
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelenophosphoramide-catalyzed diamination and oxyamination of alkenes†
John R.
Tabor
,
Derek C.
Obenschain
and
Forrest E.
Michael
 *
*
University of Washington, Department of Chemistry, Box 351700, Seattle, Washington 98195-1700, USA. E-mail: michael@chem.washington.edu
First published on 26th December 2019
Abstract
A new selenophosphoramide-catalyzed diamination of terminal- and trans-1,2-disubstituted olefins is presented. Key to the success of this transformation was the introduction of a fluoride scavenger, trimethylsilyl trifluoromethanesulfonate (TMSOTf), to prevent a competitive syn-elimination pathway, as was the use of a phosphoramide ligand on selenium to promote the desired substitution reaction. A screen of catalysts revealed that more electron-rich phosphine ligands resulted in higher yields of the desired product, with selenophosphoramides giving the optimal results. A broad range of substrates and functional groups were tolerated and yields were generally good to excellent. For (E)-1,2-disubstituted olefins, diastereoselectivities were always high, giving exclusively anti products. The conditions were also applied to substrates bearing internal nucleophiles such as esters and carbonates, giving rise to 1,2-aminoesters and cyclic carbonates, respectively.
Introduction
Organoselenium compounds have been useful reagents in organic synthesis for decades, enabling new reactivity and providing access to a diverse array of products.1 One powerful set of applications of these reagents is in promoting oxidative transformations of alkenes. However, the efficiency and utility of these reactions has been limited by the requirement that the organoselenium reagent be used in stoichiometric quantities, necessitating multi-step procedures to obtain the desired end products. In recent years, attention has turned to using organoselenium compounds as catalysts, and several new catalytic protocols have emerged.2,3 Despite these efforts, organoselenium catalysis still remains underexplored when compared to other catalyst classes, especially those based on transition metals. This disparity is more striking given the fact that selenium is potentially capable of promoting many of the same transformations as transition metal catalysts, but at lower cost. Consequently, it is important to continue to explore ways to expand the range of reactivity that can be achieved using selenium-based catalysts.To this end, we recently developed a new class of phosphine selenide catalysts and employed them in the aza-Heck reaction of terminal alkenes to correct issues of stereo- and regioselectivity in existing transition metal- and diphenyl diselenide-catalyzed methods (Scheme 1A).4 Key to our success was the innovative idea of treating the groups on selenium as ligands rather than substituents, allowing us to envision a variety of new selenium-based catalysts with a broad range of steric and electronic properties (Scheme 1B). Using this framework, we developed a diverse library of catalysts and observed that the ligand on selenium could indeed be used to improve reactivity and selectivity.
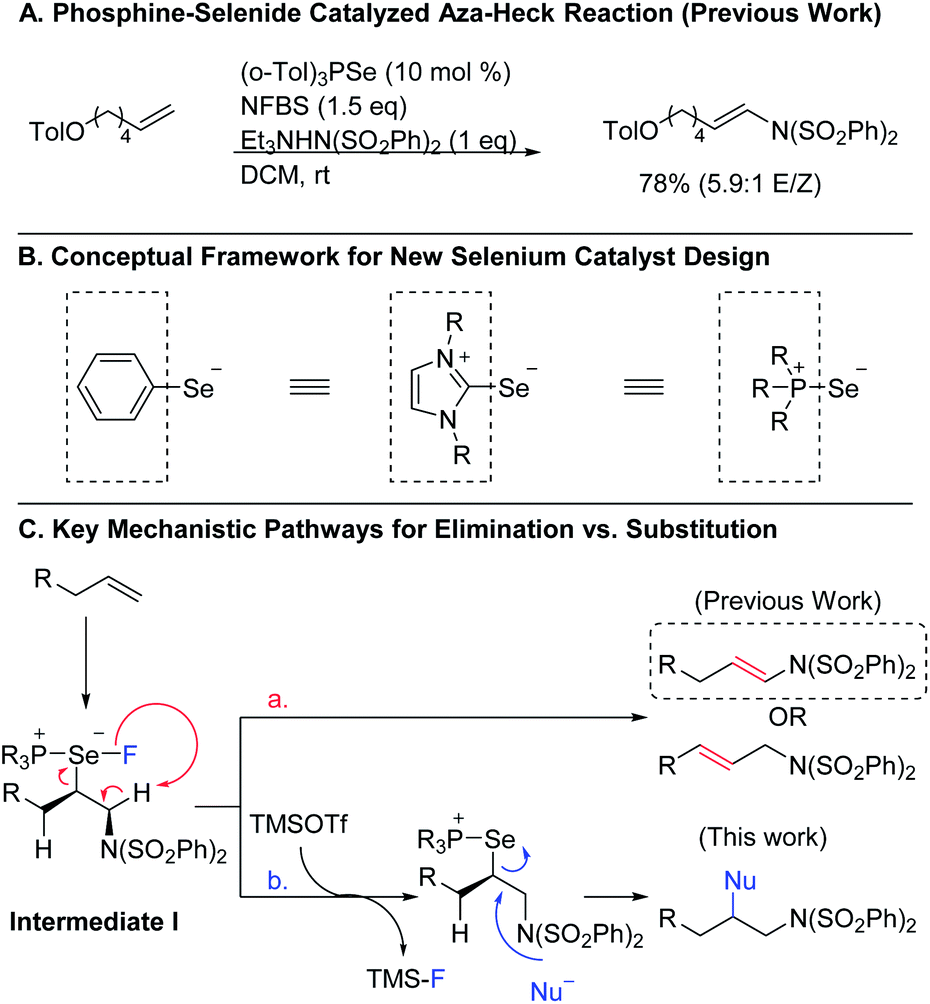 | ||
| Scheme 1 Conceptual evolution from selenium-catalyzed addition/elimination to diaddition. | ||
To expand the versatility of selenium catalysis, we imagined that it should be possible to divert the typical addition/elimination mechanism to promote a new class of transformations of alkenes. We previously established that a key step in this mechanism is the concerted syn-elimination of the selenium–fluorine bond of intermediate I to give the vinyl amine product (Scheme 1C, pathway a). This step is isoeletronic to the well-known selenoxide elimination.5 If, instead of undergoing elimination, intermediate I were intercepted by nucleophilic attack, the result would be a 1,2-difunctionalization of the alkene (Scheme 1C, pathway b). The major obstacle to this goal is the propensity for organoselenium intermediates to undergo a fast intramolecular elimination rather than the relatively slower intermolecular substitution. This is evident from the fact that the vast majority of organoselenium-promoted transformations end with this elimination step1–4 (Scheme 2A), whereas selenium-promoted 1,2-difunctionalization has remained highly elusive. There are two known methods for promoting stoichiometric substitution of organoselenides: halogenation with Cl2 or Br2 (ref. 6) and activation with PhSeOTf.7 To our knowledge, only the syn-dichlorination of alkenes reported by Denmark et al.8 has successfully employed this strategy in a catalytic reaction (Scheme 2B).9 If a more general route to substitution by non-halide nucleophiles could be realized, it would greatly expand the utility of selenium catalysis by opening up an avenue to many new classes of products.
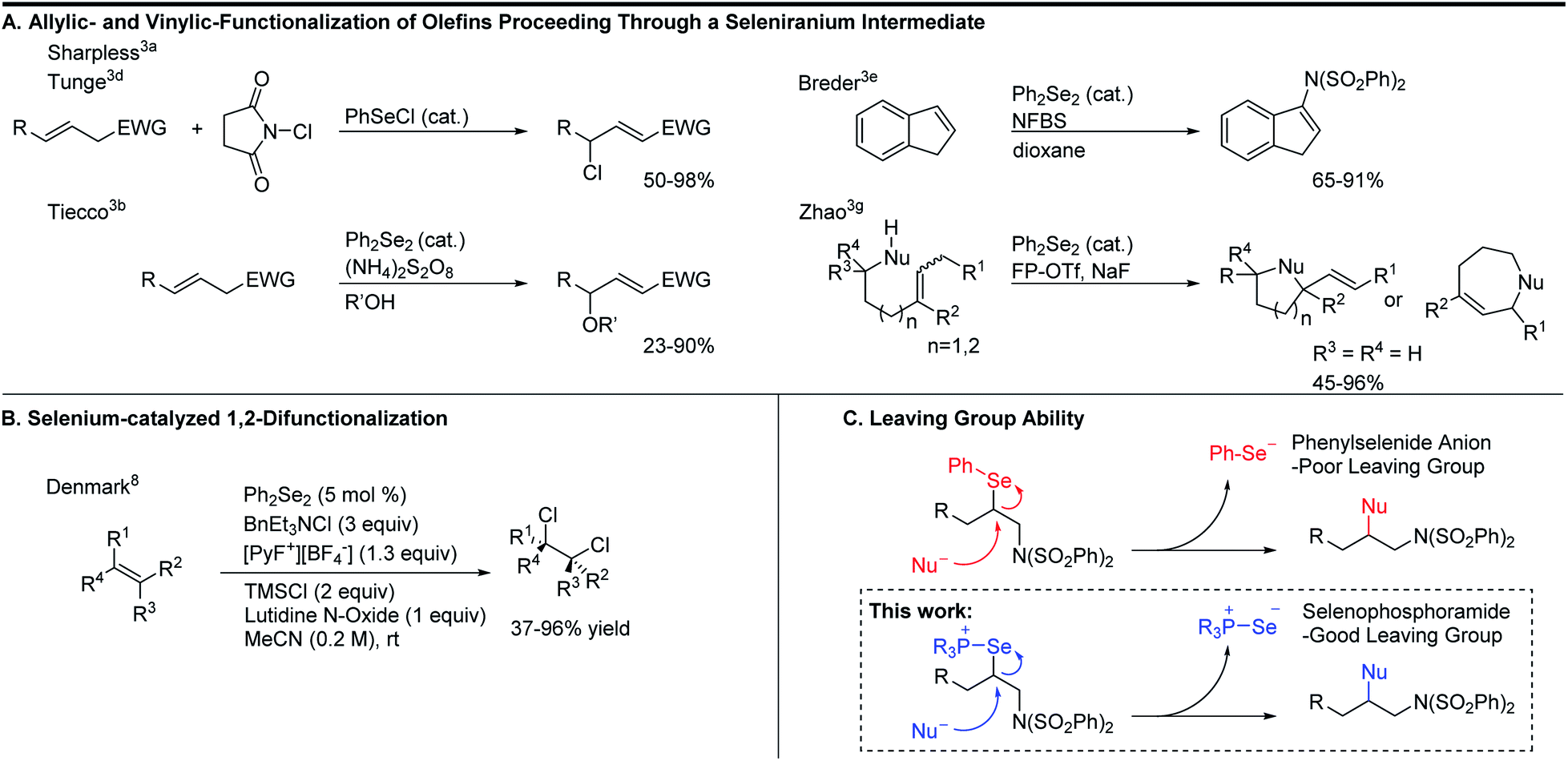 | ||
| Scheme 2 Elimination vs. substitution pathways. | ||
As a first demonstration of this principle, we imagined displacing intermediate I with a benzenesulfonimide anion, yielding a 1,2-diamination product. 1,2-Diamines are present in natural products and have widespread use in medicinal chemistry, agrochemistry, as ligands in chemical synthesis, and in many other areas.10 While several protocols exist for synthesizing 1,2-diamines from alkenes, most suffer from a limited and/or highly engineered substrate scope.11,12 There are comparatively few general and broadly applicable fully intermolecular 1,2-diamination methods.14
Analysis of the key substitution step reveals that using phosphine selenide catalysts in place of diphenyl diselenide should accelerate the desired reactivity (Scheme 2C). Instead of displacement of a phenylselenide anion, a relatively poor leaving group, substitution of the same intermediate in the phosphine selenide catalyzed system should be more facile because the phosphine selenide is a much better leaving group. However, our previous results made clear that in addition to accelerating the substitution step, the syn elimination must also somehow be inhibited.
Results and discussion
We hypothesized that the introduction of a fluoride scavenger would prevent the syn-elimination pathway by sequestering the fluoride on intermediate I, thereby allowing substitution to take place. To test this hypothesis, 4-phenyl-1-butene (1p) was subjected to catalytic phosphine selenide in presence of N-fluorobenzenesulfonimide (NFBS) and added benzenesulfonimide nucleophile (Scheme 3). As we observed previously, only elimination products were formed. However, performing the same reaction in the presence of two equivalents of trimethylsilyl trifluoromethanesulfonate (TMSOTf) suppressed the elimination, gratifyingly giving a 35% yield of the desired diamination product 3p.15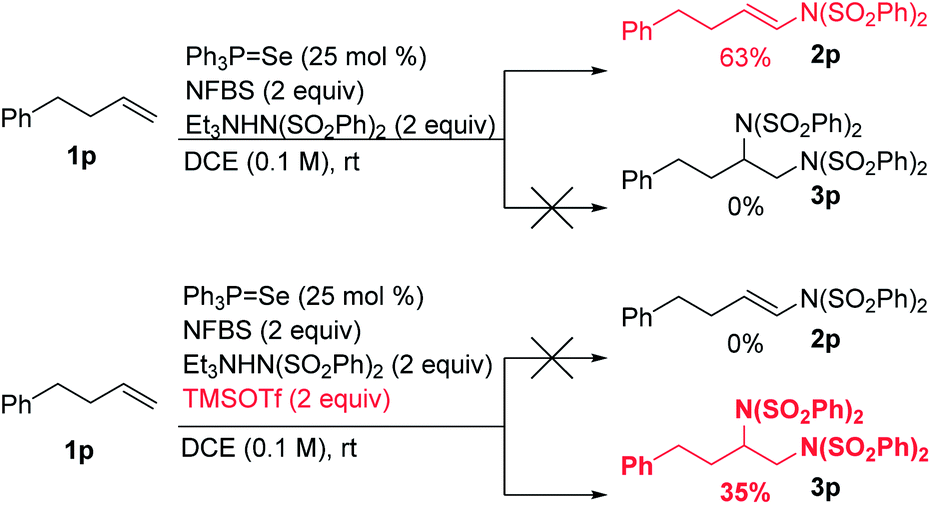 | ||
| Scheme 3 Fluoride scavenger changes reactivity. | ||
In an effort to increase the yield, a variety of sterically and electronically diverse catalysts were screened under these reaction conditions (Scheme 4). We observed a systematic trend: increasing the electron donating ability of the ligand led to higher yields of the desired product. It is important to note that neither diphenyl diselenide (Ph2Se2), the most commonly used organoselenium catalyst, nor commercially available grey selenium, gave meaningful amounts of the desired product (see ESI† for more details), even in the presence of fluoride scavenger, illustrating the key role the phosphine ligand plays in promoting this new reactivity. Ultimately, selenophosphoramide catalyst 4f provided the highest yield of 62% and was chosen as the optimal catalyst. The catalyst loading could be further reduced to 10 mol% with little impact on the yield. With the optimized conditions in hand, control experiments individually omitting each component of the reaction were performed and no desired product was observed, thus confirming the necessity of each of the reagents.
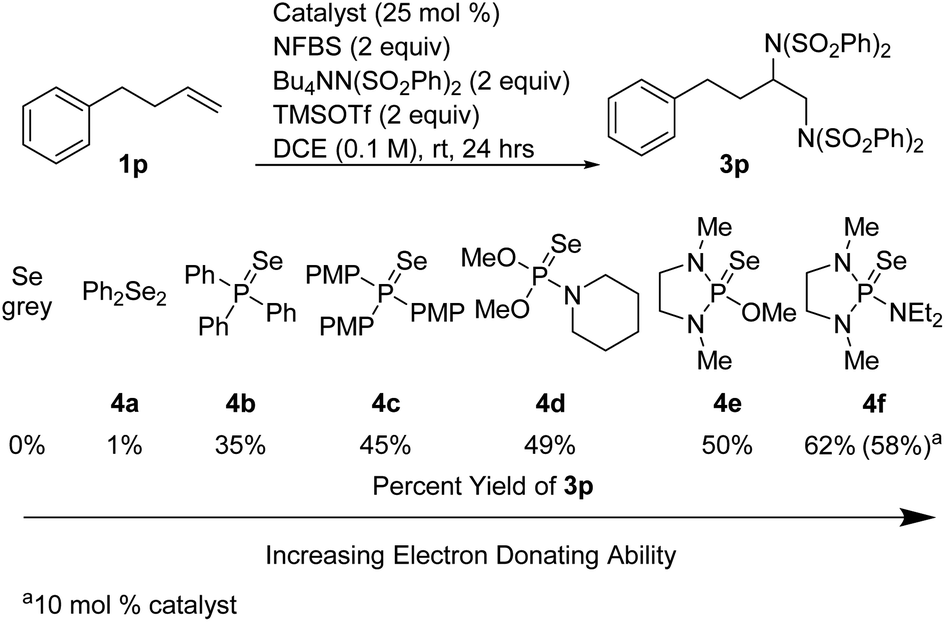 | ||
| Scheme 4 Catalyst screen. | ||
A variety of alkenes were subjected to the optimized conditions (Table 1). Both terminal- and 1,2-disubstituted olefins gave diamination products in good to excellent yields. The reaction is tolerant of silyl ethers, alkyl halides (including allyl), aryl halides, electron-rich and electron-poor aromatics, esters, nitriles, azides, sulfonates, and aldehydes. Internal olefins performed particularly well, giving yields from 74–96%. Aryl iodide 3k and heteroarene 3g were noteworthy, as these substrates are not compatible with typical metal-catalyzed protocols. All products derived from internal alkenes were formed with high diastereoselectivity, giving anti-1,2-diamines with no detectable amounts of the syn diastereomers.13 (Z)-1,2-disubstituted-olefins gave diamine products, but in poor diastereoselectivity. Tri- and tetrasubstituted olefins gave poor yields under these conditions.
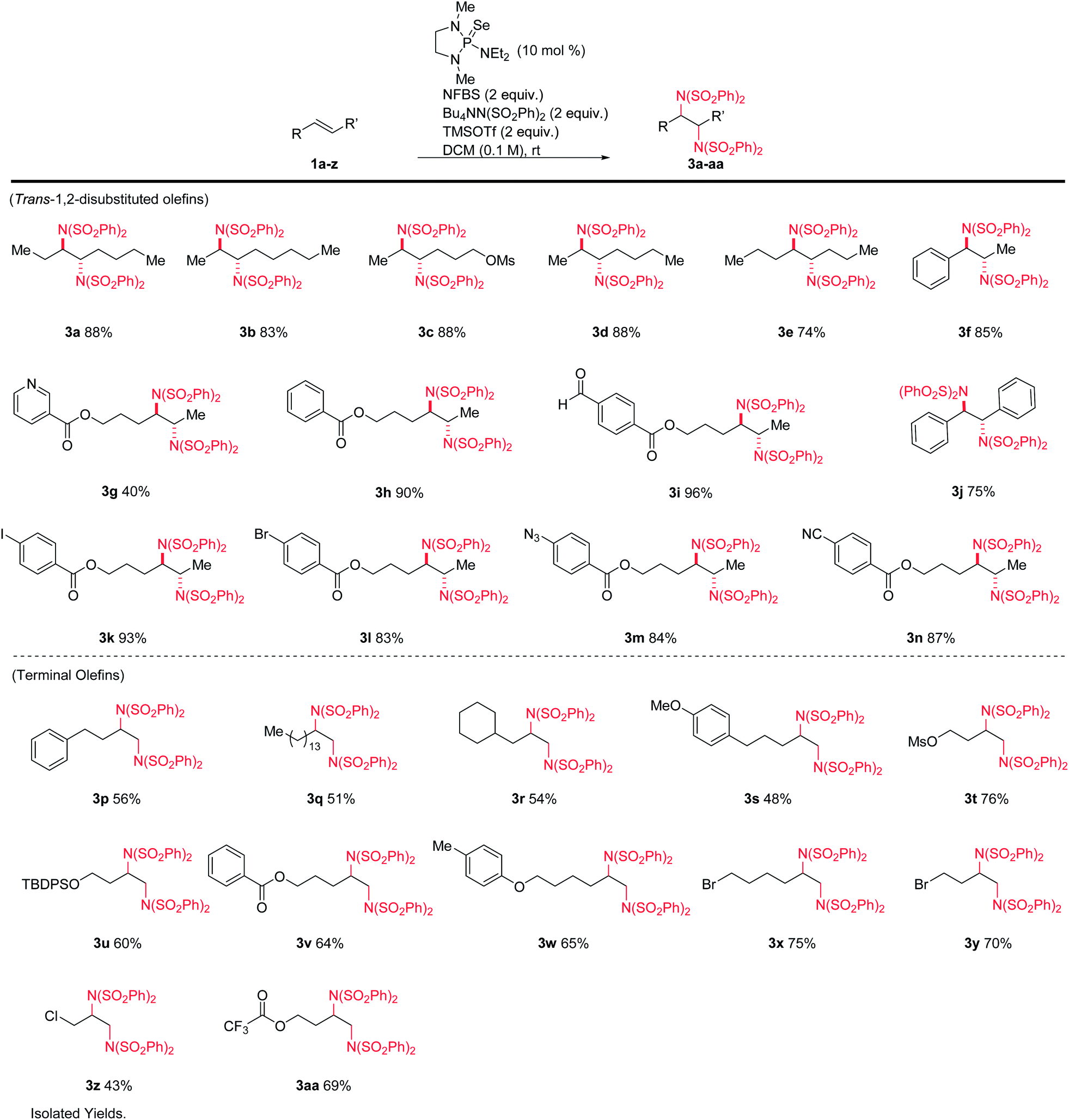
|
Interestingly, rather than a syn addition of the nitrogen nucleophiles as might be predicted by our mechanism and Denmark's dichlorination results, we observed selective formation of anti-diamines from (E)-alkenes. This indicates that an intramolecular substitution of the selenium species precedes intermolecular attack by the second benzenesulfonimide. We propose this occurs via the sulfonimide oxygen, as detailed in Scheme 5. A similar anti stereoselectivity and explanation thereof has been previously proposed by Muñiz et al. in their stoichiometric iodine(III) promoted diamination of alkenes.14a
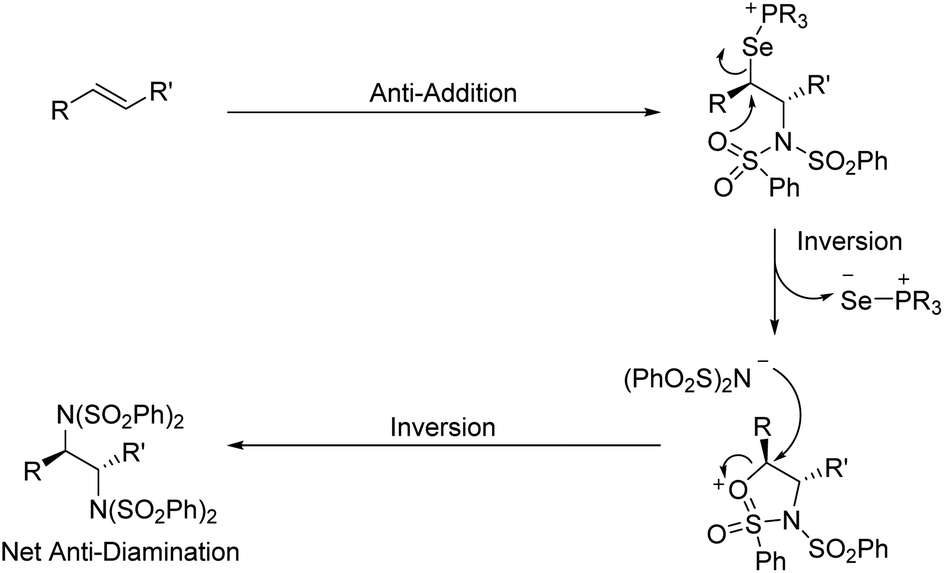 | ||
| Scheme 5 Mechanistic hypothesis for observed stereochemistry. | ||
In the course of our studies, we noticed that small amounts of an isomeric side product were consistently observed in the reaction of trifluoroacetate 1aa. We determined its structure to be rearrangement product 5aa (Scheme 6). We propose that this product is formed by attack of the ester on the initially-formed seleniranium ion, followed by ring-opening with benzenesulfonimide.
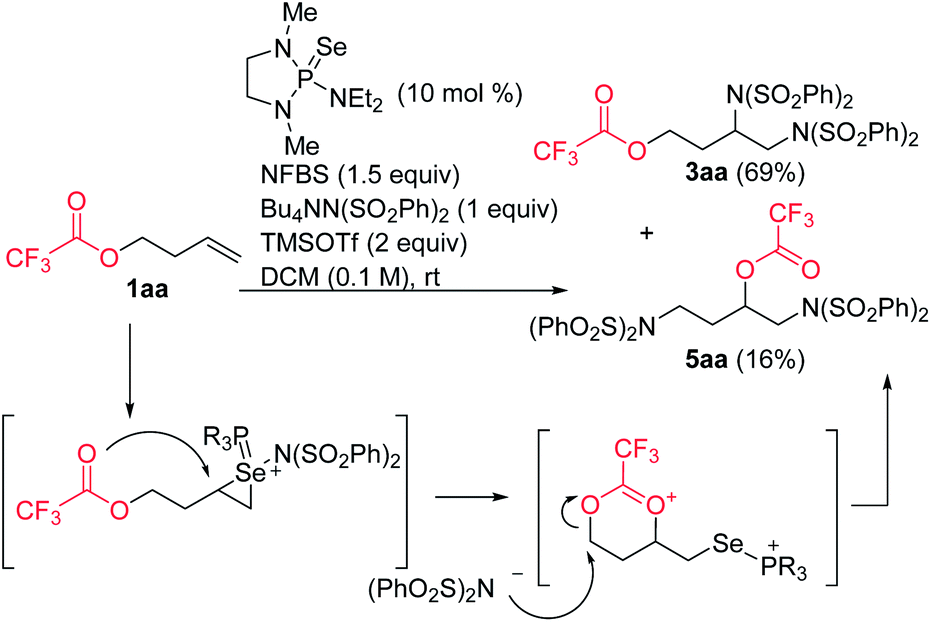 | ||
| Scheme 6 Ester rearrangement mechanism. | ||
Based on this proposed mechanism, we hypothesized that we could selectively generate either one of the isomeric products by changing the nucleophilicity of the ester oxygen. A series of electronically varied homoallyl esters (1aa–1ae) were subjected to the standard conditions and we observed that electron-rich esters gave predominantly rearrangement while electron-poor esters favored diaddition (Table 2). This is consistent with our proposed mechanism, in which an electron-rich ester would be more likely to act as a nucleophile, participating in the intramolecular cyclization before intermolecular nucleophilic attack can happen. Since the p-methoxybenzoate ester gave exclusively rearrangement product, several other substrates bearing this ester were subjected to the reaction conditions. As shown in Scheme 7, several allyl- and homoallyl esters gave rearrangement products in moderate to high yields. We found that in addition to the electronic properties of the ester, the chain length of the alkene also played a role. Allyl and homoallyl esters readily participated in the rearrangement, whereas longer chains gave little to no rearrangement (Table 1, compound 3v). These reactions are interesting because they afford 1,3-diamino-2-alcohols and 1,4-diamino-2-alcohols, both common structural units in HIV-protease inhibitors.16,17

|
 | ||
| Scheme 7 Diamination/rearrangement. | ||
Since intramolecular substitution with ester groups proceeded readily to give rearrangement products 5, we envisioned that appropriately chosen carbonates could give cyclic products via a similar intramolecular attack, followed by dealkylation of the carbonate substituent (Scheme 8). Indeed, a brief screen of carbonates (Me, t-Bu, Bn) revealed that benzyl carbonates were optimal for this purpose, giving the desired cyclic carbonates in high yields.
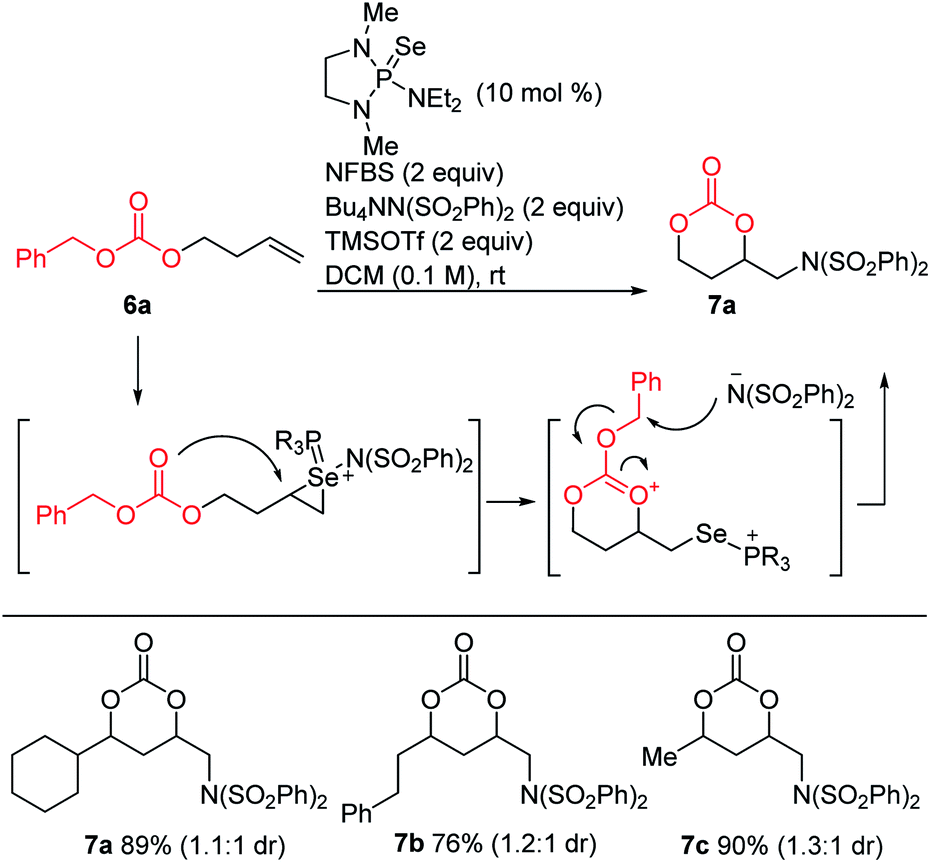 | ||
| Scheme 8 Carbonate cyclization. | ||
A variety of procedures have been reported for the deprotection of sulfonimides and sulfonamides.14a,18 To demonstrate the synthetic utility of the products, bis(sulfonimide) product 3b was mono-deprotected to give bis(sulfonamide) product 8 in high yield (Scheme 9).18d Product 8 could then be fully deprotected to the free diamine, which was isolated as bis(benzamide) 9 for ease of purification.18e
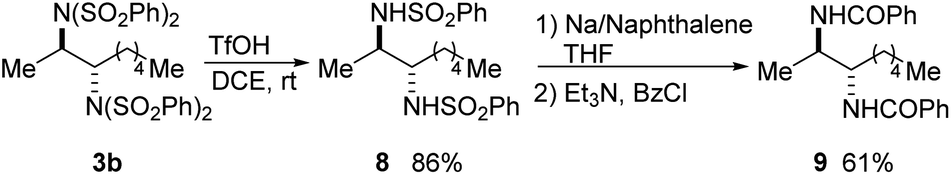 | ||
| Scheme 9 Selective sulfonamide deprotections. | ||
Conclusions
In summary, we have designed and implemented a new class of selenium-catalyzed 1,2-diaddition reactions of alkenes. This atypical mechanism was enabled by using a fluoride scavenger to shut down the typical syn-elimination pathway and by introducing a phosphine ligand on selenium to preferentially accelerate substitution reactions instead. The protocol is made successful by careful tuning of the phosphine ligand, revealing selenophosphoramides as the optimal catalysts. When an external benzenesulfonimide nucleophile was employed, 1,2-diamination of a range of terminal and 1,2-disubstituted alkenes was accomplished in high yields. The conditions tolerate a variety of functional groups. Additionally, substrates bearing appropriate internal nucleophiles, such as esters and carbonates, could be induced to undergo intramolecular substitution reactions, giving rearrangement and cyclization products. All told, the three types of diaddition reactions presented here (diamination, diamination/rearrangement, and oxyamination) allow access to a diverse set of products from simple alkene starting materials using a single set of conditions, demonstrating the utility of this new mode of catalysis. This allows access to a wide range of synthetically useful targets, including 1,2-diamines, 1,3-diamines, 1,4-diamines, and 1,2-aminocarbonates.10,16Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Washington and the National Science Foundation (CHE-1764450) for funding.Notes and references
- T. Wirth, in Organoselenium Chemistry: Synthesis and Reactions, Wiley-VCH, Weinheim, 2012 Search PubMed.
- Selenium catalysis review: F. V. Singh and T. Wirth, Catal. Sci. Technol., 2019, 9, 1073–1091 RSC.
- For select examples of selenium catalysis, see: (a) T. Hori and K. B. Sharpless, J. Org. Chem., 1979, 44, 4204–4208 CrossRef CAS; (b) M. Tiecco, L. Testaferri, M. Tingoli, L. Bagnoli and S. Santi, J. Am. Chem. Soc., 1993, 637–639 CAS; (c) T. Wirth, S. Hauptli and M. Leuenberger, Tetrahedron: Asymmetry, 1998, 9, 547–550 CrossRef CAS; (d) J. A. Tunge and S. R. Mellegaard, Org. Lett., 2004, 6, 1205–1207 CrossRef CAS PubMed; (e) J. Trenner, C. Depken, T. Weber and A. Breder, Angew. Chem., Int. Ed., 2013, 52, 8952–8956 CrossRef CAS PubMed; (f) S. E. Denmark, E. Hartmann, D. J. P. Kornfilt and H. Wang, Nat. Chem., 2014, 6, 1056–1064 CrossRef CAS PubMed; (g) Z. Deng, J. Wei, L. Liao, H. Huang and X. Zhao, Org. Lett., 2015, 17, 1834–1837 CrossRef CAS PubMed.
- T. Zheng, J. Tabor, Z. L. Stein and F. E. Michael, Org. Lett., 2018, 20, 6975–6978 CrossRef CAS PubMed.
- M. W. Young, R. F. Lauer and K. B. Sharpless, Tetrahedron Lett., 1973, 22, 1979–1982 Search PubMed.
- A. M. Morella and A. D. Ward, Tetrahedron Lett., 1985, 26, 2899–2900 CrossRef CAS.
- M. Tingoli, L. Testaferri, A. Temperini and M. Tiecco, J. Org. Chem., 1996, 61, 7085–7091 CrossRef CAS PubMed.
- (a) A. J. Cresswell, S. T.-C. Eey and S. E. Denmark, Nat. Chem., 2015, 7, 146–152 CrossRef CAS PubMed; (b) While this paper was under review, a syn-diamination of styrenes using a urea as bidentate nitrogen source was reported: Z. Tao, B. B. Gilbert and S. E. Denmark, J. Am. Chem. Soc., 2019, 141, 19161–19170 CrossRef CAS PubMed.
- Several selenium-catalyzed additions proceed via bromonium ions by simple oxidation of bromide: (a) S. R. Mellegaard and J. A. Tunge, Org. Chem., 2004, 69, 8979–8981 CrossRef CAS PubMed; (b) S. J. Balkrishna, C. D. Prasad, P. Panini, M. R. Detty, D. Chopra and S. Kumar, J. Org. Chem., 2012, 77, 9541–9552 CrossRef CAS PubMed.
- (a) D. Lucet, T. L. Gall and C. Mioskowski, Angew. Chem., Int. Ed., 1998, 37, 2580–2627 ( Angew. Chem. , 1998 , 110 , 2724–2772 ) CrossRef CAS; (b) A. Viso, R. Fernandez, A. Garcia and A. Flores, Chem. Rev., 2005, 105, 3167–3196 CrossRef CAS PubMed.
- Review: F. Cardona and A. Goti, Nat. Chem., 2009, 1, 269–275 CrossRef CAS PubMed.
- Select examples: (a) T. P. Zabawa, D. Kasi and S. R. Chemler, J. Am. Chem. Soc., 2005, 127, 11250–11251 CrossRef CAS PubMed; (b) Y. Zhu, R. G. Cornwall, H. Du, B. Zhao and Y. Shi, Chem. Res., 2014, 47, 3665–3678 CrossRef CAS PubMed; (c) E. L. Ingalls, P. A. Sibbald, W. Kaminsky and F. E. Michael, J. Am. Chem. Soc., 2013, 135, 8854–8856 CrossRef CAS PubMed; (d) J. Streuff, C. H. Hovelmann, A. Nunez and K. Muniz, Angew. Chem., Int. Ed., 2007, 46, 7125–7127 CrossRef PubMed.
- Stereochemistry assigned by analogy to ref. 14a. See ESI† for more details.
- Fully intermolecular diamination of alkenes: (a) J. A. Souto, Y. Gonzalez, A. Iglesias, D. Zian, A. Lishchynskyi and K. Muñiz, Chem. - Asian J., 2012, 7, 1103–1111 CrossRef CAS PubMed; (b) C. Martinez and K. Muñiz, Angew. Chem., Int. Ed., 2012, 51, 7031–7034 CrossRef CAS PubMed; (c) M. W. Danneman, K. B. Hong and J. N. Johnston, Org. Lett., 2015, 17, 2558–2561 CrossRef CAS PubMed; (d) K. Muñiz, L. Barreiro, R. M. Romero and C. Martinez, J. Am. Chem. Soc., 2017, 139, 4354–4357 CrossRef PubMed; (e) S.-S. Weng, K.-Y. Hsieh, Z.-J. Zeng and J.-W. Zhang, Tetrahedron Lett., 2017, 58, 670–673 CrossRef CAS; (f) N. Fu, G. S. Sauer, A. Saha, A. Loo and S. Lin, Science, 2017, 357, 575–579 CrossRef CAS PubMed; (g) B. Zhao, W. Yuan, H. Du and Y. Shi, Org. Lett., 2007, 9, 4943–4945 CrossRef CAS PubMed.
- Under these conditions there was full conversion of starting material and the remaining mass balance could not be accounted for.
- N. T. Ngoc Tam, G. Magueur, M. Ourevitch, B. Crousse, J. P. Begue and D. Bonnet-Delpon, J. Org. Chem., 2005, 70, 699–702 CrossRef PubMed.
- For selected examples of alkene oxyamination, see: (a) H. Lei and T. Rovis, J. Am. Chem. Soc., 2019, 141, 2268–2273 CrossRef CAS PubMed; (b) V. A. Schmidt and E. J. Alexanian, J. Am. Chem. Soc., 2011, 133, 11402–11405 CrossRef CAS PubMed; (c) F. C. Sequeira and S. R. Chemler, Org. Lett., 2012, 14, 4482–4485 CrossRef CAS PubMed; (d) J. Escudero, V. Bellosta and J. Cossy, Angew. Chem., Int. Ed., 2018, 57, 574–578 ( Angew. Chem. , 2018 , 130 , 583–587l ) CrossRef CAS PubMed; (e) J. Xie, Y. W. Wang, L. W. Qi and B. Zhang, J. Am. Chem. Soc., 2017, 19, 1148–1151 CAS; (f) U. Farid and T. Wirth, Angew. Chem., Int. Ed., 2012, 51, 3462–3465 CrossRef CAS PubMed.
- (a) P. A. Sibbald and F. E. Michael, Org. Lett., 2009, 11, 1147–1149 CrossRef CAS PubMed; (b) A. Iglesias, R. Alvarez, R. L. Angel and K. Muniz, Angew. Chem., Int. Ed., 2012, 51, 2225–2228 CrossRef CAS PubMed; (c) Y. Okamura, D. Sato, A. Yoshimura, V. V. Zhdankin and A. Saito, Adv. Synth. Catal., 2017, 359, 3243–3247 CrossRef CAS; (d) T. Javorskis and E. Orentas, J. Org. Chem., 2017, 82, 13423–13439 CrossRef CAS PubMed; (e) S.-S. Weng, K.-Y. Hsieh, Z.-J. Zeng and J.-W. Zhang, Tetrahedron Lett., 2017, 58, 670–673 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc05335b |
| This journal is © The Royal Society of Chemistry 2020 |