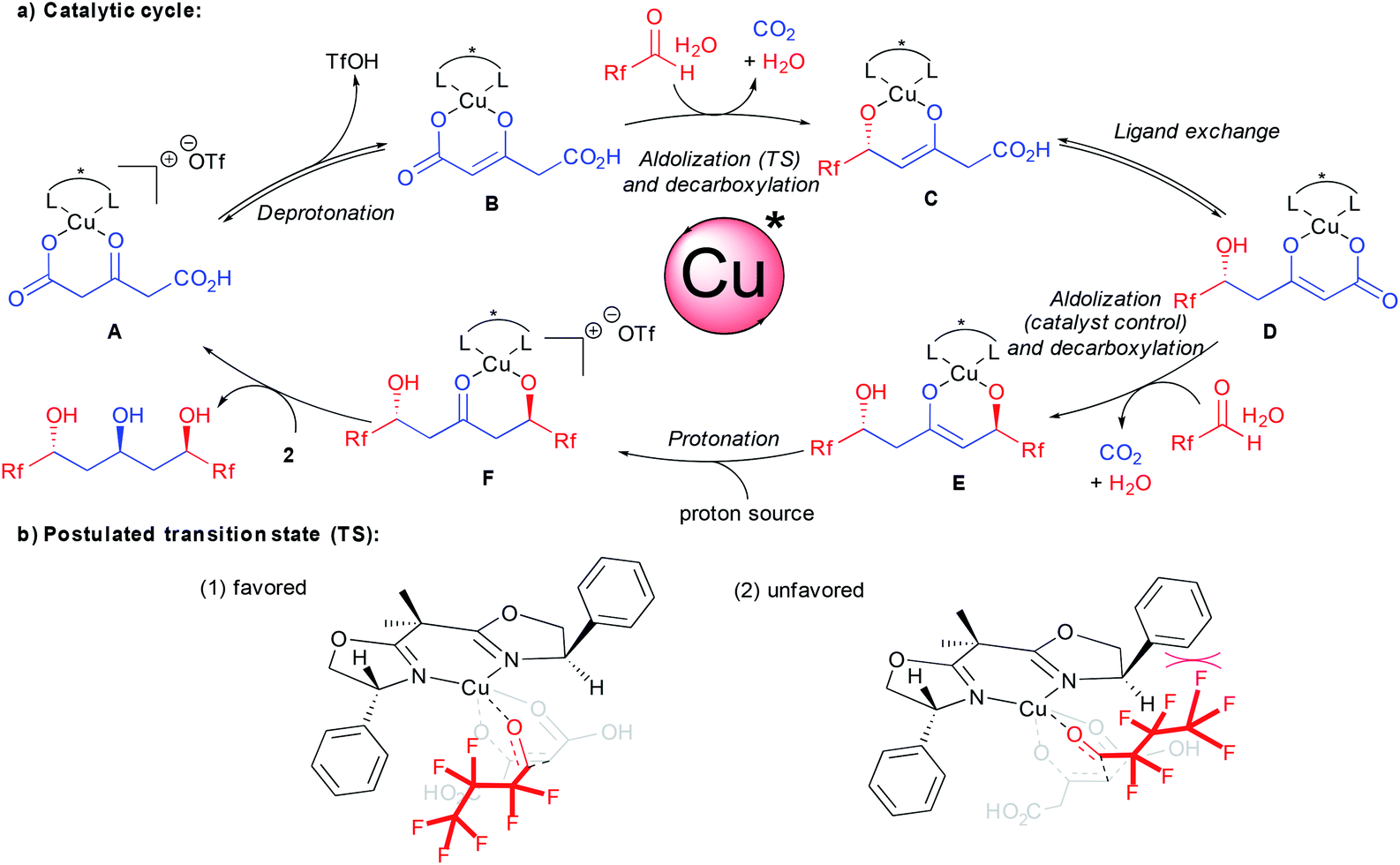 | ||
| Scheme 3 Proposed catalytic cycle and postulated transition state. | ||
With the optimized conditions in hand, different ketodiols 4a–c possessing perfluorinated chains of different lengths were prepared through the bi-directional cascade (Scheme 4). Reaction conditions tolerated the insertion of perfluorinated chains of C1 (4c), C3 (4a) or C4 (4b) length. In addition, in all cases the ketodiols were generated in good yields (54–71% yield) and good to excellent diastereo- and enantiocontrols (11.5:1 to 32:1 dr and 96–97% ee). Of particular interest, the developed conditions are amenable to scale up (25 mmol of starting 1) without tedious column chromatography requirement while tolerating the presence of water arising from the use of the commercial hydrate forms of the perfluorinated aldehydes.
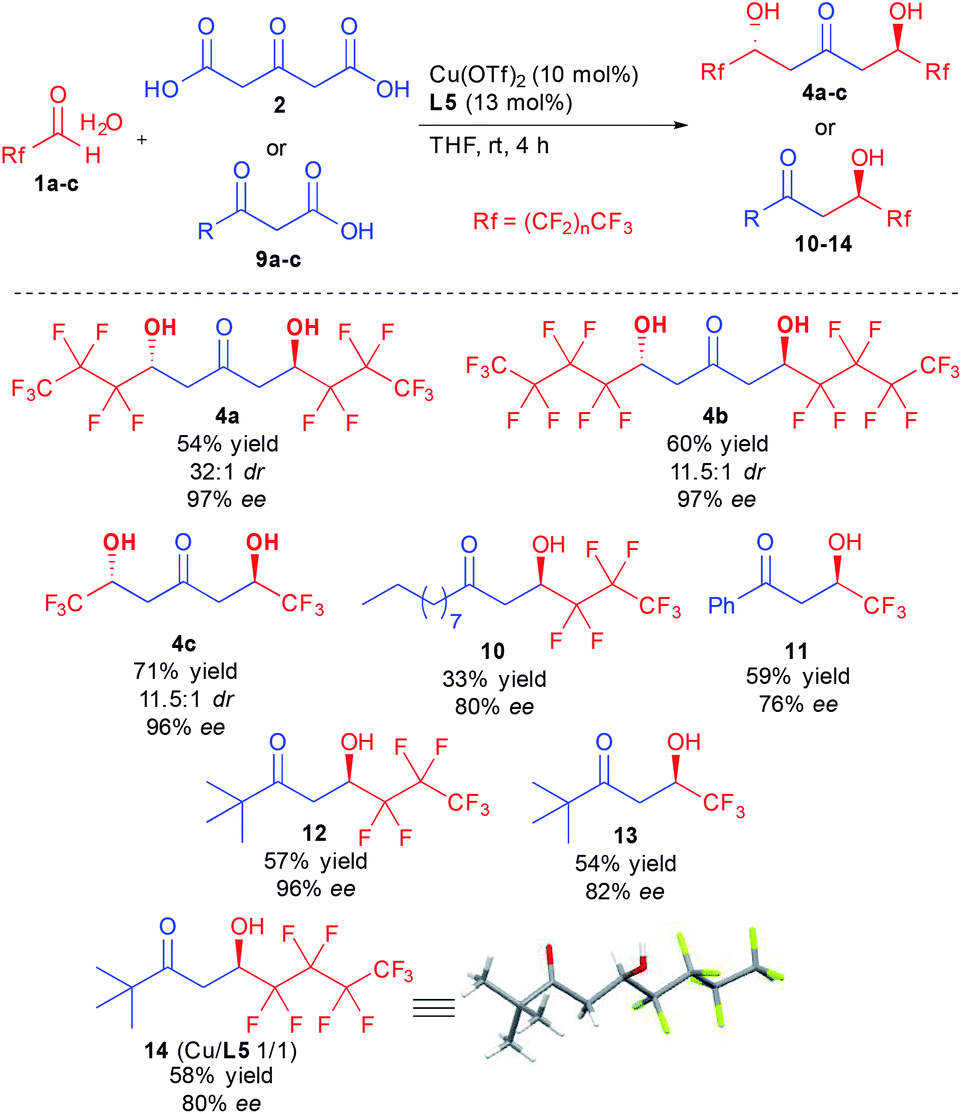 | ||
| Scheme 4 Scope of the copper-catalyzed enantioselective aldolization with perfluorinated aldehydes. | ||
In addition to the bi-directional reaction, we also tested the present conditions with different 1,3-ketoacids 9. Insertion of linear alkyl chain (10), aromatic (11) or tert-butyl groups (12–14) provided the 1,3-ketols in 76 to 96% ee. It must be pointed out that using the chiral copper complex, much higher reaction TOF is observed as compared to previous studies.12d,f Indeed, full conversion is obtained after only a few hours vs. 4–7 days for previous studies, a strong improvement leaving room for optimization of the conditions to better adjust the results to the different ketoacid structures. Finally, single crystal X-ray diffraction of 14 confirmed the absolute configuration of the obtained products.24
With a convenient method to directly access the desired enantioenriched ketodiols, simple sodium borohydride reduction provided the three different perfluorinated 1,3,5-triols 3a–c with different lateral chain lengths (Scheme 5). Of interest, all three triols were crystalline, allowing the preparation of these crucial compounds in good yield and nearly perfect stereocontrol (11.5:1 to 30:1 dr and >96% ee) by simple recrystallization. Single crystal X-ray diffraction of triol 3c also further confirmed the absolute configuration.24 Of importance, in the crystal assembly, the molecular packing is stabilized through several fluorine atoms acting as hydrogen bond acceptors, adding new evidence to the long debate on the existence of such interactions.25
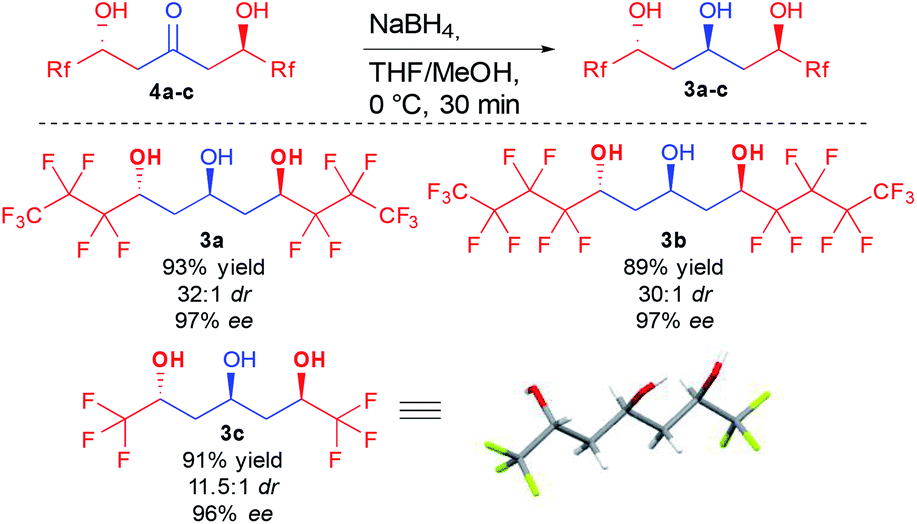 | ||
| Scheme 5 Reduction of the ketodiols and single crystal X-ray diffraction of 3c. | ||
Perfluorinated 1,3,5-triols, new tools for selective supramolecular recognition
As mentioned in the introduction, host molecules able to recognize different guests through supramolecular interactions are crucial for a wide array of applications. Most notably, anion coordination is central for sensing, extraction, catalysis, smart materials and discovery of innovative therapies.26 In this context, identifying new non-charged molecules for anion coordination is of crucial importance. For this purpose, we initially tested the anion binding properties of the three triols in CD3CN to the biologically relevant chloride anion through 1H NMR monitoring using Thodarsson method (Table 2).27|
|
|||
|---|---|---|---|
| 3a | 3b | 3c | |
|
a The addition of X− to the polyols resulted in large downfield shifts of the hydroxyl hydrogens. Association constants from triplicate experiments were obtained by fitting these spectroscopic data to 1:1 binding isotherm models, using the Thordarson method.
|
|||
| K (Cl−) 25 °C | 2100 M−1 | 3750 M−1 | 4400 M−1 |
| K (S)-15 | nd | 8100 M−1 | nd |
| K (R)-15 | nd | 1790 M−1 | nd |
| K (S)-15/K (R)-15 | nd | 4.5 | nd |
All three triols provided remarkable chloride binding ability for such poorly preorganized acyclic non-charged hosts, with 1:1 host:guest coordination constants ranging from 2100 to 4400 M−1. Most notably, the association constant did not follow a linear behavior depending on the length of the fluorinated chain, indicating that the electron-withdrawing character of the side chain is not the single responsible factor for the enhanced binding. Indeed, while the C1 (3c) and C4 (3b) perfluorinated chains provided the higher constants, the C3 (3a) chain gave lower binding.
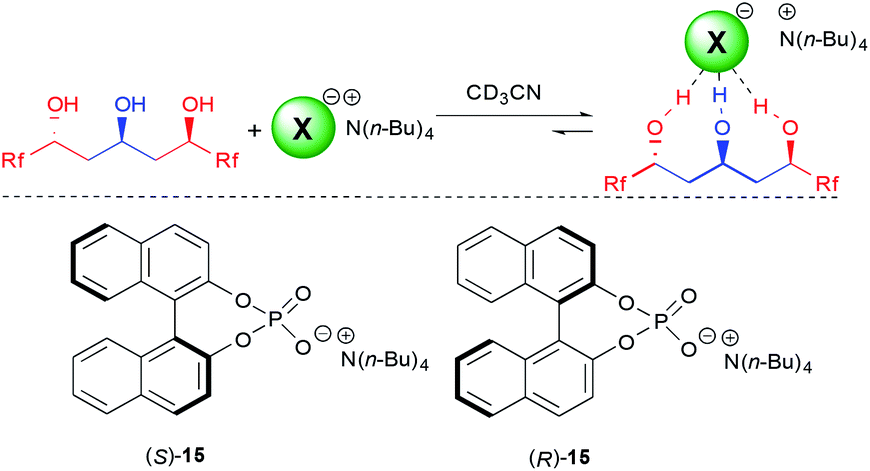
With the excellent behavior of all three triols on the model chloride anion, we next turned our attention to the more challenging coordination to chiral anions. A molecule's ability to selectively recognize one enantiomer of a chiral guest is fundamental for many important biological processes, as different enantiomers often possess totally different biological profiles but is also of importance for catalysis.28
For this purpose, we choose to study 3b, possessing one of the best binding constants and the longer perfluorinated chain for potential interactions, to test fluorinated triols' ability in chiral recognition of the model binol-phosphate derived anion 15.29 Of utmost importance, this triol was able to selectively recognize both enantiomers of the phosphate 15, providing a K(S)/K(R) of 4.5, among the highest observed for such an anion.5,6 This clearly demonstrates for the first time the efficiency of acyclic polyol scaffolds as a platform for chiral recognition. In addition, the anion binding constant with the major enantiomer is high (8100 M−1), which is a crucial parameter for further application of the synthesized triols to other technologies.
Conclusions
Identifying new scaffolds and concomitantly discovering new synthetic strategies to directly access them represents a step forward highlighting the maturity of organic chemistry. In this manuscript, we have demonstrated that chiral perfluorinated triols could be efficiently prepared diastereo- and enantio-selectively through copper-catalyzed bi-directional decarboxylative aldolization followed by reduction. The use of a phenyl substituted bis-oxazoline ligand provides unique enantiocontrol through crucial aromatic–fluorine interactions. The reaction occurs rapidly under mild conditions and tolerates the presence of water. Furthermore, the reaction could be extended to the preparation of naturally occurring hexachlorinated ketodiol of great synthetic utility.In addition, we have shown that the obtained perfluorinated 1,3,5-triols possess enhanced recognition ability. The strength of the central triol framework ensures excellent anion binding properties while the key molecules demonstrate the selective chiral anion recognition ability of synthetic 1,3-polyols.
The present study opens broad perspectives for the preparation of chiral fluorinated scaffolds. In addition, it also broadens the possibilities offered by alcohol coordination with strong implications ranging from biology to materials or catalysis.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
A patent on the preparation and properties of perfluorinated 1,3,5-triols has been submitted by Céline Sperandio, Jean Rodriguez and Adrien Quintard. Patent declaration. EP19 305862.5. Submitted 27 June 2019.Acknowledgements
The Centre National de la Recherche Scientifique (CNRS) and the Aix-Marseille Université are warmly acknowledged for financial support. All technical staff from Aix-Marseille Spectropole are acknowledged for their support.Notes and references
- S. H. Gellman, Chem. Rev., 1997, 97, 1231 CrossRef CAS PubMed.
- (a) K. Muller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (c) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308 RSC; (d) R. Berger, G. Resnati, P. Metrangolo, E. Weberd and J. Hulliger, Chem. Soc. Rev., 2011, 40, 3496 RSC; (e) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed; (f) D. Cahard and V. Bizet, Chem. Soc. Rev., 2014, 43, 135 RSC; (g) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422 CrossRef CAS PubMed; (h) M. Aufiero and R. Gilmour, Acc. Chem. Res., 2018, 51, 1701 CrossRef CAS PubMed.
- (a) J.-A. Ma and D. Cahard, Chem. Rev., 2004, 104, 6119 CrossRef CAS PubMed; (b) D. Cahard, X. Xu, S. Couve-Bonnaire and X. Pannecoucke, Chem. Soc. Rev., 2010, 39, 558 RSC; (c) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470 CrossRef CAS PubMed; (d) J. Nie, H.-C. Guo, D. Cahard and J.-A. Ma, Chem. Rev., 2011, 111, 455 CrossRef CAS PubMed; (e) X. Yang, T. Wu, R. J. Phipps and F. D. Toste, Chem. Rev., 2015, 115, 826 CrossRef CAS PubMed.
- (a) B. M. Trost, J. Org. Chem., 2014, 79, 9913 CrossRef CAS PubMed; (b) K. R. Campos, P. J. Coleman, J. C. Alvarez, S. D. Dreher, R. M. Garbaccio, N. K. Terrett, R. D. Tillyer, M. D. Truppo and E. R. Parmee, Science, 2019, 363, 6424 CrossRef.
- For a recent review, see: (a) S. A. Boer, E. M. Foyle, C. M. Thomas and N. G. White, Chem. Soc. Rev., 2019, 48, 2596 RSCFor selected examples: (b) Y. Huang and V. H. Rawal, J. Am. Chem. Soc., 2002, 124, 9662 CrossRef CAS PubMed; (c) Y. Huang, A. K. Unni, A. N. Thadani and V. H. Rawal, Nature, 2003, 424, 146 CrossRef CAS PubMed; (d) A. Shokri, J. Schmidt, X.-B. Wang and S. R. Kass, J. Am. Chem. Soc., 2012, 134, 2094 CrossRef CAS PubMed; (e) A. Shokri, A. Abedin, A. Fattahi and S. R. Kass, J. Am. Chem. Soc., 2012, 134, 10646 CrossRef CAS PubMed; (f) A. Shokri, Y. Wang, G. A. O'Doherty, X.-B. Wang and S. R. Kass, J. Am. Chem. Soc., 2013, 135, 17919 CrossRef CAS PubMed; (g) A. Shokri, X.-B. Wang, Y. Wang, G. A. O'Doherty and S. R. Kass, J. Phys. Chem. A, 2016, 120, 1661 CrossRef CAS PubMed; (h) N. Busschaert, J. Jaramillo-Garcia, M. E. Light, J. Herniman, G. J. Langley and P. A. Gale, RSC Adv., 2014, 4, 5389 RSCFor other examples in the cyclic series: (i) D. K. Smith, Org. Biomol. Chem., 2003, 1, 3874 RSC; (j) S.-I. Kondo, Y. Kobayashi and M. Unno, Tetrahedron Lett., 2010, 51, 2512 CrossRef CAS.
- For the work of Kass on acyclic flexible fluorinated diols and triols: (a) A. Shokri, X.-B. Wang and S. R. Kass, J. Am. Chem. Soc., 2013, 135, 9525 CrossRef CAS PubMed; (b) M. Samet and S. R. Kass, J. Org. Chem., 2015, 80, 7727 CrossRef CAS PubMed. For selected work on other fluorinated scaffolds, see: (c) D. Vuluga, J. Legros, B. Crousse, A. M. Z. Slawin, C. Laurence, P. Nicolet and D. Bonnet-Delpon, J. Org. Chem., 2011, 76, 1126 CrossRef CAS PubMed; (d) J. Mendizabal, P. de March, J. Recasens, A. Virgili, A. Alvarez-Larena, J. Elguero and I. Alkorta, Tetrahedron, 2012, 68, 9645 CrossRef CAS; (e) M. Samet, A. Fattahib and S. R. Kass, Org. Biomol. Chem., 2015, 13, 2170 RSC; (f) M. Samet, M. Danesh-Yazdi, A. Fattahi and S. R. Kass, J. Org. Chem., 2015, 80, 1130 CrossRef CAS PubMed; (g) C. Sperandio, G. Quintard, J. V. Naubron, M. Giorgi, M. Yemloul, J.-L. Parrain, J. Rodriguez and A. Quintard, Chem. – Eur. J., 2019, 25, 15098 CrossRef CAS PubMed.
- For a review, see: I. Colomer, A. E. R. Chamberlain, M. B. Haughey and T. J. Donohoe, Nat. Rev. Chem., 2017, 1, 0088 CrossRef CAS.
- (a) G. A. Hembury, V. V. Borovkov and Y. Inoue, Chem. Rev., 2008, 108, 1 CrossRef CAS PubMed; (b) V. Sundaresan and R. Abrol, Chirality, 2005, 17, 30 CrossRef PubMed; (c) R. J. Phipps, G. L. Hamilton and F. Dean Toste, Nat. Chem., 2012, 4, 603 CrossRef CAS PubMed.
- For examples of methods based on ketone hydrogenation: (a) S. Jeulin, S. Duprat de Paule, V. Ratovelomanana-Vidal, J.-P. Genêt, N. Champion and P. Dellis, Angew. Chem., Int. Ed., 2004, 43, 320 CrossRef CAS PubMed; (b) A. E. Cotman, D. Cahard and B. Mohar, Angew. Chem., Int. Ed., 2016, 55, 5294 CrossRef CAS PubMed. For access to one alcohol through Michael addition and subsequent derivatization: (c) K. Shibatomi, A. Narayama, Y. Abe and S. Iwasa, Chem. Commun., 2012, 48, 7380 RSC.
- For other examples generating tertiary alcohols: (a) N. Hara, R. Tamura, Y. Funahashi and S. Nakamura, Org. Lett., 2011, 13, 1662 CrossRef CAS PubMed; (b) C. G. Kokotos, J. Org. Chem., 2012, 77, 1131 CrossRef CAS PubMed; (c) K. Shibatomi, A. Narayama, Y. Abe and S. Iwasa, Chem. Commun., 2012, 48, 7380 RSC; (d) Y. Zheng, H.-Y. Xiong, J. Nie, M.-Q. Hua and J.-A. Ma, Chem. Commun., 2012, 48, 4308 RSC; (e) J. H. Park, J. H. Sim and C. E. Song, Org. Lett., 2019, 21, 4567 CrossRef CAS PubMed; (f) D.-J. B. Antúnez, M. D. Greenhalgh, A. C. Brueckner, D. M. Walden, P. Elías-Rodríguez, P. Roberts, B. G. Young, T. H. West, A. M. Z. Slawin, P. H.-Y. Cheong and A. D. Smith, Chem. Sci., 2019, 10, 6162 RSC.
- For general reviews on catalytic aldolization, see: (a) B. M. Trost and C. S. Brindle, Chem. Soc. Rev., 2010, 39, 1600 RSC; (b) Y. Yamashita, T. Yasukawa, W.-J. Yoo, T. Kitanosono and S. Kobayashi, Chem. Soc. Rev., 2018, 47, 4388 RSC.
- (a) H. Torii, M. Nakadai, K. Ishihara, S. Saito and H. Yamamoto, Angew. Chem., Int. Ed., 2004, 43, 1983 CrossRef CAS PubMed; (b) K. Funabiki, H. Yamamoto, H. Nagaya and M. Matsui, Tetrahedron Lett., 2006, 47, 5507 CrossRef CAS; (c) K. Funabiki, H. Nagaya, M. Ishihara and M. Matsui, Tetrahedron, 2006, 62, 5049 CrossRef CAS; (d) K. Funabiki, Y. Itoh, Y. Kubota and M. Matsui, J. Org. Chem., 2011, 76, 3545 CrossRef CAS PubMed; (e) K. Funabiki, Y. Furuno, Y. Yano, Y. Sakaida, Y. Kubota and M. Matsui, Chem.–Asian J., 2015, 10, 2701 CrossRef CAS PubMed; (f) Z.-Y. Yang, J.-L. Zeng, N. Ren, W. Meng, J. Nie and J.-A. Ma, Org. Lett., 2016, 18, 6364 CrossRef CAS PubMed; (g) C.-X. Yan, P.-P. Zhou, F.-L. Yang, R.-Z. Wu, X. Yang, F. Yang and X. Shao, Org. Chem. Front., 2018, 5, 2692 RSC.
- For efficient enantioselective cascade aldolizations controlling multiple alcohol stereocenters: (a) K. Mikami, S. Matsukawa, M. Nagashima, H. Funabashi and H. Morishima, Tetrahedron Lett., 1997, 38, 579 CrossRef CAS; (b) D. B. Ramachary, R. Mondal and R. Madhavachary, Org. Biomol. Chem., 2012, 10, 5094 RSC; (c) Y. Shimoda, T. Kubo, M. Sugiura, S. Kotani and M. Nakajima, Angew. Chem., Int. Ed., 2013, 52, 3461 CrossRef CAS PubMed; (d) G. Valero, J. M. Ribo and A. Moyano, Chem. – Eur. J., 2014, 20, 17395 CrossRef CAS PubMed; (e) L. Lin, K. Yamamoto, H. Mitsunuma, Y. Kanzaki, S. Matsunaga and M. Kanai, J. Am. Chem. Soc., 2015, 137, 15418 CrossRef CAS PubMed; (f) S. Kotani, K. Kai, Y. Shimoda, H. Hu, S. Gao, M. Sugiura, M. Ogasawara and M. Nakajima, Chem. – Asian J., 2016, 11, 376 CrossRef CAS PubMed; (g) S. Kotani, K. Kai, M. Sugiura and M. Nakajima, Org. Lett., 2017, 19, 3672 CrossRef CAS PubMed; (h) A. Quintard and J. Rodriguez, Org. Lett., 2019, 21, 453 CrossRef CAS PubMed.
- This molecule derived in the kg scale from citric acid was initially used by Robinson in his famous tropinone synthesis: (a) R. Robinson, J. Chem. Soc., Trans., 1917, 111, 762 RSC. For reviews on decarboxylative aldolization reactions, see: (b) Y. Pan and C.-H. Tan, Synthesis, 2011, 13, 2044 Search PubMed; (c) Z. L. Wang, Adv. Synth. Catal., 2013, 355, 2745 CrossRef CAS; (d) S. Nakamura, Org. Biomol. Chem., 2014, 12, 394 RSC.
- (a) A. Quintard and J. Rodriguez, Chem. Commun., 2015, 51, 9523 RSC; (b) A. Quintard and J. Rodriguez, Chem. – Eur. J., 2015, 21, 14717 CrossRef CAS PubMed; (c) A. Quintard and J. Rodriguez, ACS Catal., 2017, 7, 5513 CrossRef CAS; (d) A. Ricucci, J. Rodriguez and A. Quintard, Eur. J. Org. Chem., 2018, 3697 CrossRef CAS; (e) A. Quintard and J. Rodriguez, Chimia, 2018, 72, 580 CrossRef PubMed; (f) A. Quintard, C. Sperandio and J. Rodriguez, Org. Lett., 2018, 20, 5274 CrossRef CAS PubMed.
- See the ESI† for the complete catalyst and ligand optimization.
- For selected examples of Lewis acid catalyzed decarboxylative reactions see: (a) G. Lalic, A. D. Aloise and M. D. Shair, J. Am. Chem. Soc., 2003, 125, 2852 CrossRef CAS PubMed; (b) S. Orlandi, M. Benaglia and F. Cozzi, Tetrahedron Lett., 2004, 45, 1747 CrossRef CAS; (c) D. Magdziak, G. Lalic, H. M. Lee, K. C. Fortner, A. D. Aloise and M. D. Shair, J. Am. Chem. Soc., 2005, 127, 7284 CrossRef CAS PubMed; (d) K. C. Fortner and M. D. Shair, J. Am. Chem. Soc., 2007, 129, 1032 CrossRef CAS PubMed; (e) L. Yin, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 9610 CrossRef CAS PubMed; (f) H.-Y. Xiong, Z.-Y. Yang, Z. Chen, J.-L. Zeng, J. Nie and J.-A. Ma, Chem. – Eur. J., 2014, 20, 8325 CrossRef CAS PubMed; (g) C.-M. Jia, H.-X. Zhang, J. Nie and J.-A. Ma, J. Org. Chem., 2016, 81, 8561 CrossRef CAS PubMed; (h) J. Lee, S. Wang, M. Callahan and P. Nagorny, Org. Lett., 2018, 20, 2067 CrossRef CAS PubMed.
- For an excellent review on bis-oxazoline ligands, see: G. Desimoni, G. Faita and K. A. Jørgensen, Chem. Rev., 2011, 111, PR284 CrossRef CAS PubMed.
- Cu(OTf)2 and L5 must be stirred for 30 minutes at least to generate the active chiral catalyst. If the substrates are added earlier, competing coordination of the ketodiacid 2 to the copper complex (see ref. 15b) generates an achiral reactive complex thus forming 3a in lower enantiocontrol. The use of more electrophilic perfluorinated aldehydes as compared to aliphatic aldehydes might also favor rapid aldolization, avoiding this undesired slower competitive ligand decoordination process.
- R. Yu, X. Li, T. Zhu, G. Yang, Z. Li and B. Yang, Shenyang Yaoxueyuan Xuebao, 1991, 8, 113 CAS.
- R. E. Bowman and W. R. N. Williamson, J. Chem. Soc. C, 1970, 1, 101 RSC.
- (a) D. Swierczynski, R. Luboradzki, G. Dolgonos, J. Lipkowski and H.-J. Schneider, Eur. J. Org. Chem., 2005, 1172 CrossRef CAS; (b) P. Li, J. M. Maier, E. C. Vik, C. J. Yehl, B. E. Dial, A. E. Rickher, M. D. Smith, P. J. Pellechia and K. D. Shimizu, Angew. Chem., Int. Ed., 2017, 56, 7209 CrossRef CAS PubMed.
- J. P. Vigneron, M. Dhaenes and A. Horeau, Tetrahedron, 1973, 29, 1055 CrossRef CAS.
- Comparison of optical rotation of known compounds 8 and 10 also confirmed the sense of chiral induction during the catalysis. ESI†.
- See the ESI† for the structure. For discussion on these interactions, see: P. A. Champagne, J. Desroches and J.-F. Paquin, Synthesis, 2015, 47, 306 CAS.
- N. Busschaert, C. Caltagirone, W. V. Rossom and P. A. Gale, Chem. Rev., 2015, 115, 8038 CrossRef CAS PubMed.
- For the tool used, see https://supramolecular.org and (a) P. Thodarson, Chem. Soc. Rev., 2011, 40, 1305 RSC; (b) D. B. Hibbert and P. Thodarson, Chem. Commun., 2016, 52, 12792 RSC . See the ESI† for details on the anion binding.
- (a) V. Sundaresan and R. Abrol, Chirality, 2005, 17, 30 CrossRef PubMed; (b) G. A. Hembury, V. V. Borovkov and Y. Inoue, Chem. Rev., 2008, 108, 1 CrossRef CAS PubMed; (c) R. J. Phipps, G. L. Hamilton and F. D. Toste, Nat. Chem., 2012, 4, 603 CrossRef CAS PubMed.
- (a) Y. Xu and M. E. McCarroll, J. Photochem. Photobiol., A, 2007, 187, 139 CrossRef CAS; (b) J. Y. C. Lim, I. Marques, L. Ferreira, V. Félix and P. D. Beer, Chem. Commun., 2016, 52, 5527 RSC; (c) M. Vlatkovic, B. L. Feringa and S. J. Wezenberg, Angew. Chem., Int. Ed., 2016, 55, 1001 CrossRef CAS PubMed; (d) J. Y. C. Lim, I. Marques, V. Félix and P. D. Beer, J. Am. Chem. Soc., 2017, 139, 12228 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1939200 and 1939261. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc05196a |
| This journal is © The Royal Society of Chemistry 2020 |