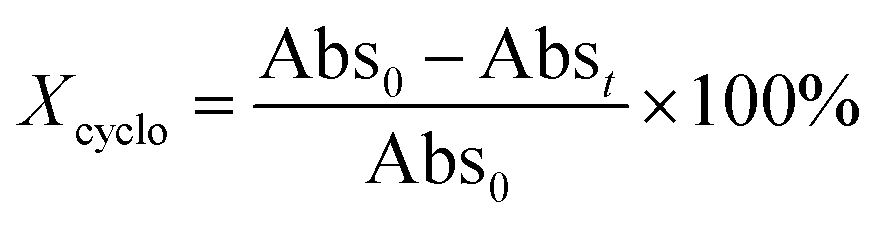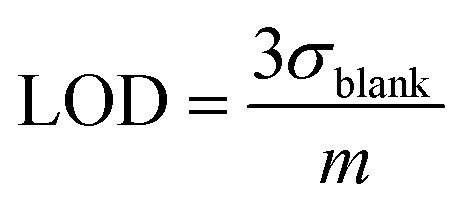DOI:
10.1039/C9SC05380H
(Edge Article)
Chem. Sci., 2020,
11, 2504-2510
Photosynergetic amplification of radiation input: from efficient UV induced cycloreversion to sensitive X-ray detection †
Received
27th October 2019
, Accepted 23rd January 2020
First published on 23rd January 2020
Abstract
Five photochromic terarylenes which show reversible photocyclisation and cycloreversion with relatively high quantum yields are presented. Some of these have been observed to undergo a highly efficient cycloreversion cascade process from their coloured, closed forms to their uncoloured open forms that leads to cycloreversion quantum yields significantly larger than unity. This cascade effect can been induced with both chemical and X-ray initiation; the limit of detection from X-ray initiation has been tested and is comparable to existing systems with detection observed at values as low as 0.3 mGy.
Introduction
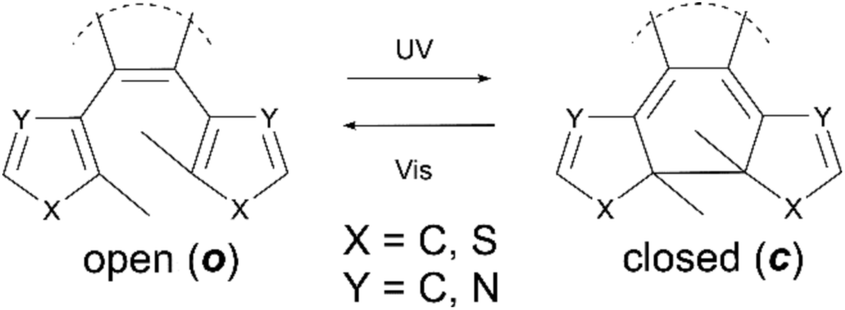
The use of a single physical stimuli such as heat or illumination to induce changes in the physical media with which they are interacting is a well-known phenomenon in both biological1,2 and materials science.3 For example the initial processes of mammalian optical vision are based on retinal-protein hybrids which regulate transport through ion-channels in cell membranes.1 These are capable of amplification above a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of ions to photons, a significant increase in the sensitivity of the system upon activation. Such a response, resulting in more than one physical output from a single input, demonstrates the principle of a synergetic cascade mechanism. In materials science many systems based on similar synergetic responses have been developed, particularly focusing on photonic applications including photo-multiplying tube (PMT) detectors4,5 and avalanche photodiodes (APD)6–9 and in which the output can be many orders of magnitude larger than the photonic input. Although these systems have been known for many years the exploitation of cascade responses on a molecular level has thus far, proved challenging. The generation of future examples requires the development of new molecules responsive to external stimuli such as heat,10,11 chemical10 or physical stress.12–15 For these applications an important requirement is to maximize the response efficiency, that is the number of changes that can be induced in the target molecules with the minimum amount of external stimuli. In the field of photochemistry a lot of focus has been placed upon the use of photo-amplifying systems, due to the ease with which they deliver large packets of energy, along with the non-invasive, rapid way they can be exploited. Molecular examples that have been reported include those of Tokumaru and Irie, who have independently studied photoisomerisation of ethenes15 and binaphothylenes16 with photochemical quantum yields amplified to higher than unity via triplet state energy transfer. Recently Garcia-Garibay reported specific quantum-chain photodecarbonylation of diarylcyclopropenones in the crystalline state.17 Here photochemical quantum yields are reported to be higher than 300%, resulting from singlet energy migration. The major drawback of all of these molecules however, is their irreversibility, meaning that unlike biological or materials examples they cannot be returned to their original state once the cascade is complete, drastically lowering their utility. To attempt to overcome this, we have investigated the use of reversibly switchable photochromic molecules including terarylenes which are known to have a controllable exchange between their two forms.15,18,19 These can undergo fully reversible structural changes through the application of selected wavelengths of light, allowing for the interchange between two isomers having different photophysical properties. In such cases the usability of the switching process depends both on the yield of the induced change and the properties of the products formed. These classes of compounds undergo the 6π-photocyclisation in the central hexatriene unit upon application of UV light from the as-synthesised open (o) form to the closed (c) form, while irradiation with visible light leads to the cycloreversion reaction (Scheme 1).
:1 ratio of ions to photons, a significant increase in the sensitivity of the system upon activation. Such a response, resulting in more than one physical output from a single input, demonstrates the principle of a synergetic cascade mechanism. In materials science many systems based on similar synergetic responses have been developed, particularly focusing on photonic applications including photo-multiplying tube (PMT) detectors4,5 and avalanche photodiodes (APD)6–9 and in which the output can be many orders of magnitude larger than the photonic input. Although these systems have been known for many years the exploitation of cascade responses on a molecular level has thus far, proved challenging. The generation of future examples requires the development of new molecules responsive to external stimuli such as heat,10,11 chemical10 or physical stress.12–15 For these applications an important requirement is to maximize the response efficiency, that is the number of changes that can be induced in the target molecules with the minimum amount of external stimuli. In the field of photochemistry a lot of focus has been placed upon the use of photo-amplifying systems, due to the ease with which they deliver large packets of energy, along with the non-invasive, rapid way they can be exploited. Molecular examples that have been reported include those of Tokumaru and Irie, who have independently studied photoisomerisation of ethenes15 and binaphothylenes16 with photochemical quantum yields amplified to higher than unity via triplet state energy transfer. Recently Garcia-Garibay reported specific quantum-chain photodecarbonylation of diarylcyclopropenones in the crystalline state.17 Here photochemical quantum yields are reported to be higher than 300%, resulting from singlet energy migration. The major drawback of all of these molecules however, is their irreversibility, meaning that unlike biological or materials examples they cannot be returned to their original state once the cascade is complete, drastically lowering their utility. To attempt to overcome this, we have investigated the use of reversibly switchable photochromic molecules including terarylenes which are known to have a controllable exchange between their two forms.15,18,19 These can undergo fully reversible structural changes through the application of selected wavelengths of light, allowing for the interchange between two isomers having different photophysical properties. In such cases the usability of the switching process depends both on the yield of the induced change and the properties of the products formed. These classes of compounds undergo the 6π-photocyclisation in the central hexatriene unit upon application of UV light from the as-synthesised open (o) form to the closed (c) form, while irradiation with visible light leads to the cycloreversion reaction (Scheme 1).
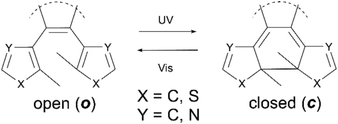 |
| | Scheme 1 Structure and photochromism in bistable terarylene organic compounds. | |
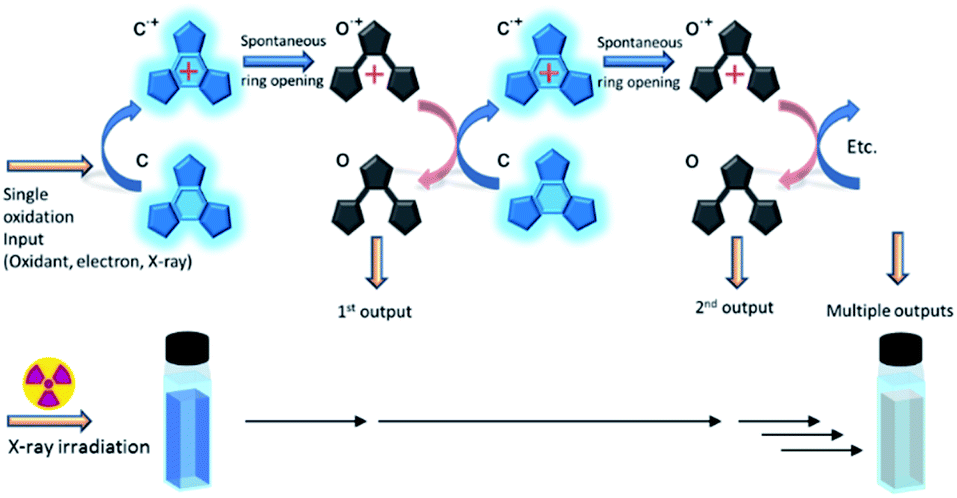
These platforms are used due to the ease with which their properties can be predictably tuned through relatively simple functionalisation. Recently, we reported a photochromic derivative harbouring atoms at strategic positions designed to hold the molecule in its most photo-reactive conformation resulting in photocyclisation quantum yields of almost unity.20 However, this high photocyclisation quantum yield comes at the cost of lower efficiencies for the cycloreversion reaction, lowering their potential towards many applications. This problem has led us to investigate an alternative ring opening method based upon an oxidative cycloreversion process (Scheme 2) which showed potential for having a response efficiency beyond unity.21–33 This proceeds via a cascade resulting from the oxidation of the ring closed form to it radical cation (c˙+) which spontaneously ring-opens to the cationic radical of the open form (o˙+). This immediately reacts with remaining neutral closed forms in solution, regenerating the closed radical cation and leading to multiple ring openings from a single oxidation source. Electron efficiencies of up to 900%, implying nine ring openings for each single oxidation source were reported,28 and ring opening turnover numbers of up to 1000 have been achieved.32 This synergetic mechanism has also been shown to be effective for some diarylethene derivatives.23,30 As it has long been known that X-rays oxidatively-generate radicals in certain halogenated solvents,34 we next decided to investigate the use of X-rays to generate the initial radical in suitable solvents in order to initiate the cascade reaction.
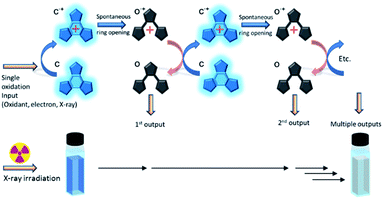 |
| | Scheme 2 Oxidative cascade reaction mechanism for cycloreversion. Inset: Schematic of the detection scheme. X-ray signals induces cycloreversion as evidenced by the discoloration of the coloured closed form. | |
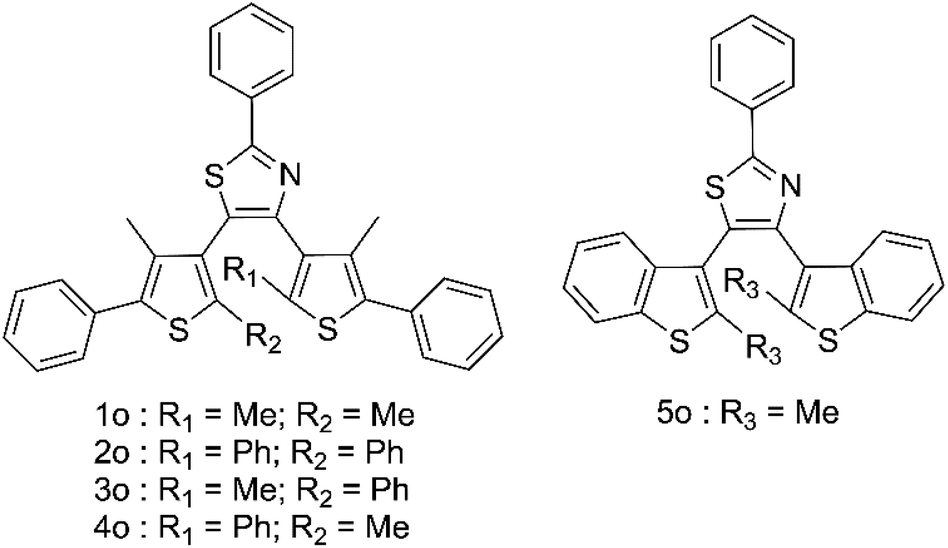
High-energy photons including X-rays offer major health risks35–37 and as such, there is a growing demand for new high sensitivity methods of their detection.35,37–41 Most existing X-ray sensing compounds are based on expensive, toxic materials, usually including metal–organic frameworks or heavy metals which are difficult to process into commercial devices.42–49 To counter this a number of organic based systems have been suggested; however, these all suffer from the difficulties of having to be paired with the solid-state electronic detection methods to produce a commercial device. Photochromic organic detectors may offer an alternative as they can report the presence of X-rays through easily observed colour changes, show relative hardness towards high energy photons, are easily recyclable and are much simpler to process. Examples of such systems have included spiropyrans and diarylethenes; however, in both these cases the organic molecules required the use of sensitisers and only offered low-level detection limits in the range of 1–0.1 Gy.50–56 This led us to propose the use of terarylenes susceptible to cascade reactions for the direct observation of the ring opening induction using X-ray induced radical generation. This method offers a highly sensitive photochromic response to low doses of X-rays, allowing for much higher sensitivities than those organic examples already reported. Five compounds 1o–5o (Fig. 1) are studied herein and have been prepared and fully characterised using NMR and mass spectroscopy (Fig. S1–S9†).
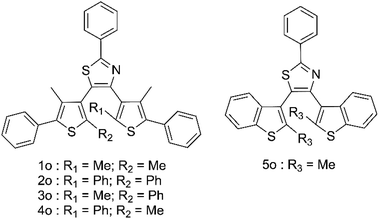 |
| | Fig. 1 Structures of compounds in this study. | |
Results and discussion
Molecular design, synthesis and characterisation
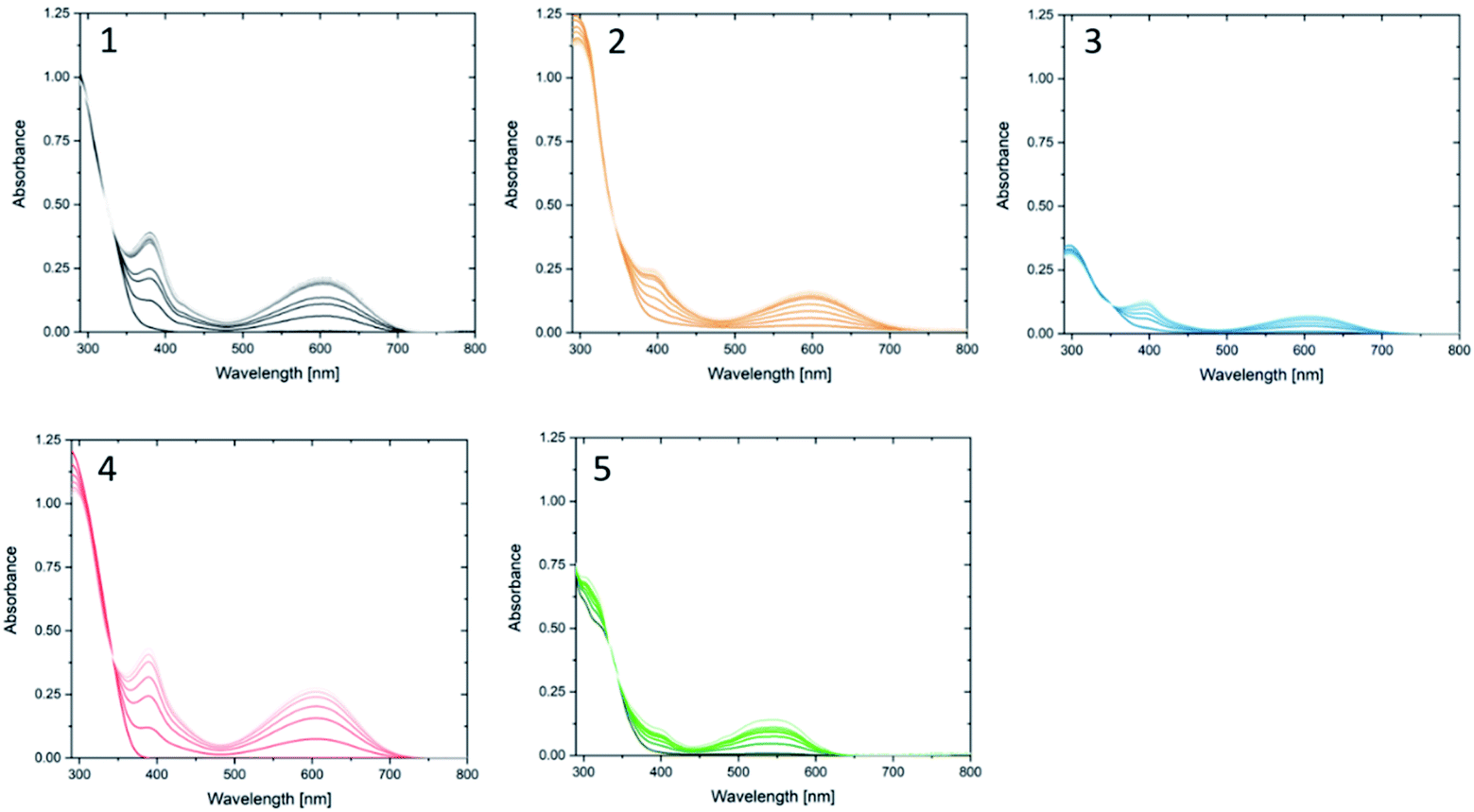
Compound 1o was taken from a family of triangle terarylenes, which was reported to show the higher photo-cyclisation quantum yield (0.6) than that of dithiazolylthiazole based one (0.4).572o was designed as a target containing phenyl rings on both photochemically reactive carbon atoms, as these rings were shown to accelerate the rate-limiting step of the oxidative cycloreversion (Scheme 2) and increase the efficiency of amplification by three orders of magnitude or more.32 However, such phenyl rings were also shown to increase the steric hindrance on the c forms which contributes to decreased thermal stability and lower photocyclisation quantum yields. For this reason, two new compounds were prepared, each having only one phenyl ring on the reactive carbons (compounds 3o and 4o) via consecutive coupling reactions, as inspired by previous works.58,59 Compound 5o containing benzothiophene moieties instead of phenylthiophene groups, in which the capability of aforementioned cycloreversion cascade process was absent,28 was also employed as a reference. First the photochromic properties of these compounds in acetonitrile as an inert media was explored, with UV irradiation (λ = 365 nm) resulting in colouration typical of diarylethenes and terarylenes for all five compounds (see Fig. 2 and S10†). Compounds 1–4 share a π-conjugated system with a similar geometry in their closed-ring forms, leading to an almost identical absorption peak in the visible range at 613 nm with photo-cyclisation quantum yields >50%, typical of terarylenes. The optical properties including the conversion ratio of c-forms at the photostationary state (PSS) are summarized in the ESI (Table S1, see also Fig. S11 and S12†). Visible light irradiation (λ > 480 nm) of the coloured solutions induced the cycloreversion reaction to afford colourless solutions with the original absorption profiles of o forms. Closed form of compound 2 exhibited spontaneous bleaching at room temperature within minutes, probably due to the steric strain in the closed ring form destabilising this isomer 2c. Photocyclisation–cycloreversion cycles were repeated for several times with the same isosbestic point, indicating typical two-component reversible photoisomerizations. No significant solvatochromism was observed with 1o/1c displaying the same spectra in hexane.56 Thermal stability of the closed-ring forms for 1c, 3c and 4c was investigated in the dark and the half-lives at room temperature are estimated to be fourteen days, six hours and six hours respectively.
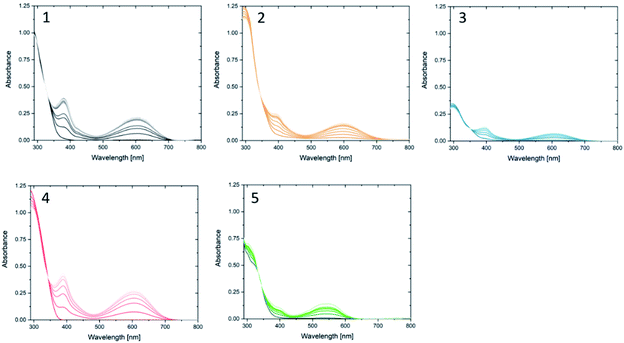 |
| | Fig. 2 New absorption bands in the visible region appear for compounds (1–5) upon irradiation with UV light at 365 nm in acetonitrile. Each spectrum was taken every after irradiation for 30 s. Visible light irradiation turns them back to the o form. Concentration of o forms = (1) 3.0 × 10−5 M, (2) 3.7 × 10−5, (3) 1.5 × 10−5, (4) 5.2 × 10−5, (5) 4.0 × 10−5. | |
Cascade reaction
Although the spontaneous bleaching of 2c to 2o within minutes prevented the further characterization of 2, the half-lives of six hours for 3c and 4c afforded the evaluation of the oxidative cycloreversion reaction cascade, most of which was complete within an hour. The half-lifetime of 5c was estimated to be 500 days in toluene. First the oxidative charge transfer chain cycloreversion for these compounds was tested using tris(bromophenyl)ammoniumyl hexachloroantimonate (TBPA) as a one-electron oxidant initiator in acetonitrile. As in previous reports,32,60 complete cycloreversion could be accomplished even with catalytic amounts of TBPA, indicating the chain reaction process. A minimum amount of 2% equivalents of TBPA was necessary to induce the complete cycloreversion of 1c, a turnover number of 200 for each initial trigger molecule (Fig. S13,† 5000% oxidant-based yield). For both 3c and 4c, oxidant equivalents as low as 0.1% was found to lead to complete decolouration, a turnover number of over 1000 for each initial trigger molecule, i.e. 100000% oxidant-based yield (Fig. S14†). This value was comparable to that of terthiazole-based compound having two phenyl moieties on the reactive carbons.32 One phenyl ring on the reactive carbon atoms appears enough to make oxidative cycloreversion faster and more efficient in the present compounds. Contrarily, the complete bleaching of 5c required an equimolar amount of TBPA, confirming the absence of a chain reaction mechanism in this case (Fig. S15†). This result might be explained by the slower cycloreversion in the radical cation state (5c˙+ → 5o˙+) together with the shorter lifetime of 5o˙+ (0.2 s) compared to those of 1, which were estimated by the electron transfer stopped-flow study28 (Fig. S16, S17 and Table S2†).
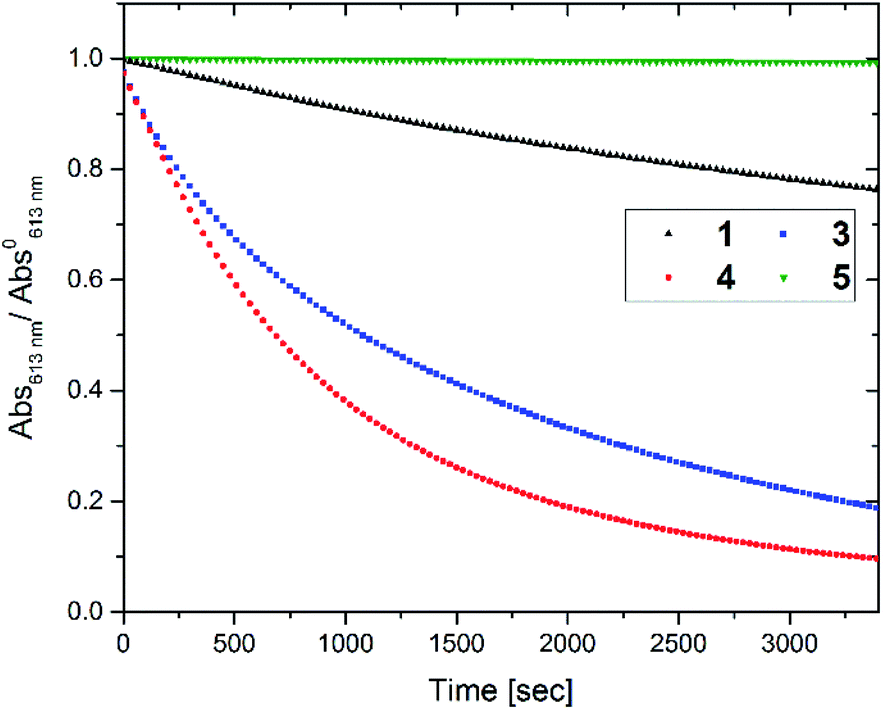
Next the oxidative cycloreversion reaction was induced by UV irradiation. The c forms of compounds were produced by illuminating 0.3 mL solutions in toluene under UV (365 nm) until the amount of c forms reached to 9.0 × 10−8 mol for all compounds. Then, the solution was diluted with 2.7 mL of chloroform to give the c form concentration of 3.0 × 10−5 M and irradiated with a controlled number of photons at 313 nm. Fig. 3 monitors the peak absorbance at 610 nm for 1, 3 and 4 and at 540 nm for 5 following the initial UV irradiation at 313 nm for 30 s. The continuous decrease in the closed-ring forms 1c, 3c and 4c, suggests that cycloreversion takes place, with absorptions typical of the o forms recovered. It can be seen that cycloreversion of 1c took around 30 min in an almost linear fashion, similar to previous results.32 Meanwhile, cycloreversion of 3c and 4c were completed within half an hour in the quasi-linear fashion, in accordance with the results of oxidative cycloreversion of derivatives having aromatic rings in the reactive centres.32 Since the ambient half-lives of 3c and 4c are six hours, the effect of spontaneous thermal cycloreversion on this experiment is much less than the photo-triggered reaction. The UV irradiation (either at 313 and 365 nm) in acetonitrile and toluene only induced the photo-cyclization (colouration) reaction. The irradiation at the absorption bands in the visible region (610 nm) did not cause such the persistent bleaching of coloured forms after closing the irradiation. Therefore, the cation radical species triggering the oxidative cycloreversion cascade could be formed in the presence of chloroform with UV irradiation. The photoinduced charge transfer between the higher excited state of c forms and chloroform molecule61 or the excitation of charge-transfer state62,63 between them were suggested to trigger the catalytic oxidation reaction cascade. Meanwhile, no decrease was observed for 5c again indicating the importance of the cascade reaction capability.
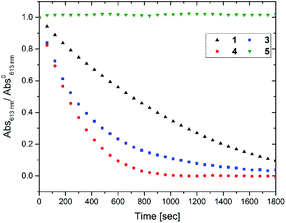 |
| | Fig. 3 Decay in absorbance at λmax after UV irradiation (313 nm), of toluene:chloroform (1:9) solutions of the c forms of different compounds. Concentration of c forms = 3.0 × 10−5 M. | |
To illustrate reversibility, consecutive photocyclisations and cycloreversions were also performed (Fig. S18†). After the first cycloreversion and recovering a clear solution of the o form, another cycle was restarted by evaporating off the chloroform until the only solvent that remained was toluene. UV irradiation resulted in a stable blue solution, without evidence of cycloreversion after several minutes. Addition of chloroform and subsequent UV irradiation led to bleaching of the colour again. This was cycled several times with no marked degradation in 1H NMR spectrum (Fig. S18 and S19†). The absorbed photon numbers at 313 nm were then counted using an absolute photoreaction quantum yield evaluation system to estimate an apparent cycloreversion quantum yield.64 For 3c and 4c, the maximum turnover number of the chain reaction process was found to be 33 to 1; that is, for every photon induced oxidation, an average of 33 molecules undergo cycloreversion. This quantum yield can thus reach up to 3300%. It is noted that the solution being irradiated with UV may contain unconverted o form that can absorb photons at 313 nm also, having an opposite effect on the decolouration. It has been reported that cycloreversion quantum yields can be improved through a similar electron transfer chain reaction mechanism with the initial oxidation induced by a photocatalyst,65,66 however in such cases quantum yields still remain below 100% due to existing side reactions. Because there is no photocatalyst in our system, no such side reactions can occur allowing for surpassing the 100% efficiency limit of the reaction.
X-ray induced cycloreversion and detection
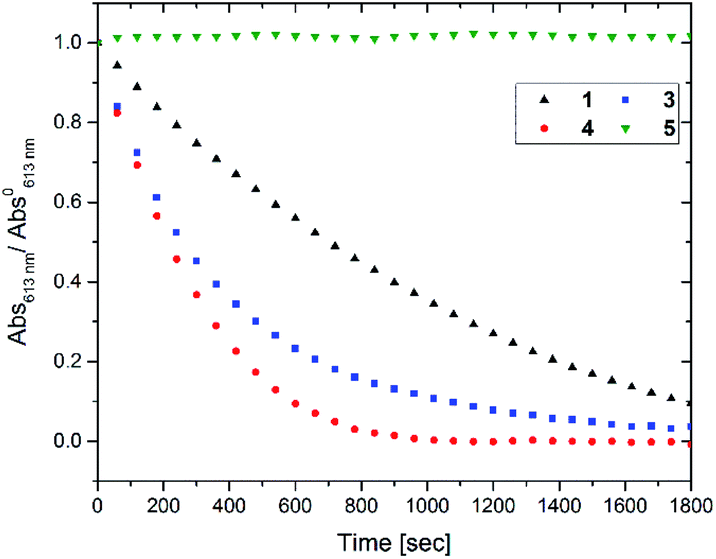
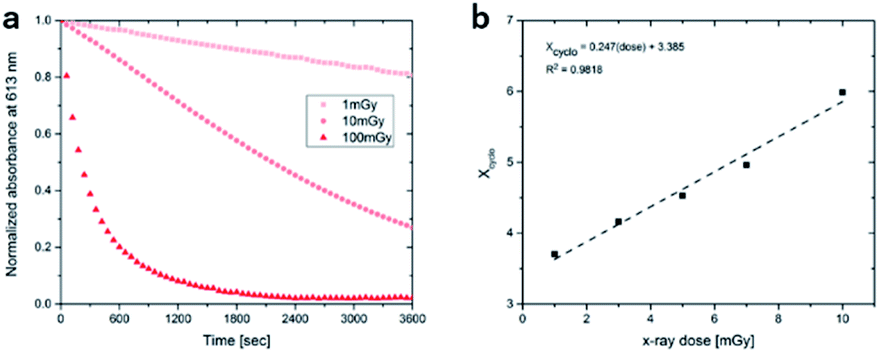
To access for X-ray induced cycloreversion, we again prepared the c form solutions in toluene/chloroform mixture in a similar manner to the UV-induced cycloreversion experiment described above. The solutions of the c form were then irradiated for 6 s under 100 mGy of overall X-ray dose through an X-ray irradiation source with a conventional X-ray tube (see the ESI† for the detailed condition) and the absorbance at 613 nm in the visible region was monitored under continued stirring. A decrease in the absorbance of the coloured forms 1c, 3c, and 4c, was observed, indicating conversion to their respective o forms (Fig. 4). The decay in absorbance of 1c was not completed within two hours while those of 3c and 4c were completed within an hour. Cycloreversion due to thermal processes may not explain this behaviour since the thermal half-lives of the c forms are considerably longer than the timescales for this X-ray induced reversion. To ensure that no significant degradation due to X-rays occurred, the o form was also recovered with 100% yield by solvent extraction procedure, which is confirmed by the unchanged 1H NMR spectrum (Fig. S20†). As no oxidant was used a simpler recovery method can be employed where the mixture was heated to 60 °C under reduced pressure leaving only the initial o form in toluene. Subsequent exposure of this solution to UV light recovered the absorbance of the c form, confirming the reversibility of the process. To the best of our knowledge, this is the first X-ray-induced cycloreversion for diarylethenes or terarylenes. The effect of X-ray radiation dosage on the bleaching reaction was then evaluated for 4c. Almost complete bleaching was observed with the X-ray irradiation of 100 mGy within 30 min, with further decreases in the dosage leading to slower decolouration. 10 mGy irradiation led to discoloration to 30% after one hour, and 80% remaining after one hour with 1 mGy dosage (Fig. 5a). X-ray irradiation to the halogenated alkanes is expected to generate radical cation species67–69 which further oxidise the neutral c forms triggering the oxidative cycloreversion reaction cascade. Even with the low efficiency in the charge transfer reaction between the radical cation species generated form chloroform molecule and the neutral c forms, the subsequent oxidative cycloreversion cascade could amplify the X-ray sensitivity. The rate of cycloreversion can be controlled by the amount of radical species oxidatively generated, which is directly related to the X-ray dosage, since the electron transfer process propagates in a chain like manner. This is in accordance with our previous report illustrating the dependence of cycloreversion kinetics on the amount of oxidised species in the process.32 In order to evaluate the decolouration process as an analytical tool, we thus conventionally define Xcyclo as the “% decrease in absorbance ten minutes after X-ray exposure” to be used as an analytical index for X-rays (eqn (1)):| | 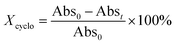 | (1) |
where Abs0 is the initial absorbance at λmax,vis of toluene:chloroform (1:9) solutions of the c forms and Abst is the absorbance at λmax,vis of toluene:chloroform (1:9) solutions of the c forms after ten minutes without irradiation. A solution of the c form of 4 was exposed to X-rays of varying doses and the absorbance decay monitored for ten minutes. A plot of Xcyclo as a function of X-ray dosage in the mGy range showed a linear response (Fig. 5b); as X-ray dosage determines the amount of cations generated this is proportional to the rate of cycloreversion and proceeds with the zero-order kinetics.32 X-ray dosage in the range of 1–10 mGy led to Xcyclo with only a slight deviation of linearity (Fig. 5b), indicating an analytical dynamic range of at least one order of magnitude. This result therefore provides a method not just for X-ray detection but also for quantification of X-ray dosage; as measurement/calculation of Xcyclo can be correlated with any X-ray dosage in the range of 1–10 mGy. Then, the limit of detection (LOD) was evaluated as follows:| | 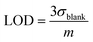 | (2) |
Here σblank is the standard deviation of Xcyclo for multiple (15) blank measurements (no X-ray irradiation) as a measure of the % decrease in absorbance due to the spontaneous cycloreversion (Table S3†), and m is the slope of the graph on Fig. 5b. We determined for 4c that the LOD is 0.3 mGy which is two orders of magnitude lower than the record set by Han and co-workers using small organic molecules34 and similar to that of Xie and co-workers for uranyl-based frameworks.70
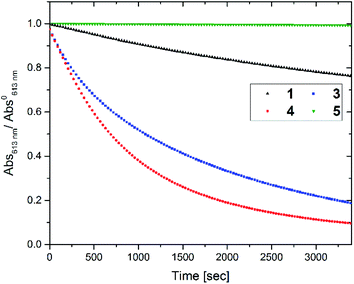 |
| | Fig. 4 Decay in absorbance at λmax,vis of toluene:chloroform (1:9) solutions of the c forms of different compounds upon irradiation of 100 mGy of X-rays. Concentration of c forms = 3.0 × 10−5 M. | |
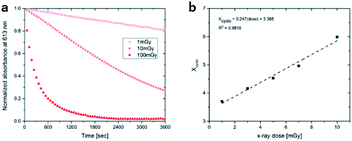 |
| | Fig. 5 (a) Decay in absorbance upon irradiation with varying doses of X-ray for 4c; (b) linear relationship between Xcyclo and X-ray dose. | |
This low LOD is due to the linear sensitivity of Xcyclo to very small changes in X-ray dosage as measured by m. The large absorbance change in response to X-ray dosage is mainly due to the highly efficient charge transfer cascade reaction mechanism for the cycloreversion. This illustrates the immense utility of an efficient photosynergetic response for highly sensitive detection. Meanwhile, σblank is minimal because of the reasonable thermal stability of the c form coupled with, of course, high precision of absorbance measurements.
Conclusions
We have prepared five photochromic terarylenes which show reversible photocyclisation and cycloreversion reactivity with relatively high photocyclisation quantum yields. Three of them further displayed a highly efficient cycloreversion response through an electron transfer cascade reaction process. Remarkable enhancement in the amplification efficiency was clearly demonstrated upon introduction of one phenyl unit on a carbon atom at the photocyclisation reaction centre. While UV irradiation in a non-halogenated solvent causes colouration to their thermally stable coloured c forms; which in the presence of chloroform can undergo a highly efficient bleaching reaction from the c form to the o form. The kinetics are indicative of the chain reaction process. We found that such a UV induced quantum chain reaction gives a boost of photochemical quantum yields significantly greater than unity (3300%). Given the cascade electro-cycloreversion efficiency up to 100000% as demonstrated by the chemical-oxidant induced reaction experiment, about 3% of photo-oxidation quantum yield can be expected to trigger the chain reaction. Similar amplified photochemical reactivity can also be expected if other halo-alkanes such as dichloromethane, dichloroethane, bromoethane and iodomethane were used. The phenyl thiophene units or diphenyl thiophene units in the present compounds would support photo-oxidation capability and efficient cascade reactivity by stabilising the cation-radical intermediate. Indeed, we observed no photo-triggered cascade reaction for our previous terthiazole derivatives of less oxidative activity. Even upon irradiation with X-rays of very low dosage (mGy levels), the fading reaction is apparently observed, with proceeding of the cycloreversion back to the colourless o form. These new compounds deliver a new approach for development of an eye-detectable radio-dosimeter with the aid of gel supports to form quasi-solid state materials, which alerts the presence of low-level radio-doses with disappearing blue color. The detection limit as low as 0.3 mGy, the highest sensitivity among organic-based X-ray colour-changing material, is already of practical interest for X-ray sensing materials.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the MEXT Program for Promoting the Enhancement of Research Universities in NAIST and also partly supported by the JSPS KAKENHI Grant (JP26107006) in Scientific Research on Innovative Areas “Photosynergetics”. RA thanks the NAIST Foundation for financial support and CJM thanks the JSPS KAKENHI Grant-in-Aid for Early-Career Scientists (19K15312).
Notes and references
-
T. L. Lentz, Cell fine structure, Philadelphia, 1971 Search PubMed.
-
B. Hille, Ionic Channels of Excitable Membranes, Oxford University Press Inc, 2001 Search PubMed.
-
V. O'Connor and D. Phillips, Time-Correlated Single Photon Counting, Academic, London, 1984 Search PubMed.
- S. Cova, M. Bertolaccini and C. Bussolati, The measurement of luminescence waveforms by single photon techniques, Phys. Status Solidi A, 1973, 18, 11–62 CrossRef CAS.
- H. Kume, K. Koyama, K. Nakatsugawa, S. Suzuki and D. Fatlowitz, Ultrafast microchannel plate photomultipliers, Appl. Opt., 1988, 27, 1170–1178 CrossRef CAS PubMed.
- R. B. Emmons, J. Appl. Phys., 1967, 38, 3705 CrossRef.
- S. Cova, A. Lacaita, M. Ghioni, G. Ripamonti and T. A. Louis, Rev. Sci. Instrum., 1989, 60, 1104–1110 CrossRef CAS.
- H. Dautet, P. Deschamps, B. Dion, A. D. MacGregor, D. MacSween, R. J. McIntyre, C. Trottier and P. P. Webb, Appl. Opt., 1993, 32, 3894–3900 CrossRef CAS PubMed.
- S. Cova, M. Ghioni, A. Lacaita, C. Samori and F. Zappa, Appl. Opt., 1996, 35, 1956–1976 CrossRef CAS PubMed.
-
G. S. Zweifel, M. H. Nantz and P. Somfai, Modern Organic Synthesis: An Introduction, John Wiley & Sons, New Jersey, 2nd edn, 2017 Search PubMed.
-
Molecular Switches, ed. B. L. Feringa and W. R. Browne, Wiley-VCH, Weinheim, 2011 Search PubMed.
-
Photon-Working Switches, ed. Y. Yokoyama and K. Nakatani, Springer, Tokyo, 2017 Search PubMed.
-
Mechanochromic Fluorescent Materials: Phenomena, Materials and Applications, ed. J. Xu and Z. Chi, Royal Society of Chemistry, Cambridge, 2014 Search PubMed.
- G. Zhang, J. Lu, M. Sabat and C. L. Fraser, J. Am. Chem. Soc., 2010, 132, 2160–2162 CrossRef CAS PubMed.
- T. Arai, T. Karatsu, H. Sakuragi and K. Tokumaru, Tetrahedron Lett., 1983, 24, 2873–2876 CrossRef CAS.
- T. Yorozu, K. Yoshida, K. Hayashi and M. Irie, J. Phys. Chem., 1981, 85, 459–462 CrossRef CAS.
- G. Kuzmanich, M. N. Gard and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2009, 131, 11606–11614 CrossRef CAS PubMed.
-
New Frontiers in Photochromism, ed. M. Irie, Y. Yokoyama and T. Seki, Springer Japan, Tokyo, 2013 Search PubMed.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed.
- S. Fukumoto, T. Nakashima and T. Kawai, Angew. Chem., Int. Ed., 2011, 50, 1565–1568 CrossRef CAS PubMed.
- T. Koshido, T. Kawai and K. Yoshino, J. Phys. Chem., 1995, 99, 6110–6114 CrossRef CAS.
- A. Peters and N. R. Branda, J. Am. Chem. Soc., 2003, 125, 3404–3405 CrossRef CAS PubMed.
- Y. Moriyama, K. Matsuda, N. Tanifuji, S. Irie and M. Irie, Org. Lett., 2005, 7, 3315–3318 CrossRef CAS PubMed.
- W. R. Browne, J. J. D. de Jong, T. Kudernac, M. Walko, L. N. Lucas, K. Uchida, J. H. van Esch and B. L. Feringa, Chem.–Eur. J., 2005, 11, 6414–6429 CrossRef CAS PubMed.
- B. Gorodetsky and N. R. Branda, Adv. Funct. Mater., 2007, 17, 786–796 CrossRef CAS.
- G. Guirado, C. Coudret and J.-P. Launay, J. Phys. Chem. C, 2007, 111, 2770–2776 CrossRef CAS.
- A. Staykov, J. Areephong, W. R. Browne, B. L. Feringa and K. Yoshizawa, ACS Nano, 2011, 5, 1165–1178 CrossRef CAS PubMed.
- T. Nakashima, Y. Kajiki, S. Fukumoto, M. Taguchi, S. Nagao, S. Hirota and T. Kawai, J. Am. Chem. Soc., 2012, 134, 19877–19883 CrossRef CAS PubMed.
- S. Lee, Y. You, K. Ohkubo, S. Fukuzumi and W. Nam, Angew. Chem., Int. Ed., 2012, 51, 13154–13158 CrossRef CAS PubMed.
- M. Akita, Organometallics, 2011, 29, 43–51 CrossRef.
- Q. Ai, S. Pang and K.-H. Ahn, Chem.–Eur. J., 2016, 22, 656–662 CrossRef CAS PubMed.
- J. P. Calupitan, T. Nakashima, Y. Hashimoto and T. Kawai, Chem.–Eur. J., 2016, 22, 10002–10008 CrossRef CAS PubMed.
- A. Mulas, X. He, Y.-M. Hervault, L. Norel, S. Rigaut and C. Lagrost, Chem.–Eur. J., 2017, 23, 10205–10214 CrossRef CAS PubMed.
- J.-M. Han, M. Xu, B. Wang, N. Wu, X. Yang, H. Yang, B. J. Salter and L. Zang, J. Am. Chem. Soc., 2014, 136, 5090–5096 CrossRef CAS PubMed.
- R. Fazel, H. M. Krumholz, Y. Wang, J. S. Ross, J. Chen, H. H. Ting, N. D. Shah, K. Nasir, A. J. Einstein and B. K. Nallamothu, N. Engl. J. Med., 2009, 361, 849–857 CrossRef CAS PubMed.
- D. J. Brenner and E. J. Hall, N. Engl. J. Med., 2007, 357, 2277–2284 CrossRef CAS PubMed.
- K. Kamiya, K. Ozasa, S. Akiba, O. Niwa, K. Kodama, N. Takamura, E. K. Zaharieva, Y. Kimura and R. Wakeford, Lancet, 2015, 386, 469–478 CrossRef CAS.
-
P. A. Rodnyi, Physical Processes in Inorganic Scintillators, CRC Press, Boca Raton, 1997 Search PubMed.
-
G. Shani, Radiation Dosimetry: Instrumentation and Methods, CRC Press, Boca Raton, 2nd edn, 2000 Search PubMed.
- T. Yanagida, Opt. Mater., 2013, 35, 1987–1992 CrossRef CAS.
- T. Yanagida, J. Lumin., 2016, 169, 544–548 CrossRef CAS.
- J. M. Han, M. Xu, B. Wang, N. Wu, X. Yang, H. Yang, B. J. Salter and L. Zang, J. Am. Chem. Soc., 2014, 136, 5090–5096 CrossRef CAS PubMed.
- X. Dong, F. Hu, Z. Liu, G. Zhang and D. Zhang, Chem. Commun., 2015, 51, 3892–3895 RSC.
- J. M. Han, N. Wu, B. Wang, C. Wang, M. Xu, X. Yang, H. Yang and L. Zang, J. Mater. Chem. C, 2015, 3, 4345–4351 RSC.
- Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 112, 1126–1162 CrossRef CAS PubMed.
- F. P. Doty, C. A. Bauer, A. J. Skulan, P. G. Grant and M. D. Allendorf, Adv. Mater., 2009, 21, 95–101 CrossRef CAS.
- Y. Wang, X. Yin, W. Liu, J. Xie, J. Chen, M. A. Silver, D. Sheng, L. Chen, J. Diwu, N. Liu, Z. Chai, T. E. Albrecht-Schmitt and S. Wang, Angew. Chem., Int. Ed., 2018, 57, 7883–7887 CrossRef CAS PubMed.
- Y. C. Kim, K. H. Kim, D. Y. Son, D. N. Jeong, J. Y. Seo, Y. S. Choi, I. T. Han, S. Y. Lee and N. G. Park, Nature, 2017, 550, 87–91 CrossRef CAS PubMed.
- S. Shrestha, R. Fischer, G. J. Matt, P. Feldner, T. Michel, A. Osvet, I. Levchuk, B. Merle, S. Golkar, H. Chen, S. F. Tedde, O. Schmidt, R. Hock, M. Rührig, M. Göken, W. Heiss, G. Anton and C. J. Brabec, Nat. Photonics, 2017, 11, 436–440 CrossRef CAS.
- S. Irie, M. S. Kim, T. Kawai and M. Irie, Bull. Chem. Soc. Jpn., 2004, 77, 1037–1040 CrossRef CAS.
- K. Asai, M. Koshimizu, Y. Fujimoto and K. Asai, Radiat. Meas., 2017, 106, 166–169 CrossRef CAS.
- K. Takahashi, M. Koshimizu, Y. Fujimoto and K. Asai, Nucl. Instrum. Methods Phys. Res., Sect. A, 2019 DOI:10.1016/j.nima.2019.01.054.
- K. Kinashi, Y. Miyashita, K. Ishida and Y. Ueda, J. Phys. Org. Chem., 2012, 25, 427–430 CrossRef CAS.
- K. Kinashi, Y. Miyamae, R. Nakamura, W. Sakai, N. Tsutsumi, H. Yamane, G. Hatsukano, M. Ozaki, K. Jimbo and T. Okabe, Chem. Commun., 2015, 51, 11170–11173 RSC.
- K. Kinashi, T. Iwata, H. Tsuchida, W. Sakai and N. Tsutsumi, ACS Appl. Mater. Interfaces, 2018, 10, 11926–11932 CrossRef CAS PubMed.
- Y. B. Su, Y. Q. Wei, L. Z. Cai, P. X. Li, M. S. Wang and G. C. Guo, Chem. Commun., 2018, 54, 12349–12352 RSC.
- T. Nakashima, K. Atsumi, S. Kawai, T. Nakagawa, Y. Hasegawa and T. Kawai, Eur. J. Org. Chem., 2007, 2007, 3212–3218 CrossRef.
- O. Galangau, T. Nakashima, F. Maurel and T. Kawai, Chem.–Eur. J., 2015, 21, 8471–8482 CrossRef CAS PubMed.
- J. P. D. C. Calupitan, O. Galangau, O. Guillermet, R. Coratger, T. Nakashima, G. Rapenne and T. Kawai, Eur. J. Org. Chem., 2017, 2017, 2451–2461 CrossRef.
- T. Nakashima, Y. Kajiki, S. Fukumoto, M. Taguchi, S. Nagao, S. Hirota and T. Kawai, J. Am. Chem. Soc., 2012, 134, 19877–19883 CrossRef CAS PubMed.
- E. A. Fitzgerald Jr, P. Wuelfing Jr and H. Richtol, J. Phys. Chem., 1971, 75, 2737–2741 CrossRef.
- W. C. Meyer, J. Phys. Chem., 1970, 74, 2118–2121 CrossRef CAS.
- K. M. C. Davis and M. F. Farmer, J. Chem. Soc. B, 1967, 28–32 RSC.
- T. Nakashima, K. Tsuchie, R. Kanazawa, R. Li, S. Iijima, O. Galangau, H. Nakagawa, K. Mutoh, Y. Kobayashi, J. Abe and T. Kawai, J. Am. Chem. Soc., 2015, 137, 7023–7026 CrossRef CAS PubMed.
- S. Lee, Y. You, K. Ohkubo, S. Fukuzumi and W. Nam, Angew. Chem., Int. Ed., 2012, 51, 13154–13158 CrossRef CAS PubMed.
- S. Lee, Y. You, K. Ohkubo, S. Fukuzumi and W. Nam, Chem. Sci., 2014, 5, 1463 RSC.
- J. Sohma, H. Kashiwabara, T. Komatsu and T. Takahashi, Bull. Fac. Eng., Hokkaido Univ., 1964, 35, 511–519 Search PubMed.
- R. S. Alger, T. H. Anderson and L. A. Webb, J. Chem. Phys., 1959, 30, 695–706 CrossRef CAS.
- P. B. Ayscough and C. Thomson, Trans. Faraday Soc., 1962, 58, 1477–1494 RSC.
- J. Xie, Y. Wang, W. Liu, X. Yin, L. Chen, Y. Zou, J. Diwu, Z. Chai, T. E. Albrecht-Schmitt and G. Liu,
et al.
, Angew. Chem., Int. Ed., 2017, 56, 7500–7504 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional UV-vis and X-ray data along with experimental data for compounds 1 to 5. See DOI: 10.1039/c9sc05380h |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Jan Patrick
Calupitan
ab,
Jan Patrick
Calupitan