
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric pyrone Diels–Alder reactions enabled by dienamine catalysis†
Charles J. F.
Cole
 ,
Lilia
Fuentes
and
Scott A.
Snyder
*
,
Lilia
Fuentes
and
Scott A.
Snyder
*
Department of Chemistry, University of Chicago, 5735 S. Ellis Avenue, Chicago, IL 60637, USA. E-mail: sasnyder@uchicago.edu
First published on 26th December 2019
Abstract
Despite the proven value in utilizing pyrone dienes to create molecular complexity via Diels–Alder reactions with varied dienophiles, few examples of effective catalytic, asymmetric variants of this process have been developed. Herein, we show that the use of Jørgensen–Hayashi-type catalysts can convert an array of α,β-unsaturated aldehydes into chiral dienamines that can formally add in a Diels–Alder fashion to a number of electron-deficient pyrones of the coumalate-type to generate optically active [2.2.2]-bicyclic lactones. In most cases, the reactions proceed with good to excellent diastereo- and enantiocontrol (up to 99% ee). Models to explain that stereoselectivity, as well as several additional transformations of the resultant products, are also presented.
Introduction
Over the course of the past several years, pyrone-based Diels–Alder reactions have served as a valuable tool to generate diverse molecules, including structurally complex natural products.1,2 Indeed, not only can they lead directly to the [2.2.2]-bicyclic lactones found in targets such as basiliolide C (1, Fig. 1)3 and scholarisine A (2),4 but their functionality can also be converted into a number of other patternings. Those changes include the highlighted domains of α-lycorane (3),5 chatancin (4),6 and gracilamine (5)7 as achieved through ring openings, as well as the aromatized systems of rufescine (6)8 and cavicularin (7)9 formed via retro-Diels–Alder reactions expelling CO2.10 Despite this collated potential, however, none of the cycloadditions leading to these diverse products served as the enantiodetermining step in their sequences. That result likely reflects the fact that few catalytic, asymmetric variants have been developed to date,11 perhaps due to the pyrone unit itself precluding traditional strategies that activate and facially differentiate dienophiles. Key exceptions shown in Scheme 1 include (1) work by the Posner group12 using stoichiometric TADDOL Lewis acid complexes to achieve asymmetric inverse demand Diels–Alder reactions between 3-carbomethoxy-2-pyrones and aryl vinyl ethers and (2) precedent by Deng,13 built upon by Wang,14 in which the 3-hydroxy motif of pyrones 8 and 10 enabled hydrogen-bonding catalysis with cinchona-derived amino alcohol or amino thiourea promoters to deliver 9 and 11 with high stereochemical control in a normal demand version. Herein, we highlight a new approach to achieve catalytic, enantioselective pyrone Diels–Alder chemistry with distinct substrate scope fueled by activating α,β-unsaturated aldehydes (19) as their dienamine counterparts. This process affords an array of structurally useful [2.2.2]-bicyclic lactones with good to excellent diastereo- and enantiocontrol.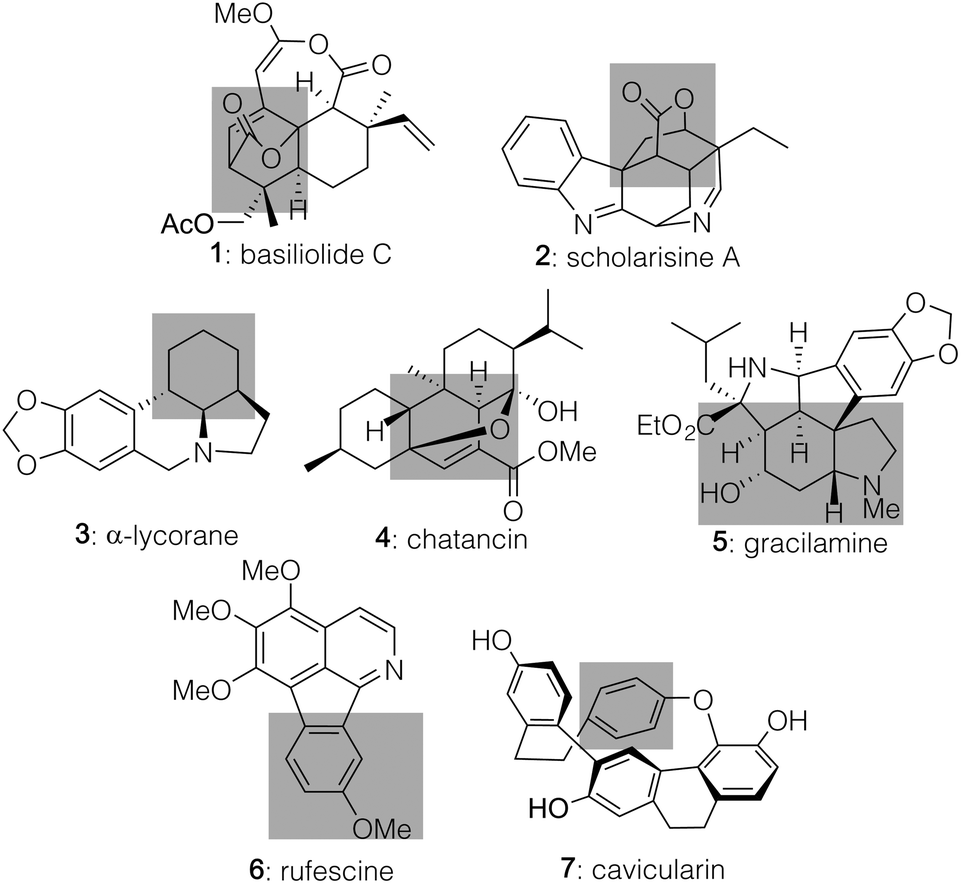 | ||
| Fig. 1 Selected natural products synthesized through pyrone Diels–Alder reactions. | ||
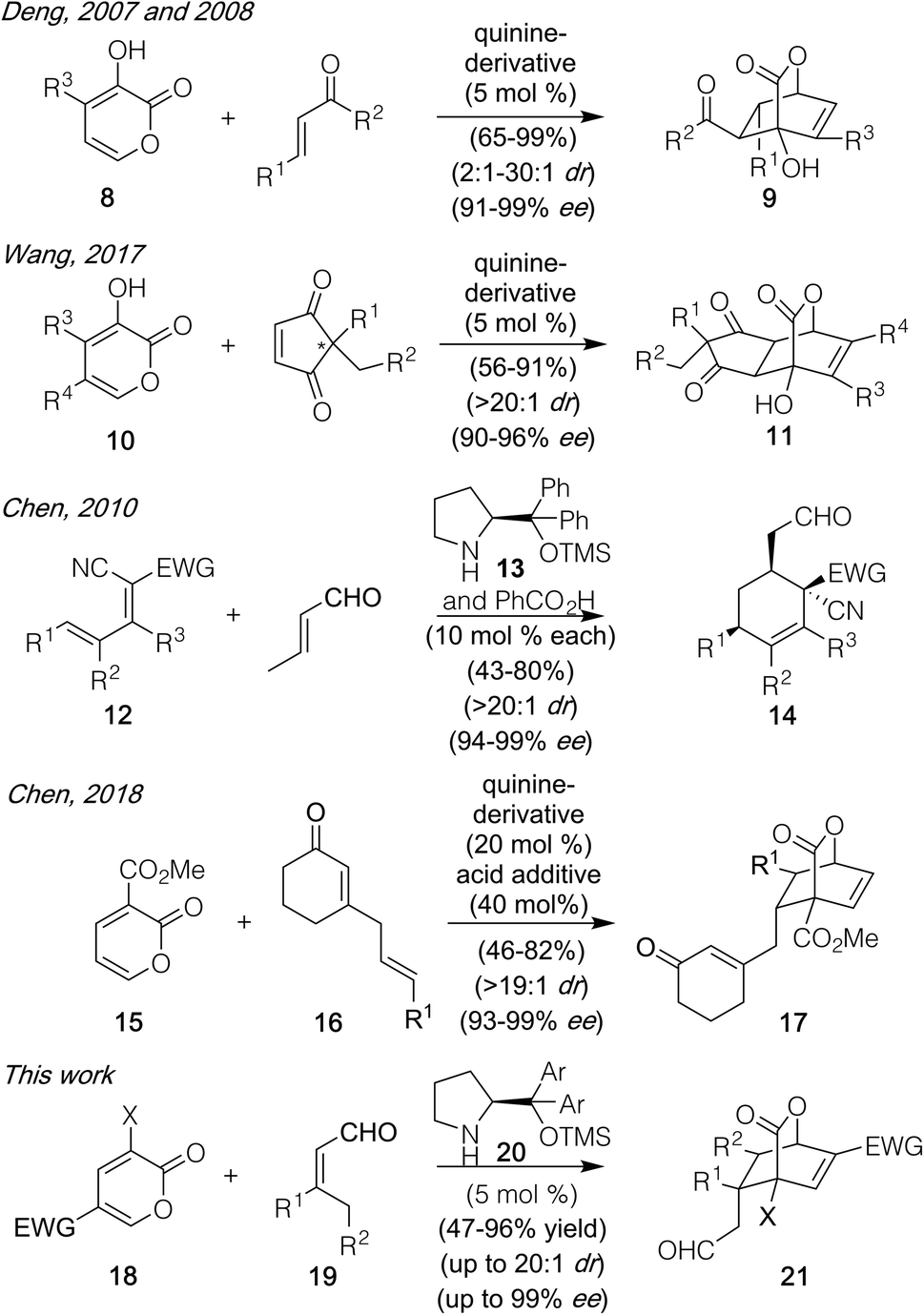 | ||
| Scheme 1 Examples of asymmetric pyrone Diels–Alder reactions, and the development of a new variant of such a process using dienamine catalysis. | ||
Recent precedent from Chen and co-workers15,16 showed that the use of the proline-derived promoter 13 (Scheme 1)17 was able to effect Diels–Alder reactions between a variety of substrates, including the electron deficient dienes and crotonaldehyde, to forge highly substituted cyclohexene systems. We wondered whether mechanistically similar chemistry could be achieved with an array of substituted pyrones (18) with diverse aldehyde-derived dienophiles (19) to generate chiral [2.2.2]-bicyclic products. If so, then arguably greater reaction scope could be achieved versus existing precedent, particularly in terms of the pyrone partner, since the specific structural requirements critical to the Posner, Deng, and Wang designs would not be required given the distinct mode of activation. Questioning that assumption was a report18 issued after our studies began which revealed that one such Diels–Alder process could be achieved, though only under more forcing conditions16 (50 °C, 20 mol% promoter, 40 mol% acid) and restricted to a single pyrone (15) and dienophile class in which a cyclic enone coupled with an allyl substituent (in the form of 16) was needed to generate the competent reaction partner. Herein, we show that it is possible to achieve significant breadth in this reaction process, under milder conditions which allow for low catalyst loadings (25 °C, 5 mol% promoter, no additive), using an array of pyrones (18) and simple α,β-unsaturated materials (19) as the dienophile precursor.
Results and discussion
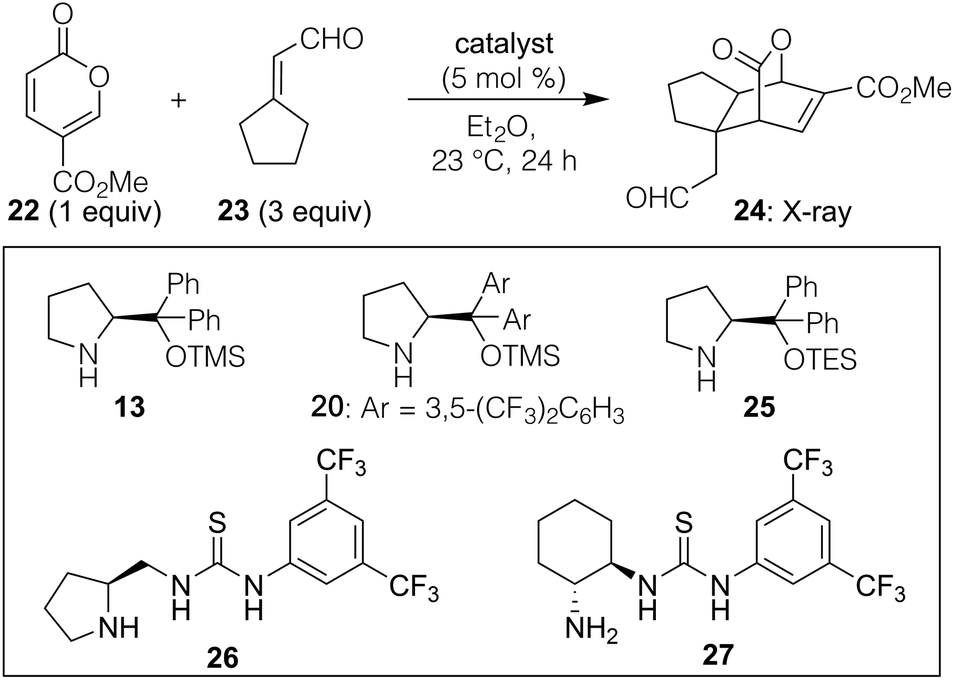
Our explorations began by using methyl coumalate (22, Table 1) as the pyrone-based diene given its documented success in Diels–Alder chemistry19 along with a variety of α,β-unsaturated aldehydes to determine what patterning, if any, could afford reasonably high reactivity and stereoselectivity. That screen showed that in Et2O as solvent at 23 °C,20 3,3′-disubstituted enals were by far the most effective partners (see ESI† for full details) when used as the model dienophile for further optimizations. As shown in Table 1, we found that Chen's original catalyst (13) in combination with equimolar benzoic acid additive could afford some of the desired endo product (24) containing a fused ring and a new quaternary carbon (entry 1; structure verified by X-ray analysis)21 with fairly good selectivity over its undrawn exo alternative. However, using the 3,5-bis(trifluoromethyl) phenyl variant 20 at only a 5 mol% loading (entry 2), improved yield (56%) and selectivity [5.7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr, 93% ee for the major diastereomer drawn, ee values determined by HPLC analysis of the UV-active homologated methyl ester formed via reaction of the aldehyde with methyl (triphenylphosphoranylidene)acetate] were obtained. Given that most reactions involving dienamines typically require catalyst loadings around 20 mol%,16 we next tested whether the acidic or basic additives, which are often used to ensure high conversion and stereoselectivity, were needed. As indicated in entry 4, that turned out not to be the case, as the absence of such a promoter using catalyst 20 led to a significant increase in yield to 93% and an improved dr of 6.7:1.22 No other catalyst screened, including several thioureas,23 provided superior results. It should be noted that upon resubjection of the products to the reaction conditions (5 mol% 20 in Et2O), no change in dr was observed, suggesting that cycloreversion/equilibration is not taking place.
:1 dr, 93% ee for the major diastereomer drawn, ee values determined by HPLC analysis of the UV-active homologated methyl ester formed via reaction of the aldehyde with methyl (triphenylphosphoranylidene)acetate] were obtained. Given that most reactions involving dienamines typically require catalyst loadings around 20 mol%,16 we next tested whether the acidic or basic additives, which are often used to ensure high conversion and stereoselectivity, were needed. As indicated in entry 4, that turned out not to be the case, as the absence of such a promoter using catalyst 20 led to a significant increase in yield to 93% and an improved dr of 6.7:1.22 No other catalyst screened, including several thioureas,23 provided superior results. It should be noted that upon resubjection of the products to the reaction conditions (5 mol% 20 in Et2O), no change in dr was observed, suggesting that cycloreversion/equilibration is not taking place.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Additive | Catalyst | Yield (%) | dr | eec (%) |
| a 10 mol% loading of both catalyst and benzoic acid. b 0.1 equiv. added. c Measured for major diastereomer. | |||||
| 1a | PhCO2Ha | 13 | 52 | 4.9:1 |
87 |
| 2 | PhCO2Hb | 20 | 56 | 5.7:1 |
93 |
| 3 | Et3Nb | 20 | 13 | 4.9:1 |
n.d. |
| 4 | — | 20 | 93 | 6.7:1 |
94 |
| 5 | — | 13 | 70 | 2.3:1 |
90 |
| 6 | — | 25 | 64 | 3.5:1 |
92 |
| 7 | — | 26 | 10 | 3.0:1 |
20 |
| 8 | — | 27 | — | n.d. | n.d. |
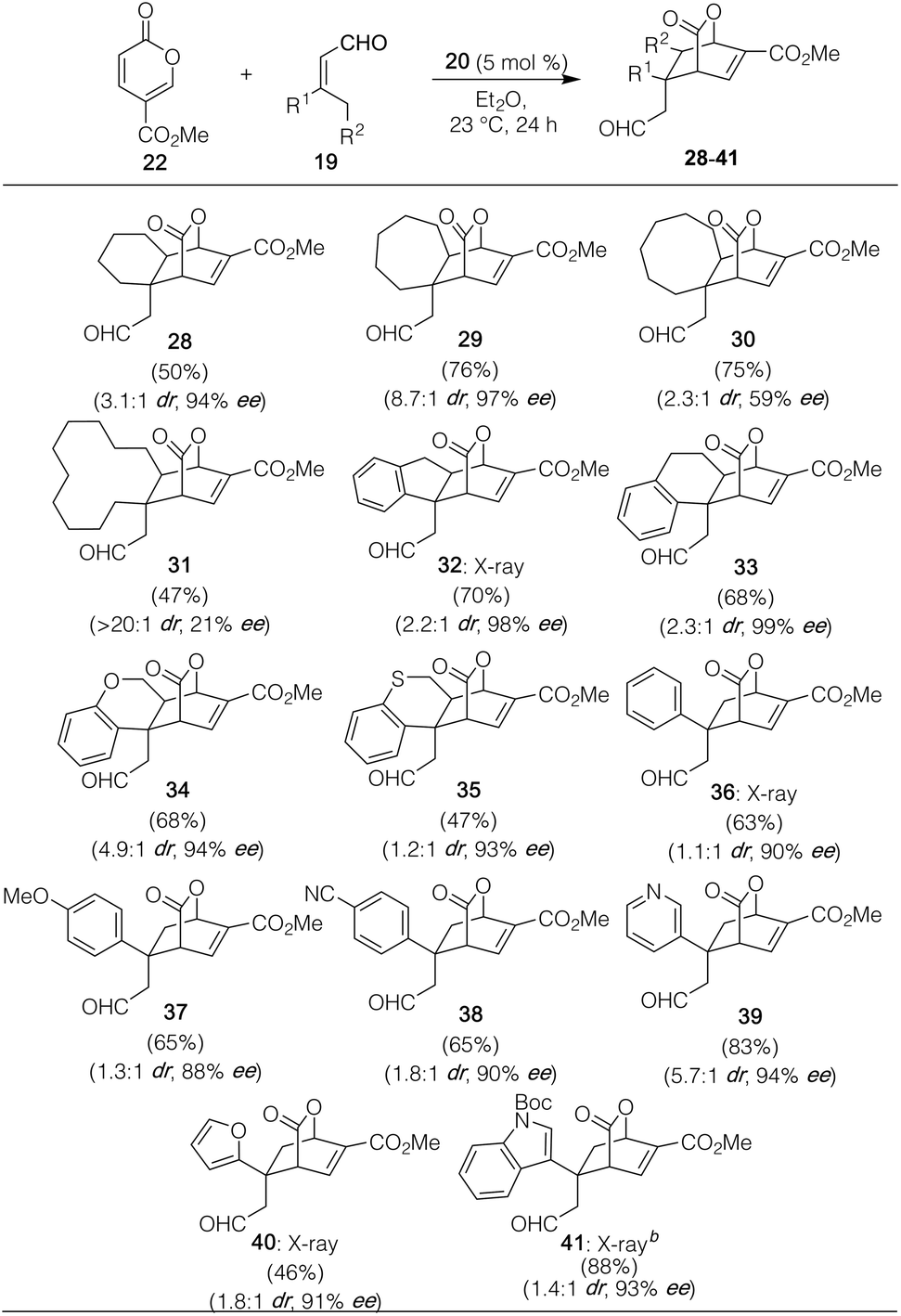
With these optimized conditions in hand, we then probed the dienophile scope with a number of cyclic and acyclic 3,3′-disubstituted enals. As shown in Table 2, rings of varied size, including 6-, 7-, and 8-membered carbocycles, could be readily fused onto pyrone 22 to deliver 28–30. Quite pleasingly, the ring size could even be expanded to include the 12-membered carbocycle of 31, albeit with modest enantioselectivity, though in near perfect diastereoselectivity. Similarly, a number of aryl-containing products, arising from indanone, tetralone, chromanone, thiochromanone, and a variety of aryl and heteroaryl-containing precursors succeeded smoothly as well irrespective of being electron-rich or electron-poor. In all of these cases, enantioselectivity for the major drawn diastereomer (with several such as 32, 36, 40, and 41 verified via X-ray crystallographic analysis) was high, though diastereocontrol was more modest than the fully aliphatic examples (vide infra).24 In the case of the furanyl substituted enal leading to 40, we attribute the slightly lower yield as being the result of the reactive furan moiety itself serving as a diene partner. Of note, the use of 3,3′-disubstituted enals is essential to the reaction's success; concurrent with our studies, Liu and Zu showed that under similar modes of catalysis with pyrone 22, a variety of differentially substituted 3-aryl α,β-unsaturated aldehydes participated in Rauhut–Currier reactions instead.25
We next explored variations in the pyrone partner in its reaction with cyclopentylidene carboxaldehyde (23). As shown in Table 3, both 5-substituted ketone and nitrile-containing substrates provided the desired products (42 and 43), the latter with far superior dr (9.8:1) versus the other counterparts probed. A 4,5-diester variant also succeeded to afford 44. A 3-bromomethyl coumalate starting material, as well as its phenyl ketone analog, worked as well to deliver 45 and 46; their bridgehead bromide atom is of potential synthetic use to enable further functionalizations. Finally, 2-pyrone substrates containing a single methyl ester group at different positions on the ring were also explored; however, besides methyl coumalate, only the 3-carbomethoxy-2-pyrone was capable of undergoing the desired transformation to provide 47, albeit with reduced enantioselectivity. It should be noted that a similar trend was observed by Liu and Zu.25 These final examples (45–47) collectively reveal the ability of the method to construct optically active products containing two fully substituted vicinal carbon atoms in a single step.26
| a Reactions were performed with pyrones (0.3 mmol) and 23 (0.9 mmol) in Et2O (0.2 M) open to air. |
|---|

|
With this body of results in hand, a transition state analysis to account for the stereoselection observed for the process is provided in Fig. 2; here, we have focused in particular on explaining the variations in dr observed based on the structural differences in the two respective partners. For aliphatic-containing substrates, we reason that transition state endo 48, leading to the observed major diastereomer in each case, is favored over transition state exo 48, due to secondary orbital overlap between the 4,5-position of the pyrone and the formed enamine. When aryl-containing substrates are deployed, that diastereoselectivity bias is eroded due to competing secondary orbital overlap present in both transition states. By contrast, when a nitrile group is attached to the pyrone at the 5-position instead of an ester, the geometry imposed by its sp hybridization prevents effective secondary orbital overlap with added aryl rings in transition state exo 50, and generally would seem to favor transition state endo 50 through the similar stabilization of the 4,5-positions on the diene partner even if the dienophile contains only aliphatic substituents.
 | ||
| Fig. 2 Proposed transition state analysis to account for observed dr trends.27 | ||
In support of that assertion, both products 51 and 52 were formed in significantly superior levels of dr versus the corresponding analogs generated with pyrone 22 at the price of a very small drop in enantioselection; thus, if high dr is a significant concern over enantiocontrol, the use of a nitrile-containing pyrone reactant might be the better choice given these observations. Finally, for dienophile precursors where both kinetic and thermodynamic dienamines could potentially react, as in substrate 53, the conditions allow for the preferential reaction of the thermodynamic dienamine given the formation of 55 in an ∼4:1 ratio with the separable 54.
Finally, to further illustrate the value of the developed reaction and its overall scope, a number of additional transformations were effected along the lines indicated in Scheme 2. First, as shown with [2.2.2]-bicycle 32, a Stetter reaction15b promoted by triazolium salt 56 can enable coupling of the enoate with the pendant aldehyde on the quaternary carbon to generate the new fused ring system of 57 in 91% yield as a single diastereomer about the ester. This example highlights the asymmetric construction of a fully substituted cyclohexane ring in 2 steps, thus demonstrating the ability of the approach to rapidly build congested molecular complexity.
 | ||
| Scheme 2 Selected transformations of pyrone Diels–Alder products to afford additional compounds of value. (a) 56 (20 mol%), NaOAc (1.2 equiv.), CHCl3, 40 °C, 30 min, 91%, 96% ee; (b) toluene, 155 °C, 24 h, sealed tube, 67%, 95% ee; (c) 1,2-bis(trimethoxysilyl)ethane (2.0 equiv.), TMSOTf (0.1 equiv.), 0 °C, 1 h, 98%; (d) NaOMe (2.0 equiv.), MeOH, 0 °C, 2 h; BzCl (2.0 equiv.), Et3N (2.0 equiv.), 4-DMAP (2.0 equiv.), CH2Cl2, 23 °C, 1 h, 67% over 2 steps, 95% ee; (e) Me3SnOH (3.0 equiv.), ClCH2CH2Cl, 80 °C, 16 h; (f) SOCl2 (5.0 equiv.), toluene, 23 °C, 24 h; AIBN (0.1 equiv.), mercaptopyridine N-oxide sodium salt (1.1 equiv.), BrCCl3, 100 °C, 1 h, 38% over 3 steps, 92% ee. | ||
Second, upon subjection to thermal conditions, the strained bicyclic lactone of 24 underwent a retro-Diels–Alder reaction expelling CO2 that generated an electron-poor diene with functionality suitable for further elaboration in the form of 58; the pre-generated quaternary carbon of the initial product prevented aromatization to allow access to this atypical, pyrone-derived material. Third, following protection of the aldehyde within 24 as its ethylene glycol acetal (to generate 59), methanolysis of the lactone followed by in situ benzoyl protection of the resultant alcohol afforded a highly-substituted hydrindane core (60) in 67% yield over two steps. Alternatively, it is possible to differentially functionalize the exocyclic methyl ester over the potentially more strained lactone via controlled methanolysis of 59 as effected with Me3SnOH.28 This event was followed by a Hunsdiecker-type reaction29 using a Barton thiohydroximate ester intermediate and CBrCl3 to generate 61 in 38% yield over three steps. Not only is the resultant bromide a handle for further diversification, but the material formally reflects the Diels–Alder product of 5-bromo-2-pyrone,30 a notoriously challenging diene to engage in cycloaddition chemistry to afford products in racemic format, let alone the asymmetric version obtained here.31
Conclusions
In conclusion, we have developed a novel catalytic asymmetric inverse electron demand pyrone Diels–Alder reaction that has a unique scope relative to previously developed alternatives; indeed, to the best of our knowledge, all compounds prepared constitute new molecular entities.32 The reaction process is operationally simple, non-sensitive to air or moisture, and can be conducted with very low catalyst loadings (5 mol%) in the absence of additional additives typical of other transformations involving dienamines. As highlighted by the products formed, an array of synthetically valuable and structurally distinct [2.2.2]-bicyclic lactones can be accessed in moderate to good yields with effective diastereo- and high enantiocontrol. Applications to the synthesis of complex natural products and other materials, as well as the extension of the general protocol to other substrate types, is the subject of current endeavors.Author contributions
S. A. S. and C. J. F. C. conceived the project. S. A. S directed the research, and S. A. S. and C. J. F. C. composed the manuscript and the ESI† section; all authors commented on the manuscript. C. J. F. C. developed the initial reaction and explored substrate scope, while L. F. completed the synthesis of several pyrone starting materials and contributed significantly to the substrate tables.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Alexander Filatov and Mr Andrew McNeece for X-ray analysis of our crystalline intermediates, and Dr Antoni Jurkiewicz and Dr C. Jin Qin for assistance with NMR and mass spectrometry, respectively. We also thank Ms Lindsey Jay and Ms Jenna Reisler for the preparation and purification of some of the starting materials. Financial support for this work came from the University of Chicago (start-up funds and graduate fellowships to C. J. F. C.) and CONACyT (postdoctoral fellowship to L. F.).Notes and references
- For selected reviews of the Diels–Alder reaction, see: (a) E. J. Corey, Angew. Chem., Int. Ed., 2002, 41, 1650 CrossRef CAS; (b) K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668 CrossRef CAS.
- For reviews of pyrone Diels–Alder reactions, see: (a) K. Afarinkia, V. Vinader, T. D. Nelson and G. H. Posner, Tetrahedron, 1992, 48, 9111 CrossRef CAS; (b) B. T. Woodard and G. H. Posner, Adv. Cycloaddit., 1999, 5, 47 CAS.
- J. R. Gordon, H. M. Nelson, S. C. Virgil and B. M. Stoltz, J. Org. Chem., 2014, 79, 9740 CrossRef CAS PubMed.
- M. W. Smith and S. A. Snyder, J. Am. Chem. Soc., 2013, 135, 12964 CrossRef CAS PubMed.
- Y.-G. Jung, S.-C. Lee, H.-K. Cho, N. B. Darvatkar, J.-Y. Song and C.-G. Cho, Org. Lett., 2012, 15, 132 CrossRef PubMed.
- Y. M. Zhao and T. J. Maimone, Angew. Chem., Int. Ed., 2015, 54, 1223 CrossRef CAS PubMed.
- P. Gan, M. W. Smith, N. R. Braffman and S. A. Snyder, Angew. Chem., Int. Ed., 2016, 55, 3625 CrossRef CAS PubMed.
- (a) D. L. Boger and C. E. Brotherton, J. Org. Chem., 1984, 49, 4050 CrossRef CAS; (b) D. L. Boger and K. Takahashi, J. Am. Chem. Soc., 1995, 117, 12452 CrossRef CAS.
- P. Zhao and C. M. Beaudry, Org. Lett., 2013, 15, 402 CrossRef CAS PubMed.
- For other examples of pyrone Diels–Alder reactions in total synthesis, see: (a) G. H. Posner and D. G. Wettlaufer, J. Am. Chem. Soc., 1986, 108, 7373 CrossRef CAS; (b) G. H. Posner and C. M. Kinter, J. Org. Chem., 1990, 55, 3967 CrossRef CAS; (c) H. Shimizu, H. Okamura, T. Iwagawa and M. Nakatani, Tetrahedron, 2001, 57, 1903 CrossRef CAS; (d) P. S. Baran and N. Z. Burns, J. Am. Chem. Soc., 2006, 128, 3908 CrossRef CAS PubMed; (e) I.-J. Shin, E.-S. Choi and C.-G. Cho, Angew. Chem., Int. Ed., 2007, 46, 2303 CrossRef CAS PubMed; (f) N. T. Tam and C.-G. Cho, Org. Lett., 2008, 10, 601 CrossRef CAS PubMed; (g) H. M. Nelson, K. Murakami, S. C. Virgil and B. M. Stoltz, Angew. Chem., Int. Ed., 2011, 50, 3688 CrossRef CAS PubMed; (h) L. Min, Y. Zhang, X. Liang, J. Huang, W. Bao and C.-S. Lee, Angew. Chem., Int. Ed., 2014, 53, 11294 CrossRef CAS PubMed; (i) X. Yu, L. Xiao, Z. Wang and T. Luo, J. Am. Chem. Soc., 2019, 141, 3440 CrossRef CAS PubMed.
- For examples utilizing chiral auxiliaries, see: (a) I. E. Markó and G. R. Evans, Tetrahedron Lett., 1994, 35, 2767 CrossRef; (b) H. Okamura, K. Morishige, T. Iwagawa and M. Nakatani, Tetrahedron Lett., 1998, 39, 1211 CrossRef CAS; (c) G. H. Posner and D. G. Wettlaufer, Tetrahedron Lett., 1986, 27, 667 CrossRef CAS.
- (a) G. H. Posner, J.-C. Carry, J. K. Lee, D. S. Bull and H. Dai, Tetrahedron Lett., 1994, 35, 1321 CrossRef CAS; (b) I. E. Markó, G. R. Evans and J.-P. Declercq, Tetrahedron, 1994, 50, 4557 CrossRef; (c) G. H. Posner, H. Dai, D. S. Bull, J.-K. Lee, F. Eydoux, Y. Ishihara, W. Welsh, N. Pryor and S. Petr, J. Org. Chem., 1996, 61, 671 CrossRef CAS PubMed; (d) I. E. Markó, I. Chellé-Regnaut, B. Leroy and S. L. Warriner, Tetrahedron Lett., 1997, 38, 4269 CrossRef; (e) Y. Hashimoto, R. Abe, N. Morita and O. Tamura, Org. Biomol. Chem., 2018, 16, 8913 RSC; (f) X.-W. Liang, Y. Zhao, X.-G. Si, M.-M. Xu, J.-H. Tan, Z.-M. Zhang, C.-G. Zheng, C. Zheng and Q. Cai, Angew. Chem., Int. Ed., 2019, 58, 14562 CrossRef CAS PubMed.
- (a) Y. Wang, H. Li, Y.-Q. Wang, Y. Liu, B. M. Foxman and L. Deng, J. Am. Chem. Soc., 2007, 129, 6364 CrossRef CAS PubMed; (b) R. P. Singh, K. Bartelson, Y. Wang, H. Su, X. Lu and L. Deng, J. Am. Chem. Soc., 2008, 130, 2422 CrossRef CAS PubMed.
- L.-M. Shi, W.-W. Dong, H.-Y. Tao, X.-Q. Dong and C.-J. Wang, Org. Lett., 2017, 19, 4532 CrossRef CAS PubMed.
- (a) J.-L. Li, T.-R. Kang, S.-L. Zhou, R. Li, L. Wu and Y.-C. Chen, Angew. Chem., Int. Ed., 2010, 49, 6418 CrossRef CAS PubMed; (b) J.-L. Li, S.-L. Zhou, P.-Q. Chen, L. Dong, T.-Y. Liu and Y.-C. Chen, Chem. Sci., 2012, 3, 1879 RSC.
- For recent reviews of dienamine catalysis in organic transformations, see: (a) S. Mukherjee, J. W. Yang, S. Hoffman and B. List, Chem. Rev., 2007, 107, 5471 CrossRef CAS PubMed; (b) A. Moyano and R. Rios, Chem. Rev., 2011, 111, 4703 CrossRef CAS PubMed; (c) J.-L. Li, T.-Y. Liu and Y.-C. Chen, Acc. Chem. Res., 2012, 45, 1491 CrossRef CAS PubMed; (d) E. Arceo and P. Melchiorre, Angew. Chem., Int. Ed., 2012, 51, 2012 CrossRef PubMed; (e) D. B. Ramachary and Y. V. Reddy, Eur. J. Org. Chem., 2012, 865 CrossRef CAS; (f) I. D. Jurberg, I. Chatterjee, R. Tannert and P. Melchiorre, Chem. Commun., 2013, 49, 4869 RSC; (g) H. Jiang, Ł. Albrecht and K. A. Jørgensen, Chem. Sci., 2013, 4, 2287 RSC; (h) X. Jiang and R. Wang, Chem. Rev., 2013, 113, 5515 CrossRef CAS PubMed; (i) V. Marcos and J. Alemán, Chem. Soc. Rev., 2016, 45, 6812 RSC; (j) F. E. Held and S. B. Tsogoeva, Catal. Sci. Technol., 2016, 6, 645 RSC; (k) L. Klier, F. Tur, P. Poulsen and K. A. Jørgensen, Chem. Soc. Rev., 2017, 46, 1080 RSC. For reviews on computational studies of dienamine catalyzed transformations, see: (l) K. Halskov, B. S. Donslund, B. M. Paz and K. A. Jørgensen, Acc. Chem. Res., 2016, 49, 974 CrossRef CAS PubMed; (m) P. Renzi, J. Hioe and R. M. Gschwind, Acc. Chem. Res., 2017, 50, 2936 CrossRef CAS PubMed.
- (a) Y. Hayashi, H. Gotoh, T. Hayashi and M. Shoji, Angew. Chem., Int. Ed., 2005, 44, 4212 CrossRef CAS PubMed; (b) J. Franzen, M. Marigo, D. Fielenbach, T. C. Wabnitz, A. Kjærsgaard and K. A. Jørgensen, J. Am. Chem. Soc., 2005, 127, 18296 CrossRef CAS PubMedFor reviews, see: (c) A. Mieglo and C. Palomo, Chem.–Asian J., 2008, 3, 922 CrossRef PubMed; (d) K. L. Jensen, G. Dickmeiss, H. Jiang, Ł. Albrecht and K. A. Jørgensen, Acc. Chem. Res., 2012, 45, 248 CrossRef CAS; (e) B. S. Donslund, T. K. Johansen, P. H. Poulsen, K. S. Halskov and K. A. Jørgensen, Angew. Chem., Int. Ed., 2015, 54, 13860 CrossRef CAS PubMed; (f) G. J. Reyes-Rodríguez, N. M. Rezayee, A. Vidal-Albalat and K. A. Jørgensen, Chem. Rev., 2019, 119, 4221 CrossRef PubMed.
- Y. Zhou, Z. Zhou, W. Du and Y.-C. Chen, Acta Chim. Sin., 2018, 76, 382 CrossRef CAS.
- (a) T. Imagawa, N. Sueda and M. Kawanisi, Tetrahedron, 1974, 30, 2227 CrossRef CAS; (b) M. E. Jung, L. J. Street and Y. Usui, J. Am. Chem. Soc., 1986, 108, 6810 CrossRef CAS; (c) I. E. Markó and G. R. Evans, Tetrahedron Lett., 1993, 34, 7309 CrossRef; (d) G. A. Kraus and S. Wang, RSC Adv., 2017, 89, 56760 RSC; (e) H. Yu and G. A. Kraus, Tetrahedron Lett., 2018, 59, 4008 CrossRef CAS.
- Although reactions were conducted under an argon atmosphere, strict exclusion of air and water was not essential to its success.
- All crystals obtained for diffraction analysis in this study were racemates; no ee measurements were taken of the resultant mother liquors to determine the degree of enhancement. The absolute configuration of 24 as drawn was determined using a copper source.
- Based on crude NMR analysis, acidic additives seem to promote undesired pathways, as reaction profiles in their presence were never clean. For a review on the importance of additives in such transformations, see: L. Hong, W. Sun, D. Yang, G. Li and R. Wang, Chem. Rev., 2016, 116, 4006 CrossRef CAS PubMed.
- (a) S. Bertelsen, M. Marigo, S. Brandes, P. Diner and K. A. Jørgensen, J. Am. Chem. Soc., 2006, 128, 12973 CrossRef CAS PubMed; (b) Ł. Albrecht, G. Dickmeiss, C. F. Weise, C. Rodríguez-Escrich and K. A. Jørgensen, Angew. Chem., Int. Ed., 2012, 51, 13109 CrossRef; (c) C. F. Weise, V. H. Lauridsen, R. S. Rambo, E. H. Iversen, M.-L. Olsen and K. A. Jørgensen, J. Org. Chem., 2014, 79, 3537 CrossRef CAS PubMed; (d) B. S. Donslund, A. Monleón, J. Larsen, L. Ibsen and K. A. Jørgensen, Chem. Commun., 2015, 51, 13666 RSC.
- All ee values for the minor diastereomers are provided in the ESI† section. In addition, we were able to obtain an X-ray crystal structure of exo 41 which enabled us to assign its configuration in relative terms. Given the generally low ee values for the exo products, we have been unable to obtain a suitable crystal structure to determine absolute configuration despite significant effort.
- Q. Liu and L. Zu, Angew. Chem., Int. Ed., 2018, 57, 9505 CrossRef CAS PubMed.
- G. H. Posner, T. D. Nelson, C. M. Kinter and K. Afarinkia, Tetrahedron Lett., 1991, 32, 5295 CrossRef CAS.
- Note that the s-cis conformation presented in the proposed transition state analysis is based on the absolute stereochemistry of the products as obtained by X-ray crystallographic analysis of endo 41 and exo 41. It is believed that the s-cis configuration is preferred due to the A1,3 steric interaction present in the s-trans conformation as well as the additional frontier molecular orbital stabilization when in the s-cis conformation. We thank a referee for his/her comments which provoked further thought on the proposed transition state structures.
- K. C. Nicolaou, A. A. Estrada, M. Zak, S. H. Lee and B. S. Safina, Angew. Chem., Int. Ed., 2005, 44, 1378 CrossRef CAS PubMed.
- D. H. R. Barton, D. Crich and W. B. Motherwell, Tetrahedron, 1985, 41, 3901 CrossRef CAS.
- K. Afarinkia and G. H. Posner, Tetrahedron Lett., 1992, 33, 7839 CrossRef CAS.
- In all cases, the ee of the initial Diels–Alder products was retained in the subsequent transformations.
- As judged by using standard search engines in early November, 2019.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1965037–1965044. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc05738b |
| This journal is © The Royal Society of Chemistry 2020 |