
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWavelength-gated photoreversible polymerization and topology control†
Hendrik
Frisch‡
 a,
Kai
Mundsinger‡
a,
Berwyck L. J.
Poad
b,
Stephen J.
Blanksby
b and
Christopher
Barner-Kowollik
*ac
a,
Kai
Mundsinger‡
a,
Berwyck L. J.
Poad
b,
Stephen J.
Blanksby
b and
Christopher
Barner-Kowollik
*ac
aCentre for Materials Science, School of Chemistry and Physics, Queensland University of Technology (QUT), 2 George Street, Brisbane, QLD 4000, Australia. E-mail: christopher.barnerkowollik@qut.edu.au
bCentral Analytical Research Facility, Institute for Future Environments, Queensland University of Technology (QUT), 2 George Street, Brisbane, QLD 4000, Australia
cMacromolecular Architectures, Institut für Technische Chemie und Polymerchemie, Karlsruhe Institute of Technology (KIT), Engesserstrasse 18, 76131 Karlsruhe, Germany
First published on 12th February 2020
Abstract
We exploit the wavelength dependence of [2 + 2] photocycloadditions and -reversions of styrylpyrene to exert unprecedented control over the photoreversible polymerization and topology of telechelic building blocks. Blue light (λmax = 460 nm) initiates a catalyst-free polymerization yielding high molar mass polymers (Mn = 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1), which are stable at wavelengths exceeding 430 nm, yet highly responsive to shorter wavelengths. UVB irradiation (λmax = 330 nm) induces a rapid depolymerization affording linear oligomers, whereas violet light (λmax = 410 nm) generates cyclic entities. Thus, different colors of light allow switching between a depolymerization that either proceeds through cyclic or linear topologies. The light-controlled topology formation was evidenced by correlation of mass spectrometry (MS) with size exclusion chromatography (SEC) and ion mobility data. Critically, the color-guided topology control was also possible with ambient laboratory light affording cyclic oligomers, while sunlight activated the linear depolymerization pathway. These findings suggest that light not only induces polymerization and depolymerization but that its color can control the topological outcomes.
000 g mol−1), which are stable at wavelengths exceeding 430 nm, yet highly responsive to shorter wavelengths. UVB irradiation (λmax = 330 nm) induces a rapid depolymerization affording linear oligomers, whereas violet light (λmax = 410 nm) generates cyclic entities. Thus, different colors of light allow switching between a depolymerization that either proceeds through cyclic or linear topologies. The light-controlled topology formation was evidenced by correlation of mass spectrometry (MS) with size exclusion chromatography (SEC) and ion mobility data. Critically, the color-guided topology control was also possible with ambient laboratory light affording cyclic oligomers, while sunlight activated the linear depolymerization pathway. These findings suggest that light not only induces polymerization and depolymerization but that its color can control the topological outcomes.
Introduction
Ever since Staudinger established the existence of covalent bonds between the monomeric building blocks of polymers,1 synthetic polymers have revolutionized our daily life. To govern the formation of covalent bonds between monomeric units, an impressive toolbox of chemistries has been developed, enabling control over chain lengths,2–6 sequences7–10 and topology of synthetic polymers11 (including linear, branched,12–14 crosslinked,15–17 cyclic18–23 or polycyclic polymers). The majority of those polymers and architectures are of a static nature as they emerge from non-dynamic covalent links.Inspired by the metamorphic capabilities of biochemical systems in nature – most famously the butterfly – the design of metamorphic macromolecules, which are dynamic not only in conformation but also in the bonding of their fundamental building blocks,24 hold potential for a new class of soft materials: materials whose formation, decomposition and mechanical properties can be remotely manipulated by light. Key in achieving such control is the interplay of chemical bonds with their physical, chemical and biological environment.
The toolbox of dynamic covalent chemistry25 provides significant potential to exert control over stable covalent bonds, which translates into remotely controllable macromolecular architectures.26,27 Classically, chemical triggers including pH,28–30 oxidizing or reducing conditions,31 catalysts and reagents32–34 are used to control the formation and cleavage of covalent bonds within polymers. Moving away from chemical triggers, physical forces such as thermal35–37 or mechanical38 energy allow direct, external manipulation of structure and bonding with electromagnetic fields offering the highest level of spatiotemporal control.39
Herein, we harness the potential of light-gated dynamic covalent chemistry to break new ground in catalyst free polymer chemistry on three levels: (i) introducing the first visible light induced [2 + 2] photopolymerization that yields high degrees of polymerization (DP) in the liquid state under ambient conditions; (ii) enabling an unprecedented depolymerization with visible light and under ambient conditions; and critically (iii) exerting control over the topology of the depolymerization outcome, allowing switching between cyclic (violet light) or linear topologies (UVB light).
Results and discussion
Cyclization and linearization
To give access to wavelength controlled dynamic covalent bonds, the photochemistry of the stilbene derivative styrylpyrene was exploited (Scheme 1A).40–43 By introducing carboxystyrylpyrene units at the termini of polyethylene glycol (PEG), a linear, telechelic macromolecular building block (L1) was obtained. Telechelic L1 can undergo [2 + 2] photocycloaddition of the photoreactive termini resulting in either intramolecular macrocyclization or intermolecular ligation inducing a step growth polymerization (Scheme 1C). To elucidate the wavelength at which either cycloaddition or cycloreversion is predominantly triggered, we recently investigated the wavelength dependence of conversion for both reactions after irradiation with the same number of photons at different wavelengths.42,44 The resulting action plot serves as a blueprint for the wavelength dependent reactivity of the two competing reactions (Scheme 1B). To exclusively initiate intramolecular cyclisation, a solution of L1 (0.05 mg mL−1) was irradiated with a blue LED at wavelengths >430 nm, where only cycloaddition is triggered (Scheme 1C, blue). The reduction in hydrodynamic volume upon cyclization of L1 was monitored by a characteristic shift in elution volume of the size exclusion chromatography (SEC) trace (Fig. 1A). After 1 min, a distinct shoulder towards lower elution volumes was detected. After 120 min, the initial trace of L1 had vanished and only the cyclic building block (C1) was observed. In addition to the macromolecular observation, the photocycloaddition was monitored on the level of the photoreactive groups by UV/vis spectroscopy (Fig. S1.1.1†). By tracking the intensity decrease of the styrylpyrene absorption maximum (λmax = 383 nm) upon aromatic decoupling and the concomitant intensity increase of the absorption maxima characteristic for the resulting unconjugated pyrene substituent (λmax = 333 and 352 nm),40–43 the reaction was found to be complete after 120 min.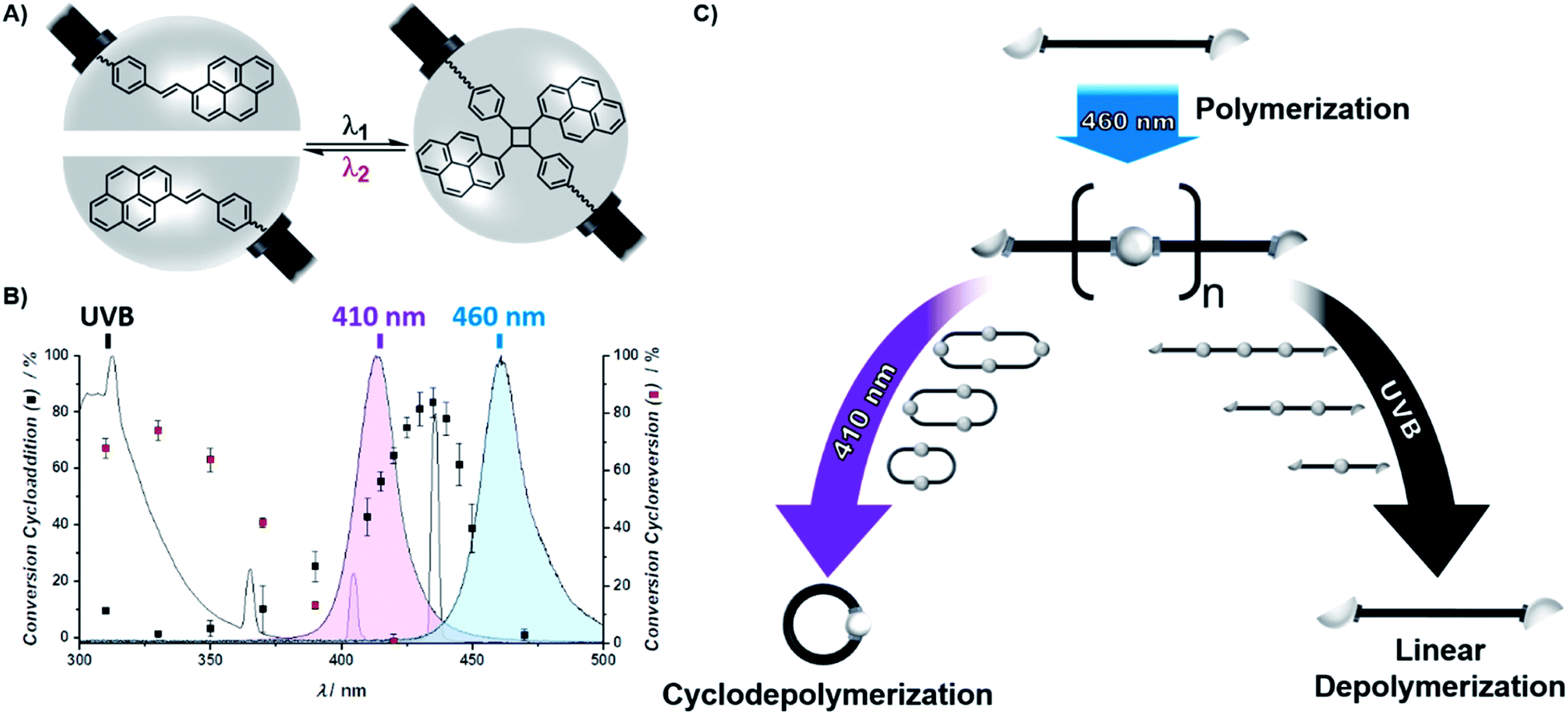 | ||
| Scheme 1 (A) Chemical structure of the reversible [2 + 2] photocycloaddition of styrylpyrene used as a wavelength gated binding site of telechelic polymers. (B) Action plot for the conversion of the photocycloaddition (black cubes) and the photocycloreversion of the cycloadduct (red cubes) after irradiation with the same number of photons at different wavelengths, as previously reported for the styrylpyrene derivative hydroxyl-styrylpyrene.42 The normalized emission spectra of the herein utilized UVB lamp (grey) and LEDs (violet, λmax = 410 nm and blue, λmax = 460 nm) indicate which reaction is triggered in the respective photonic field. (C) Schematic representation of the polymerization and depolymerization of L1 comprising two terminal styrylpyrene units. Irradiation of L1 with blue light induces its photopolymerization yielding P1, which can be either cyclodepolymerized under violet light or linearly depolymerized under UVB light. | ||
 | ||
| Fig. 1 (A) SEC trace of the cyclization of L1 (black) into the macrocycle C1 (blue) using blue light irradiation. (B) Cycloreversion of C1 (grey) to L1 (black) under UVB irradiation. | ||
To initiate the transition of cyclic C1 back into its linear topology, C1 was irradiated with UVB light, a wavelength regime dominated by the cycloreversion (Fig. 1A, S1.1.1 and S1.2.1†). The SEC trace shows that the majority of L1 is rapidly restored after 20 s (Fig. 1B, S1.2.1 and S1.2.2†). After 60 s, the SEC trace of the cyclic topology has vanished. These conditions are the mildest reported to date for a reversible topology change that is controlled exclusively by different wavelengths.22,23 Such topology transitions from linear to cyclic can affect the physical properties of polymers drastically including melting and crystallization point,45 melt rheology,46 yet also chemical properties such as their degradation behavior.47 The reported remote control over the polymer topology provides a facile access to explore such property changes in both materials science and biomedical applications, where the potential of cyclic polymers remains largely untapped.48
Photopolymerization
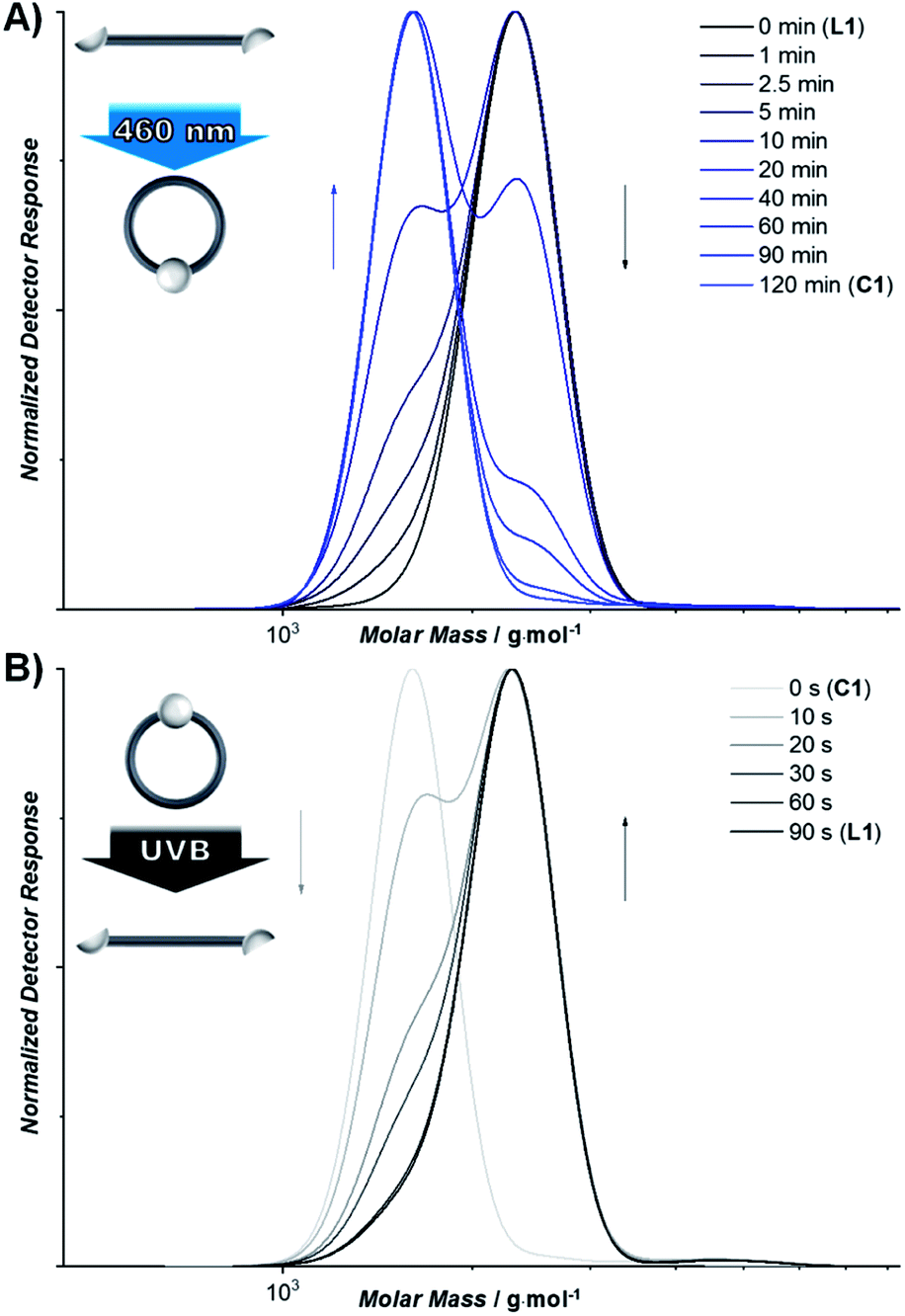
To induce photopolymerization of L1, the intermolecular reaction has to be favored over intramolecular cyclization. Consequently, the concentration of L1 was increased to 266 mg mL−1 and the reaction solution deoxygenated to suppress triplet state quenching and to increase the lifetime of the excited species (refer to ESI chapter 3.3†). Following irradiation with a blue LED, successful polymerization affording P1 was observed by SEC, indicating a Mw of 100000 g mol−1 over the entire sample (Fig. 2A and S1.3.2†). On a molecular level, the polymerization induced by [2 + 2] photocycloaddition of the styrylpyrene units was observed through a decrease of the absorbance of styrylpyrene in the UV-vis spectra (Fig. S1.3.1†) and the characteristic resonances of the cycloadducts of different stereochemistries in the 1H-NMR spectra between δ = 4–6 ppm (Fig. S1.3.3†).42 In addition to the formed polymer (Mn = 60000 g mol−1, DP = 38, Đ = 2.2), smaller cyclic oligomers were observed. The high molar mass fraction exhibits a high degree of polymerization and low dispersity compared to previous examples of step-growth polymerizations of telechelic building blocks exploiting the widely applied copper(I)-catalyzed azide–alkyne cycloaddition (Mn = 21500 g mol−1, DP = 8, Đ = 4.85).49 Using SEC hyphenated with electrospray ionization mass spectrometry (ESI-MS), the smaller oligomers were identified as 1 to 5 membered cycloadducts (Fig. 2B–F). While photocycloadditions are a powerful tool to induce chemical bonds in the absence of catalysts, they require an overlap of the involved bonds according to Schmidt's topochemical postulate.50–52 As a consequence, previously reported polymerization reactions induced by photocycloadditions only occurred efficiently in the crystalline state upon precise alignment via crystal engineering.53,54 We propose that the polymerization reaction described here proceeds efficiently for two reasons: (i) the combination of large aromatic systems with a polar PEG linker lead to a prearrangement of the styrylpyrene units in THF, favored by dispersion interactions between the large aromatic systems. (ii) The relatively low photon energies of the applied wavelengths (>430 nm) prevent non-specific photodamage of the reactive groups and polymeric backbone. In addition to organic solvents, the photopolymerization can be carried out at lower concentrations in aqueous systems (50 mg mL−1 in water containing 20% DMSO). Irradiation of the resulting suspension overnight affords a molar mass distribution (Mw = 95000 g mol−1 over the entire sample) very similar to P1 obtained in THF. The polymer formed in aqueous suspension (P1H2O, Mn = 54000 g mol−1, DP = 34, Đ = 1.9, refer to Fig. S1.3.4†) had a slightly lower molar mass than P1.
 | ||
| Fig. 2 (A) SEC trace of the photopolymerization of L1 (grey) to P1 (black, Mn = 60 kDa, Đ = 2, DP = 38), cyclic oligomers (DP = 1–5) denoted by black arrows. The different cyclic oligomers were identified by SEC-ESI-MS and the recorded mass spectra are plotted along with the simulated isotopic patterns of the respective sum formula. (B) Mass spectrum of the first eluting species assigned to a monomeric species (m/z of the most abundant ion = 1626.76763, simulated for C92H114O24Na1+ = 1626.76233, Δm/z = 3.26 ppm). (C) Mass spectrum of the second eluting species assigned to a dimeric species (m/z of the most abundant ion = 1626.76917, simulated for C184H228O48Na22+ = 1626.76233, Δm/z = 4.20 ppm). (D) Mass spectrum of the third eluting species assigned to a trimeric species (m/z of the most abundant ion = 1626.77647, simulated for C276H342O72Na33+ = 1626.76233, Δm/z = 8.69 ppm). (E) Mass spectrum of the fourth eluting species assigned to a tetrameric species (m/z of the most abundant ion = 2161.37567, simulated for C368H456O96Na33+ = 2161.35383, Δm/z = 10.10 ppm). (F) Mass spectrum of the fifth eluting species assigned to a pentameric species (m/z of the most abundant ion = 1374.16201, simulated for C464H578O122Na66+ = 1374.14239, Δm/z = 11.38 ppm). | ||
Wavelength-gated depolymerization
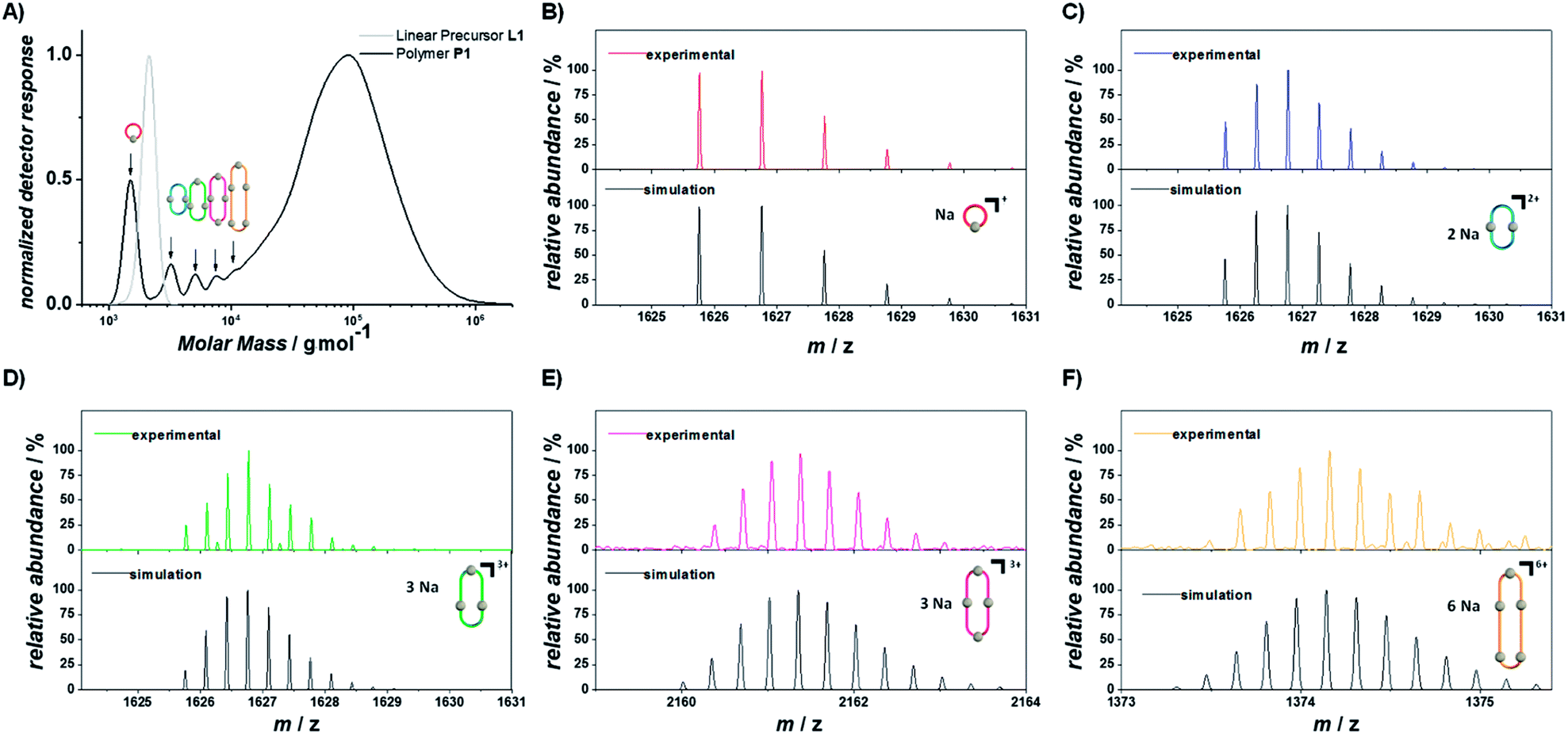
To depolymerize P1, the cyclobutane moieties within the polymer main chain have to be opened through a photocycloreversion. As photocycloaddition and photocycloreversion are triggered with different efficiencies at different wavelengths (Scheme 1B), it is possible to control which reaction is predominant through the selected color of light. If P1 is irradiated with blue light >430 nm, cycloreversion is not triggered and P1 showed no depolymerization after irradiation for 90 min under dilute conditions (0.05 mg mL−1, Fig. S1.4.1†). The concentration of 0.05 mg mL−1, also used for the cyclization and decyclization of L1, was chosen as it allowed monitoring the reaction in situ using UV/vis spectroscopy. When the irradiation wavelength was switched under otherwise identical conditions to UVB light in order to favor the cycloreversion, a rapid depolymerization was observed (Fig. 3 left column, S1.5.1–S1.5.3†). After 10 s only linear oligomers were observed in the SEC trace and after 10 min the polymer was transformed into its monomeric building blocks with only traces of the dimer detected. The depolymerization of P1 was also evidenced by the restored absorption band of styrylpyrene (Fig. S1.5.1†). | ||
| Fig. 3 Depolymerization of P1 under UVB (left columns) and cyclodepolymerization under violet light (right columns). Selected SEC traces of the depolymerization of P1 after different irradiation times, with highlighted characteristic molar mass ranges of the cyclic and linear oligomers (DP = 1–6). Refer to chapter 1.5 and 1.6 of the ESI† for the full depolymerization kinetics. XIC traces of the oligomers (DP = 1–5) extracted from the SEC-ESI-MS (bottom). Refer to the ESI chapter 1.10† for the full mass spectra of the oligomers and the selected ions. | ||
In addition to those two extreme scenarios, completely dominated by either cycloaddition or -reversion, their wavelength dependency also allows the triggering of both reactions dynamically simply by adjusting the color of light. Irradiation with a violet LED centered around 410 nm, which also emits low intensity light below 400 nm, allows both reactions to be initiated simultaneously, albeit with drastically different efficiencies. Irradiation of P1 with a violet LED therefore also induced an efficient depolymerization yielding only cyclic oligomers after 30 min of irradiation (Fig. 3 right column, S1.6.2 and S1.6.3†). Since the violet LED emits at wavelengths which predominantly initiate the cycloaddition, the dynamic equilibrium lies strongly on the side of the cycloadduct, as evidenced by the UV/vis spectra (Fig. S1.6.1†) featuring the characteristic absorption maxima of the photoadduct throughout the reactions (Fig. S1.6.1†). The depolymerization of P1 is thus inherently different under violet light than under UVB light. All bonds opened throughout the depolymerization under violet light are either dynamically reformed, not affecting the degree of polymerization, or react with the other polymer chain end to form a smaller cyclic polymer. It can be expected that the resulting cyclodepolymerization rate becomes slower over the course of the reaction, as two photodimers of the same macrocycle must be cleaved simultaneously to enable the separation into smaller oligomers, which becomes less likely as the number of styrylpyrene adducts reduces with each successive cycle. Owing to the different depolymerization mechanisms, cyclodepolymerization of P1 under violet light irradiation is inherently slower than linear depolymerization under UVB light, even though the employed irradiation setups provide a significantly higher irradiance under the violet LED irradiation. Under these irradiation conditions, cyclic oligomers of DP = 1, 2 and 3 are obtained after 24 h. Cyclodepolymerizations have already been described by Carothers as a synthetic route towards macrocyclic oligomers on the basis of polyesters55 and usually require high temperatures20 and catalysts.33,56 To date, however, photochemically induced cyclodepolymerizations have not been reported.20
Owing to the different depolymerization conditions of P1 under violet or UVB light, the observed oligomers have different topologies and elution times in the SEC. To correlate the differences in elution time with the oligomer topology, samples of P1 were investigated after irradiation for either 30 min under violet light or 20 s under UVB light irradiation. Ion-mobility measurements monitoring the arrival-time distributions (ATDs) of [C86H102O21Na]+ (m/z = 1493) and oligomers of the same m/z ratio, show distinctly different ATDs for the two differently irradiated samples, resulting from the respective difference in collision cross section (Fig. S1.9.1†). Irradiation with UVB light yielded a major distribution centered at 13.7 ms and a less abundant distribution at 8.5 ms. Comparison with the ion mobility of the non-irradiated linear precursor of L1, which showed only one ATD at 13.7 ms, indicates that this feature is associated with the linear topology of the singly-charged oligomer with DP = 1 (L1), while the distribution at 8.5 ms results from a doubly-charged oligomer of DP = 2 (L2). In contrast, the ion mobility measurements of the sample after irradiation with violet light yielded ATDs shifted to later arrival times of 14.1 ms and 8.7 ms for the singly charged oligomer of DP = 1 (C1) and doubly charged oligomer of DP = 2 (C2), respectively, along with additional features at shorter arrival times. Although cyclic polymers often exhibit earlier ATDs compared to their linear analogues,57 the ATDs of the smaller oligomers appear to be dominated by the cycloadduct of the terminal styrylpyrene moieties. While the large aromatic groups of styrylpyrene are flat and able to closely align, the aromatic rings of the photoproduct are constrained by the cyclobutane ring, giving rise to a sterically bulkier structure. As isomerization can also drastically effect drift times,58 the shoulder at later arrival time in the ATD of C1 is likely to arise from the different stereoisomers of the cycloadducts.42
To investigate the topology of larger oligomers (DP = 2–6), extracted ion chromatograms (XICs)59,60 were obtained from the SEC-ESI-MS analysis of the same samples as used in the previously discussed ion mobility measurements. As the cycloaddition reaction does not affect the molar mass of the oligomers, the isolation of discrete mass-to-charge ratios from the SEC-ESI-MS data allows comparison between the elution times of oligomers and the exact same molar masses and DP. Consequently, any observed differences in the elution time exclusively arise from different hydrodynamic volumes in THF, as a result of their topology. It was possible to extract ions of oligomers from the depolymerization, via either UVB (20 s) or blue light (30 min) irradiation of P1 from DP = 1 up to DP = 5 (Fig. 3, bottom row). Following identical m/z ranges, XICs of the oligomers resulting from the depolymerization under UVB light display longer elution times for all DPs compared to the violet light induced cyclodepolymerization. The elution time distribution of the isolated ions is well aligned with the normalized detector response of the SEC traces (Fig. S1.10.1†), demonstrating that both depolymerization pathways are highly selective in topology.
Whereas the dilute reaction conditions (0.05 mg mL−1) used for the cyclic and linear depolymerization allowed monitoring the reaction directly with SEC and UV/vis spectroscopy, the dilute concentration regime is no prerequisite to control the topology of the depolymerization. Both depolymerization reactions were also carried out at 20 mg mL−1, with either UVB or violet light to selectively yield cyclic (Fig. S1.6.4†) or linear depolymerization products (Fig. S1.5.4†). The only adjustment for higher concentrations under identical irradiation is the necessity of stirring and prolonged reaction times. The higher concentrated solutions feature less light penetration and require a higher number of photons to achieve quantitative conversion.
Utilizing synthetically convenient higher concentrations and the dynamic nature of the investigated system, it is readily possible to conduct multiple polymerization and depolymerization cycles. After 800 s UVB irradiation of P1 in THF (20 mg mL−1) only linear oligomers were observed (Fig. S1.5.4†). These oligomers were subsequently polymerized (266 mg mL−1) yielding P1′, with a molar mass that reached up to 106 g mol−1. Irradiating P1′ with UVB light yields linear oligomers again. Following a third photopolymerization cycle, the obtained P1′′ was irradiated for 100 min with UVB light to obtain L1 along with linear oligomers.
Depolymerization under ambient conditions
While the color of light gates selectively between depolymerization and cyclodepolymerization of P1, the depolymerization pathway of P1 was finally investigated under ambient conditions. When solutions of P1 at otherwise identical conditions (0.05 mg mL−1, ambient temperature) were placed in an elevated position in the laboratory, away from direct sunlight but exposed to laboratory fluorescent lights, a slow depolymerization was observed (Fig. S1.8.1 and S1.8.2†). After one week, the majority of the polymer is consumed, and a mixture of cyclic and linear oligomers remained. The longer reaction time results from the overall lower light intensity, which is furthermore predominantly outside of the wavelength regime that gates the photoreaction of styrylpyrene (<450 nm).42,43 The reduced selectivity results from insufficient energy provided by the ambient light at wavelengths required for the photocycloaddition to induce the cyclization of the depolymerized fragments. If subsequently irradiated with blue light, however, all linear oligomers can be transformed into their cyclic analogues within 2 h (Fig. S1.8.3 and S1.8.4†). Exposed solely to ambient laboratory conditions for 25 d, the cyclic C1 of a DP = 1 is the most populated species, while in the dark the polymeric P1 remained stable (Fig. S1.8.2†). These results highlight that a suitable photonic field exerts not only control over the topology of the final depolymerization product, but throughout the entire depolymerization process. Such a highly selective photonic field can be readily accessed, if samples of P1 (0.05 mg mL−1, ambient temperature) are placed into direct sunlight (Fig. 4). After 5 s, a rapid depolymerization was observed, restoring the absorption band of styrylpyrene (Fig. S1.7.1†) and leaving only linear oligomers (Fig. S1.7.2†). After 4 min, the smallest linear oligomer of L1 (DP = 1) was the most abundant species according to the SEC trace. The high selectivity observed under sunlight irradiation is likely to result from the high intensity of UV light between 300–400 nm and the kinetically favored monomolecular cycloreversion. Furthermore, the presence of oxygen quenches the lifetime of excited triplet states, making the bimolecular photocycloaddition even less favorable. The reported polymer P1 can hence be efficiently depolymerized under the ambient conditions inside or outside of the laboratory marking styrylpyrene as a powerful tool for dynamic covalent chemistry.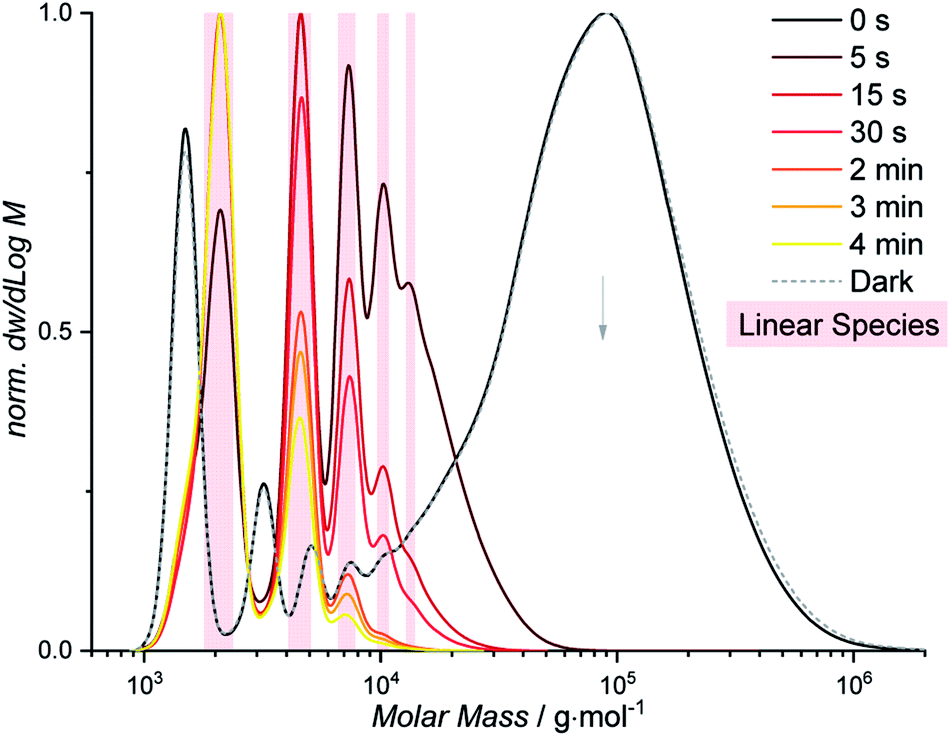 | ||
| Fig. 4 Depolymerization of P1 (black) into linear oligomers (highlighted in red) within 4 min of sunlight irradiation. | ||
Conclusions
The telechelic functionalization of macromolecular building blocks with styrylpyrene provides a unique route to control the formation, disintegration as well as the resulting topologies of polymers. In dilute solution, the wavelength dependence of the [2 + 2] photocycloaddition and photocycloreversion of styrylpyrene was used to cyclize the macromolecules under blue light (λmax = 460 nm) and reopen the cycles under UVB irradiation into linear topologies.At higher concentrations, the catalyst free polymerization of the building blocks was induced by blue light (λmax = 460 nm). The resulting high molecular weight polymer (P1) is stable at wavelength > 430 nm, yet highly and specifically responsive towards different colors of light in its depolymerization behavior. By choice of irradiation wavelength, it is possible to gate the depolymerization of P1 into linear (UVB light, λmax = 330 nm) or cyclic topologies (violet light, λmax = 410 nm), marking it the first reported photocyclodepolymerization reaction.
In addition to the mild stimuli of LEDs, the depolymerization of P1 was induced by the ambient light of the laboratory and within 5 s of sun light irradiation, while retaining control over the depolymerization topology. The highly specific responsiveness of the photoreactive building blocks to the color of light, and thus the resulting polymers, holds potential for the design of materials that are adaptable not only in their bonding state but also the resulting topology.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
H. F., B. L. J. P., S. J. B. and C. B.-K. acknowledge continued support from the Queensland University of Technology (QUT) through the Centre for Materials Science. C. B.-K. acknowledges support by the Australian Research Council (ARC) in the form of a Laureate Fellowship enabling his photo-chemical research program. Some of the data reported in this paper were obtained at the Central Analytical Research Facility operated by the Institute for Future Environments (QUT). Access to CARF is supported by generous funding from the Science and Engineering Faculty (QUT).References
- H. Staudinger, Über Polymerisation, Ber. Dtsch. Chem. Ges. A, 1920, 53(6), 1073–1085, DOI:10.1002/cber.19200530627.
- M. Szwarc, ‘Living’ Polymers, Nature, 1956, 178(4543), 1168–1169, DOI:10.1038/1781168a0.
- G. Moad, E. Rizzardo and S. H. Thang, Radical Addition–fragmentation Chemistry in Polymer Synthesis, Polymer, 2008, 49(5), 1079–1131, DOI:10.1016/j.polymer.2007.11.020.
- M. Kamigaito, T. Ando and M. Sawamoto, Metal-Catalyzed Living Radical Polymerization, Chem. Rev., 2001, 101(12), 3689–3746, DOI:10.1021/cr9901182.
- C. J. Hawker, A. W. Bosman and E. Harth, New Polymer Synthesis by Nitroxide Mediated Living Radical Polymerizations, Chem. Rev., 2001, 101(12), 3661–3688, DOI:10.1021/cr990119u.
- K. Matyjaszewski and J. Xia, Atom Transfer Radical Polymerization, Chem. Rev., 2001, 101(9), 2921–2990, DOI:10.1021/cr940534g.
- J.-F. Lutz, M. Ouchi, D. R. Liu and M. Sawamoto, Sequence-Controlled Polymers, Science, 2013, 341(6146), 628, DOI:10.1126/science.1238149.
- N. G. Engelis, A. Anastasaki, R. Whitfield, G. R. Jones, E. Liarou, V. Nikolaou, G. Nurumbetov and D. M. Haddleton, Sequence-Controlled Methacrylic Multiblock Copolymers: Expanding the Scope of Sulfur-Free RAFT, Macromolecules, 2018, 51(2), 336–342, DOI:10.1021/acs.macromol.7b01987.
- M. A. R. Meier and C. Barner-Kowollik, A New Class of Materials: Sequence-Defined Macromolecules and Their Emerging Applications, Adv. Mater., 2019, 31(26), 1806027, DOI:10.1002/adma.201806027.
- J. O. Holloway, K. S. Wetzel, S. Martens, F. E. Du Prez and M. A. R. Meier, Direct Comparison of Solution and Solid Phase Synthesis of Sequence-Defined Macromolecules, Polym. Chem., 2019, 10(28), 3859–3867, 10.1039/c9py00558g.
- Y. Tezuka and H. Oike, Topological Polymer Chemistry: Systematic Classification of Nonlinear Polymer Topologies, J. Am. Chem. Soc., 2001, 123(47), 11570–11576, DOI:10.1021/ja0114409.
- D. Konkolewicz, M. J. Monteiro and S. Perrier, Dendritic and Hyperbranched Polymers from Macromolecular Units: Elegant Approaches to the Synthesis of Functional Polymers, Macromolecules, 2011, 44(18), 7067–7087, DOI:10.1021/ma200656h.
- G. Polymeropoulos, G. Zapsas, K. Ntetsikas, P. Bilalis, Y. Gnanou and N. Hadjichristidis, 50th Anniversary Perspective: Polymers with Complex Architectures, Macromolecules, 2017, 50(4), 1253–1290, DOI:10.1021/acs.macromol.6b02569.
- T. Pelras, C. S. Mahon, Nonappa, O. Ikkala, A. H. Gröschel and M. Müllner, Polymer Nanowires with Highly Precise Internal Morphology and Topography, J. Am. Chem. Soc., 2018, 140(40), 12736–12740, DOI:10.1021/jacs.8b08870.
- J. Pomposo, Single-Chain Polymer Nanoparticles: Synthesis, Characterization, Simulations, and Applications, Wiley-VCH, Weinheim, Germany, 2017, DOI:10.1002/9783527806386.
- A. M. Hanlon, C. K. Lyon and E. B. Berda, What Is Next in Single-Chain Nanoparticles?, Macromolecules, 2016, 49(1), 2–14, DOI:10.1021/acs.macromol.5b01456.
- H. Frisch, F. R. Bloesser and C. Barner-Kowollik, Controlling Chain Coupling and Single-Chain Ligation by Two Colours of Visible Light, Angew. Chem., Int. Ed., 2019, 58(11), 3604–3609, DOI:10.1002/anie.201811541.
- Z. Jia and M. J. Monteiro, Cyclic Polymers: Methods and Strategies, J. Polym. Sci., Part A: Polym. Chem., 2012, 50(11), 2085–2097, DOI:10.1002/pola.25999.
- D. Pasini and D. Takeuchi, Cyclopolymerizations: Synthetic Tools for the Precision Synthesis of Macromolecular Architectures, Chem. Rev., 2018, 118(18), 8983–9057, DOI:10.1021/acs.chemrev.8b00286.
- P. Hodge, Cyclodepolymerization as a Method for the Synthesis of Macrocyclic Oligomers, React. Funct. Polym., 2014, 80, 21–32, DOI:10.1016/j.reactfunctpolym.2013.12.008.
- C. D. Roland, H. Li, K. A. Abboud, K. B. Wagener and A. S. Veige, Cyclic Polymers from Alkynes, Nat. Chem., 2016, 8(8), 791–796, DOI:10.1038/nchem.2516.
- S. Honda, M. Oka, H. Takagi and T. Toyota, Topology-Reset Execution: Repeatable Postcyclization Recyclization of Cyclic Polymers, Angew. Chem., Int. Ed., 2019, 58(1), 144–148, DOI:10.1002/anie.201809621.
- T. Yamamoto, S. Yagyu and Y. Tezuka, Light- and Heat-Triggered Reversible Linear–Cyclic Topological Conversion of Telechelic Polymers with Anthryl End Groups, J. Am. Chem. Soc., 2016, 138(11), 3904–3911, DOI:10.1021/jacs.6b00800.
- H. Sun, C. P. Kabb, Y. Dai, M. R. Hill, I. Ghiviriga, A. P. Bapat and B. S. Sumerlin, Macromolecular Metamorphosis via Stimulus-Induced Transformations of Polymer Architecture, Nat. Chem., 2017, 9(8), 817–823, DOI:10.1038/nchem.2730.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Dynamic Covalent Chemistry, Angew. Chem., Int. Ed., 2002, 41(6), 898–952, DOI:10.1002/1521-3773(20020315)41:6<898::aid-anie898>3.0.co;2-e.
- Y. Jin, Q. Wang, P. Taynton and W. Zhang, Dynamic Covalent Chemistry Approaches Toward Macrocycles, Molecular Cages, and Polymers, Acc. Chem. Res., 2014, 47(5), 1575–1586, DOI:10.1021/ar500037v.
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Using the Dynamic Bond to Access Macroscopically Responsive Structurally Dynamic Polymers, Nat. Mater., 2011, 10(1), 14–27, DOI:10.1038/nmat2891.
- W. L. A. Brooks and B. S. Sumerlin, Synthesis and Applications of Boronic Acid-Containing Polymers: From Materials to Medicine, Chem. Rev., 2016, 116(3), 1375–1397, DOI:10.1021/acs.chemrev.5b00300.
- S. P. Black, J. K. M. Sanders and A. R. Stefankiewicz, Disulfide Exchange: Exposing Supramolecular Reactivity through Dynamic Covalent Chemistry, Chem. Soc. Rev., 2014, 43(6), 1861–1872, 10.1039/c3cs60326a.
- M. E. Belowich and J. F. Stoddart, Dynamic Imine Chemistry, Chem. Soc. Rev., 2012, 41(6), 2003–2024, 10.1039/c2cs15305j.
- A. P. Vogt and B. S. Sumerlin, Temperature and Redox Responsive Hydrogels from ABA Triblock Copolymers Prepared by RAFT Polymerization, Soft Matter, 2009, 5(12), 2347–2351, 10.1039/b817586a.
- N. Giuseppone, J.-L. Schmitt, E. Schwartz and J.-M. Lehn, Scandium(III) Catalysis of Transimination Reactions. Independent and Constitutionally Coupled Reversible Processes, J. Am. Chem. Soc., 2005, 127(15), 5528–5539, DOI:10.1021/ja042469q.
- M. J. Marsella, H. D. Maynard and R. H. Grubbs, Template-Directed Ring-Closing Metathesis: Synthesis and Polymerization of Unsaturated Crown Ether Analogs, Angew. Chem., Int. Ed. Engl., 1997, 36(10), 1101–1103, DOI:10.1002/anie.199711011.
- S. W. Sisco, B. M. Larson and J. S. Moore, Relaxing Conformational Constraints in Dynamic Macrocycle Synthesis, Macromolecules, 2014, 47(12), 3829–3836, DOI:10.1021/ma500673x.
- P. Reutenauer, E. Buhler, P. J. Boul, S. J. Candau and J.-M. Lehn, Room Temperature Dynamic Polymers Based on Diels–Alder Chemistry, Chem.–Eur. J., 2009, 15(8), 1893–1900, DOI:10.1002/chem.200802145.
- X. A. Chen, Thermally Re-Mendable Cross-Linked Polymeric Material, Science, 2002, 295(5560), 1698–1702, DOI:10.1126/science.1065879.
- K. K. Oehlenschlaeger, J. O. Mueller, J. Brandt, S. Hilf, A. Lederer, M. Wilhelm, R. Graf, M. L. Coote, F. G. Schmidt and C. Barner-Kowollik, Adaptable Hetero Diels–Alder Networks for Fast Self-Healing under Mild Conditions, Adv. Mater., 2014, 26(21), 3561–3566, DOI:10.1002/adma.201306258.
- A. M. Belenguer, G. I. Lampronti, D. J. Wales and J. K. M. Sanders, Direct Observation of Intermediates in a Thermodynamically Controlled Solid-State Dynamic Covalent Reaction, J. Am. Chem. Soc., 2014, 136(46), 16156–16166, DOI:10.1021/ja500707z.
- H. Frisch, D. E. Marschner, A. S. Goldmann and C. Barner-Kowollik, Wavelength-Gated Dynamic Covalent Chemistry, Angew. Chem., Int. Ed., 2018, 57(8), 2036–2045, DOI:10.1002/anie.201709991.
- N. P. Kovalenko, A. Abdukadirov, V. I. Gerko and M. V. Alfimov, Some Peculiarities of Diarylethylenes with 3-Pyrenyl Fragments, J. Appl. Spectrosc., 1980, 32(6), 607–612, DOI:10.1007/bf00604286.
- V. X. Truong, F. Li, F. Ercole and J. S. Forsythe, Wavelength-Selective Coupling and Decoupling of Polymer Chains via Reversible [2 + 2] Photocycloaddition of Styrylpyrene for Construction of Cytocompatible Photodynamic Hydrogels, ACS Macro Lett., 2018, 464–469, DOI:10.1021/acsmacrolett.8b00099.
- D. E. Marschner, H. Frisch, J. T. Offenloch, B. T. Tuten, C. R. Becer, A. Walther, A. S. Goldmann, P. Tzvetkova and C. Barner-Kowollik, Visible Light [2 + 2] Cycloadditions for Reversible Polymer Ligation, Macromolecules, 2018, 51(10), 3802–3807, DOI:10.1021/acs.macromol.8b00613.
- H. Frisch, J. P. Menzel, F. R. Bloesser, D. E. Marschner, K. Mundsinger and C. Barner-Kowollik, Photochemistry in Confined Environments for Single-Chain Nanoparticle Design, J. Am. Chem. Soc., 2018, 140(30), 9551–9557, DOI:10.1021/jacs.8b04531.
- D. E. Fast, A. Lauer, J. P. Menzel, A.-M. Kelterer, G. Gescheidt and C. Barner-Kowollik, Wavelength-Dependent Photochemistry of Oxime Ester Photoinitiators, Macromolecules, 2017, 50(5), 1815–1823, DOI:10.1021/acs.macromol.7b00089.
- C. W. Bielawski, D. Benitez and R. H. Grubbs, An Endless Route to Cyclic Polymers, Science, 2002, 297(5589), 2041–2044, DOI:10.1126/science.1075401.
- Y. Doi, K. Matsubara, Y. Ohta, T. Nakano, D. Kawaguchi, Y. Takahashi, A. Takano and Y. Matsushita, Melt Rheology of Ring Polystyrenes with Ultrahigh Purity, Macromolecules, 2015, 48(9), 3140–3147, DOI:10.1021/acs.macromol.5b00076.
- J. N. Hoskins and S. M. Grayson, Synthesis and Degradation Behavior of Cyclic Poly(ε-Caprolactone), Macromolecules, 2009, 42(17), 6406–6413, DOI:10.1021/ma9011076.
- X.-Y. Tu, M.-Z. Liu and H. Wei, Recent Progress on Cyclic Polymers: Synthesis, Bioproperties, and Biomedical Applications, J. Polym. Sci., Part A: Polym. Chem., 2016, 54(11), 1447–1458, DOI:10.1002/pola.28051.
- N. V. Tsarevsky, B. S. Sumerlin and K. Matyjaszewski, Step-Growth “Click” Coupling of Telechelic Polymers Prepared by Atom Transfer Radical Polymerization, Macromolecules, 2005, 38(9), 3558–3561, DOI:10.1021/ma050370d.
- M. D. Cohen and G. M. J. Schmidt, 383. Topochemistry. Part I. A Survey, J. Chem. Soc., 1964, 1996–2000, 10.1039/jr9640001996.
- M. D. Cohen, G. M. J. Schmidt and F. I. Sonntag, 384. Topochemistry. Part II. The Photochemistry of Trans-Cinnamic Acids, J. Chem. Soc., 1964, 2000–2013, 10.1039/jr9640002000.
- G. M. J. Schmidt, 385. Topochemistry. Part III. The Crystal Chemistry of Some Trans-Cinnamic Acids, J. Chem. Soc., 1964, 2014–2021, 10.1039/jr9640002014.
- P. Johnston, C. Braybrook and K. Saito, Topochemical Photo-Reversible Polymerization of a Bioinspired Monomer and Its Recovery and Repolymerization after Photo-Depolymerization, Chem. Sci., 2012, 3(7), 2301–2306, 10.1039/c2sc20380d.
- L. Dou, Y. Zheng, X. Shen, G. Wu, K. Fields, W.-C. Hsu, H. Zhou, Y. Yang and F. Wudl, Single-Crystal Linear Polymers Through Visible Light–Triggered Topochemical Quantitative Polymerization, Science, 2014, 343(6168), 272–277, DOI:10.1126/science.1245875.
- J. W. Hill and W. H. Carothers, Studies of polymerization and ring formation. Xiv. A linear superpolyanhydride and a cyclic dimeric anhydride from sebacic acid, J. Am. Chem. Soc., 1932, 54(4), 1569–1579, DOI:10.1021/ja01343a050.
- S. W. Sisco and J. S. Moore, Directional Cyclooligomers via Alkyne Metathesis, J. Am. Chem. Soc., 2012, 134(22), 9114–9117, DOI:10.1021/ja303572k.
- J. N. Hoskins, S. Trimpin and S. M. Grayson, Architectural Differentiation of Linear and Cyclic Polymeric Isomers by Ion Mobility Spectrometry-Mass Spectrometry, Macromolecules, 2011, 44(17), 6915–6918, DOI:10.1021/ma2012046.
- A. Galanti, J. Santoro, R. Mannancherry, Q. Duez, V. Diez-Cabanes, M. Valášek, J. De Winter, J. Cornil, P. Gerbaux and M. Mayor, et al., A New Class of Rigid Multi(Azobenzene) Switches Featuring Electronic Decoupling: Unravelling the Isomerization in Individual Photochromes, J. Am. Chem. Soc., 2019, 141(23), 9273–9283, DOI:10.1021/jacs.9b02544.
- T. Gruendling, M. Guilhaus and C. Barner-Kowollik, Fast and Accurate Determination of Absolute Individual Molecular Weight Distributions from Mixtures of Polymers via Size Exclusion Chromatography–Electrospray Ionization Mass Spectrometry, Macromolecules, 2009, 42(17), 6366–6374, DOI:10.1021/ma900755z.
- T. Nitsche, J. Steinkoenig, K. De Bruycker, F. R. Bloesser, S. J. Blanksby, J. P. Blinco and C. Barner-Kowollik, Mapping the Compaction of Discrete Polymer Chains by Size Exclusion Chromatography Coupled to High-Resolution Mass Spectrometry, Macromolecules, 2019, 52(6), 2597–2606, DOI:10.1021/acs.macromol.9b00203.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional experiments, experimental details, photochemical and synthetic procedures (PDF). See DOI: 10.1039/c9sc05381f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |