
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSerine is the molecular source of the NH(CH2)2 bridgehead moiety of the in vitro assembled [FeFe] hydrogenase H-cluster†
Guodong
Rao
 ,
Lizhi
Tao
and
R. David
Britt
*
,
Lizhi
Tao
and
R. David
Britt
*
Department of Chemistry, University of California, Davis, CA 95616, USA. E-mail: rdbritt@ucdavis.edu
First published on 18th December 2019
Abstract
The active site of [FeFe] hydrogenase, the H-cluster, consists of a canonical [4Fe–4S]H subcluster linked to a unique binuclear [2Fe]H subcluster containing three CO, two CN− and a bridging azadithiolate (adt, NH(CH2S−)2) ligand. While it is known that all five diatomic ligands are derived from tyrosine, there has been little knowledge as to the formation and installation of the adt ligand. Here, by using a combination of a cell-free in vitro maturation approach with pulse electronic paramagnetic resonance spectroscopy, we discover that serine donates the nitrogen atom and the CH2 group to the assembly of the adt ligand. More specifically, both CH2 groups in adt are sourced from the C3 methylene of serine.
Hydrogenases catalyze the reversible reactions of H2 oxidation and proton reduction, and are involved in many microbial metabolic pathways.1 [FeFe] hydrogenases in particular are hyper-efficient, with turnover rates up to 104/s.2 This has led to intense focus on [FeFe] hydrogenases for sustainable production of H2 and the design of fuel cells.3 The active site of [FeFe] hydrogenases is a six-iron cofactor called the H-cluster (Scheme 1), which consists of a canonical cuboid [4Fe–4S]H subcluster linked through a bridging cysteine (Cys) residue to a binuclear [2Fe]H subcluster in which the two iron ions are coordinated by three CO, two CN− and an azadithiolate (adt, NH(CH2S−)2) bridging ligand. The [2Fe]H subcluster has been proposed to be the site for H2 binding and hydride formation,4,5 which serves as a natural blueprint for designing small molecule catalysts for hydrogen evolution reactions.6 The unique structure and catalytic activity has thus raised much interest in the biosynthesis of the H-cluster, which poses a great challenge in cofactor assembly that involves toxic ligands, oxygen sensitivity and an organic adt ligand that has little inherent stability.
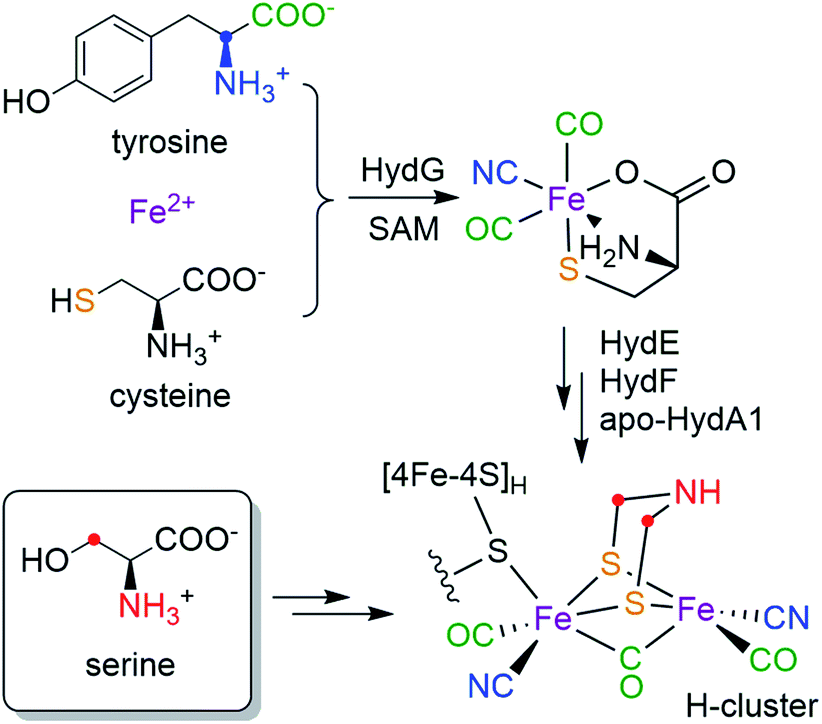 | ||
| Scheme 1 Bioassembly of the H-cluster highlighting the source of each moiety. | ||
While the [4Fe–4S]H subcluster in the H-cluster can be formed by the housekeeping gene products that are used to assemble such standard Fe–S clusters, the in vivo bioassembly of the unique [2Fe]H subcluster requires three special Fe–S “maturase” proteins: HydE, HydF, and HydG.7,8 Although the functions of HydE and HydF have not been fully elucidated,9–12 recent studies indicate that HydG is a bifunctional 4Fe–4S radical S-adenosyl-L-methionine (SAM) enzyme which lyses tyrosine to generate CO and CN− and forms a [(Cys)Fe(CO)2(CN)] organometallic precursor to the H-cluster on a dangler Fe(Cys) site in HydG.13–16 More recently, by using a synthetic [(Cys)Fe(CO)2(CN)] carrier we have shown that the two sulfur atoms in the adt ligand are derived from the precursor-bound Cys, but that the CH2NHCH2 component is not.17 Taken together, the biosynthetic origins of the [Fe2S2(CO)3(CN)2] part of the [2Fe]H subcluster are depicted in Scheme 1: all five diatomic ligands are tailored from tyrosine by HydG;18 the two sulfur atoms and the two Fe atoms are from the dangler Fe(Cys) site in HydG (which can be reconstituted with Fe2+ and free Cys in solution19). Remarkably, these components are all delivered to the binuclear cluster assembly in the form of the [(Cys)Fe(CO)2(CN)] product of HydG. Given these recent advances, the only missing part of the puzzle is the crucial NH(CH2)2 moiety: what are its molecular precursors? It has been hypothesized that HydE, which is also a 4Fe–4S radical SAM enzyme, may be involved in the formation of adt, though its physiological substrate and reaction mechanism remains under investigation.9,10 As for any enzymatic reaction, knowing the actual substrate(s) for the reaction is crucial for unraveling the ultimate mechanism. Therefore, determining of molecular sourcing of the CH2NHCH2 component of the adt bridge, currently unknown, is the focus of this work.
Assembly of the H-cluster in the lab can be achieved by semi-synthetic and biochemical approaches other than directly co-expressing hydA, hydE, hydF and hydG genes in cells. One very useful method alleviates the need for HydG, HydE, and in some cases, HydF, by using a synthetic [Fe2(adt)(CO)4(CN)2] complex as a direct donor to the [2Fe]H subcluster assembly.20–22 Another “cell free synthesis” approach uses HydE/F/G in an in vitro H-cluster maturation reaction developed by the Swartz group.23,24 The specific in vitro maturation reaction used in our current investigation contains a mixture of E. coli cell lysate containing separately overexpressed HydE, HydF, HydG (all from Shewanella oneidensis), apo-HydA1 (from Chlamydomonas reinhardtii) that harbors the [4Fe-4S]H subcluster, and a cocktail of low molecular weight cofactors and precursors.23 This biochemical approach gives us the opportunity to use the same set of enzymes that build the H-cluster in cells, but also enables us to determine the molecular source of each of the components in the H-cluster by using isotope-labeled cofactors/precursors, a procedure that would be very difficult to carry out and fully control in vivo. For example, by supplementing 1-13C-Tyr or 2-13C-Tyr into the in vitro maturation reaction, the CO or CN− ligands to the diiron subcluster of the maturated HydA1 are respectively labeled with 13C.25,26 The presence of these 13C labels can then in turn be detected and analyzed by using advanced electron paramagnetic resonance (EPR) spectroscopy to measure the hyperfine couplings between the magnetic 13C nuclei and the unpaired electron spin distributed over the H-cluster in its redox-poised paramagnetic states. In this work, we now search for the source(s) of the CH2NHCH2 moiety by using a similar strategy of in vitro maturation coupled to high resolution EPR to screen the assembly products formed with various isotopically labeled small molecule candidates. The presence of nitrogen element in the CH2NHCH2 fragment suggests an amino acid origin as one possibility. A systematic screening by pulse EPR of the in vitro maturation products generated with 13C, 15N, and 2H-labeled amino acids reveals that serine (Ser) serves as a molecular source for the NH(CH2)2 moiety of the H-cluster.
Results
To test whether one or more amino acids may provide the source for the CH2NHCH2 moiety, we performed the maturation reaction supplemented with a fully 13C, 15N-labeled amino acids mixture (see methods for details27). As expected, the maturation reaction afforded holo-HydA1 with the characteristic Hox EPR spectrum when redox-poised in the Hox state via thionine-oxidation (Fig. 1a and S1†). We then employed pulse EPR spectroscopy to examine whether the adt ligand is labeled, i.e., whether adt is formed from any of the labeled amino acids. The hyperfine coupling of 15N in the adt ligand, previously labeled by using the synthetic [Fe2(15N-adt)(CO)4(CN)2] complex, has been investigated by X-band HYSCORE spectroscopy, revealing a hyperfine tensor of A15N = [1.9, 1.6, 1.6] MHz.28 This doublet of 15N peaks (accompanied by the disappearance of some 14N features), centered at the Larmor frequency of 15N (ν15N = 1.02 MHz at 333 mT) and separated by 1.9 MHz, is exactly found in the HYSCORE spectrum recorded at g = 2.103, g1 of the Hox state of HydA1 maturated from labeled amino acids mixture (Fig. 2), strongly suggesting that the adt of this HydA1 sample is labeled with 15N. Moreover, the same HYSCORE spectrum also exhibits cross-peaks centered at the 13C Larmor frequency (ν13C = 3.56 MHz at 333 mT) and not present in the native HydA1 sample, with a hyperfine value of ca. 3 MHz. This value is consistent with the recently reported ENDOR study of the 13C-adt labeled by using the synthetic [Fe2(13C-adt)(CO)4(CN)2] complex,29 and different from those of the five CO and CN− ligands,25,26 proving that the adt of the maturated HydA1 must also be labeled with 13C. Thus, these HYSCORE results demonstrate that nitrogen and carbon atoms in the adt ligand indeed originate from one or more amino acids.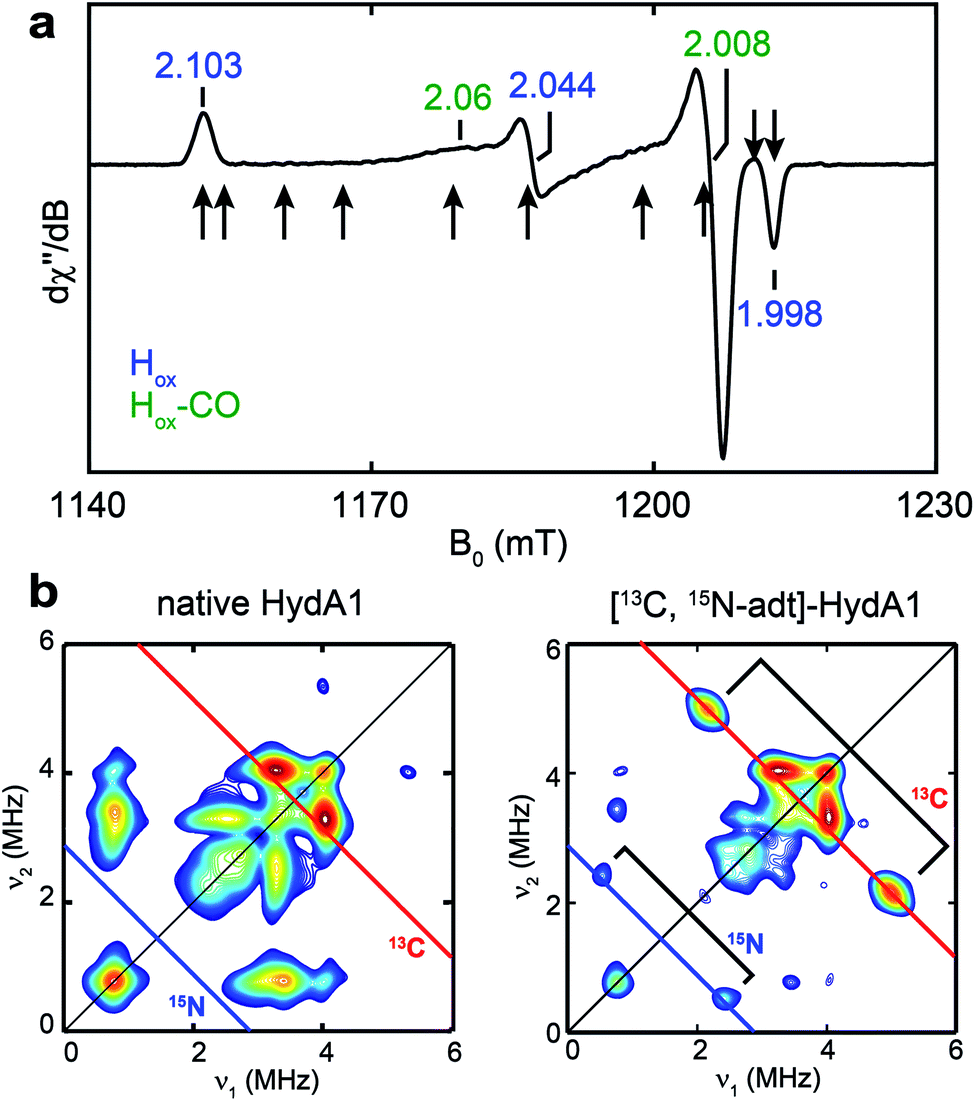 | ||
| Fig. 1 EPR and HYSCORE spectra of [13C, 15N-adt]-HydA1. (a) Q-band echo-detected EPR spectrum of a typical HydA1 sample poised in the Hox state via thionine oxidation. The field positions where the 13C Mims ENDOR spectra were collected (Fig. 3b) are indicated by arrows. See ESI† for spectral simulation. (b) X-band HYSCORE spectra of native HydA1 (left) and HydA1 maturated from a reaction supplemented with 13C3, 15N-Ser (right), both recorded at g = 2.103, g1 of Hox. Blue and red antidiagonal lines indicate the Larmor frequencies of 15N and 13C, respectively. Conditions: frequency = 9.8 GHz; temperature = 15 K; τ = 140 ns; π/2 = 16 ns. | ||
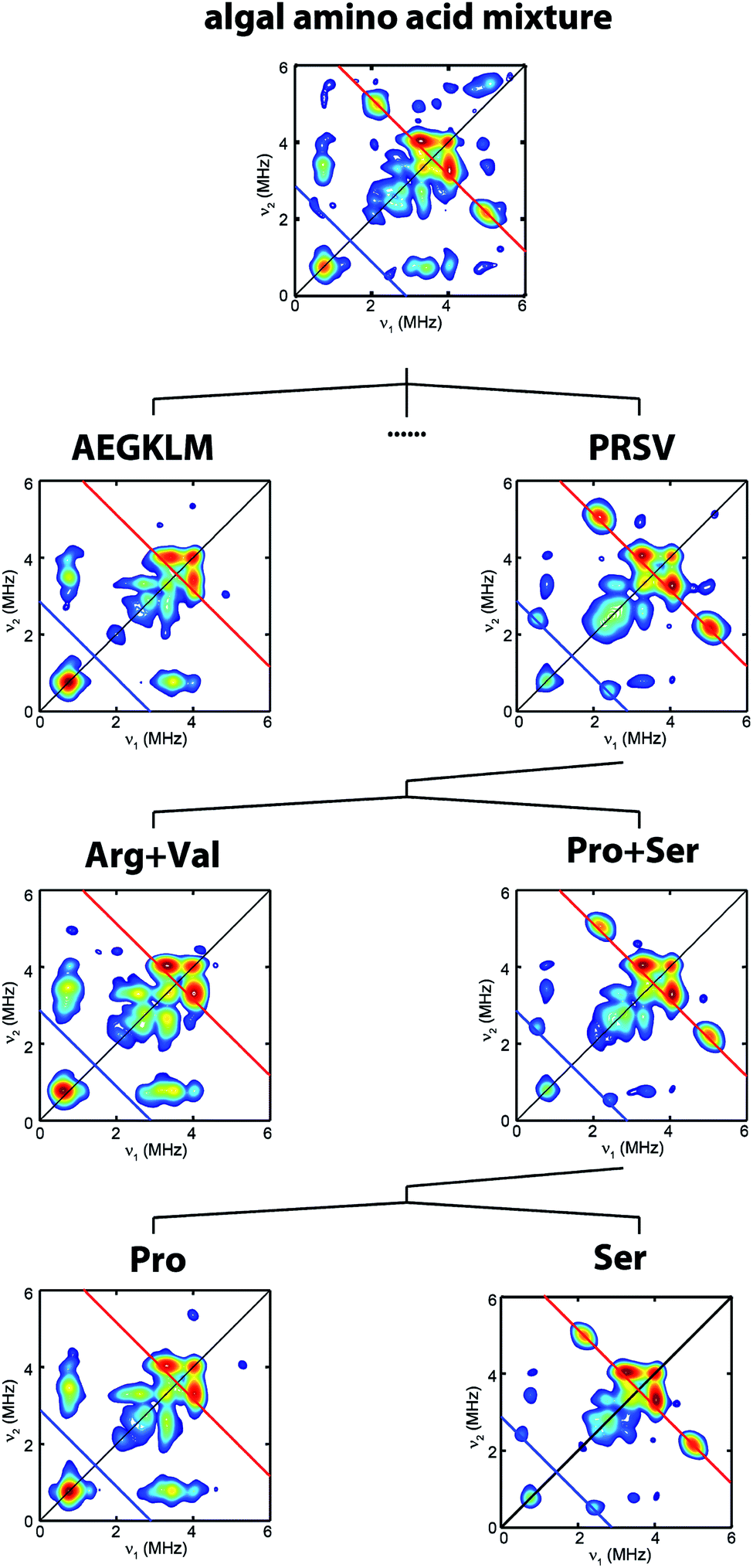 | ||
| Fig. 2 Process to identify serine as the source of the NH(CH2)2 moiety. HYSCORE spectra of CrHydA1 Hox samples isolated from maturation reactions supplemented with different sets of 13C, 15N-amino acid mixtures. Amino acid codes follow their standard abbreviations. Cys and Tyr have been previously tested.17,25 Eight amino acids (Asp, Phe, His, Ilv, Asn, Gln, Thr, Trp) have not been individually tested. Spectrometer settings are as indicated in Fig. 1. | ||
To clarify which amino acid(s) gives rise to the 15N and 13C HYSCORE features, we performed additional maturation reactions supplemented with different sets of amino acids mixture to stepwise identify possible candidates. As shown in Fig. 2, this process leads to the discovery that the bridge 13C and 15N HYSCORE signals are present when 13C3, 15N-Ser alone is supplemented into the maturation reaction, hence revealing that both the nitrogen and the carbon atoms in the adt ligand are derived from serine.
The hyperfine couplings of both 15N and 13C in the adt ligand are further analyzed by Q-band field-dependent Mims ENDOR spectroscopy (Fig. 3). The 15N hyperfine tensor of A15N = [1.90, 1.57, 1.63] MHz (aiso15N = 1.70 MHz) extracted from the ENDOR simulations is almost identical to that determined from the previous HYSCORE study with the synthetic subcluster labeling,28 further validating our assignment of 15N labeling in the adt ligand as sourced from the amino group of serine (Fig. 3a). Simulation of the field-dependent 13C ENDOR spectra requires two distinct 13C hyperfine tensors of +[3.40, 1.35, 1.37] MHz (aiso13C = 2.04 MHz) and −[0.28, 1.32 1.38] MHz (aiso13C = −0.99 MHz), with their signs determined by various mixing-time ENDOR experiments (Fig. 3b, S2 and Table 1). The ENDOR spectra near g2 also have contributions from the Hox-CO state, which is analyzed in Fig. S3.† In comparison, the hyperfine tensors of the 13C nuclei in the recent ENDOR study of HydA1 maturated with [Fe2(13C-adt)(CO)4(CN)2] complex are +[3.30, 1.30, 1.00] and −[0.45, 1.75, 1.49] MHz (Table 1).29 Both the magnitudes and signs of the parallel sets of 13C hyperfine tensors agree quite well, despite that the 13C atoms in the adt ligand are introduced with completely different approaches. We note that the second of the two inequivalent carbon ENDOR signals is not distinguished in the HYSCORE spectrum, because the corresponding crosspeaks overlap with the strong 14N features. It is interesting that the two adt carbons have such different hyperfine tensors given the inherent symmetry of the adt ligand. While our isotope labeling experiments are not able to assign which 13C hyperfine interaction corresponds to which specific adt carbon, a specific assignment has been made by quantum chemical approaches in the recent report, which indicates that the different signs of these two 13C hyperfine couplings are related to the asymmetric coordination of the distal iron site with its CO and CN− ligands.29 The two methylene groups are also geometrically inequivalent as seen from the X-ray crystallographic structure of the H-cluster (Fig. S4†).30 This asymmetry is proposed to be relevant for catalysis.29
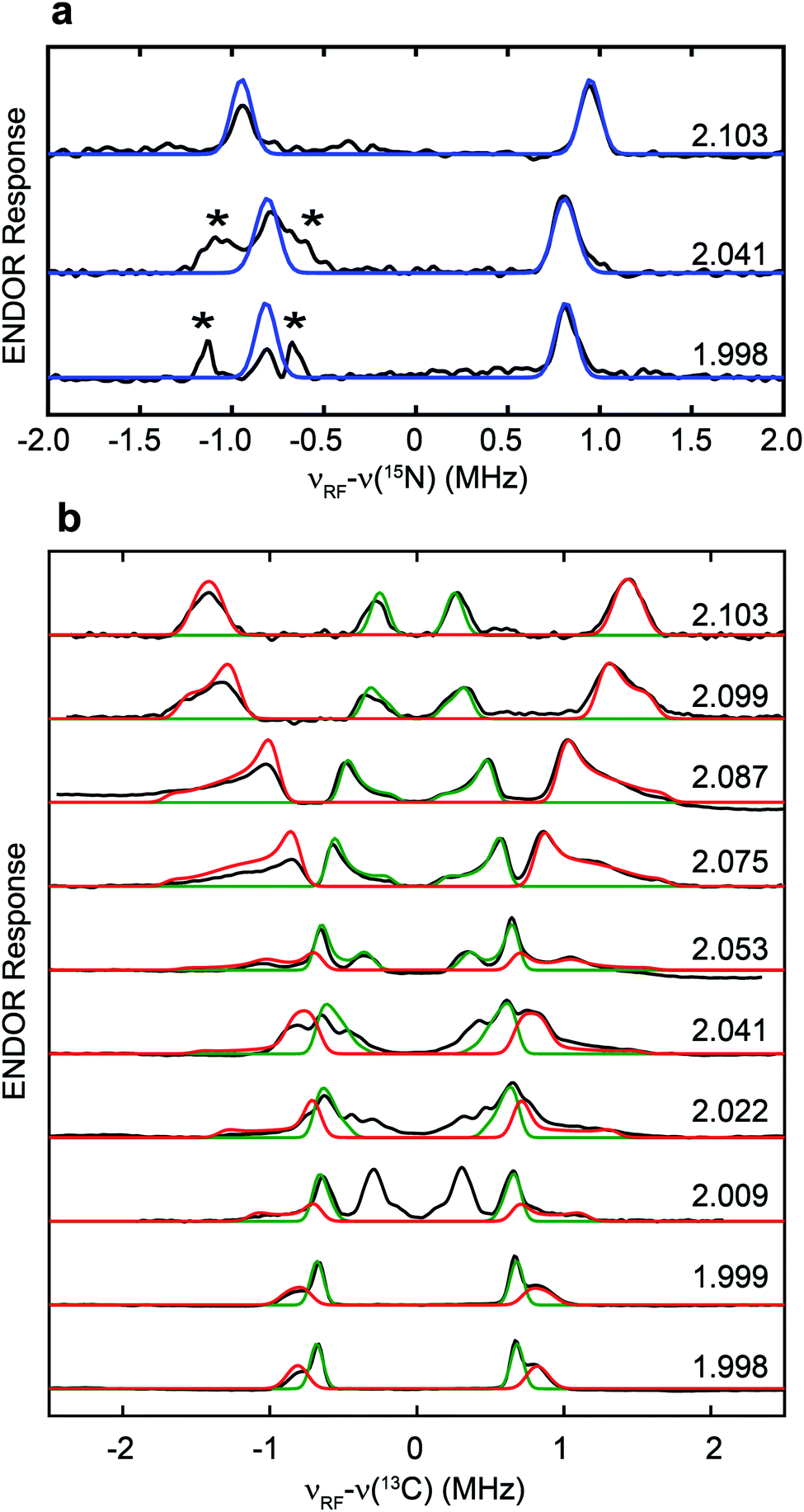 | ||
| Fig. 3 Q-band field-dependent 15N (a) and 13C (b) Mims ENDOR spectra of the [13C, 15N-adt]-HydA1 Hox sample. Black traces: experimental data. Blue traces: simulation for 15N with A15N = [1.90, 1.57, 1.63] MHz, Euler angle = [0, 0, 0]°. Red and green traces: simulations for 13C with A13C = +[3.40, 1.35, 1.37] MHz, Euler angle = [21, 21, 0]° and A13C = −[0.28, 1.32, 1.38] MHz, Euler angle = [25, 6, 0]°, respectively. ENDOR features marked by asterisks in panel (a) correspond to the third order harmonics of 13C ENDOR signals shown in panel (b). Conditions: frequency = 34.1 GHz; temperature = 15 K; τ = 260 ns; π/2 pulse = 12 ns; RF pulse = 30 µs. | ||
To determine which carbon of Ser is transferred into adt, and to probe whether both adt carbons have the same source, we performed the maturation reaction supplemented with specific labeled Ser: 1-13C-Ser, 2-13C-Ser or 3-13C-Ser. As shown in Fig. 4a, the 13C Mims ENDOR spectrum of [3-13C-Ser]-HydA1 is identical to that of [13C3, 15N-Ser]-HydA1, indicating that both carbons in the adt ligand are probably sourced from Ser C3 carbon. We note that we cannot yet rigorously exclude the possibility that only one carbon is derived from 3-13C-Ser, with another coming from a different source, and that in the ENDOR spectra we observe an equal mix of two isotopologues, resulting in each carbon half 13C-labeled from serine. Experiments are ongoing to distinguish between these two possibilities.
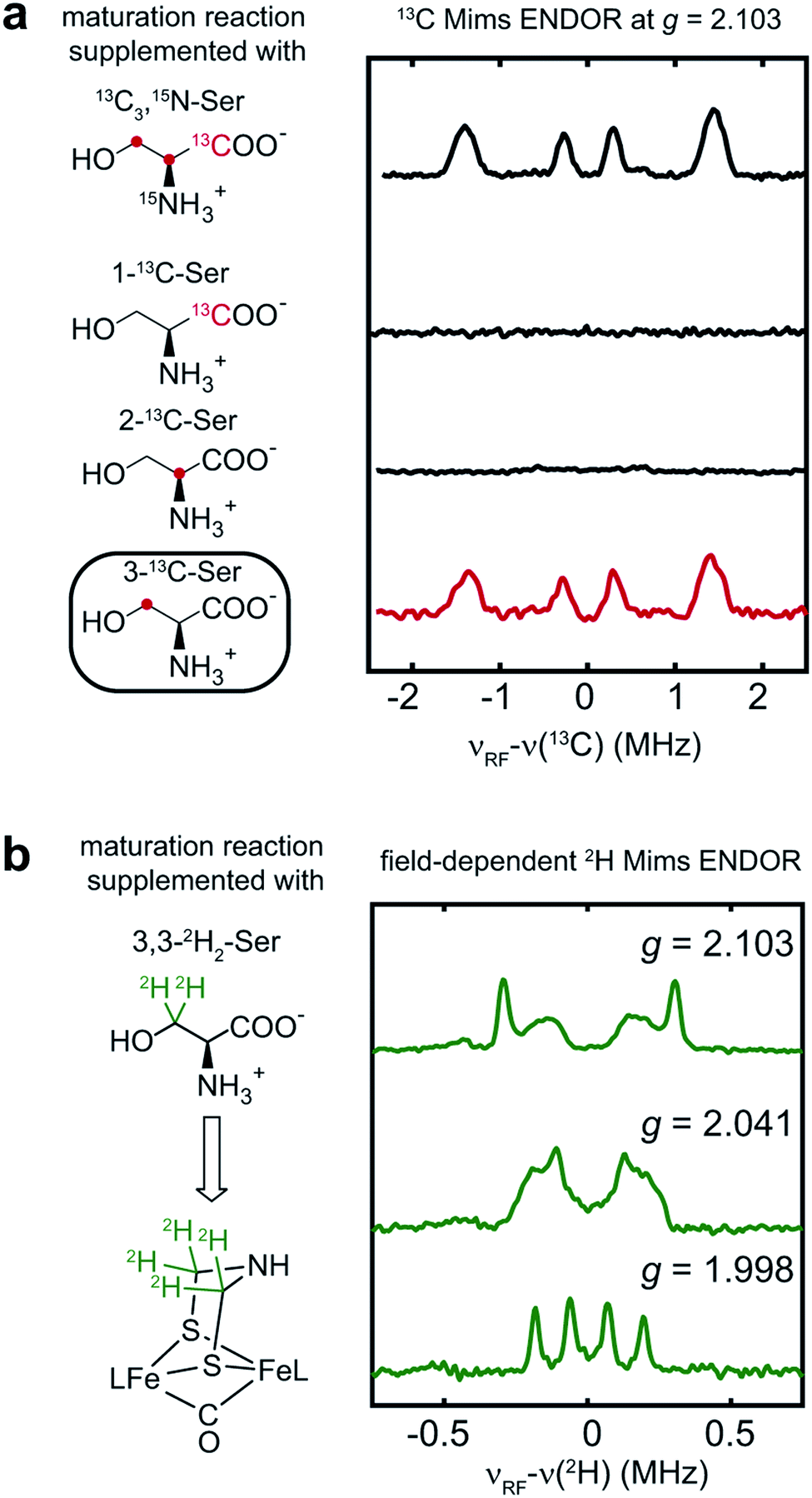 | ||
| Fig. 4 Serine donates the C3 methylene group to the H-cluster. (a) Comparison of 13C Mims ENDOR spectra of HydA1 samples maturated from reactions supplemented with differently labeled Ser. (b) Q-band field-dependent 2H Mims ENDOR spectra of HydA1 samples maturated from reaction supplemented with 3,3-2H2-Ser. ENDOR conditions are indicated in Fig. 3. | ||
In addition to the 3-13C-Ser labeling experiment, we performed the maturation reaction supplemented with 3,3-2H2-Ser. As expected, 2H incorporation into the H-cluster is revealed by the 2H Mims ENDOR spectra which disclose overlapping hyperfine couplings of ca. 0.13–0.60 MHz (Fig. 4b). These 2H signals must be attributed to the C2H2 groups in the CH2NHCH2 moiety since they are the only non-exchangeable hydrogens in the [2Fe]H subcluster. Combined with the 13C ENDOR results, we can therefore conclude that the entire CH2 group is transferred as a unit from Ser to the adt ligand.
Discussion
The combination of the in vitro maturation approach with high resolution pulse EPR has allowed us to screen a number of amino acid precursors and to determine that the atoms of the bridgehead CH2NHCH2 moiety of the H-cluster are derived from serine, specifically the amino nitrogen and carbons and hydrogens of the 3-CH2 group. Our results provide new and significant insights in the bioassembly of the H-cluster. Using results from previous studies and this work, the biosynthetic origins of all the parts of the [2Fe]H subcluster are now completely determined—it can be built from three amino acids, Tyr, Cys (in the form of a dangler Fe(Cys) site) and Ser (Scheme 1). While it is well-established how Tyr is cleaved by the radical SAM enzyme HydG to generate the diatomic ligands,14 it is not yet clear how Cys and Ser are metabolized and what specific enzymes are involved. Our recent semi-synthesis study indicates that Cys of the [(Cys)Fe(CO)2(CN)] HydG product donates its sulfur atom to the H-cluster, with the rest of Cys converted into pyruvate, and presumably ammonia and water.17 Here we show that only the C3 carbon/hydrogens and the nitrogen atoms in Ser are incorporated into the H-cluster, but there remain open questions as to how the CH2 group and the amino group insertions are carried out, and what exact roles HydE and HydF may play in this process. Certainly, having now assigned molecular sources of the atoms in the binuclear Fe subcluster will serve as a crucial basis for future determination of the detailed enzymology required in the cluster assembly.It should be possible for Ser to be involved in the reactions catalyzed by HydE (or HydF), although it has been proposed that HydE chemistry is related to sulfur-containing substrates.9,10 While we are further exploring this direction, we also note that pyridoxal-5′-phosphate (PLP) is one of the necessary cofactor in the in vitro maturation reaction to form the H-cluster.31 Noting such a requirement for cysteine was one of the lines of evidence that ultimately led to the assignment of cysteine coordination to the dangler Fe(II) in the resting state of HydG.19 However, the sequences of HydE, HydF or HydG do not contain any clear PLP-binding domains, nor has there been experimental evidence showing that these enzymes bind PLP. This observation leads us to end this article with some speculation that some PLP-dependent enzyme present in the E. coli lysate may be involved in the maturation cascade. If so, presumably this enzyme is also then present in the native [FeFe] hydrogenase-producing organisms. Since most PLP-dependent enzymes use some amino acids as their substrates,32 it is possible that the serine identified in the current study could be the substrate of some PLP-dependent enzyme that generates a downstream precursor for the adt ligand. An examination of the PLP-dependent enzymes in E. coli metabolic pathways reveals two such candidates that use Ser as their substrate: serine dehydratase and serine hydroxymethyltransferase. The former enzyme cleaves Ser into pyruvate and ammonia,33 while the latter enzyme transfers the C3 carbon of Ser onto tetrahydrofolate (THF) to form 5,10-methylene-THF which is a cofactor for several biochemical reactions where it serves as one-carbon carrier.34,35 This carbon transfer mechanism of serine hydroxymethyltransferase is possibly consistent with our observation that the C3 carbon of Ser is transferred into the H-cluster. In this regard, a possible mechanism of adt formation is discussed in the ESI.†
We are left with some open questions regarding the roles of Ser, HydE and HydF, and possible enzymes present in the lysate, that need be addressed in future studies regarding the late-stages of H-cluster bioassembly. We anticipate that further use of pulse EPR combined with isotopic editing via the in vitro maturation process will help define these reactions, particularly now with these current results that we finally know all of the molecular sources needed to assemble the H-cluster (Scheme 1).
Methods
General consideration
Individual 13C, 15N and 2H-labeled amino acids and a full 13C, 15N-labeled amino acid mixture (algal source, 90% amino acid) were purchased from Cambridge Isotope Inc. All other chemicals were purchased from Sigma-Aldrich unless specified. All handling of Fe–S cluster proteins, including purification of HydA1, preparation of HydE, HydF and HydG lysate, maturation of HydA1, and preparation of EPR samples, were carried out in an anaerobic chamber with O2 level <1 ppm.In vitro maturation of CrHydA1
The maturation reaction requires apo-Chlamydomonas reinhardtii HydA1 that harbors the [4Fe–4S]H subcluster, lysate of cells overexpressing Shewanella oneidensis HydE, HydF and HydG, and necessary small molecules. Purification of apo-CrHydA1 with a N-terminal Strep-II tag, and preparation of untagged HydE, HydF and HydG lysate, followed previous procedures.23,26 Apo-CrHydA1 was first desalted by using a PD-10 desalting column (GE Healthcare) to remove the desthiobiotin introduced during protein purification. For the maturation of CrHydA1, each 25 mL reaction contained 12.5 mL HydG lysate, 4 mL HydF lysate, 1.5 mL HydE lysate, 4 mM DTT, 1 mM Fe2+, 1 mM Cys, 2 mM S-adenosyl-L-methionine (SAM), 1 mM pyridoxal phosphate (PLP), 20 mM guanosine triphosphate (GTP), 2 mM natural abundance tyrosine, 2 mM sodium dithionite, ∼10 µM apo-CrHydA1, as well as desired labeled amino acid mixture (3 mM for each amino acids). When maturation reaction was performed with the labeled amino acid mixture added, natural abundance Tyr was added to 10 mM to suppress labeling of CO/CN− in the H-cluster. Tyr is the donor to the CO and CN− ligand but not the adt ligand as previously examined.25 The pH of all the components were adjusted to ∼7.5 before added into the cell lysate to prevent precipitation. The reaction mixture was incubated at room temperature in an anaerobic chamber containing 2% H2 for ∼16 h, and then clarified by centrifugation. Maturated CrHydA1 was re-purified from the supernatant by using a Strep-Tactin chromatography as described previously.26 Fractions containing CrHydA1 were pooled and concentrated to ∼300 µM.For EPR experiments, as-purified holo-CrHydA1 was transferred into a glovebox containing 100% N2, oxidized by using 2 mM thionine, quickly transferred into the EPR tube, and frozen for storage.
EPR spectroscopy
EPR spectroscopy was performed in the CalEPR center in Department of Chemistry, University of California at Davis. Pulse X-band (9.8 GHz) HYSCORE and Q-band (34 GHz) field-swept EPR and ENDOR experiments were performed on the Bruker Biospin EleXsys 580 spectrometer. X-band HYSCORE spectra were recorded using a split-ring MS5 resonator. Q-band experiments were performed using a R. A. Isaacson cylindrical TE011 resonator.36 All experiments were performed at 15 K, and cryogenic temperatures were achieved and controlled with an Oxford Instrument CF935 cryostat. Q-band Mims ENDOR experiments employed a 10 W microwave amplifier with an attenuator (0–60 dB) and a 1 kW RF (radio frequency) amplifier with an RF range of 0.3–35 MHz. An RF low-pass filter with a cutoff range of 35 MHz was used to removed unwanted RF signals at higher frequencies that causes high order harmonics in the low-frequency range. The pulse sequences employed were as follows: electron spin-echo detected field swept EPR (π/2-τ-π-τ-echo), hyperfine sublevel correlation (HYSCORE; π/2-τ-π/2-t1-π-t2-π/2-τ-echo), and Mims electron nuclear double resonance (Mims ENDOR; π/2-τ-π/2-RF-π/2-τ-echo). Simulations of EPR spectra were performed in Matlab 2014a (MathWorks, Inc.) and EasySpin 5.2.13 toolbox.37 Euler angles correlates A tensors to g tensors and follow z–y–z convention.Mims ENDOR is a pulse EPR technique that resolves small hyperfine couplings with high sensitivity. For nuclei with I = 1/2 (15N and 13C in this study), the ENDOR transitions for the ms = ±1/2 are detected. To first order approximation, two NMR transitions are observed centered at the Larmor frequency of the nuclei (νN) and split by orientation-dependent hyperfine coupling (A), ν± = νN ± A/2, in the weak-coupling regime. The intensities of Mims-ENDOR response are modulated by the response factor (R) which is a function of A and the time interval between the first and the second π/2 microwave pulse in the three-pulse sequence (τ): R ∼ [1 − cos(2πAτ)]. As a consequence, intensity of Mims-ENDOR vanishes at τ = n/A (n = 1, 2, 3…), the so-called “Mims holes”. The first Mims hole occurs at τ = 1/A. In our Q-band instrument, the shortest τ that can be used is ∼250 ns to allow the echo to be separated from the ring-down, which means Mims ENDOR can detect A up to ∼4 MHz without significant Mims holes effect. The effect of Mims holes on the ENDOR lineshape is also considered in the spectral simulation by specifying the τ value in the experimental parameters. For nuclei with I = 1 (2H in our study), each NMR transition is further split by the nuclear quadrupole interaction (usually quite small for 2H, ∼50 kHz, and not well-resolved).
The signs of the 13C hyperfine tensors are determined by various mixing time Mims ENDOR (VMT ENDOR) spectroscopy (see ESI† for the pulse sequence). With longer mixing-time after the RF pulse, relaxation of the α electron spin manifold (ms = +1/2, ν+1/2 = νN − A/2) relative to the β manifold (ms = −1/2, ν−1/2 = νN + A/2) decreases the relative ENDOR signal intensity within this α manifold. The α manifold corresponds to the low-frequency ENDOR peak when A > 0, and to the high-frequency ENDOR peak when A < 0. Therefore, if the high-frequency ENDOR peak has decreased relative signal intensity at longer mixing time, then A < 0, and vice versa.
Author contribution
G. R., L. T. and R. D. B. designed research. G. R. and L. T. performed research and analyzed data. G. R., L. T. and R. D. B. wrote the paper.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work is funded by the National Institutes of Health (1R35GM126961 to RDB). We thank James Swartz (Stanford) for providing the E. coli strains used in this study.References
- W. Lubitz, H. Ogata, O. Rudiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- K. Pandey, S. T. Islam, T. Happe and F. A. Armstrong, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 3843–3848 CrossRef CAS PubMed.
- R. M. Evans, B. Siritanaratkul, C. F. Megarity, K. Pandey, T. F. Esterle, S. Badiani and F. A. Armstrong, Chem. Soc. Rev., 2019, 48, 2039–2052 RSC.
- D. W. Mulder, Y. Guo, M. W. Ratzloff and P. W. King, J. Am. Chem. Soc., 2017, 139, 83–86 CrossRef CAS PubMed.
- E. J. Reijerse, C. C. Pham, V. Pelmenschikov, R. Gilbert-Wilson, A. Adamska-Venkatesh, J. F. Siebel, L. B. Gee, Y. Yoda, K. Tamasaku, W. Lubitz, T. B. Rauchfuss and S. P. Cramer, J. Am. Chem. Soc., 2017, 139, 4306–4309 CrossRef CAS PubMed.
- T. B. Rauchfuss, Acc. Chem. Res., 2015, 48, 2107–2116 CrossRef CAS PubMed.
- E. M. Shepard, F. Mus, J. N. Betz, A. S. Byer, B. R. Duffus, J. W. Peters and J. B. Broderick, Biochemistry, 2014, 53, 4090–4104 CrossRef CAS PubMed.
- J. B. Broderick, A. S. Byer, K. S. Duschene, B. R. Duffus, J. N. Betz, E. M. Shepard and J. W. Peters, J. Biol. Inorg Chem., 2014, 19, 747–757 CrossRef CAS PubMed.
- J. N. Betz, N. W. Boswell, C. J. Fugate, G. L. Holliday, E. Akiva, A. G. Scott, P. C. Babbitt, J. W. Peters, E. M. Shepard and J. B. Broderick, Biochemistry, 2015, 54, 1807–1818 CrossRef CAS PubMed.
- R. Rohac, P. Amara, A. Benjdia, L. Martin, P. Ruffié, A. Favier, O. Berteau, J.-M. Mouesca, J. C. Fontecilla-Camps and Y. Nicolet, Nat. Chem., 2016, 8, 491 CrossRef CAS PubMed.
- E. M. Shepard, S. E. McGlynn, A. L. Bueling, C. S. Grady-Smith, S. J. George, M. A. Winslow, S. P. Cramer, J. W. Peters and J. B. Broderick, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 10448–10453 CrossRef CAS PubMed.
- G. Caserta, L. Pecqueur, A. Adamska-Venkatesh, C. Papini, S. Roy, V. Artero, M. Atta, E. Reijerse, W. Lubitz and M. Fontecave, Nat. Chem. Biol., 2017, 13, 779–784 CrossRef CAS PubMed.
- J. M. Kuchenreuther, W. K. Myers, T. A. Stich, S. J. George, Y. Nejatyjahromy, J. R. Swartz and R. D. Britt, Science, 2013, 342, 472–475 CrossRef CAS PubMed.
- J. M. Kuchenreuther, W. K. Myers, D. L. Suess, T. A. Stich, V. Pelmenschikov, S. A. Shiigi, S. P. Cramer, J. R. Swartz, R. D. Britt and S. J. George, Science, 2014, 343, 424–427 CrossRef CAS PubMed.
- D. L. Suess, C. C. Pham, I. Burstel, J. R. Swartz, S. P. Cramer and R. D. Britt, J. Am. Chem. Soc., 2016, 138, 1146–1149 CrossRef CAS PubMed.
- G. Rao, L. Tao, D. L. M. Suess and R. D. Britt, Nat. Chem., 2018, 10, 555–560 CrossRef CAS PubMed.
- G. Rao, S. A. Pattenaude, K. Alwan, N. J. Blackburn, R. D. Britt and T. B. Rauchfuss, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 20850–20855 CrossRef CAS PubMed.
- J. M. Kuchenreuther, S. J. George, C. S. Grady-Smith, S. P. Cramer and J. R. Swartz, PLoS One, 2011, 6, e20346 CrossRef CAS PubMed.
- D. L. Suess, I. Burstel, L. De La Paz, J. M. Kuchenreuther, C. C. Pham, S. P. Cramer, J. R. Swartz and R. D. Britt, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11455–11460 CrossRef CAS PubMed.
- R. Gilbert-Wilson, J. F. Siebel, A. Adamska-Venkatesh, C. C. Pham, E. Reijerse, H. Wang, S. P. Cramer, W. Lubitz and T. B. Rauchfuss, J. Am. Chem. Soc., 2015, 137, 8998–9005 CrossRef CAS PubMed.
- G. Berggren, A. Adamska, C. Lambertz, T. R. Simmons, J. Esselborn, M. Atta, S. Gambarelli, J. M. Mouesca, E. Reijerse, W. Lubitz, T. Happe, V. Artero and M. Fontecave, Nature, 2013, 499, 66–69 CrossRef CAS PubMed.
- J. Esselborn, C. Lambertz, A. Adamska-Venkatesh, T. Simmons, G. Berggren, J. Noth, J. Siebel, A. Hemschemeier, V. Artero, E. Reijerse, M. Fontecave, W. Lubitz and T. Happe, Nat. Chem. Biol., 2013, 9, 607 CrossRef CAS PubMed.
- J. M. Kuchenreuther, S. A. Shiigi and J. R. Swartz, Methods Mol. Biol., 2014, 1122, 49–72 CrossRef CAS PubMed.
- M. E. Boyer, J. A. Stapleton, J. M. Kuchenreuther, C. W. Wang and J. R. Swartz, Biotechnol. Bioeng., 2008, 99, 59–67 CrossRef CAS.
- W. K. Myers, T. A. Stich, D. L. M. Suess, J. M. Kuchenreuther, J. R. Swartz and R. D. Britt, J. Am. Chem. Soc., 2014, 136, 12237–12240 CrossRef CAS PubMed.
- G. Rao and R. D. Britt, Inorg. Chem., 2018, 57, 10935–10944 CrossRef CAS PubMed.
- Excess natural abundance Tyr was added to ∼10 mM in this case to suppress labeling of CO/CN in the H-cluster.
- A. Adamska-Venkatesh, S. Roy, J. F. Siebel, T. R. Simmons, M. Fontecave, V. Artero, E. Reijerse and W. Lubitz, J. Am. Chem. Soc., 2015, 137, 12744–12747 CrossRef CAS PubMed.
- E. J. Reijerse, V. Pelmenschikov, J. A. Birrell, C. P. Richers, M. Kaupp, T. B. Rauchfuss, S. P. Cramer and W. Lubitz, J. Phys. Chem. Lett., 2019, 6794–6799, DOI:10.1021/acs.jpclett.9b02354.
- J. Esselborn, N. Muraki, K. Klein, V. Engelbrecht, N. Metzler-Nolte, U. P. Apfel, E. Hofmann, G. Kurisu and T. Happe, Chem. Sci., 2016, 7, 959–968 RSC.
- J. M. Kuchenreuther, R. D. Britt and J. R. Swartz, PLoS One, 2012, 7, e45850 CrossRef CAS PubMed.
- T. P. Begley and M. D. Toney, in Wiley Encyclopedia of Chemical Biology, 2008, pp. 1–6 Search PubMed.
- G. Litwack, in Human Biochemistry, ed. G. Litwack, Academic Press, Boston, 2018, pp. 359–394 Search PubMed.
- N. V. Bhagavan and C.-E. Ha, in Essentials of Medical Biochemistry, ed. N. V. Bhagavan and C.-E. Ha, Academic Press, San Diego, 2nd edn, 2015, pp. 227–268 Search PubMed.
- R. Y. Wang, W. R. Wilcox and S. D. Cederbaum, in Emery and Rimoin's Principles and Practice of Medical Genetics, ed. D. Rimoin, R. Pyeritz and B. Korf, Academic Press, Oxford, 2013, pp. 1–42 Search PubMed.
- R. Calvo, E. C. Abresch, R. Bittl, G. Feher, W. Hofbauer, R. A. Isaacson, W. Lubitz, M. Y. Okamura and M. L. Paddock, J. Am. Chem. Soc., 2000, 122, 7327–7341 CrossRef CAS.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional EPR spectra and analysis, further discussion of Ser role. See DOI: 10.1039/c9sc05900h |
| This journal is © The Royal Society of Chemistry 2020 |