DOI:
10.1039/C9SC05321B
(Edge Article)
Chem. Sci., 2020,
11, 3016-3021
Visualizing intracellular particles and precise control of drug release using an emissive hydrazone photochrome†
Received
22nd October 2019
, Accepted 5th February 2020
First published on 25th February 2020
Abstract
The spatiotemporal control over the structure of nanoparticles while monitoring their localization in tumor cells can improve the precision of controlled drug release, thus enhancing the efficiency of drug delivery. Here, we report on a photochromic nanoparticle system (LSNP), assembled from fluorescent bistable hydrazone photoswitch-modified amphiphilic copolymers. The intrinsic emission of the hydrazone switch allows for the visualization of particle uptake, as well as their intracellular distribution. The Z → E photoswitching of the hydrazone switch within the nanoparticle leads to the expansion of the nanoparticles (i.e., drug release) accompanied by emission quenching, the degree of which can function as an internal indicator for the amount of drug released. The bistability of the switch enables the kinetic trapping of particles of different sizes as a function of irradiation time, and allows for the exhibition of light-dependent cell cytotoxicity in MDA-MB-231 cells using LSNP loaded with doxorubicin.
Introduction
Stimuli-responsive nanoplatforms whose structures and properties are sensitive to environmental factors can be used for the on-demand release of drugs to pathological sites.1 The precise control of drug release using nanoparticles that respond to physiological stimuli (i.e., pH, enzymes, and reductant), remains challenging however because of the complexity of the human physiology. To this end, various drug delivery systems (DDSs) that respond to external stimuli (i.e., light, ultrasound, electrical and magnetic fields) have been developed.2 Among these, photoresponsive systems stand out, because light enables the remote and noninvasive control over targeted release with high spatiotemporal resolution.3,4 Usually external light is used to influence the chemical structure and/or polarity of photosensitive moieties, such as azobenzenes,5 spiropyrans6 and diarylethenes7 that are embedded within the nanoparticles, thereby disrupting the nanostructure of the carriers and inducing the leakage of cargo.8 Such systems though usually require ultraviolet (UV) light in their activation, which limits their biomedical application.9 Generally speaking, visible and near-infrared light are more suitable for such applications compared to UV10,11 because they have deeper tissue penetration12 and lower phototoxicity.13
In addition to stimuli responsiveness, it is also important to track the DDS for spatial control over drug release. Visualization of nanoparticles by fluorescent labels has become a useful approach in drug delivery research as it enables real-time and non-invasive monitoring of particle localization, motion, or interactions with biomolecules in living systems.14 In most reported DDSs, the tracking of nanocarriers relies on additional loading/labeling of fluorophores (i.e., fluorescent probes,15 carbon dots16 and aggregation-induced emission (AIE) reagents17) and/or Förster resonance energy transfer (FRET)-mediated monitoring,18,19 which over-complicate the delivery system. DDSs with inherent and tunable fluorescence emission can simplify the process but have been far less explored.
Recently, we developed a new family of photochromic hydrazone switches that are characterized by their extremely long thermal relaxation half-lives (up to 5300 years) and visible light Z → E isomerization.20,21 The introduction of dimethylamine (NMe2) to the switch renders it emissive in the Z but not in the E form allowing for switching its emission ON/OFF using light in both solution and the solid-state.22 We envisioned that the coupling of the light-induced switching of configuration and fluorescence properties of the hydrazone switch would allow us to simultaneously tune the geometry of nanoparticles, and visualize their cellular uptake and distribution using a single photochrome. Herein, we describe the functionalization of an amphiphilic copolymer with NMe2-substituted photochromic hydrazone, which can assemble into a core/shell nanostructure (LSNPs) (Scheme 1). Switching the hydrazone from the Z to E form expands the particle leading to drug release. The bistable nature of the hydrazone switch allows for the kinetic trapping of particles in variable sizes as a function of irradiation time, i.e., controlled release of drugs. Meanwhile, the cellular uptake of the nanoparticles can be visualized by the green emission coming from the embedded hydrazone photochrome, and the incremental quenching of its emission during Z → E photoswitching within cells indicates the degree of particle expansion and thus the amount of drug released. With this combined release and visualization function of light-switchable nanoparticles (LSNP), we are able to trace the uptake of the doxorubicin (DOX)-loaded nanoparticles and achieve a light-dependent cell cytotoxicity in MDA-MB-231 tumor cells.
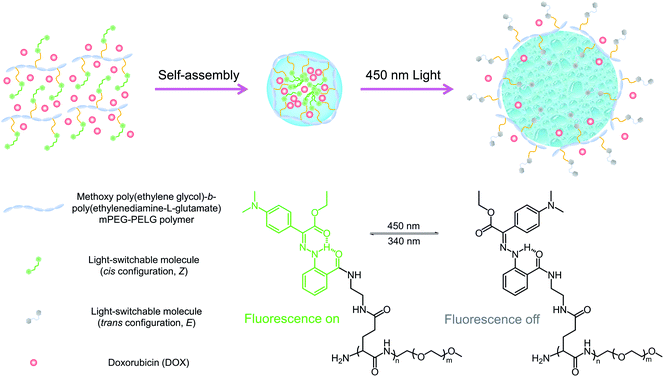 |
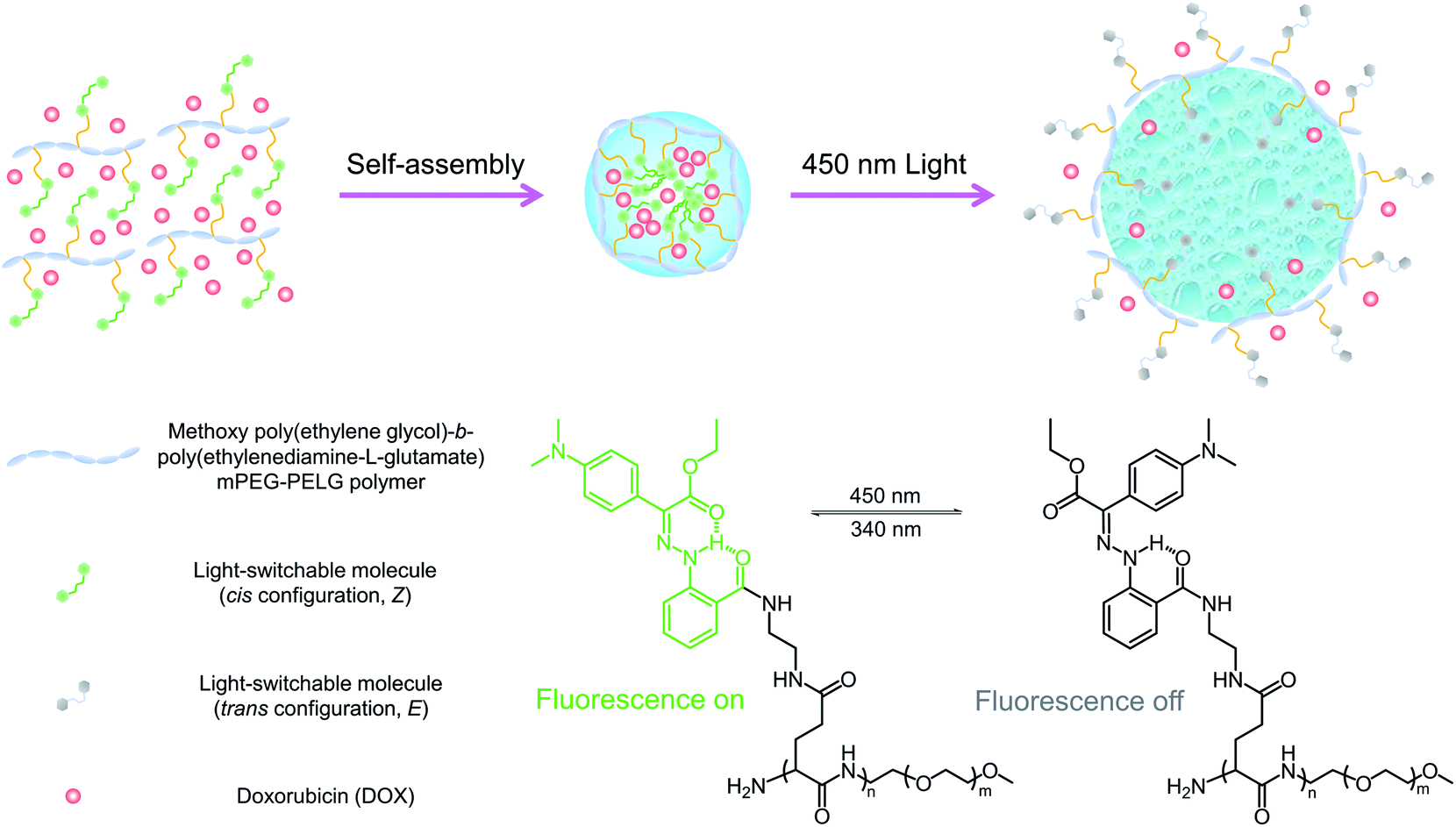
| | Scheme 1 Schematic illustration showing the photoisomerization of LSNP. Upon 450 nm irradiation, the side-chain switchable molecules in the Z configuration are converted to the E form, leading to the disruption of the nanoparticles, and subsequent triggering of drug release. | |
Results and discussion
Synthesis and characterization of LSP
Emissive hydrazone switch 1 (Scheme 2) was synthesized (71% yield) through condensation of ethyl 2-(4-(dimethylamino)phenyl)-2-oxoacetate and 2-hydrazineylbenzoic hydrochloride (Scheme S1 in the ESI†). Model compound 2, which was designed to help with the characterization of the photophysical properties of the switch, was obtained by reacting 1 with n-butylamine. The chemical structures of 1 and 2 were confirmed using NMR spectroscopy and mass spectrometry (Fig. S1–S8 in the ESI†). The light-switchable polymer (LSP) was synthesized using a three-step reaction (Scheme S1 in the ESI†). First, the methoxy poly(ethylene glycol)-b-poly(benzyl-L-glutamate) (mPEG-PBLG) copolymer was obtained via ring-opening polymerization of (S)-benzyl 3-(2,5-dioxooxazolidin-4-yl) propanoate (BLG-NCA) initiated by mPEG-NH2.23 Next, mPEG-PBLG was reacted with excess ethylenediamine to get mPEG-b-poly(ethylenediamine-L-glutamate) (mPEG-PELG) through ammonolysis.24 Finally, the emissive hydrazone switch 1 was attached to the side chains of mPEG-PELG through amidation, catalyzed by EDC/NHS, to afford LSP. The chemical structures of mPEG-PBLG (Fig. S9 in the ESI†), mPEG-PELG (Fig. S10 in the ESI†) and LSP (Fig. S11 in the ESI†) were confirmed by 1H NMR spectroscopy, and the molecular weight of LSP was calculated to be 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da.
000 Da.
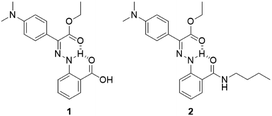 |
| | Scheme 2 Chemical structures of emissive hydrazone 1 and model compound 2. | |
Photostationary state studies of LSP
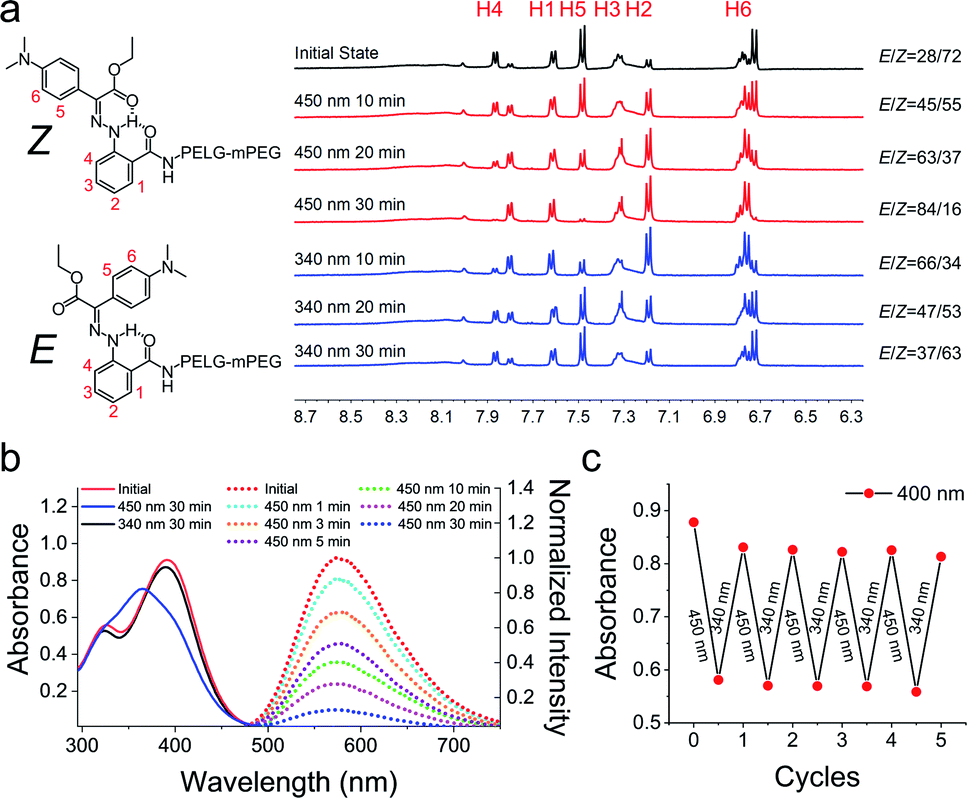
We studied the photoisomerization and emission properties of the hydrazone-modified polymer LSP using UV-Vis, 1H-NMR and fluorescence spectroscopies. Irradiation of LSP (Z:E = 72:28) in DMSO-d6 with visible light (λ = 450 nm) induces the Z → E photoisomerization, yielding a photostationary state (PSS450) of 84% E (Fig. 1a). The reverse E → Z process can be triggered by ultraviolet light (λ = 340 nm) and yields a PSS340 consisting of 63% Z isomer. The DMSO solution of LSP shows an absorption maximum (λmax) at 391 nm, which shifts hypsochromically by 28 nm to λmax = 365 nm upon irradiation with 450 nm light (Fig. 1b). The reverse E → Z isomerization can be triggered using a 340 nm light source, and the E/Z photoisomerization can be cycled multiple times by alternating the irradiation wavelength between 450 and 340 nm (Fig. 1c) with no signs of photofatigue. The model compound 2 was used to measure the photoisomerization quantum yields of the system (ΦZ→E = 14.0 ± 1.2%) and (ΦE→Z = 8.3 ± 0.2%) and the E → Z thermal relaxation half-life (2476 ± 122 years; Fig. S21 in the ESI†). Upon excitation at 430 nm (λex), the DMSO solution of LSP exhibits an emission band (λem) at 575 nm (Fig. 1b), with a fluorescence quantum yield of 1.1 ± 0.1%.25 Exposing the Z-rich solution of LSP to visible light (λ = 450 nm) causes emission to recede over time (Fig. 1a), which is consistent with what we observed for the model hydrazone 2 (Fig. S12b in the ESI†).
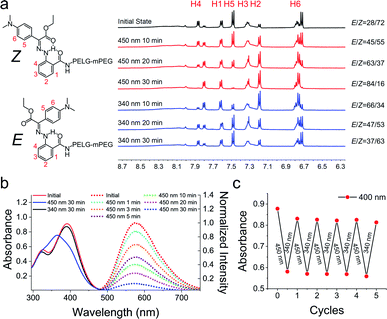 |
| | Fig. 1 (a) 1H NMR spectra showing the photoisomerization of LSP upon 450 nm and 340 nm irradiation. The irradiation-time dependent isomer ratio is also shown. (b) The solid lines are the UV-Vis spectra of LSP (10 μg mL−1) in DMSO; the dashed lines are the fluorescence emission spectra of LSP in DMSO before and after 450 nm irradiation (λex = 430 nm). (c) Isomerization cycles of LSP in DMSO upon alternating the irradiation wavelength between 450 and 340 nm (the absorbance change at λ = 400 nm was monitored). | |
Light-triggered particle expansion, emission quenching and drug release
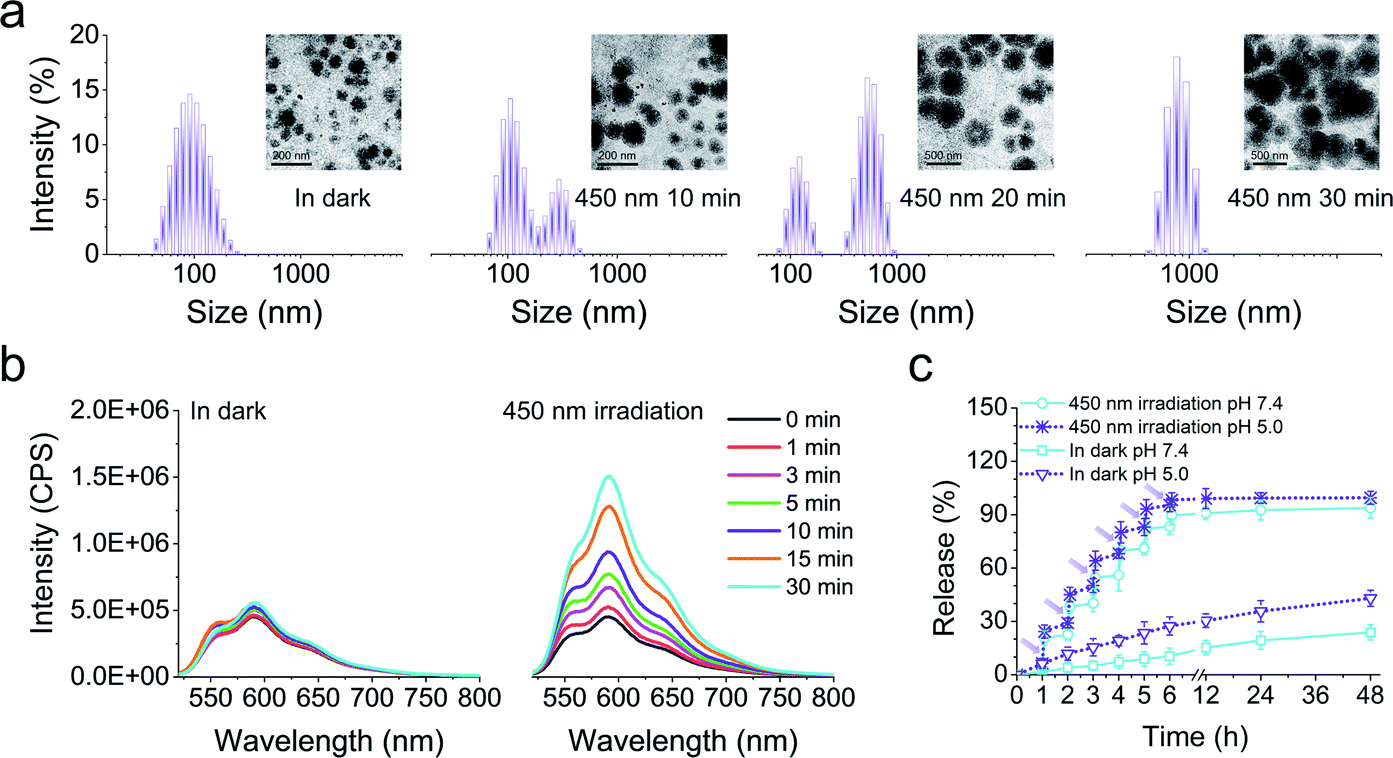
Next, we fabricated the core/shell nanoparticles (LSNPs), which spontaneously self-assembled from the amphiphilic polymer LSP in aqueous solutions. Transmission electron microscopy (TEM) and dynamic light scattering (DLS) were used to test the photo-induced structure/morphology change of LSNPs. The initial nanoparticles show a spherical shape with an average size of ∼90 nm (Fig. 2a). Irradiation of the LSNPs with 450 nm light led to a gradual expansion of the particles and the average size of the LSNPs at PSS450 increased to ∼860 nm. It is important to point out that the bistability of the switch enables us to kinetically trap all the intermediate structures and sizes of the nanoparticles just by controlling the irradiation time, and hence, the E/Z isomer distribution (Fig. 1a). We speculate that the expansion of the nanoparticles stems from swelling that is driven by the enhanced interactions between the solvent and the hydrazone NH proton that becomes available for H-bonding in the E form. Unfortunately, this process is not reversible and eventually leads to the disintegration of the self-assembled structure. Nonetheless, such a big increase in particle size (ca. 10-fold) along with the irradiation-time dependent size modulation, highlights the feasibility of using LSNP to deliver drugs in an on-demand and quantitative manner. To evaluate this property, DOX-loaded LSNPs with encapsulation efficiency of 68% were prepared and their light-triggered response studied. A similar size expansion was observed in comparison with LSNPs by themselves, indicating that loading the particles with drugs does not interfere with swelling. The drug-loaded particles were then placed into ultrafiltration tubes with PBS buffer at pH 7.4, and their emission properties were studied. Low DOX fluorescence intensity was initially observed from the tubes that slightly increases after being left in the dark for 30 min, whereas a strong emission appeared upon irradiation of the tubes with visible light for the same amount of time (Fig. 2b) implying that DOX is being released from the nanoparticles upon photoisomerization. Concurrently the emission of LSP is quenched (Fig. 1b) thus establishing a correlation between hydrazone emission quenching and drug release. The long thermal relaxation half-life of the hydrazone switch ensures the fidelity of the fluorescence emission/quenching process, and so the degree of emission quenching can be used as an internal indicator for the amount of drug released (Fig. S23 in the ESI†). In this vein, quantitative release of the drug at pH 7.4 was studied by irradiating the tube for 5 minutes (min) per hour for a span of 6 hours (h). After each irradiation sequence a burst of drug release was observed (e.g., 20.3%, 16.33% and 14.4% of the drug was released in the first three irradiation cycles as see in Fig. 2c). This sequential irradiation resulted in the on-demand and controlled release of ∼90% DOX after six rounds of irradiation. The total release amount increases to 94% by the end of a 48 h run. By contrast, the release rate is very slow in the dark, as only ∼20% of DOX is released from the nanocarriers within 48 h. To evaluate if the DOX release rate is affected by a decrease in pH level, the DOX release kinetics at pH 5.0 solution was also investigated to mimic the pH of subcellular compartments. The results show that there is enhanced DOX release from the LSNPs incubated at pH 5.0 even without light irradiation. More than 40% of DOX was released after 48 h at pH 5.0 in dark, which is 2-fold of that at pH 7.4. This acid-triggered drug release is attributed to the protonation of the glycosidic amine of DOX, leading to the increased solubility of DOX.26 The fastest release though was determined when incubating the DOX-loaded LSNPs at pH 5.0 and applying light irradiation, resulting in the complete DOX release after six rounds of irradiation (ca. 30 min).
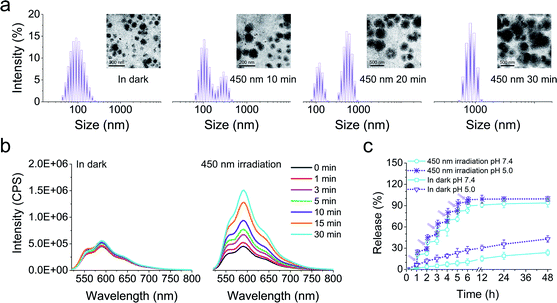 |
| | Fig. 2 (a) DLS and TEM (inset) analyses of LSNPs in the dark and after applying 450 nm irradiation for different time intervals. (b) Fluorescence spectra of DOX-loaded LSNPs left in the dark, and after irradiation with 450 nm light for 30 min. (c) In vitro drug release profiles of DOX-loaded LSNPs at pH 7.4 or pH 5.0 left in the dark and after irradiation with 450 nm light (irradiation for 5 min per hour for the first 6 h). | |
Intracellular photoisomerization of LSNPs
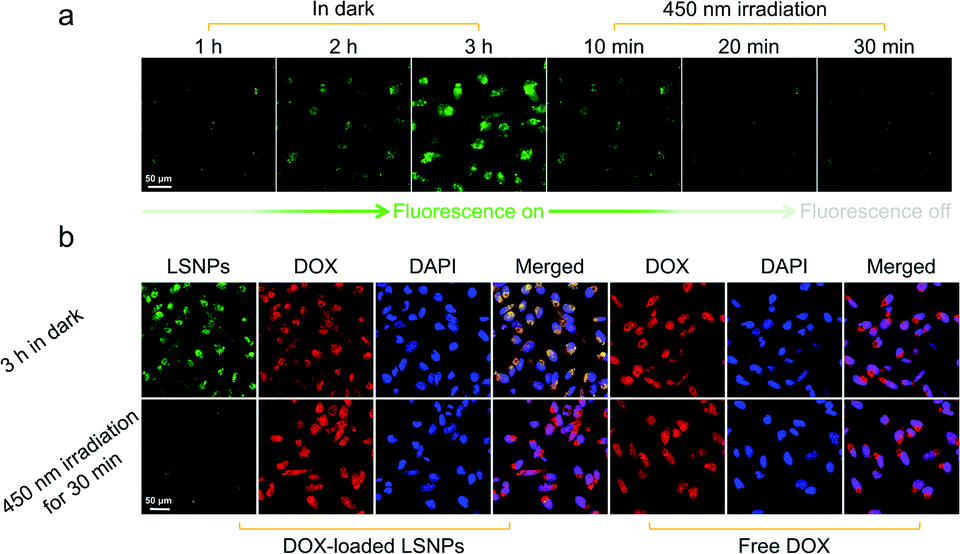
Driven by the promising in vitro results, we further investigated the intracellular structure and fluorescence switching of LSNPs in MDA-MB-231 cell lines. The internalization and photoswitching of LSNPs in living cells were assessed by following the emission intensity changes upon photoisomerization using confocal laser scanning microscopy (CLSM). Prior to light irradiation, LSNPs was incubated with MDA-MB-231 cells for 3 h in the dark to allow for cellular uptake. A gradual enhancement of green fluorescence was observed in the cells over the incubation time implying an efficient cellular internalization of LSNPs (Fig. 3a). When visible light was shed on the cells, the green fluorescence faded with time as a result of the Z → E photoisomerization of the hydrazone switch.
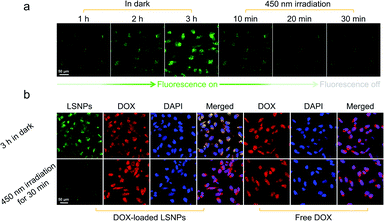 |
| | Fig. 3 (a) Confocal images showing the cellular internalization in the dark, and the light-induced emission quenching of LSNPs after 450 nm irradiation in MDA-MB-231 cells. (b) Confocal images showing the intracellular drug release of DOX-loaded LSNPs in MDA-MB-231 cells. After 3 h incubation in the dark, both LSNPs and DOX showed significant cellular uptake. The green fluorescence was quenched while the red fluorescence increased when applying 450 nm irradiation for 30 min. Nuclear localization of DOX indicates the intracellular drug release from LSNPs after 450 nm irradiation. | |
Light-triggered cellular uptake and intracellular drug release
We then studied the extracellular drug release by using the DOX-loaded LSNPs incubated with MDA-MB-231 cells. The red fluorescence of the DOX is barely observable from the cells that were incubated in the dark for 1 h (Fig. S24 in the ESI†), which is consistent with the cellular uptake efficiency of LSNPs (Fig. 3a). On the other hand, a relatively strong red fluorescence was found in the cells (mainly in the nucleus) upon 450 nm light irradiation, the intensity of which increased with irradiation time, indicating that DOX was photo-released from LSNPs and enriched in the cell nucleus. To explore the intracellular drug release, we initially incubated DOX-loaded LSNPs with MDA-MB-231 cells in the dark for 3 h to ensure cell uptake, we also incubated free DOX as a control. Prominent uptake of the DOX-loaded LSNPs was obtained as expected and the overlapping orange color demonstrates the colocalization of LSNPs with DOX, indicating that minimal drug release occurs within the cells in the dark (Fig. 3b). The overlapping pink color in the nucleus (see merged figure) indicates that DOX primarily accumulates in the cytoplasm. Irradiating the cells for 30 min with 450 nm light quenches the fluorescence of LSNPs and releases DOX from the particles, which subsequently enter the nucleus, leading to the observed enrichment of red fluorescence in the nucleus. In contrast DOX by itself distributes uniformly within the cells and shows comparable red fluorescence in the nucleus to DOX-loaded LSNPs after light irradiation (Fig. S25 in the ESI†). These results show the robustness of this hydrazone-driven drug delivery system. Unfortunately, the quantitative correlation between the degree of emission quenching and the amount of released drug cannot be established because of the difficulty in distinguishing between the released drug from the loaded one.
Cytocompatibility and cell cytotoxicity
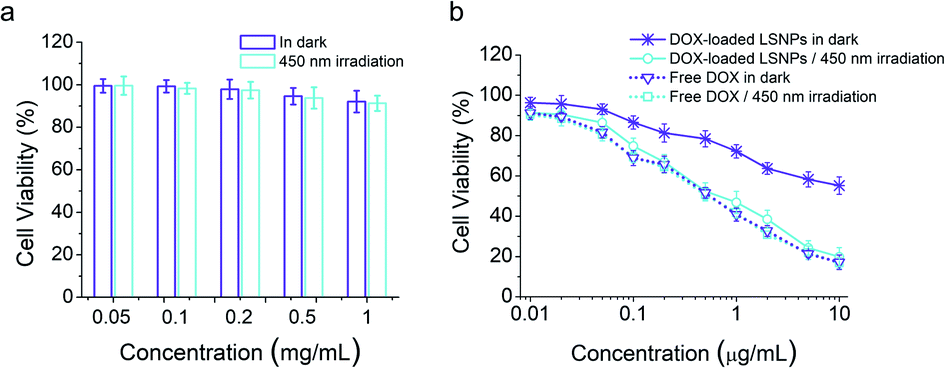
Cytocompatibility is essential if drug carriers are to be used in clinical applications. Hence, AlamarBlue assay on LSNPs in the MDA-MB-231 cells was conducted, and the data illustrate that the blank nanoparticles possess a high cell viability (over 90%) in the concentration range of 0.05–1 mg mL−1 regardless of light exposure (Fig. 4a), indicating the low toxicity and high-level of safety of LSNPs. We also assessed the light-dependent cell cytotoxicity of DOX-loaded LSNP, to explore the potential of the system for antitumor therapy. MDA-MB-231 cells were treated with DOX-loaded LSNPs and free DOX in the dark or with 450 nm light irradiation for 5 min per hour for a duration of 6 h followed by further incubation in the dark for a total of 48 h. The cell viability of DOX-loaded LSNPs in the dark is ∼55% at a concentration of 10 μg mL−1 (Fig. 4b). This dark cytotoxicity is attributed to the acid-triggered drug release within the acidic organelles (e.g., endosomes and lysosomes) (Fig. 2c). Moreover, after exposure to visible light for a total of 30 min, the cell viability decreased dramatically to around 20% at the same concentration. The half maximal inhibitory concentration (IC50) of the nanotherapeutics was determined to be around 0.65 μg mL−1 upon irradiation, which is much lower than that in the dark (>10 μg mL−1). This indicates that a 4-fold increase in dox concentration upon light irradiation is enough for antitumor therapy, while the 20% leakage of dox under dark is not that harmful (Fig. 2c). Besides, the cytotoxicity of nanotherapeutics upon irradiation was comparable to free DOX (0.59 μg mL−1 in the dark and 0.55 μg mL−1 with light). The similar cytotoxicities of light-triggered DOX-loaded LSNPs and free DOX can be attributed to the complete drug release from the particles after illuminating at acidic conditions (Fig. 2c). To distinguish the cytotoxicity between DOX released from the particles outside of the cells from ones taken up by the cells, we incubated the cells with samples for 3 h to allow for cellular uptake first, then washed them with PBS, irradiated them with and incubated them for another 45 h. The data indicate that free DOX shows similar cytotoxicity as before, because it can be quickly taken up by the cells, and so 3 h is enough for the majority of DOX to enter the cells (Fig. S26 in the ESI†). Besides, higher viabilities can be found in the LSNPs groups, indicating that the cytotoxicity in Fig. 4b can be attributed to both extracellular and intracellular DOX.
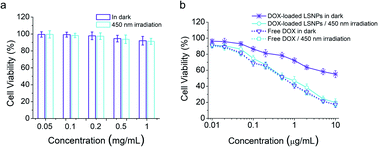 |
| | Fig. 4 Viability of MDA-MB-231 cells after treating with LSNPs (a) or free DOX/DOX-loaded LSNPs (b) in the dark or irradiation with 450 nm light (irradiation for 5 min per hour for the first 6 h) at different concentrations (n = 5). | |
Conclusion
In summary, photochromic nanoparticles exhibiting visible-light triggered fluorescence switching were fabricated from mPEG-PELG diblock copolymers conjugated with photoswitchable hydrazones. The hydrazone moieties within the self-assembled nanoparticles undergo reversible photoisomerization between emissive Z- and non-emissive E isomers. Interestingly, photoinduced Z → E transition leads to particle enlargement and eventual disintegration resulting in spatiotemporally controlled release of drugs and enhanced cellular internalization. The intracellular particle location along with the drug release process was monitored using fluorescence switching of the LSNPs upon 450 nm light irradiation. The cell cytotoxicity study confirmed the efficient light-triggered antitumor capacity of DOX-loaded LSNPs. This hydrazone-based system featuring visible light-induced drug release and fluorescence switching paves the way for the development of advanced DDSs for biomedical applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
X. G. acknowledges the support from the National Natural Science Foundation of China (Grant No. 51603172) and the Fundamental Research Funds for the Central Universities (Grant No. A1920502051907-12). I. A. would like to acknowledge the Natural Science Foundation for the generous support (CHE-1807428). S. Z. would like to acknowledge the National Natural Science Foundation of China (Grant No. 21574105 and 51725303). Z.C. acknowledges the support from The Branco Weiss Fellowship – Society in Science, administered by ETH Zürich and Dartmouth startup fund.
Notes and references
- Y. Dai, C. Xu, X. Sun and X. Chen, Chem. Soc. Rev., 2017, 46, 3830 RSC.
- Y. Wang and D. S. Kohane, Nat. Rev. Mater., 2017, 2, 17020 CrossRef CAS.
- E. R. Ruskowitz and C. A. DeForest, Nat. Rev. Mater., 2018, 3, 17087 CrossRef CAS.
- Y. Qin, L. Chen, F. Dong, S. Jiang, G. Yin, X. Li, Y. Tian and H. Yang, J. Am. Chem. Soc., 2019, 141, 8943 CrossRef CAS PubMed.
- T. Eom, W. Yoo, S. Kim and A. Khan, Biomaterials, 2018, 185, 333–347 CrossRef CAS PubMed.
- X. Wang, J. Hu, G. Liu, J. Tian, H. Wang, M. Gong and S. Liu, J. Am. Chem. Soc., 2015, 137, 15262 CrossRef CAS PubMed.
- K. Higashiguchi, G. Taira, J. I. Kitai, T. Hirose and K. Matsuda, J. Am. Chem. Soc., 2015, 137, 2722 CrossRef CAS PubMed.
- D. F. Costa, L. P. Mendes and V. P. Torchilin, Adv. Drug Delivery Rev., 2019, 138, 105 CrossRef CAS PubMed.
- S. K. Sahoo, R. Misra and S. Parveen, Nanomedicine, 2012, 8, 147 CrossRef PubMed.
- F. Ding, Z. Chen, W. Kim, A. Sharma, C. Li, Q. Ouyang, H. Zhu, G. Yang, Y. Sun and J. S. Kim, Chem. Sci., 2019, 10, 7023–7028 RSC.
- S. Jia, W. K. Fong, B. Graham and B. J. Boyd, Chem. Mater., 2018, 30, 2873 CrossRef CAS.
- R. Weissleder, Nat. Biotechnol., 2001, 19, 316 CrossRef CAS PubMed.
-
C. Raulin and S. Karsai, Laser and IPL Technology in Dermatology and Aesthetic Medicine, Springer Science & Business Media., 2011 Search PubMed.
- D. Vinciguerra, S. Denis, J. Mougin, M. Jacobs, Y. Guillaneuf, S. Mura, P. Couvreur and J. Nicolas, J. Controlled Release, 2018, 286, 425 CrossRef CAS PubMed.
- H. Zhao, J. Duan, Y. Xiao, G. Tang, C. Wu, Y. Zhang, Z. Liu and W. Xue, Chem. Mater., 2018, 30, 3438 CrossRef CAS.
- Y. Yang, L. Wang, H. Cao, Q. Li, Y. Li, M. Han, H. Wang and J. Li, Nano Lett., 2019, 19, 1821 CrossRef CAS PubMed.
- F. Hu, D. Mao, X. Cai, W. Wu, D. Kong and B. Liu, Angew. Chem., Int. Ed., 2018, 130, 10339 CrossRef.
- X. Guo, L. Wang, K. Duval, J. Fan, S. Zhou and Z. Chen, Adv. Mater., 2018, 30, 1705436 CrossRef PubMed.
- T. Senthilkumar, L. Zhou, Q. Gu, L. Liu, F. Lv and S. Wang, Angew. Chem., Int. Ed., 2018, 57, 13114 CrossRef CAS PubMed.
- H. Qian, S. Pramanik and I. Aprahamian, J. Am. Chem. Soc., 2017, 139, 9140 CrossRef CAS PubMed.
- Q. Li, H. Qian, B. Shao, R. P. Hughes and I. Aprahamian, J. Am. Chem. Soc., 2018, 140, 11829 CrossRef CAS PubMed.
- B. Shao, M. Baroncini, H. Qian, L. Bussotti, M. Donato, A. Credi and I. Aprahamian, J. Am. Chem. Soc., 2018, 140, 12323 CrossRef CAS PubMed.
- J. Yue, R. Wang, S. Liu, S. Wu, Z. Xie, Y. Huang and X. Jing, Soft Matter, 2012, 8, 7426 RSC.
- F. Tong, X. Tang, X. Li, W. Xia and D. Liu, Int. J. Nanomed., 2016, 11, 1717 CAS.
- The fluorescence quantum yield was measured using model compound 2.
- M. Prabaharan, J. J. Grailer, S. Pilla, D. A. Steeber and S. Gong, Biomaterials, 2009, 30, 3009 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc05321b |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b and
Zi
Chen
*b and
Zi
Chen