
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAutomated linkage of proteins and payloads producing monodisperse conjugates†
Igor
Dovgan
a,
Alexandre
Hentz
a,
Oleksandr
Koniev
b,
Anthony
Ehkirch
c,
Steve
Hessmann
c,
Sylvain
Ursuegui
 a,
Sébastien
Delacroix
b,
Margaux
Riomet
d,
Frédéric
Taran
d,
Sarah
Cianférani
c,
Sergii
Kolodych
*b and
Alain
Wagner
*a
a,
Sébastien
Delacroix
b,
Margaux
Riomet
d,
Frédéric
Taran
d,
Sarah
Cianférani
c,
Sergii
Kolodych
*b and
Alain
Wagner
*a
aBiofunctional Chemistry Laboratory, UMR 7199, LabEx Medalis, University of Strasbourg, France. E-mail: alwag@unistra.fr
bSyndivia SAS, 650 Boulevard Gonthier d'Andernach, 67400 Illkirch, France. E-mail: sk@syndivia.com
cLaboratoire de Spectrométrie de Masse BioOrganique, Université de Strasbourg, CNRS, IPHC UMR 7178, 67000 Strasbourg, France
dService de Chimie Bio-organique et Marquage DRF/JOLIOT - CEA, Université Paris-Saclay, F-91191, Gif-sur-Yvette, France
First published on 2nd January 2020
Abstract
Controlled protein functionalization holds great promise for a wide variety of applications. However, despite intensive research, the stoichiometry of the functionalization reaction remains difficult to control due to the inherent stochasticity of the conjugation process. Classical approaches that exploit peculiar structural features of specific protein substrates, or introduce reactive handles via mutagenesis, are by essence limited in scope or require substantial protein reengineering. We herein present equimolar native chemical tagging (ENACT), which precisely controls the stoichiometry of inherently random conjugation reactions by combining iterative low-conversion chemical modification, process automation, and bioorthogonal trans-tagging. We discuss the broad applicability of this conjugation process to a variety of protein substrates and payloads.
Applications of protein conjugates are limitless, including imaging, diagnostics, drug delivery, and sensing.1–4 In many of these applications, it is crucial that the conjugates are homogeneous.5 The site-selectivity of the conjugation process and the number of functional labels per biomolecule, known as the degree of conjugation (DoC), are crucial parameters that define the composition of the obtained products and are often the limiting factors to achieving adequate performance of the conjugates. For instance, immuno-PCR, an extremely sensitive detection technique, requires rigorous control of the average number of oligonucleotide labels per biomolecule (its DoC) in order to achieve high sensitivity.6 In optical imaging, the performance of many super-resolution microscopy techniques is directly defined by the DoC of fluorescent tags.7 For therapeutics, an even more striking example is provided by antibody–drug conjugates, which are prescribed for the treatment of an increasing range of cancer indications.8 A growing body of evidence from clinical trials indicates that bioconjugation parameters, DoC and DoC distribution, directly influence the therapeutic index of these targeted agents and hence must be tightly controlled.9
Standard bioconjugation techniques, which rely on nucleophile–electrophile reactions, result in a broad distribution of different DoC species (Fig. 1a), which have different biophysical parameters, and consequently different functional properties.10
 | ||
| Fig. 1 Schematic representation of the types of protein conjugates. | ||
To address this key issue and achieve better DoC selectivity, a number of site-specific conjugation approaches have been developed (Fig. 1b). These techniques rely on protein engineering for the introduction of specific motifs (e.g., free cysteines,11 selenocysteines,12 non-natural amino acids,13,14 peptide tags recognized by specific enzymes15,16) with distinct reactivity compared to the reactivity of the amino acids present in the native protein. These motifs are used to simultaneously control the DoC (via chemo-selective reactions) and the site of payload attachment. Both parameters are known to influence the biological and biophysical parameters of the conjugates,11 but so far there has been no way of evaluating their impact separately.
The influence of DoC is more straightforward, with a lower DoC allowing the minimization of the influence of payload conjugation on the properties of the protein substrate. The lowest DoC that can be achieved for an individual conjugate is 1 (corresponding to one payload attached per biomolecule). It is noteworthy that DoC 1 is often difficult to achieve through site-specific conjugation techniques due to the symmetry of many protein substrates (e.g., antibodies). Site selection is a more intricate process, which usually relies on a systematic screening of conjugation sites for some specific criteria, such as stability or reactivity.17
Herein, we introduce a method of accessing an entirely new class of protein conjugates with multiple conjugation sites but strictly homogenous DoCs (Fig. 1c). To achieve this, we combined (a) iterative low conversion chemical modification, (b) process automation, and (c) bioorthogonal trans-tagging in one workflow.
The method has been exemplified for protein substrates, but it is applicable to virtually any native bio-macromolecule and payload. Importantly, this method allows for the first time the disentangling of the effects of homogeneous DoC and site-specificity on conjugate properties, which is especially intriguing in the light of recent publications revealing the complexity of the interplay between payload conjugation sites and DoC for in vivo efficacy of therapeutic bioconjugates.18 Finally, it is noteworthy that this method can be readily combined with an emerging class of site-selective bioconjugation reagents to produce site-specific DoC 1 conjugates, thus further expanding their potential for biotechnology applications.19
Results and discussion
Since a truly universal method cannot make use of any individual structural features of the proteins, we chose to exploit a very general mathematical feature of all statistical distributions of species obtained following conjugation of multisite macromolecules. Indeed, the abundance of DoC species in a given bioconjugate can be approximated by a Poisson distribution model.20 It is noteworthy that, according to the Poisson model, the DoC distribution narrows as the average DoC decreases (Fig. 2a). However, reducing the average DoC inevitably increases the proportion of unconjugated protein. For instance, while classical bioconjugation of a protein with an average DoC of 3 yields a mixture of eight detectable (over 1% abundance) species with different DoCs, and 5% unconjugated species, targeting an average DoC of 0.05 produces a mixture composed of only two detectable species (DoC 0 and 1), although the unconjugated species will account for around 95% of all species. Obviously, this capacity of low conversion reactions to generate monodisperse mixtures of conjugates can only be practically exploited if the product can readily be separated from the starting material, and if the unconverted protein can be recycled and reused in a subsequent conjugation cycle. Ideally, the process should be automated and compatible with any payload.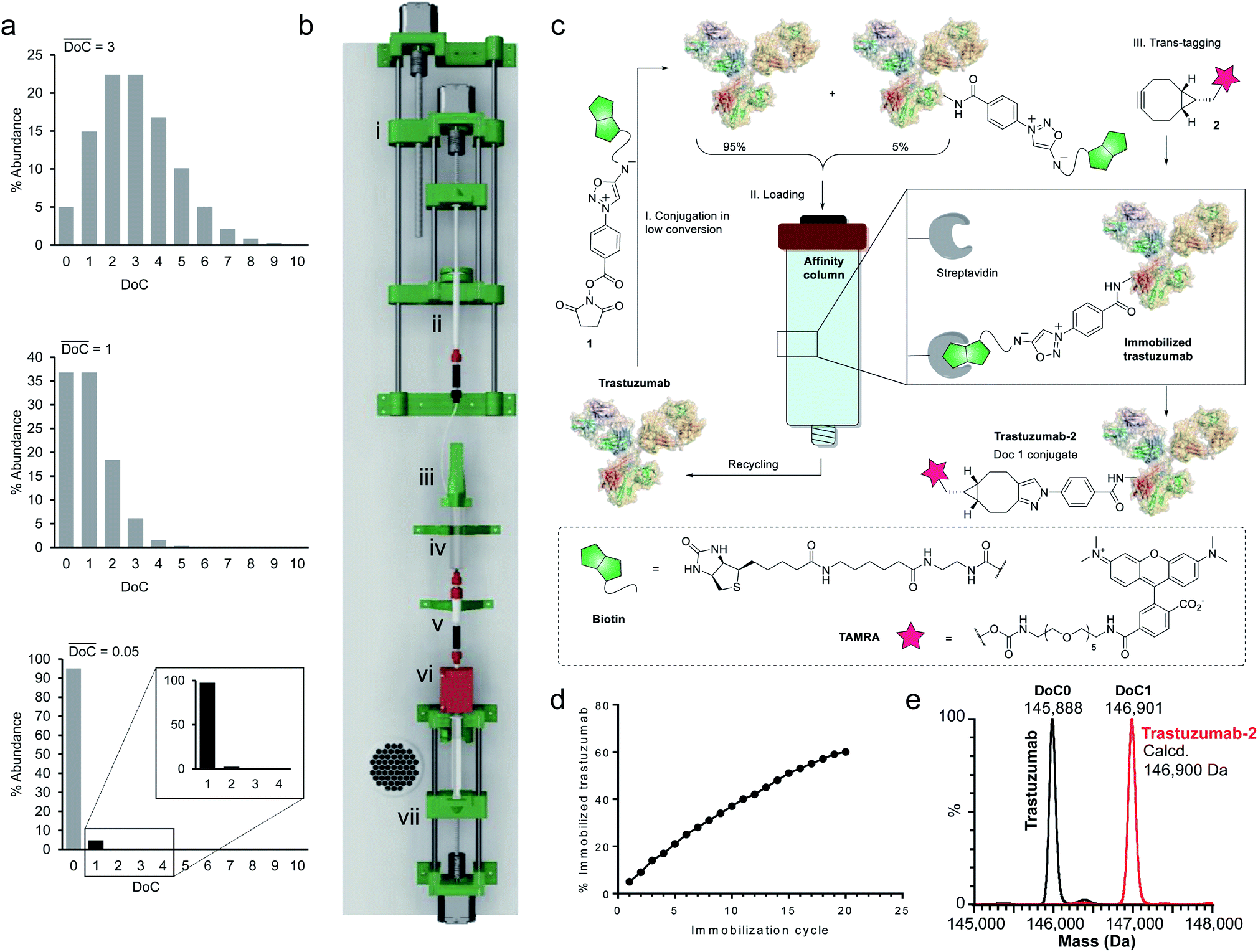 | ||
| Fig. 2 Development of equimolar native chemical tagging (ENACT). (a) Simulated Poisson distributions for conjugates with an average DoC of 3 (top), 1 (middle), and 0.05 (bottom). (b) 3D CAD model of the automation device, comprising a syringe pump for the addition of reagent 1 (i); a 1 mL syringe containing solution of 1 (ii); a reactor cap with a stirring motor (iii); the body of a 10 mL syringe used as a reactor (iv); a streptavidin column (v); a UV detector (vi); and a 10 mL syringe pump controlling the flow of the reaction mixture through the streptavidin column (vii). (c) Illustration of the ENACT process, including conjugation of compound 1 with trastuzumab at low conversion (Step I); capture of the conjugated antibody and recycling of the unconjugated fraction (Step II); bioorthogonal trans-tagging (Step III). (d) Total fraction of immobilized trastuzumab after each immobilization cycle measured by UV absorbance. (e) Deconvoluted native MS spectra of the trastuzumab and trastuzumab-2 conjugate. | ||
To separate the functionalized protein from the substrate, classical affinity purification can be used. We selected a well-known affinity tag (biotin), since it has been extensively described and is frequently used due to its extremely high affinity for streptavidin.21 This approach allows efficient immobilization and separation of biotinylated proteins on readily available streptavidin columns. In order to make the process compatible with any payload, we applied a recently discovered cycloaddition of iminosydnones and strained alkynes22 as a bioorthogonal trans-tagging reaction to replace the affinity tag by the payload and simultaneously release the protein from the affinity support. This trans-tagging reaction is an ideal candidate for this application, since it has a high rate constant (k up to 8.1 M−1 s−1) in aqueous buffers and does not interfere with complex biological molecules.23 A trifunctional molecular construct 1 was thus synthesized, comprising a reactive function for protein conjugation (N-hydroxysuccinimide ester), a bioorthogonal trans-tagging function (iminosydnone), and an affinity tag (biotin). N-hydroxysuccinimide (NHS) ester was selected as the reactive group as it is one of the most widely used functions in bioconjugation. This group is known to produce a wide statistical DoC distribution profile due to the presence of multiple primary amine residues in most proteins and was therefore ideal for the validation of the method.
Since only a small fraction of protein is immobilized during each conjugation/loading cycle, the process has to be repeated multiple times in order to reach acceptable protein conversion. However, this repetitive process of column loading can be readily automated by developing a simple device capable of performing multiple conjugation–immobilization cycles without human intervention (Fig. 2b). Inspired by the advances of 3D-printing technology in the field of automated chemical synthesis,24,25 we chose to introduce this promising technology into the field of bioconjugation and use it for fabrication of the device. We used polylactic acid (PLA) to 3D-print the main structural components of the device, which were complemented with low-cost electronic hardware. The device consists of an upgraded version of the 10 mL open-source syringe pump26 to control flow through the streptavidin affinity column, a reactor with a mechanical stirrer and a 1 mL syringe pump for reagent addition, mounted on a moving platform. The body of a 10 mL syringe served as a single-use reactor in this setup. Reagent 1 is added via a PEEK tube (i.d. 0.15 mm) immersed in the protein solution when the 1 mL syringe pump moves downward. The system is actuated by three step motors and one DC motor. The device is controlled by a Raspberry Pi computer running GNU/Linux, and complemented with a UV detector to monitor protein concentrations throughout the process.
For our first demonstration, we used a well-known therapeutic monoclonal antibody (trastuzumab) as the model substrate. The first step of the process (Fig. 2c, Step I) involved reaction of the activated ester (1) with excess trastuzumab. In the second step (Fig. 2c, Step II) the conjugated antibody was captured on a streptavidin column, and the unconjugated protein was eluted ready for a subsequent coupling cycle. We used the device to perform 20 coupling/immobilization cycles on trastuzumab with an average conversion rate of 5% per cycle (Fig. 2d) and an overall conversion of 60%. In the final trans-tagging step (Fig. 2c, Step III), the affinity column was equilibrated with a buffered solution of a cyclooctyne derivative (2). Following incubation of 2 in the column, pure DoC-1 trastuzumab-2 conjugate was eluted (Fig. 2e).
To investigate the scope of the new conjugation process, named equimolar native chemical tagging (ENACT), we first used a trastuzumab-loaded column to evaluate different strained alkynes, including BCN (3), DBCO-COOH (4) and TMTH (5), in the trans-tagging step. All strained alkynes were compatible with the trans-tagging process and yielded the corresponding trastuzumab conjugates (Fig. 3).
 | ||
| Fig. 3 Examples of the ENACT for protein functionalization. (a) Structures of payloads used for the exemplification of the ENACT process, including a series of strained alkynes (3, 4 and 5), a fluorophore (6), cytotoxic drugs (7 and 8), and an oligonucleotide (BCN-ON). (b) Deconvoluted native MS spectra for the unconjugated proteins (black) and corresponding DoC 1 conjugates (red). MS spectra of trastuzumab conjugated with BCN-ON and annealed with a complimentary DNA strand (cON) is depicted in blue. | ||
We subsequently tested ENACT process using a range of proteins, including lysozyme (14 kDa), human serum albumin (HSA, 67 kDa), trastuzumab (146 kDa) and bevacizumab (149 kDa). The proteins were loaded onto streptavidin columns according to the process described for trastuzumab and were submitted to the trans-tagging reaction with the BCN derivative of the Cy5 fluorophore (6). Native MS analysis of the conjugates confirmed clean formation of the DoC 1 species in all cases, thus validating the applicability of the process to proteins of a wide variety of sizes and structures. We then used the ENACT process to prepare conjugates of lysozyme and trastuzumab with the derivatives of two highly potent cytotoxic agents used in targeted therapeutics: MMAE and MMAF. In these experiments, the protein-loaded columns were equilibrated and incubated with strained alkynes 7 and 8, and yielded the corresponding DoC 1 conjugates lysozyme-7 and trastuzumab-8. Finally, we evaluated the applicability of the ENACT process to the conjugation of proteins with oligonucleotides (ON). To this end, immobilized trastuzumab was reacted with a BCN-functionalized oligonucleotide (BCN-ON) in the trans-tagging step under standard conditions. The resulting DoC 1 trastuzumab-ON conjugate was eluted from the column and purified by gel filtration. Upon incubation in PBS, trastuzumab-ON readily annealed with the complimentary DNA strand (cON), as shown by native MS (Fig. 3b).
In order to identify the conjugation sites in a DoC 1 conjugate produced using the ENACT process, we performed a peptide-mapping experiment. To this end, we prepared trastuzumab-4 using ENACT with a trans-tagging reagent 4. Both trastuzumab and the resulting trastuzumab-4 DoC 1 conjugate were digested, and the digests were separated by RP chromatography, followed by UV and MS analysis in tandem for peptide identification. A total of 12 lysine residues (7 on the heavy chain and 5 on the light chain) were identified as being conjugated (Fig. 4).
 | ||
| Fig. 4 Map of conjugation sites in trastuzumab-4. Red spheres represent conjugated lysine residues. | ||
We subsequently applied ENACT to introduce a single stable arylpropiolonitrile (APN) electrophilic handle onto a protein for further reaction with thiol-containing payloads.27–29 using trans-tagging reagent 9, bearing an APN function (Fig. 5a). The conjugate was then reacted with glutathione (10a) and with a thiol derivative of TAMRA (10b), to produce trastuzumab-10a and trastuzumab-10b conjugates respectively (Fig. 5b). These results demonstrated that ENACT can be used as part of a plug-and-play approach, yielding reactive protein derivatives for subsequent simple, one-step post-modifications.30
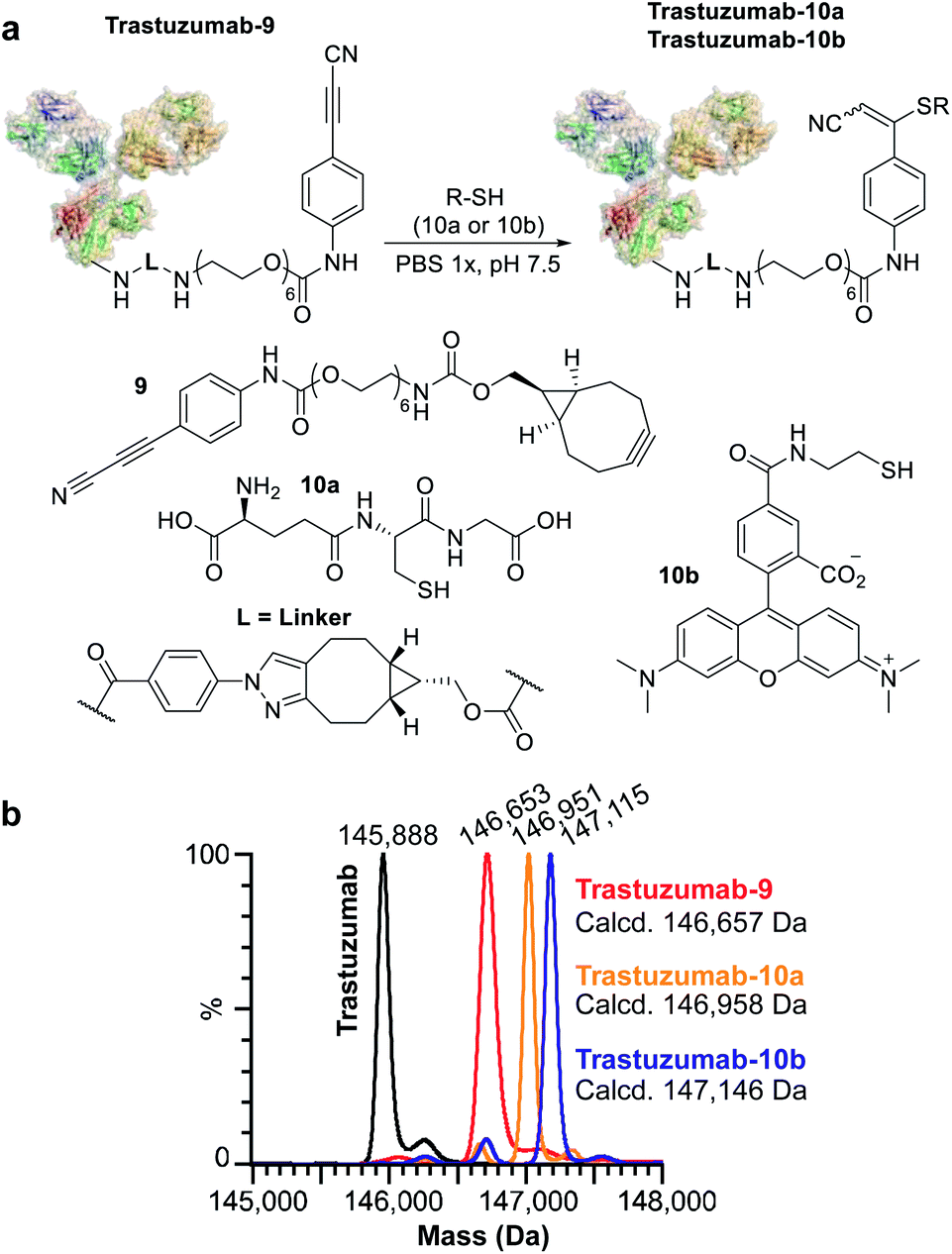 | ||
| Fig. 5 Application of ENACT for the introduction of reactive handles. (a) Placement of the APN reactive handle on trastuzumab and post-modification with glutathione (10a) and a fluorescent probe (10b). (b) Deconvoluted native MS spectra for trastuzumab; trastuzumab-9 containing an APN reactive handle; trastuzumab-10a; and trastuzumab-10b obtained via post-modification of trastuzumab-9. | ||
Finally, we investigated the applicability of ENACT for the preparation of DoC 2 conjugates through immobilization and subsequent trans-tagging of previously prepared DoC 1 conjugates. Thus, trastuzumab-4 (Fig. 6a) was immobilized on the streptavidin column using standard ENACT procedure. The column was then equilibrated and incubated with the solution of 4 to yield trastuzumab-4-4 with two DBCO derivatives attached per each biomolecule (Fig. 6b). By repeating the procedure and using 2 in the trans-tagging step we obtained trastuzumab-4-2 with two different payloads per antibody. Another example of dual–payload conjugate was obtained by using DBCO-PEG12-Me (11) in the trans-tagging step, yielding trastuzumab-4-11. These examples demonstrate that higher DoC conjugates with different combinations of payloads can be accessed through iterative ENACT process.
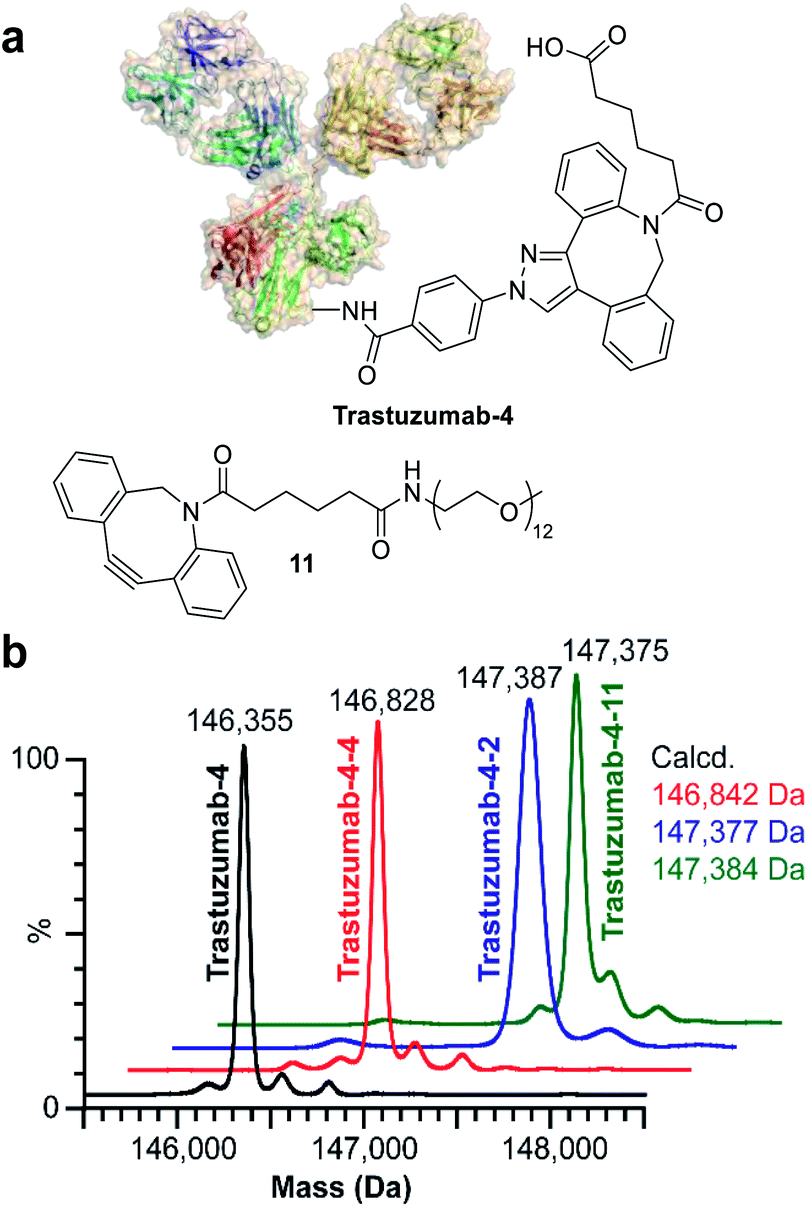 | ||
| Fig. 6 Preparation of DoC 2 conjugates using ENACT. (a) Structures of trastuzumab-4 and payload 11. (b) Deconvoluted native MS spectra for trastuzumab-4 substrate and the derived DoC 2 conjugates: trastuzumab-4-4, trastuzumab-4-2 and trastuzumab-4-11. | ||
Conclusions
In conclusion, we have developed a general process enabling on-demand preparation of monodisperse protein conjugates having precise stoichiometry yet variable spatial configuration, thus for the first time decoupling DoC and site-specificity of conjugation. The process was automated using a custom-made 3D-printed device and exemplified on a series of proteins (lysozyme, HSA, IgGs) and complex payloads (fluorescent probes, drugs, oligonucleotides, reactive handles). Future work will explore the combination of ENACT with an emerging class of site-selective bioconjugation reagents and will focus on comparing monodisperse therapeutic conjugates with their site-specific analogues.Conflicts of interest
O. K., A. E., S. D. and S. K. are employees of Syndivia SAS.Acknowledgements
This work was supported by the CNRS, the University of Strasbourg, the iCFRC, the “Agence Nationale de la Recherche” (project ClickReal; ANR-14-CE06-0004) and the French Proteomic Infrastructure (ProFI; ANR-10-INBS-08-03). The authors thank GIS IBiSA and Région Alsace for financial support toward purchasing a Synapt G2 HDMS instrument. A. E. acknowledges the “Association Nationale de la Recherche et de la Technologie” (ANRT) and Syndivia for PhD funding. I. D. acknowledges Région Alsace for PhD funding.Notes and references
- E. M. Milczek, Chem. Rev., 2018, 118, 119–141 CrossRef CAS.
- K. C. Nicolaou and S. Rigol, Angew. Chem., Int. Ed., 2019, 58, 11206–11241 CrossRef CAS PubMed.
- J. H. Ko and H. D. Maynard, Chem. Soc. Rev., 2018, 47, 8998–9014 RSC.
- I. Dovgan, O. Koniev, S. Kolodych and A. Wagner, Bioconjugate Chem., 2019, 30, 2483–2501 CrossRef CAS PubMed.
- S. Panowski, S. Bhakta, H. Raab, P. Polakis and J. R. Junutula, mAbs, 2014, 6, 34–45 CrossRef PubMed.
- A. V. Maerle, M. A. Simonova, V. D. Pivovarov, D. V Voronina, P. E. Drobyazina, D. Y. Trofimov, L. P. Alekseev, S. K. Zavriev and D. Y. Ryazantsev, PLoS One, 2019, 14, e0209860 CrossRef CAS PubMed.
- J. Schnitzbauer, M. T. Strauss, T. Schlichthaerle, F. Schueder and R. Jungmann, Nat. Protoc., 2017, 12, 1198–1228 CrossRef CAS PubMed.
- A. Beck, L. Goetsch, C. Dumontet and N. Corvaïa, Nat. Rev. Drug Discovery, 2017, 16, 315–337 CrossRef CAS PubMed.
- K. J. Hamblett, P. D. Senter, D. F. Chace, M. M. C. Sun, J. Lenox, C. G. Cerveny, K. M. Kissler, S. X. Bernhardt, A. K. Kopcha, R. F. Zabinski, D. L. Meyer and J. A. Francisco, Clin. Cancer Res., 2004, 10, 7063–7070 CrossRef CAS PubMed.
- X. Sun, J. F. Ponte, N. C. Yoder, R. Laleau, J. Coccia, L. Lanieri, Q. Qiu, R. Wu, E. Hong, M. Bogalhas, L. Wang, L. Dong, Y. Setiady, E. K. Maloney, O. Ab, X. Zhang, J. Pinkas, T. A. Keating, R. Chari, H. K. Erickson and J. M. Lambert, Bioconjugate Chem., 2017, 28, 1371–1381 CrossRef CAS PubMed.
- J. R. Junutula, H. Raab, S. Clark, S. Bhakta, D. D. Leipold, S. Weir, Y. Chen, M. Simpson, S. P. Tsai, M. S. Dennis, Y. Lu, Y. G. Meng, C. Ng, J. Yang, C. C. Lee, E. Duenas, J. Gorrell, V. Katta, A. Kim, K. McDorman, K. Flagella, R. Venook, S. Ross, S. D. Spencer, W. Lee Wong, H. B. Lowman, R. Vandlen, M. X. Sliwkowski, R. H. Scheller, P. Polakis and W. Mallet, Nat. Biotechnol., 2008, 26, 925–932 CrossRef CAS PubMed.
- T. Hofer, J. D. Thomas, T. R. Burke and C. Rader, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 12451–12456 CrossRef CAS PubMed.
- J. Y. Axup, K. M. Bajjuri, M. Ritland, B. M. Hutchins, C. H. Kim, S. a Kazane, R. Halder, J. S. Forsyth, A. F. Santidrian, K. Stafin, Y. Lu, H. Tran, A. J. Seller, S. L. Biroc, A. Szydlik, J. K. Pinkstaff, F. Tian, S. C. Sinha, B. Felding-Habermann, V. V Smider and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16101–16106 CrossRef CAS PubMed.
- E. S. Zimmerman, T. H. Heibeck, A. Gill, X. Li, C. J. Murray, M. R. Madlansacay, C. Tran, N. T. Uter, G. Yin, P. J. Rivers, A. Y. Yam, W. D. Wang, A. R. Steiner, S. U. Bajad, K. Penta, W. Yang, T. J. Hallam, C. D. Thanos and A. K. Sato, Bioconjugate Chem., 2014, 25, 351–361 CrossRef CAS PubMed.
- D. Rabuka, J. S. Rush, G. W. DeHart, P. Wu and C. R. Bertozzi, Nat. Protoc., 2012, 7, 1052–1067 CrossRef CAS PubMed.
- R. R. Beerli, T. Hell, A. S. Merkel and U. Grawunder, PLoS One, 2015, 10, e0131177 CrossRef PubMed.
- R. Ohri, S. Bhakta, A. Fourie-O’Donohue, J. dela Cruz-Chuh, S. P. Tsai, R. Cook, B. Wei, C. Ng, A. W. Wong, A. B. Bos, F. Farahi, J. Bhakta, T. H. Pillow, H. Raab, R. Vandlen, P. Polakis, Y. Liu, H. Erickson, J. R. Junutula and K. R. Kozak, Bioconjugate Chem., 2018, 29, 473–485 CrossRef CAS PubMed.
- N. C. Yoder, C. Bai, D. Tavares, W. C. Widdison, K. R. Whiteman, A. Wilhelm, S. D. Wilhelm, M. A. McShea, E. K. Maloney, O. Ab, L. Wang, S. Jin, H. K. Erickson, T. A. Keating and J. M. Lambert, Mol. Pharm., 2019, 16, 3926–3937 CrossRef CAS PubMed.
- K. Yamada and Y. Ito, ChemBioChem, 2019, 20, 2729–2737 CrossRef CAS PubMed.
- M. T.-J. Kim, Y. Chen, J. Marhoul and F. Jacobson, Bioconjugate Chem., 2014, 25, 1223–1232 CrossRef CAS PubMed.
- P. Weber, D. Ohlendorf, J. Wendoloski and F. Salemme, Science, 1989, 243, 85–88 CrossRef CAS PubMed.
- S. Bernard, D. Audisio, M. Riomet, S. Bregant, A. Sallustrau, L. Plougastel, E. Decuypere, S. Gabillet, R. A. Kumar, J. Elyian, M. N. Trinh, O. Koniev, A. Wagner, S. Kolodych and F. Taran, Angew. Chem., Int. Ed., 2017, 56, 15612–15616 CrossRef CAS PubMed.
- M. Riomet, E. Decuypere, K. Porte, S. Bernard, L. Plougastel, S. Kolodych, D. Audisio and F. Taran, Chem.–Eur. J., 2018, 24, 8535–8541 CrossRef CAS PubMed.
- P. J. Kitson, G. Marie, J.-P. Francoia, S. S. Zalesskiy, R. C. Sigerson, J. S. Mathieson and L. Cronin, Science, 2018, 359, 314–319 CrossRef CAS PubMed.
- M. D. Symes, P. J. Kitson, J. Yan, C. J. Richmond, G. J. T. Cooper, R. W. Bowman, T. Vilbrandt and L. Cronin, Nat. Chem., 2012, 4, 349–354 CrossRef CAS PubMed.
- B. Wijnen, E. J. Hunt, G. C. Anzalone and J. M. Pearce, PLoS One, 2014, 9, e107216 CrossRef PubMed.
- O. Koniev, G. Leriche, M. Nothisen, J.-S. Remy, J.-M. Strub, C. Schaeffer-Reiss, A. Van Dorsselaer, R. Baati and A. Wagner, Bioconjugate Chem., 2014, 25, 202–206 CrossRef CAS PubMed.
- S. Kolodych, O. Koniev, Z. Baatarkhuu, J.-Y. Bonnefoy, F. Debaene, S. Cianférani, A. Van Dorsselaer and A. Wagner, Bioconjugate Chem., 2015, 26, 197–200 CrossRef CAS PubMed.
- O. Koniev, S. Kolodych, Z. Baatarkhuu, J. Stojko, J. Eberova, J.-Y. Bonnefoy, S. Cianférani, A. Van Dorsselaer and A. Wagner, Bioconjugate Chem., 2015, 26, 1863–1867 CrossRef CAS PubMed.
- A. Maruani, M. E. B. Smith, E. Miranda, K. A. Chester, V. Chudasama and S. Caddick, Nat. Commun., 2015, 6, 6645 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc05468e |
| This journal is © The Royal Society of Chemistry 2020 |