
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnravelling the effect of the E545K mutation on PI3Kα kinase†
Ioannis
Galdadas
 a,
Francesco Luigi
Gervasio
*abc and
Zoe
Cournia
*d
a,
Francesco Luigi
Gervasio
*abc and
Zoe
Cournia
*d
aDepartment of Chemistry, University College London, London WC1E 6BT, UK. E-mail: f.l.gervasio@ucl.ac.uk
bInstitute of Structural and Molecular Biology, University College London, London WC1E 6BT, UK
cPharmaceutical Sciences, University of Geneva, Geneva CH-1211, Switzerland
dBiomedical Research Foundation, Academy of Athens, Athens 11527, Greece. E-mail: zcournia@bioacademy.gr
First published on 26th February 2020
Abstract
PI3Kα controls several cellular processes and its aberrant signalling is implicated in tumorigenesis. One of its hotspot mutations, E545K, increases PI3Kα lipid kinase activity, but its mode of action is only partially understood. Here, we perform biased and unbiased molecular dynamics simulations of PI3Kα and uncover, for the first time, the free energy landscape of the E545K PI3Kα mutant. We reveal the mechanism by which E545K leads to PI3Kα activation in atomic-level detail, which is considerably more complex than previously thought.
Introduction
Members of the phosphoinositide-3 kinase (PI3K) family control several cellular responses. PI3Kα, a class IA PI3K isoform, catalyses the phosphorylation of the inositol lipid PIP2 at the 3′ position of the inositol ring, generating the signalling molecule PIP3, which controls cellular activities including proliferation, survival, and motility. Elevated PI3Kα signalling can contribute to oncogenesis and is a hallmark of human cancer.1A number of structural and biophysical studies, motivated by the role of PI3Kα in cancer development, have shed light on its architecture and the regulation of its activity.2 PI3Kα is comprised of a catalytic subunit, p110α, and a regulatory subunit, p85α (Fig. 1). The p110α subunit of PI3Kα associates with the p85α subunit, which contains two SH2 domains (nSH2, cSH2) flanking a region known as the inter-SH2 (iSH2) domain (Fig. 1). The catalytic and regulatory subunits form contacts that maintain the enzyme in a low activity state in the absence of a signal. Binding of phosphotyrosine (pY)-containing peptides to the nSH2 relieves these inhibitory contacts and positions the dimer near the plasma membrane, where it can access and phosphorylate the lipid substrate PIP2 in the presence of ATP as well as receive further inputs from Ras and other signalling components.3–5
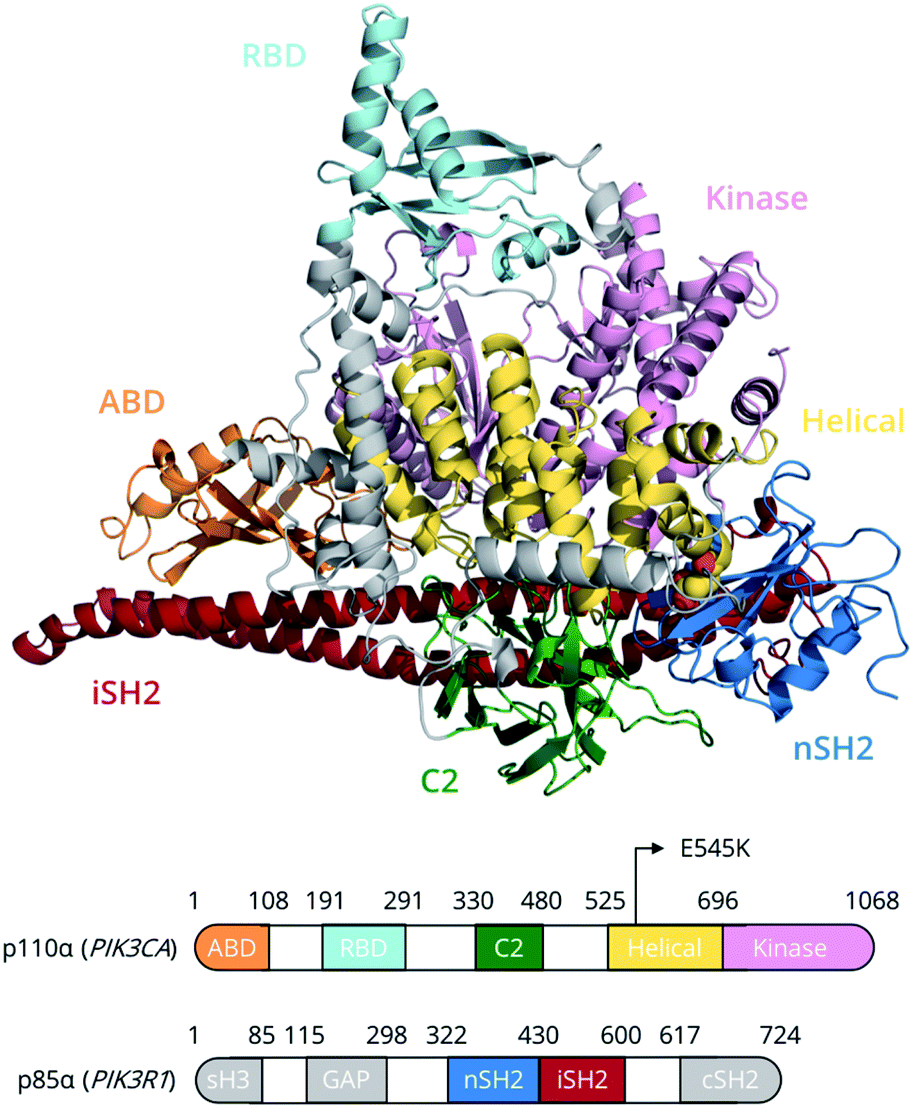 | ||
| Fig. 1 PI3Kα domain organisation of the p110α (catalytic) and p85α (regulatory) subunits. The E545K (yellow van der Waals spheres) point mutation lies at the interface between the nSH2 (p85α) and helical (p110α) domains. Grey regions in p85α have not been simulated due to the absence of structural information. | ||
PI3Kα is the most commonly mutated kinase in cancer, and PIK3α-activating mutations have been identified in human tumours, clustering in two hotspot mutations, H1047R and E545K.6 The hotspot mutation E545K in the helical domain of p110α has been found to increase PI3Kα lipid kinase activity.7 In this study, we unravel, for the first time, the conformational free energy landscape of E545K PI3Kα and elucidate the effect of the charge reversal mutation E545K on the dynamics and function of PI3Kα using the largest enhanced sampling calculations to date.8–10 The results suggest that, despite the progress that has been made in elucidating the effect of the E545K in atomic level detail,11–14 the mechanism of action of this mutation is more complicated than previously thought.
Results and discussion
Unbiased MD simulations
In the unbiased MD simulations, Glu545 in the WT PI3Kα forms salt-bridges with Lys379p85α and Lys548p110α, a backbone hydrogen bond with Leu380p85α, and participates in hydrophobic interactions with the side chain of Ile381p85α through the aliphatic part of its side chain (Fig. S1†). Lys379p85α is part of a highly conserved phosphopeptide binding surface on nSH2, and a charge reversal mutation of Lys379p85α (K379E) has been reported to induce increased lipid kinase activity, highlighting the importance of the interactions it participates in.13The repulsive potential between the positively charged side chains of Lys545 and Lys379p85α in the E545K PI3Kα is expected to trigger the disruption of the nSH2–helical contacts, including the backbone hydrogen bond between Lys545 and Leu380p85α, which was shown to be the last point of contact between the two domains in our previous work,12 and lead to the detachment of the nSH2 from the helical domain. Indeed, in previous work, we observed a spontaneous rotation of the nSH2 domain away from the helical domain driven by the E545K mutation,12 which did not have an immediate effect on the conformation of the kinase domain. Interestingly, in this work, in two out of the three independent unbiased mutant simulations, we observe nSH2 moving away from the kinase and C2 domains (Fig. S2†), which drives the release of the iSH2 regulatory contacts from the activation loop, without actually detaching from the helical domain. In fact, the interaction of Lys545 with Asp421p85α compensates for the loss of the nSH2–helical interaction that the Glu545–Lys349p85α provides in the WT, and prevents nSH2 from detaching. Asp421p85α is located on the loop that links the nSH2 with the iSH2 domain. The interaction of Lys545 with Asp421p85α leads to the gradual detachment of the C-terminal region of the iSH2 and its iα3 helix from the activation loop of the kinase domain that together keep it to the inactive conformation (Fig. S3A†). The third helix of the iSH2 domain (p85α residues 587–598), iα3, forms in the WT an interface with the activation loop that consists of hydrophobic interactions between Leu598p85α and Phe945p110α, and a hydrogen bond between Gln591p85α and Lys948p110α (Fig. S3B†). This interface is expected to stabilise the activation loop to the inactive conformation15 and deletions (Δ583–602), or truncations (p85-572STOP) of this section of the iSH2 domain are known to be oncogenic.16 In the mutant, the iα3 helix moves further down the iSH2 domain retaining the hydrogen bond of Gln591p85α with Lys948p110α.
By extending our analysis of the unbiased MD simulations further away from the 545 position, a closer look on the salt-bridges formed between the charged residues on the surface of nSH2 and its neighbouring domains (Fig. S4†) shows that the introduction of Lys545 disrupts additionally the helical–nSH2 interaction Glu542p110α–Arg358p85α, as well as the nSH2–kinase interaction Glu342p85α–Lys948p110α. The latter, presumably, is one of the key interactions that keep the activation loop in the inactive conformation. The activation loop of the kinase domain is responsible for the recognition of PIP2 by PI3Kα,17 and a catalytic scenario has been recently proposed, in which the 941KKKK944 polybasic stretch on the activation loop recognizes the PIP2 substrate, while His936p110α and His917p110α facilitate phosphorylation.11,25 Through analysis of the root mean square fluctuation (RMSF) of the activation loop over the course of our unbiased simulations, we see that the release of the niSH2–activation loop contacts, which we observe in two out of the three independent simulations of the E545K mutant, results in increased flexibility of the activation loop (Fig. S5†). We, thus, expect that this flexibility will allow the polybasic stretch to approach the γ-phosphate of ATP in the activated PI3Kα.
It has been established that the effects of mutations can extend well beyond their specific points of localisation, resulting in long-range allosteric effects that overall translate into increased stabilisation of different states or of different regions assuming a functional role. Dynamical decomposition analysis has been used in the past to provide a simple yet valuable description of proteins dynamics, which can, in turn, determine the response to mutations.18,19 The results of the dynamical domain decomposition analysis of the WT PI3Kα performed herein suggest that, at a subdomain level, the movement of the ABD domain is highly correlated with the C-terminal region of the iSH2 domain, while the N-lobe of the kinase domain is highly coupled with the C-terminal region of the helical domain (Fig. S6A and B†). In the case of the E545K PI3Kα, the clustering of residues into eight subdomains suggests that the mutation increases the coupling of the motion of the C2 domain with that of the helical domain, and especially with the interfacial region between the nSH2 and helical domains (Fig. S6B†). Interestingly, when the residues are clustered into 11 subdomains, which is the number of domains with the second maximum quality score in the replicas where the mutation drives the release of the iSH2–activation loop regulatory contacts (rep0, rep2, Fig. S6A†), the residues of the Ser501p110α–Asp527p110α loop cluster together and independently of the helical domain (Fig. S6C†). This is in agreement with the observed sliding of the nSH2 that is seen in this set of simulations, which in turn increases the flexibility of the Ser501p110α–Asp527p110α loop. In the WT, decomposition into 11 domains leads to a clear division of the helical domain into two subdomains; one where the nSH2 domain binds to, and one where the N-lobe of the kinase domain is attached to (Fig. S6C†).
Multiple-walkers metadynamics simulations
The unbiased MD results suggest a mutant-driven activation, where the mutation leads to disruption of the iSH2 regulation of the conformational flexibility of the activation-loop of the kinase domain. Even at 1000 ns, however, MD simulations are too short to capture significant conformational changes. Since 1 μs of unbiased MD was insufficient to sample large conformational changes, we used multiple-walkers metadynamics (metaD) simulations to enhance the sampling. The main free energy minima have been fully explored and a clear qualitative picture of the PI3Kα mechanism of E545K action can be drawn.The free energy surface of the WT exhibits two minima that are equally populated within the error bar. Basin α1 (Fig. 2, Video S1†) corresponds to an ensemble of conformations that resembles the crystal structure. In this ensemble, the nSH2 is in contact with the helical domain even though the backbone hydrogen bond between Glu545 and Leu380p85α is not present in all conformations of the ensemble, and Glu545 interacts with Lys548p110α and Lys379p85α, and occasionally with Lys573p110α as well (Fig. S7A†). In this ensemble, the interaction of Lys419p85α with Asp549p110α of the loop that connects the nSH2 with the iSH2 domain, keeps the nSH2 bound to the helical domain even when the Glu545–Lys379p85α salt-bridge is broken. The activation loop is maintained in the inactive conformation due to salt-bridges between the side chains of Lys948p110α and Glu342p85α or Glu345p85α, and through a network of hydrogen bonds between Gln591p85α, and Arg949p110α or Tyr947p110α (Fig. S7B†).
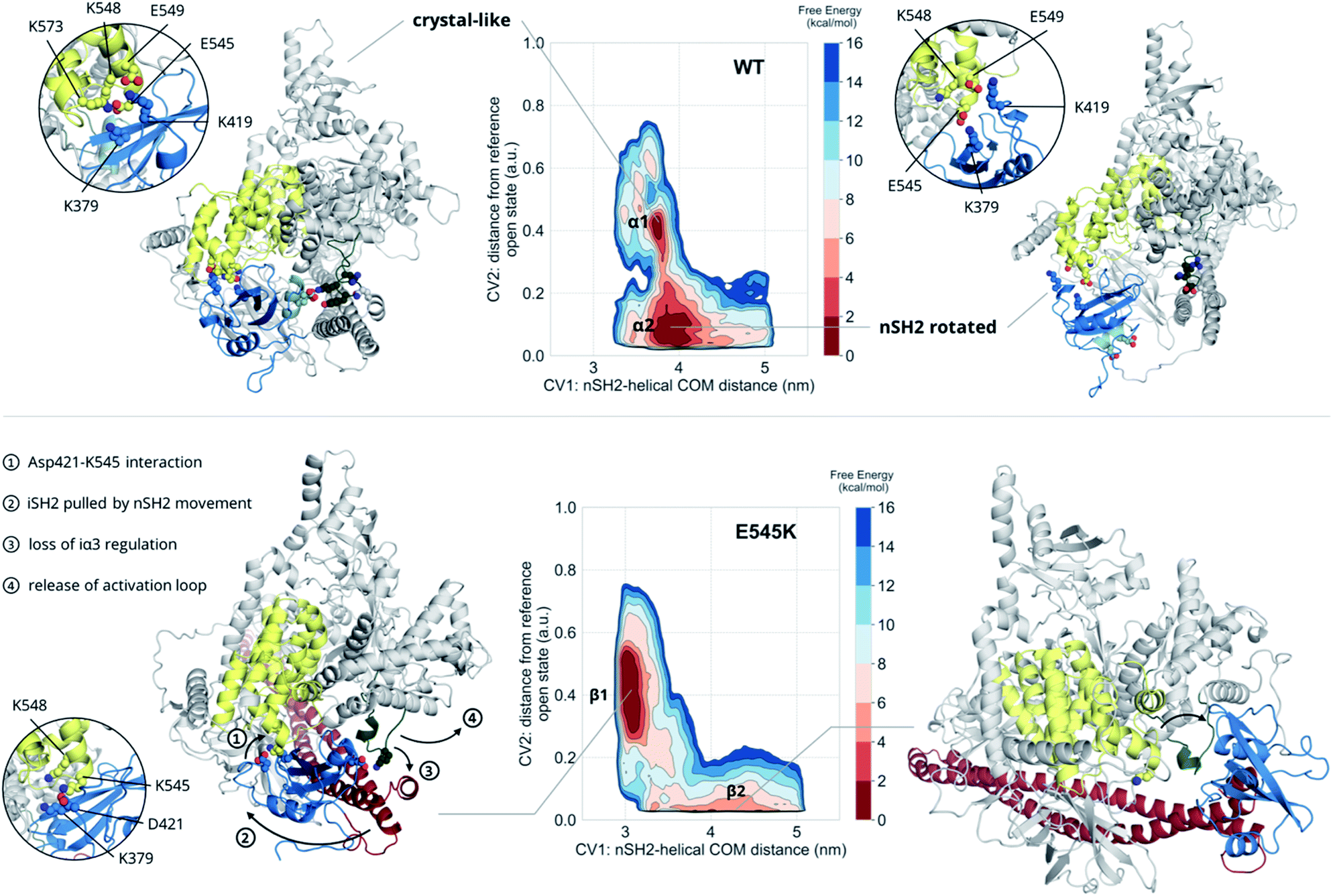 | ||
| Fig. 2 Free energy surfaces of the WT and E545K as a function of CV1 and CV2. The contour lines are drawn every 2 kcal mol−1. Helix A (residues 339–347) of the nSH2 domain (blue) is depicted in light blue to showcase the rotation of the domain relative to the helical domain (yellow). | ||
Surprisingly, the second basin in the free energy of the WT (basin α2, Fig. 2, Video S2†) corresponds to a previously unreported ensemble of conformations, where nSH2 has rotated around the helical domain (Fig. S7C†), as can be seen from the exposure of helix A (residues 339–347) to the solvent. This rotation makes biological sense as it may facilitate the binding of pY peptides or other tyrosine-phosphorylated signalling proteins since the cluster of residues (Arg340p85α, Arg358p85α, Ser361p85α, Thr369p85α) that stabilise the phosphotyrosine becomes solvent-exposed and therefore accessible for binding (Fig. S7C†). It should be noted that the rotation of the nSH2 domain does not affect the conformation of the activation loop in the kinase domain, which is held in the inactive conformation by the iSH2.
The main minimum on the free energy surface of the E545K mutant corresponds to a very broad energy basin in the defined contact map space (basin β1, Fig. 2, Video S3†). In this basin, the E545K PI3Kα is found in two main conformations. In the first conformation, the loop that connects helix B of nSH2 (residues 400–411p85α) with the iSH2 domain is flexible, Lys545 interacts mainly with Glu549p110α of the helical domain, and the salt bridge between Glu342p85α of the nSH2 domain and Lys948p110α of the activation loop is formed, keeping helix A in its original position. In the second conformation, Lys545 interacts with Asp421p85α while Lys419p85α points towards Glu579p110α, similar to the conformation found in the unbiased MD. The interactions engaged by Lys545 may be the trigger of a conformational change in which the loop that connects helix B of nSH2 with the iSH2 domain pulls the iSH2 away from the activation loop. Specifically, the pulling effect bends the N-terminal part of the iSH2, and the subsequent disruption of the inhibitory interactions between the iα3 helix and the activation loop weakens the niSH2 domains-mediated regulation of the kinase activity. It should be noted that in the conformations of this basin, the backbone hydrogen bond between Lys545 and Leu380p85α is present, indicating that PI3Kα E545K can be activated without the nSH2 domain detaching from the helical domain.
The breakthrough discovery here is that nSH2 does not need to detach from the catalytic domain to release the niSH2 regulatory contacts with the activation loop upon the E545K mutation, and thus activate the kinase. The release of the iα3 – activation-loop contacts and the bending of the N-terminal part of the iSH2 along with a higher flexibility of the activation loop, expose helix A to the solvent (Fig. S8†), as also seen in the HDX-MS experiments.13 At the same time, the stronger nSH2–helical interaction in the predicted conformation (basin β1, Fig. 2) hinders pY binding, which could explain the reported insensitivity of E545K PI3Kα to pY activation.2
The second minimum in the free energy surface of the E545K (basin β2, Fig. 2, Video S4†) corresponds to an ensemble of conformations, where the nSH2 domain has detached from the helical domain and moved away from the activation loop, in agreement with previous proposals.2 The nSH2 domain moves as a rigid body maintaining its overall folding and secondary structure elements and slides along the surface of the catalytic subunit rather than completely detaching from the protein surface into the solvent as was previously thought. It should be noted that in the absence of a pY peptide or a phosphorylated signalling protein in the simulated system, the nSH2 is stabilised only by non-specific interactions with the iSH2 domain, and therefore this basin is not highly populated.
Conclusions
Our simulations and free energy calculations show two different displacement paths resulting from the E545K mutation (Fig. 3). One is the detachment of the nSH2 domain from the helical domain, and the other is the sliding along the helical domain. The first was also observed in our previous MD simulations and, as discussed above, might be the main event in the presence of the pY peptide that is expected to stabilise the detached nSH2 domain. When the nSH2 domain is detached from the helical domain, the activation-loop stays intact because its interactions with the iSH2 domain are maintained, in agreement with HDX-MS experiments in the presence of pY, where E545K does not affect the catalytic subunit.14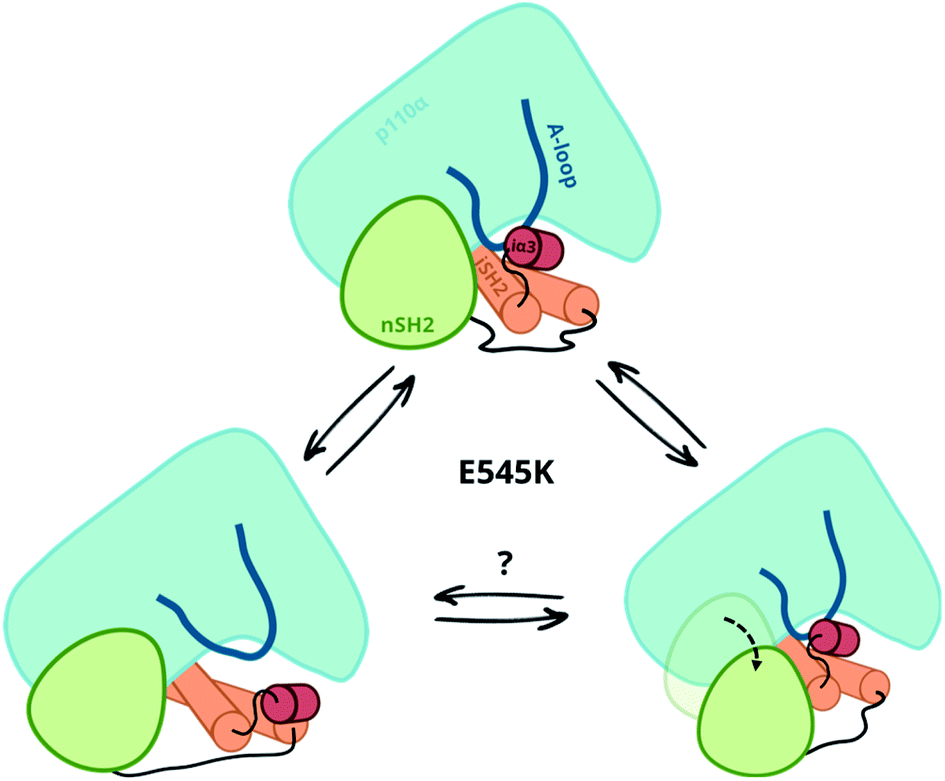 | ||
| Fig. 3 Observed displacement paths upon the E545K mutation. | ||
The other effect of E545K is more surprising and directly affects the catalytic subunit. The newly formed contacts of the Lys545 with residues of the linker that connects the nSH2 with the iSH2 domain trigger a conformational change in the iSH2 domain that results in the release of the iSH2–activation loop regulatory contacts and affect the activity allosterically. The perturbation around E545K is propagated to the iSH2 domain through the loop that connects the two domains, and the breaking of the inhibitory contacts between iSH2 and the activation-loop “unlocks” the activation-loop, which is expected to be free to adopt active-like conformations, especially in the presence of PIP2. Such a mode of action is in line with recent observations in several multi-domain proteins, showing that linkers can propagate mechanical signals allosterically between domains and facilitate correlated movement.20
Both events, that is the sliding of nSH2 around the helical domain and its detachment from the helical domain result from the E545K mutation (Fig. 3). However, the first effect is probably the most relevant in terms of the deregulation of the catalytic activity as it exerts an allosteric effect on the catalytic subunit triggering the release of the niSH2 regulatory contacts. This does not exclude the possibility of both events happening sequentially; first, the release of the iSH2–activation loop regulatory contacts and then the detachment of nSH2, although more experimental data is needed to validate this hypothesis. The proposed mechanism and the new conformation reported here provide also a different and more complete interpretation of existing experimental data.
Methods
The structure of the WT PI3Kα heterodimer was retrieved from the Protein Data Bank (PDB ID: 4OVU15). Missing residues and the E545K mutation were modelled using Prime21 (Schrödinger suite). Following system setup, the WT and mutant proteins were solvated and equilibrated as described in the ESI.† The CHARMM36m force field8 was used for the protein and the CHARMM TIP3P model8 for the water molecules. A total of 2 μs of unbiased MD simulations were performed for each system with the GROMACS 2016.4 MD engine22 to characterise the local dynamics.Subsequently, we used multiple walkers metadynamics23 simulations to characterise the effect of the E545K mutation to the nSH2-mediated autoinhibition and calculate the corresponding free energy surfaces (FESs). The FESs of the WT and mutant PI3Kα were reconstructed as a function of two collective variables (CVs) after 400 ns of simulation time per system. CV1 is the distance between the centres of mass of the nSH2 and helical domains, and CV2 is the distance in terms of contact maps from a reference state in which the nSH2 domain has detached from the helical domain (see ESI† for more details on the setup). A funnel-shaped potential around the nSH2 domain was used to aid the convergence of the free energy (Fig. S9†).
The quasi-rigid dynamic domain decomposition was performed using the SPECTRUS method, a dimensionality reduction approach (spectral clustering) that allows the identification of dynamical domains in protein complexes.19,24 The clustering algorithm computes the inter-residue distance fluctuation matrix on frames extracted from an MD simulate, which is used to define the subdivision into domains. Then, a quality score is used to measure the robustness of the domain separation, i.e., how much the calculated clusters are overlapping and distinct from each other. This score results from computing the ratio between the distance of each residue to its own cluster centre and the distance to the second closest cluster centre. The local maxima of the reciprocal of this quantity, averaged over all residues, mark the decomposition into well-defined clusters and allow the a posteriori identification of the optimal number of clusters (domains) to be considered. Here, we used the default setup of the SPECTRUS method with 1000 iterations of the k-medoids algorithm and 10 Å for nearest neighbours cutoff.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank D. Dellis, C. Athanasiou and T. Heinemann for helpful discussions. I. G. is funded by Astra Zeneca-EPSRC case studentship award to F. L. G. Z. C. and F. L. G. would like to acknowledge funding from the European Union's Horizon 2020 Framework Programme for Research and Innovation under Grant Agreement No. 785907 (Human Brain Project SGA2). F. L. G. acknowledges EPSRC [grant no EP/P022138/1; EP/P011306/1; EP/M013898/1] for financial support. We acknowledge PRACE for computer time on CURIE at GENCI@CEA, France. Computational resources from project s847@PizDiant of the Swiss National Supercomputing Center (CSCS) in Lugano are gratefully acknowledged. This work was further supported by computational time granted from the Greek Research & Technology Network (GRNET) in the National HPC facility ARIS, under project IDs pr005036/pi3ka-mut and pr006019/e545k. All data is accessible at: https://kg.ebrains.eu/search/instances/Model/7f44abeb3068cdc74506bac6e72a8802.Notes and references
- D. A. Fruman, H. Chiu, B. D. Hopkins, S. Bagrodia, L. C. Cantley and R. T. Abraham, Cell, 2017, 170, 605–635 CrossRef CAS PubMed.
- J. E. Burke, O. Perisic, G. R. Masson, O. Vadas and R. L. Williams, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15259–15264 CrossRef CAS PubMed.
- M. Zhang, H. Jang, V. Gaponenko and R. Nussinov, Biophys. J., 2017, 113, 1956–1967 CrossRef CAS PubMed.
- R. Nussinov, G. Wang, C.-J. Tsai, H. Jang, S. Lu, A. Banerjee, J. Zhang and V. Gaponenko, Trends Cancer, 2017, 3, 214–224 CrossRef CAS PubMed.
- D. A. Fruman and C. Rommel, Nat. Rev. Drug Discovery, 2014, 13, 140–156 CrossRef CAS PubMed.
- J. E. Burke, Mol. Cell, 2018, 71, 653–673 CrossRef CAS PubMed.
- J. D. Carson, G. Van Aller, R. Lehr, R. H. Sinnamon, R. B. Kirkpatrick, K. R. Auger, D. Dhanak, R. A. Copeland, R. R. Gontarek, P. J. Tummino and L. Luo, Biochem. J., 2008, 409, 519–524 CrossRef CAS PubMed.
- J. Huang, S. Rauscher, G. Nawrocki, T. Ran, M. Feig, B. L. De Groot, H. Grubmüller and A. D. MacKerell, Nat. Methods, 2016, 14, 71–73 CrossRef PubMed.
- A. Kuzmanic, R. B. Pritchard, D. F. Hansen and F. L. Gervasio, J. Phys. Chem. Lett., 2019, 10, 1928–1934 CrossRef CAS PubMed.
- P. Robustelli, S. Piana and D. E. Shaw, Proc. Natl. Acad. Sci. U. S. A., 2018, 0260, 201800690 Search PubMed.
- M. Zhang, H. Jang and R. Nussinov, Chem. Sci., 2019, 10, 3671–3680 RSC.
- H. Leontiadou, I. Galdadas, C. Athanasiou and Z. Cournia, Sci. Rep., 2018, 8, 15544 CrossRef PubMed.
- J. E. Burke and R. L. Williams, Adv. Biol. Regul., 2013, 53, 97–110 CrossRef CAS PubMed.
- J. E. Burke, O. Perisic, G. R. Masson, O. Vadas and R. L. Williams, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15259–15264 CrossRef CAS PubMed.
- M. S. Miller, O. Schmidt-Kittler, D. M. Bolduc, E. T. Brower, D. Chaves-Moreira, M. Allaire, K. W. Kinzler, I. G. Jennings, P. E. Thompson, P. A. Cole, L. M. Amzel, B. Vogelstein and S. B. Gabelli, Oncotarget, 2014, 5, 5198–5208 CrossRef PubMed.
- H. Wu, S. C. Shekar, R. J. Flinn, M. El-Sibai, B. S. Jaiswal, K. I. Sen, V. Janakiraman, S. Seshagiri, G. J. Gerfen, M. E. Girvin and J. M. Backer, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20258–20263 CrossRef CAS PubMed.
- L. Pirola, M. J. Zvelebil, G. Bulgarelli-Leva, E. Van Obberghen, M. D. Waterfield and M. P. Wymann, J. Biol. Chem., 2001, 276, 21544–21554 CrossRef CAS PubMed.
- A. Paladino, F. Marchetti, L. Ponzoni and G. Colombo, J. Chem. Theory Comput., 2018, 14, 1059–1070 CrossRef CAS PubMed.
- D. Granata, L. Ponzoni, C. Micheletti and V. Carnevale, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E10612–E10621 CrossRef CAS PubMed.
- E. Papaleo, G. Saladino, M. Lambrughi, K. Lindorff-Larsen, F. L. Gervasio and R. Nussinov, Chem. Rev., 2016, 116, 6391–6423 CrossRef CAS PubMed.
- M. P. Jacobson, D. L. Pincus, C. S. Rapp, T. J. F. Day, B. Honig, D. E. Shaw and R. A. Friesner, Proteins: Struct., Funct., Genet., 2004, 55, 351–367 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindah, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- P. Raiteri, A. Laio, F. L. Gervasio, C. Micheletti and M. Parrinello, J. Phys. Chem. B, 2006, 110, 3533–3539 CrossRef CAS PubMed.
- L. Ponzoni, G. Polles, V. Carnevale and C. Micheletti, Structure, 2015, 23, 1516–1525 CrossRef CAS PubMed.
- S. Maheshwari, M. S. Miller, R. O'Meally, R. N. Cole, L. M. Amzel and S. B. Gabelli, J. Biol. Chem., 2017, 292, 13541–13550 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc05903b |
| This journal is © The Royal Society of Chemistry 2020 |