
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective oxygenation of exocyclic methylene groups by a manganese porphyrin catalyst with a chiral recognition site†
Finn
Burg
 ,
Stefan
Breitenlechner
,
Christian
Jandl
and
Thorsten
Bach
*
,
Stefan
Breitenlechner
,
Christian
Jandl
and
Thorsten
Bach
*
Department Chemie, Catalysis Research Center (CRC), Technische Universität München, 85747 Garching, Germany. E-mail: thorsten.bach@ch.tum.de; Fax: +49 89 28913315; Tel: +49 89 28913330
First published on 14th January 2020
Abstract
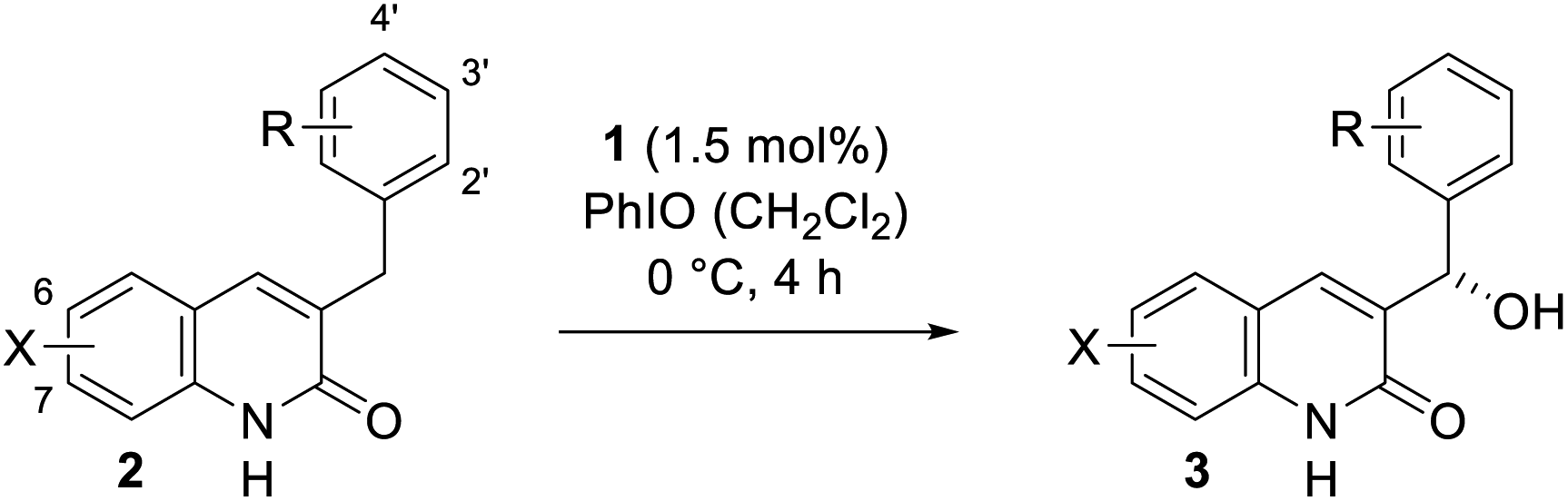
The natural enzyme cytochrome P450 is widely recognised for its unique ability to catalyse highly selective oxygen insertion reactions into unactivated C–H bonds under mild conditions. Its exceptional potential for organic synthesis served as an inspiration for the presented biomimetic hydroxylation approach. Via a remote hydrogen bonding motif a high enantioselectivity in the manganese-catalysed oxygenation of quinolone analogues (27 examples, 18–64% yield, 80–99% ee) was achieved. The site-selectivity was completely altered in favour of a less reactive but more accessible position.
Introduction
The direct functionalisation of carbon hydrogen (C–H) bonds unambiguously belongs to the most coveted transformations in modern organic synthesis.1 The C–H bond activation of sp3 carbon centres presents a significant challenge due to the inert nature of hydrocarbons and due to the desired distinction between the various aliphatic C–H bonds embodied in organic molecules.2 In this context, the hydroxylation of prochiral methylene compounds poses an additional conundrum, as the corresponding secondary alcohol should ideally be formed as a single enantiomer without subjecting the newly generated stereogenic centre to an additional oxidation step.3,4 Inspired by the remarkable efficiency of enzymatic oxygenations catalysed by cytochrome P450,5 there are numerous examples of transition metal complexes, containing salen,6,7 porphyrin,8,9 and aminopyridine ligands10,11 which have been employed in the oxygenation of C–H bonds. Since in many cases the chirality is transferred by a bulky ligand, it is perhaps not surprising that such a sterically demanding environment can be repulsive thus resulting in an insufficient conversion. Unlike its natural occurring congeners, these systems lack a favourable substrate–catalyst orientation which in turn facilitates an enantioselective approach to the substrate while maintaining a high turnover.In recent years, it has been realised that the quest for enantio- and site-selective oxygenation reactions at remote positions (remote functionalisation) can be successfully tackled by catalysts with a non-covalent recognition element.12 After seminal work by Breslow and co-workers on selective oxidation and oxygenation reactions,13 notable contributions have been made among others by the groups of Groves,14 Crabtree,15 and Costas.16 An early example that demonstrated how recognition elements can control selectivity in epoxidation reactions was reported by the group of Breslow (Fig. 1a).13b They used a manganese salen complex, that was functionalised by a chelating dipyridine moiety inviting coordination of other metals. In a competing experiment of a nicotinyl- and a benzoyl-substituted olefin, a very high selectivity (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was achieved for the epoxidation of the former derivative in the presence of Cu2+ ions indicating that pre-coordination via complexation of the pyridine residues enhanced the reaction.
:1) was achieved for the epoxidation of the former derivative in the presence of Cu2+ ions indicating that pre-coordination via complexation of the pyridine residues enhanced the reaction.
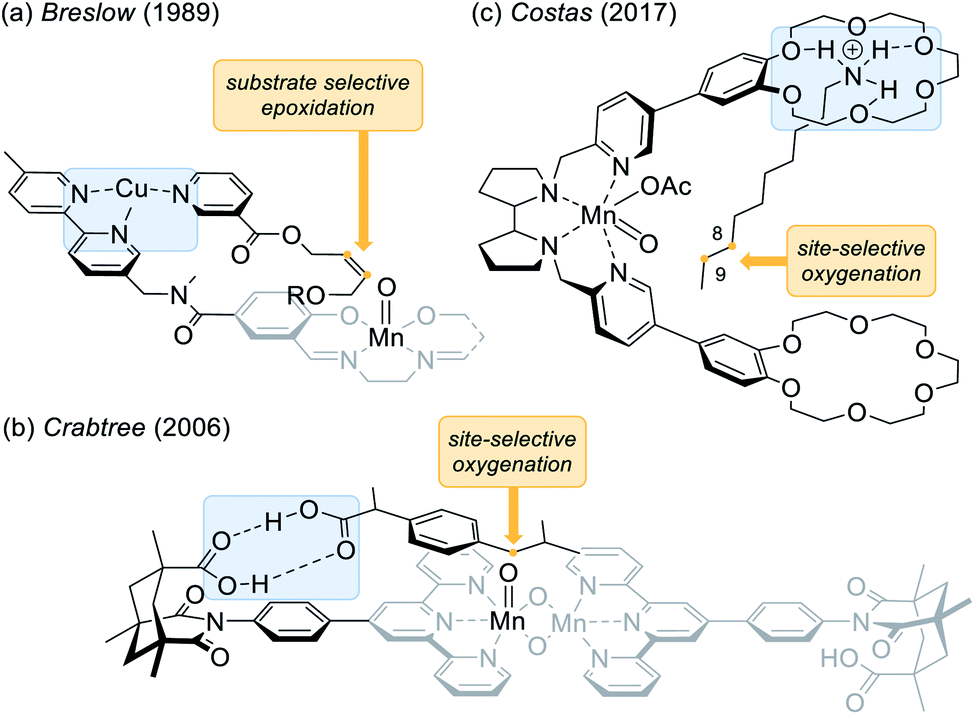 | ||
| Fig. 1 Controlling selectivity in oxidation and oxygenation reactions via supramolecular recognition. (a) Substrate selective epoxidation of nicotinic acid derivatives controlled by complexation of copper ions. (b) Site-selective oxygenation of ibuprofen mediated by two-point hydrogen bonding between two carboxylic acids. (c) Site-selective oxygenation of ndecylamine directed by a 18-crown-6 functional group. | ||
A contribution by Crabtree15a and co-workers (Fig. 1b) made intentional use of hydrogen bonds as an operative tool to control selectivity in oxygenation reactions. In this specific case, a di-μ-oxo dimanganese centre is flanked by two terpyridine ligands, which in turn are connected to a molecular recognition unit derived from Kemp's triacid.17,18 The presented site-selective oxygenation of ibuprofen can thus be rationalised by a two-point hydrogen bonding between the two carboxylic acids. In a more recent example, the group of Costas employed a manganese aminopyridine ligand that was functionalised with two 18-crown-6 ether units (Fig. 1c).16b The remote crown ether effectively anchors the ndecylamine at the terminal ammonium ion whereupon a selective oxygenation at carbon atoms C-8 and C-9 is induced.
Although it was beyond the intended scope of the above-mentioned catalysts to differentiate between enantiotopic hydrogen atoms, there have been several reports in which hydrogen bonding was invoked as the key element to induce enantioselectivity in catalysis.19,20 However, examples remain scarce in which both the site- and the enantioselectivity of oxygenation reactions were successfully addressed.21 In recent years, our group has designed and synthesised bifunctional ligands with which a catalytically active metal centre is tailored to a chiral lactam22 recognition unit. Originally conceived for applications in photochemistry the concept of two-point hydrogen bonding23 has successfully been applied to enantioselective olefin addition24 and C–H functionalisation25 reactions. As a recent supplement to this type of catalysts we prepared the chiral manganese porphyrin complex 125c for an application to enantioselective oxygenation reactions (Fig. 2, left).
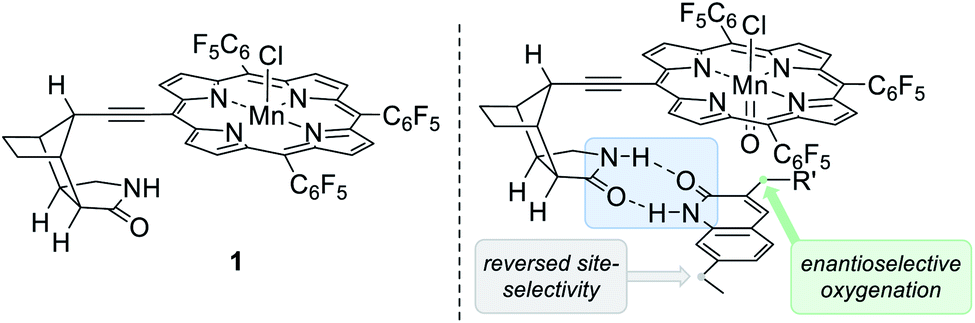 | ||
| Fig. 2 Structure of the manganese porphyrin complex 1 exhibiting a chiral lactam recognition unit (left). Proposed transition state for the oxygenation of exocyclic methylene groups in quinolone analogues where hydrogen bonding effectively induces enantioface differentiation (right). | ||
In the present study we report a general bioinspired approach towards the site- and enantioselective oxygenation of a vast array of 3-benzyl and 3-alkyl substituted quinolones displaying an exocyclic methylene group (Fig. 2, right). Despite the free rotation around the quinolone–methylene bond C–H oxygenations occurred with high enantioselectivities (up to 99% ee). Some of the quinolone substrates were also probed in a racemic oxygenation reaction using manganese(III)-5,10,15,20-tetrakis(pentafluorophenyl)porphyrin chloride (Mn[TPFPP]Cl), whereupon the conversion decreased drastically, suggesting that the non-covalent hydrogen bonding interactions have also a rate accelerating effect. Most importantly, in the case of a second reactive benzylic position, the site-selectivity of the racemic reaction was reverted compared to the hydrogen bond-mediated system. This observation indicates that hydrogen bonds direct the oxygenation event to a previously inaccessible carbon centre, while the intrinsically more reactive position remains intact.
Results and discussion
Our work commenced with an investigation into the enantioselective oxygenation of the literature-known 3-(4′-methylbenzyl)quinolone (2a)25a under our previously optimised reaction conditions (Scheme 1, t = 16 h).25c We were delighted to observe that the enantiomeric excess in the exocyclic oxygenation was exceptionally high (95% ee) when employing 2.0 mol% of porphyrin 1 and iodosobenzene as a stoichiometric oxidant. The reaction delivered the desired alcohol 3a, which was accompanied by ketone 4a, the product of over-oxidation. It was quickly realised, though, that the catalytic activity decreased significantly before even less than half of the starting material 2a was consumed. The alcohol to ketone ratio (3a/4a = 70/30) after 4 h was relatively low compared to the ratio we had observed in related studies and accordingly we sought for ways to improve the mass balance while concomitantly increasing the catalytic turnover.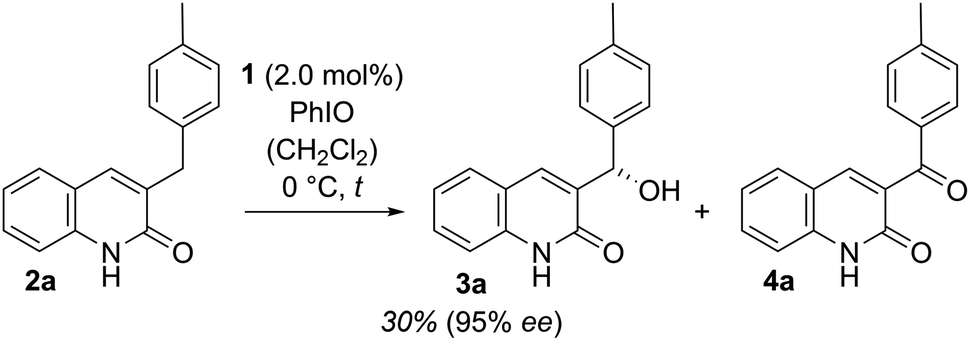 | ||
| Scheme 1 Enantioselective oxygenation of 3-(4′-methylbenzyl)quinolone (2a) to alcohol 3a and ketone 4a catalysed by the chiral manganese porphyrin 1 under previously reported conditions (initial experiment, see also Fig. 3 and the narrative).25c | ||
One promising approach that caught our attention was to use an excess of the lactam 2a relative to the oxidant. In this instance, an even lower catalyst loading of 1.0 mol% was sufficient to generate a significantly higher alcohol concentration (Fig. 3), and simultaneously the alcohol to ketone ratio improved notably (3a/4a = 80/20).
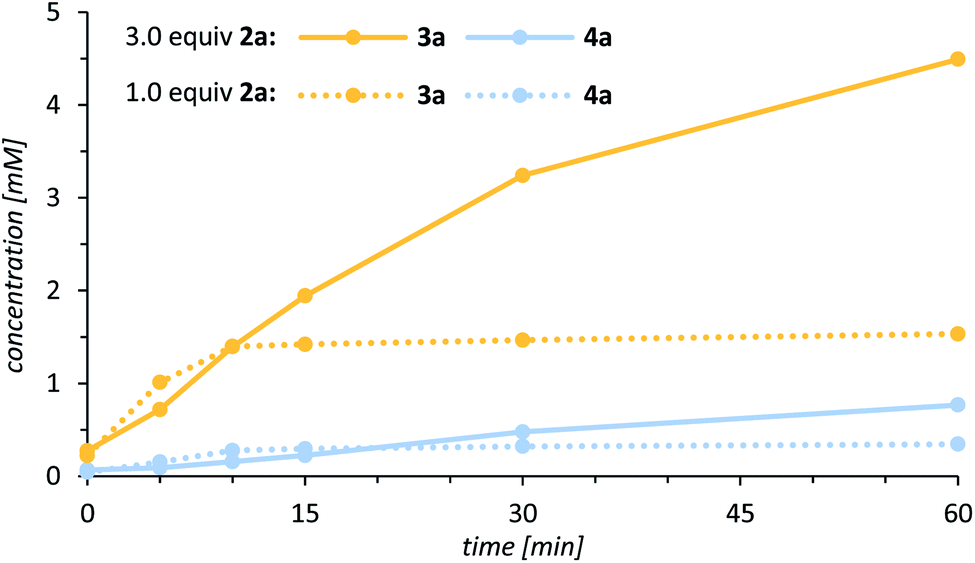 | ||
| Fig. 3 Rate profile for the oxygenation of 3-(4′-methylbenzyl)quinolone (2a) to alcohol 3a (yellow) and ketone 4a (blue) catalysed by 1.0 mol% of the chiral manganese porphyrin complex 1 [0 °C, CH2Cl2, solid line = 3.0 equiv. of 2a, 1.0 equiv. PhIO (c = 10 mM); dashed line = 1.0 equiv. of 2a (c = 10 mM), 2.0 equiv. PhIO]. | ||
We continued with our optimisation using the oxidant iodosobenzene as the limiting reagent in a dichloromethane solution and varied the amount of quinolone 2a applied (Table 1, entries 1 and 2). With 2.0 equivalents, the alcohol to ketone ratio deteriorated (3a/4a = 76/24) and it seemed warranted to keep the concentration of the substrate – which was readily available on gram scale – high. An increase or decrease of the substrate concentration c led to a lower enantioselectivity (entries 3 and 4), while a higher catalyst loading turned out to be almost inconsequential for the outcome of the reaction (entry 5). Neither a solvent variation (entries 6 and 7) nor prolonged reaction times (entry 8) were found to be beneficial to conversion and enantioselectivity. A slightly higher yield of alcohol 3a was achieved by a portion-wise addition of the oxidant (entry 9). Under these conditions a final variation of the catalyst loading (entries 10 and 11) was performed from which eventually a catalyst loading of 1.5 mol% turned out to be ideal (entry 11).
| Entrya | c [mM] | 1 [mol%] | Solvent | 3a [%] | % eed | 4a [%] |
|---|---|---|---|---|---|---|
| a All reactions were run with 3.0 equiv. of 2a (180 μmol) under the indicated conditions (0 °C, t = 4 h) and were initiated by addition of the oxidant PhIO (1.0 equiv.) to a pre-cooled solution of 2a and 1 in the given solvent. b Concentration of quinolone 2a in the solution. c All yields were determined by GLC-FID analysis using ndodecane as a stoichiometric internal standard. d Enantiomeric excess (% ee) as determined by HPLC analysis on a chiral stationary phase. e 2.0 equiv. of 2a were employed. f The reaction was performed at 23 °C. g The reaction time was 16 h. h PhIO was added in three portions. i Not determined. | ||||||
| 1 | 30 | 1.0 | CH2Cl2 | 48 | 91 | 11 |
| 2e | 20 | 1.0 | CH2Cl2 | 42 | 93 | 13 |
| 3 | 20 | 1.0 | CH2Cl2 | 39 | 86 | 11 |
| 4 | 60 | 1.0 | CH2Cl2 | 50 | 87 | 12 |
| 5 | 30 | 2.0 | CH2Cl2 | 48 | 91 | 10 |
| 6f | 30 | 1.0 | PhH | 30 | 85 | 3 |
| 7 | 30 | 1.0 | DCE | 42 | 88 | 12 |
| 8g | 30 | 1.0 | CH2Cl2 | 45 | 85 | 12 |
| 9h | 30 | 1.0 | CH2Cl2 | 52 | 93 | 17 |
| 10h | 30 | 0.5 | CH2Cl2 | 37 | —i | 10 |
| 11h | 30 | 1.5 | CH2Cl2 | 56 | 95 | 18 |
Under optimised conditions the enantiomerically enriched alcohol 3a was isolated in 60% yield and with an enantiomeric excess of 96% ee (Table 2). Since a clean separation from both the starting material 2a and the ketone 4a was feasible the yield based on recovered starting material 2a could be readily determined (80%). We were interested in studying the electronic and steric effects of the benzyl substituent and accordingly prepared a series of various 3-benzylquinolones 2 which were consequentially subjected to the enantioselective oxygenation protocol. The yields in Table 2 refer to isolated material but are based on the oxidant as the limiting reagent. The yields based on substrate conversion were determined by clean reisolation of the starting material 2 and are given in brackets.
|
|
|||||
|---|---|---|---|---|---|
| Entrya | 2 | X | R | 3 [%] | % eec |
| a All reactions were conducted at a iodosobenzene concentration of 10 mM in dichloromethane employing 1.0 equiv. of PhIO (60 μmol), 3.0 equiv. of 2 (180 μmol) and 1.5 mol% of 1 (0.9 μmol). b All yields refer to isolated material. Yields in brackets are based on reisolated starting material 2. c Enantiomeric excess (% ee) as determined by HPLC analysis on a chiral stationary phase. | |||||
| 1 | 2a | H | 4′-Me | 60 (80) | 96 |
| 2 | 2b | H | 3′-Me | 58 (82) | 99 |
| 3 | 2c | H | 2′-Me | 64 (92) | 97 |
| 4 | 2d | H | 3′,4′-Me2 | 59 (68) | 92 |
| 5 | 2e | H | H | 61 (84) | 95 |
| 6 | 2f | H | 4′-OMe | 53 (74) | 93 |
| 7 | 2g | H | 4′-F | 53 (60) | 91 |
| 8 | 2h | H | 4′-Cl | 18 (62) | 98 |
| 9 | 2i | H | 3′-CF3 | 26 (54) | 92 |
| 10 | 2j | 6-Me | 4′-Me | 60 (71) | 98 |
| 11 | 2k | 7-Me | 4′-Me | 53 (65) | 96 |
Altering the position of the methyl group to a meta- or ortho-substitution gave similar results in terms of yield (58% 3b, 64% 3c) and the enantiomeric excess for the corresponding alcohols 3b and 3c remained exceptionally high (99% ee and 97% ee). The 3′,4′-dimethylbenzyl substituted quinolone 2d reacted almost analogously (59%), but the enantiomeric excess decreased slightly to 92% ee. The transformation of unsubstituted 3-benzylquinolone (2e) delivered the oxygenated product 3e in a yield of 61% with 95% ee. An electron donating substituent (MeO) in the para position of the aromatic ring (3f, 53% yield, 93% ee) was tolerated as was an inductively electron withdrawing substituent (3g, 53% yield, 91% ee). Indeed, the outcome of the oxygenation seemed consistent upon variation of functional groups on the benzyl ring. A deviation was observed when 3-(4′-chlorobenzyl)quinolone (2h) was taken into the reaction and resulted in a low yield of only 18% (62% based on recovered starting material). While the enantioselectivity remained outstanding (98% ee), the lower conversion was attributed to the poor solubility of substrate 2h in dichloromethane. Substrate 2i with the strongly electron withdrawing m-(trifluoromethyl)aryl substituent gave rise to the corresponding alcohol 3i in a yield of 26% (54% based on recovered starting material) and an enantiomeric excess of 92% ee. Finally, substituents on the quinolone core were introduced into the substrates and the 6- and 7-methyl-substituted quinolones 2j and 2k were probed in the oxygenation event. Both substrates gave a clean conversion to the corresponding alcohols 3j and 3k in satisfactory yields (60% and 53%) and with remarkable enantioselectivity (98% ee and 96% ee).
In order to establish the absolute configuration of the oxygenated products, the enantiomerically enriched alcohol 3h was subjected to a chemoselective N-methylation using potassium carbonate and methyl iodide in N,N-dimethylformamide (DMF) whereupon compound 5 was obtained in almost quantitative yield (Scheme 2). Colorless crystals were obtained by slow evaporation from dichloromethane which were suited for X-ray crystallographic analysis and the absolute configuration as determined by anomalous diffraction was in agreement with the proposed transition state (Fig. 2, right).
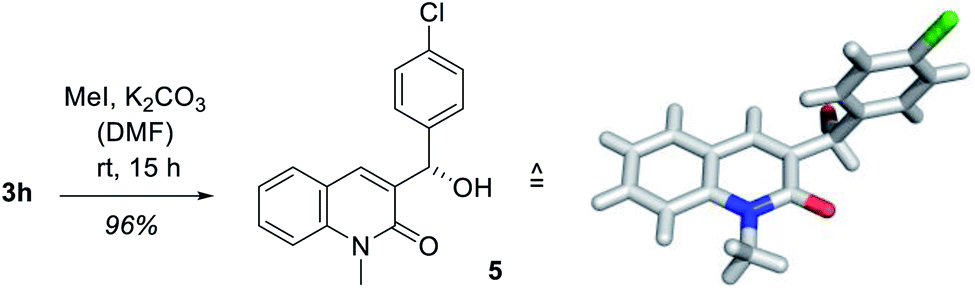 | ||
| Scheme 2 Synthesis of the N-methylated alcohol 5 derived from 3-(4′-chlorobenzyl)-quinolone (3h) by chemoselective methylation and its crystal structure. The absolute configuration was determined by anomalous X-ray diffraction. | ||
Apart from the high enantioselectivity, a most notable feature of the oxygenation reaction was its exquisite site-selectivity. When promoted by achiral Mn(TPFPP)Cl as the catalyst (2.0 mol%), substrates exhibiting a methylene group in 4′-position of the benzyl substituent [R = 4′-ethyl (2l); 4′-isopropyl (2m)] underwent a sluggish but site-selective (r.r. = regioisomeric ratio) oxygenation (Scheme 3) to the remote alcohols rac-2n (27%) and 2o (20%). In stark contrast, catalyst 1 (1.5 mol%) guided the oxygenation reaction almost exclusively to the benzylic carbon atom adjacent to the quinolone core whereupon the oxygenated products 3l (57%) and 3m (50%) were obtained with excellent enantioselectivity (93% ee and 91% ee). The reversal of site-selectivity supports a pre-coordination of quinolone substrates 2 to the catalyst which in turn leads to a defined exposure of a single C–H towards the active oxomanganese26 complex. Accordingly, non-covalent hydrogen bonding completely alters the site of the oxygenation reaction to a position that seems inaccessible by conventional oxygenation methods.
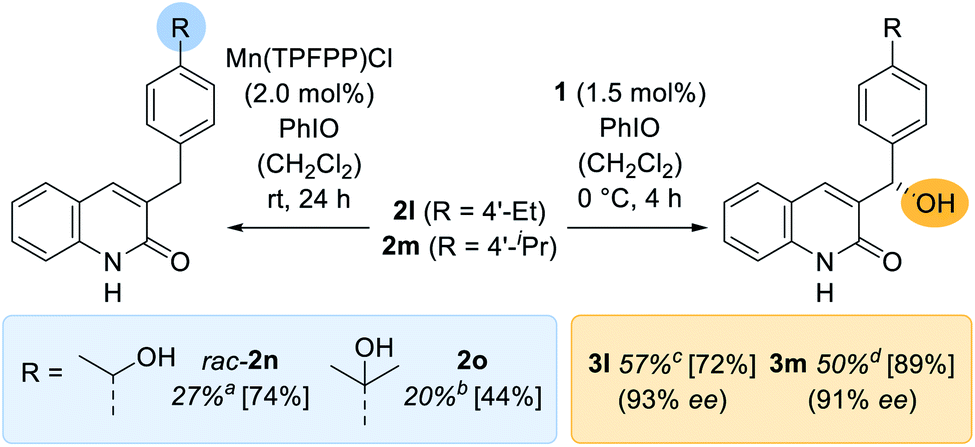 | ||
| Scheme 3 Enantioselective and racemic oxygenation of quinolones 2l and 2m bearing an additional reactive methylene site. The racemic reaction was promoted by Mn(TPFPP)Cl (left) and the enantioselective reaction by the chiral manganese porphyrin complex 1 (right). Yields in square brackets are based on recovered starting material 2 (ar.r. = 93/7, br.r. = 82/18, cr.r. = 94/6, dr.r. = 97/3). | ||
The scope of the reaction was substantially expanded by varying the size and nature of the quinolone substituent in position C-3. A brief screening of reaction conditions based on the previously developed protocol indicated that even simple 3-alkylsubstituted quinolones underwent the desired transformation (Table 3) and that an aryl group was not necessary to activate the prostereogenic methylene group. A slightly higher catalyst loading of 2.0 mol% was required to transform 3-ethylquinolone (6a) into its oxygenated analogue 7a in 56% yield and with an outstanding enantiomeric excess of 95% ee. The specific rotation of product 7a was compared with literature data,24a allowing an assignment of its absolute configuration. It was interesting to see, that the formation of the corresponding ketone 8a was less prominent (7a/8a = 88/12) than for the benzylic substrate 2a, resulting in a high yield of 90% based on recovered starting material. Encouraged by these results, a more extensive set of 3-alkylsubstituted quinolones was prepared and the effect of quinolone substitution (variation of X) was examined. Upon elongation of the alkyl chain the yield remained constant [R′ = Et (53%), nPr (50%), iBu (48%)], but a minor decrease in enantioselectivity was observed [7b (88% ee), 7c (86% ee) 7d (88% ee)]. This effect became even more evident when a tertiary carbon atom was installed adjacent to the oxygenation site (entry 5). The oxygenated product 7e, derived from 3-isobutylquinolone (6e), was obtained in 48% yield, but only with 80% ee.
|
|
|||||
|---|---|---|---|---|---|
| Entrya | 6 | X | R′ | 7 [%] | % eec |
| a All reactions were conducted at a iodosobenzene concentration of 10 mM in dichloromethane employing 1.0 equiv. of PhIO (60 μmol), 3.0 equiv. of 6 (180 μmol) and 2.0 mol% of 1 (1.2 μmol). b All yields refer to isolated material. Yields in brackets are based on reisolated starting material 6. c Enantiomeric excess (% ee) as determined by HPLC analysis on a chiral stationary phase. | |||||
| 1 | 6a | H | Me | 56 (90) | 95 |
| 2 | 6b | H | Et | 53 (84) | 88 |
| 3 | 6c | H | n Pr | 50 (68) | 86 |
| 4 | 6d | H | iBu | 48 (96) | 88 |
| 5 | 6e | H | iPr | 48 (89) | 80 |
| 6 | 6f | 6-Me | Me | 51 (93) | 94 |
| 7 | 6g | 7-Me | Me | 58 (94) | 96 |
| 8 | 6h | 6,7-Me2 | Me | 51 (94) | 95 |
| 9 | 6i | 6-OMe | Me | 21 (38) | 96 |
| 10 | 6j | 7-OMe | Me | 62 (73) | 95 |
| 11 | 6k | 7-Cl | Me | 34 (95) | 95 |
| 12 | 6l | 7-F | Me | 42 (84) | 96 |
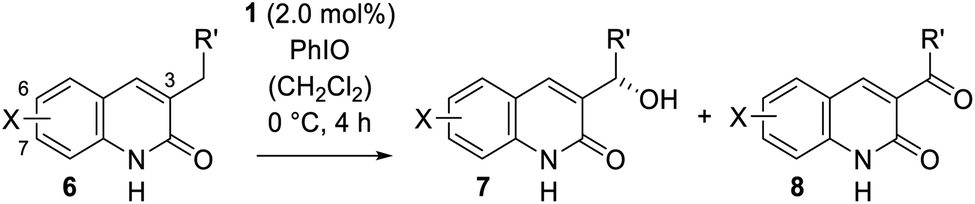
When simple methyl substituents were implemented into the aromatic quinolone core the yield of isolated product remained constant for both the C-6 and the C-7 position [X = 6-Me (51%), 7-Me (58%), 6,7-Me2 (51%)] and the enantiomeric excess was continuously high [7f (94% ee), 7g (96% ee) 7h (95% ee)]. An electron donating methoxy substituent at the C-6 carbon atom led to a diminished yield (21%), but high enantioselectivity (96% ee) possibly due to oxidative degradation of the electron rich aryl core. The same functional group was less disruptive at the deconjugated 7-position and alcohol 7j was obtained in 62% yield and with an enantiomeric excess of 95% ee. Electron deficient quinolones seem to be slightly less reactive [X = 7-Cl (34%), 7-F (42%)]. Nevertheless, both substrates gave rise to the desired alcohols 7k and 7l in again exceptional enantiomeric excess (95% ee and 96% ee).
Given the high conformational flexibility of a simple 3-ethylquinolone (6a), it might be surprising that hydrogen bonding can provide sufficient enantioface differentiation between the two pro-stereogenic hydrogen atoms. Rotation around the indicated single bond (Scheme 4) leads to a change of topicity even if an approach of the oxygenation reagent was guided by hydrogen bonding to occur from the back face of the quinolone. It appears as if the active manganese species behaves like a catalytically active cleft similar to the active site of a natural enzyme. It reserves only a very limited reactive domain around the oxomanganese porphyrin in which an oxygenation can occur by a rebound mechanism.27 As previously established for the epoxidation of 3-alkenylquinolone by a chiral ruthenium complex,24b the trajectory of the active site to the substrate requires a perfect adjustment of the rotatable single bond. In our specific case, only the pro-(S) hydrogen atom is properly positioned to engage in oxygenation while potential transition states for the pro-(R) hydrogen atom are too high in energy to be significantly populated. Based on the proposed model (Fig. 2, right), 3-ethylquinolone in its s-trans conformation is aligned in a way that the oxygenation event proceeds with high selectivity at the pro-(S) hydrogen atom.
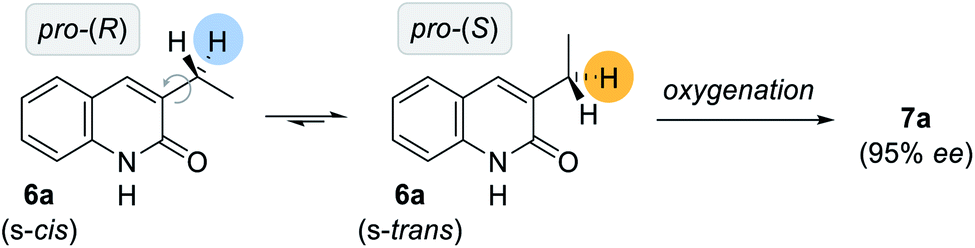 | ||
| Scheme 4 Rotation around the C–C bond between the exocyclic methylene group and the quinolone carbon skeleton. Based on the proposed model, the s-trans isomer is favoured in the coordinated transition state whereupon the (S)-configured alcohol 7a is delivered in high enantiomeric excess. | ||
Although their outstanding stereoselectivity belongs to the most admired features of enzymatic reactions, the site-selectivity is often considered as its most useful catalytic property. As previously alluded to, molecular recognition devices cannot only help to induce enantioselectivity, but also provide a powerful tool to differentiate between two potentially reactive sites in a remote functionalization approach. This aspect was further exemplified when 3,7-diethylquinolone (6m) and (3′-phenylpropyl)quinolone (6o) were subjected to the oxygenation protocol (Scheme 5). The racemic oxygenation proceeded exclusively adjacent to the phenyl ring whereupon rac-6n (20%) and rac-6p (21%) were obtained as single regioisomers. The oxygenation site was however relocated, when the manganese porphyrin complex 1 was employed. The oxygenated product 7m was isolated in 60% yield with almost perfect enantioselectivity (98% ee). The presented site-selective oxygenation even withstands a proximal more reactive benzylic position. (3′-Phenylpropyl)quinolone (6o) was converted into the corresponding alcohol 7o with reverted site-selectivity but with a slightly reduced enantioselectivity (84% ee).
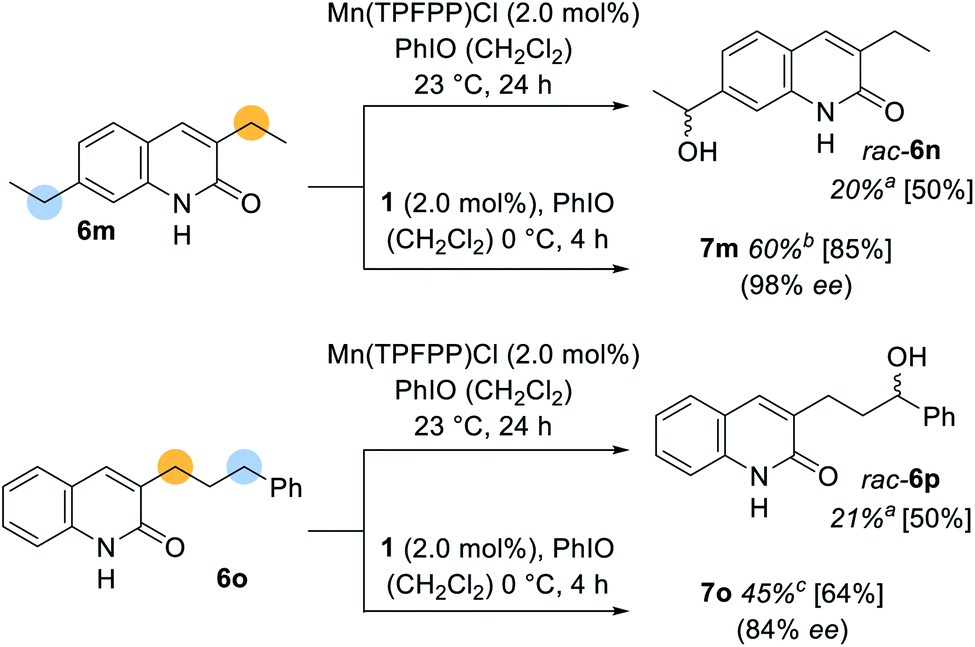 | ||
| Scheme 5 Enantioselective and racemic oxygenation of 3,7-diethylquinolone (6m) and 3-(3′-phenylpropyl)quinolone (6o) using the chiral manganese porphyrin complex 1 and Mn(TPFPP)Cl, respectively. Yields in square brackets are based on recovered starting material 6 (asingle regioisomer, br.r. = 88/12, cr.r. = 72/28). | ||
To support the hypothesis that hydrogen bonding is responsible for both the chirality transfer and for the enhanced reactivity a control experiment was performed. One hydrogen bonding site of substrate 6a was blocked by N-methylation to provide quinolone 9. When compound 9 was probed in the catalytic reaction (Scheme 6) under otherwise identical conditions its oxygenated product 10 was obtained in racemic form and a notably lower yield of 22%.
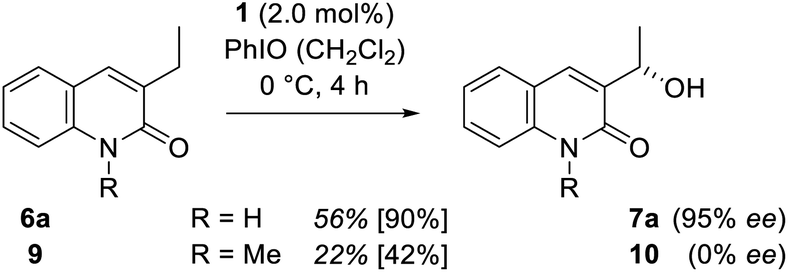 | ||
| Scheme 6 Oxygenation of 6a and the N-methylated congener 9 to alcohol 7a and 10 catalysed by manganese porphyrin 1. | ||
Further mechanistic work was devoted to study the over-oxidation of the enantiomerically enriched alcohol to its corresponding ketone and to elucidate the fate of the catalyst in the course of the reaction. Although a relatively high alcohol to ketone ratio clearly indicated that the enantioselectivity arises from a C–H activation step, we envisioned that detailed kinetic studies will provide a more comprehensive mechanistic picture of the over-oxidation step. The racemic alcohol rac-3a was subjected to the oxygenation reaction (1.0 mol% 1, 1.0 equiv. PhIO) and the formation of the ketone 4a as well as the enantiomeric excess of the remaining starting material 3a were closely monitored over time (Scheme 7). The rate profile derived from the experimental data indicated that after a conversion of 59% the remaining starting material 3a was enantiomerically enriched by 71% ee (Fig. 4).
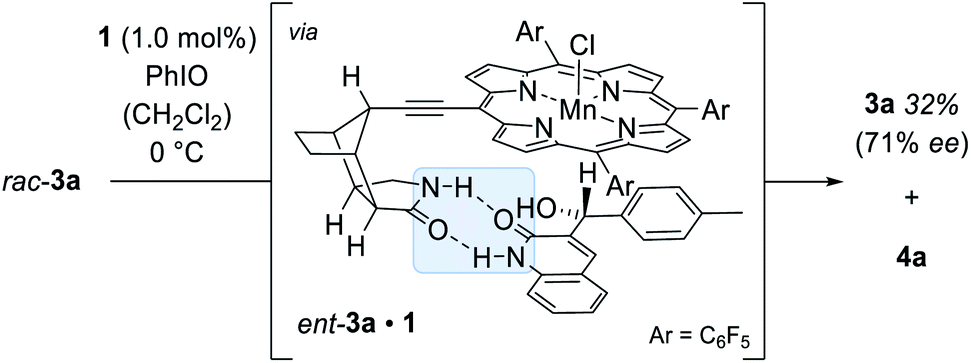 | ||
| Scheme 7 Kinetic resolution of rac-3a catalysed by the chiral manganese porphyrin 1. | ||
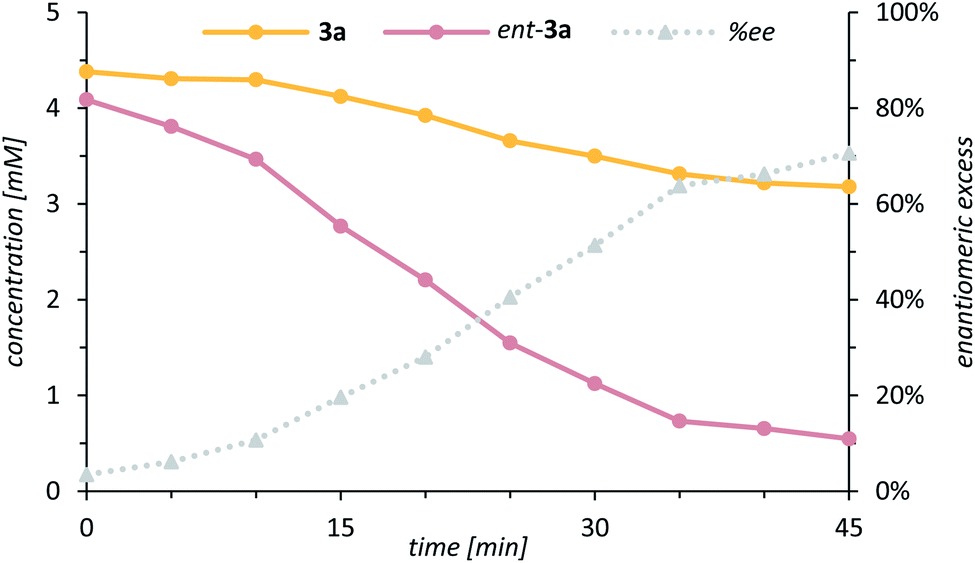 | ||
| Fig. 4 Rate profile of the two enantiomers 3a (yellow) and ent-3a (pink) in the kinetic resolution of rac-3a to ketone 4a and enantiomeric excess (grey, dashed line) of the remaining enantiomer 3a. | ||
The s-factor28kR/kS = 6.1 calculated from these data confirmed that a kinetic resolution via an over-oxidation step does not contribute significantly to the enantiomeric excess.29 Under optimised standard conditions (Table 1, entry 11) only 18% of ketone 4a were formed but 56% of alcohol 3a (95% ee). It is interesting to note, though, that the residual alcohol has the same absolute configuration as the alcohol derived from the C–H oxygenation step. Indeed, upon coordination of ent-3a to catalyst 1, the remaining hydrogen atom at the former methylene group is directly exposed towards the catalytically active metal centre thus favouring an additional oxidation step via hydrogen abstraction.
Although all experimental data were coherent, it remained uncertain what exactly caused the rapid decrease of the catalytic activity if the quinolone substrate was used as a limiting reagent (Fig. 3). Based on detailed rate profiles, we assumed that the catalytic activity might be affected by one of the oxygenation products. To verify this hypothesis, a kinetic inhibition experiment was performed, which at the same time was supposed to shine light on the robustness of the catalyst. Quinolone 2a was subjected towards the enantioselective oxygenation employing 1.0 mol% of the chiral catalyst 1 with 2.0 equivalents of the oxidant PhIO and the documented rate profile was used as a reference for further experiments. After 30 min 22% of the starting material 2a had been converted into 16% of the alcohol 3a and 5% of the ketone 4a (Fig. 5, solid lines). Within the next 90 min the concentration of all reactants remained almost constant (<5% conversion) clearly indicating that the catalytic activity had decreased drastically. A second oxygenation experiment was set up, in which the concentration of all reactants strictly followed the “same excess” approach.30 Specifically, a reaction mixture containing the exact stoichiometry of all reactants 2a, 3a and 4a after 60 min of the previous experiment was treated with 1.0 mol% of catalyst 1 and eventually the oxidant PhIO was added to the pre-cooled reaction mixture. The oxygenation reaction was expected to remain idle and no further conversion of the starting material 2a should occur if the catalytic system was inhibited either by alcohol 3a or by ketone 4a. Upon addition of the oxidant to the premixed reaction solution, however, the oxygenation was initiated and 15% of 2a were converted into 10% of alcohol 3a and 7% of ketone 4a within the next hour (Fig. 5, dashed lines).
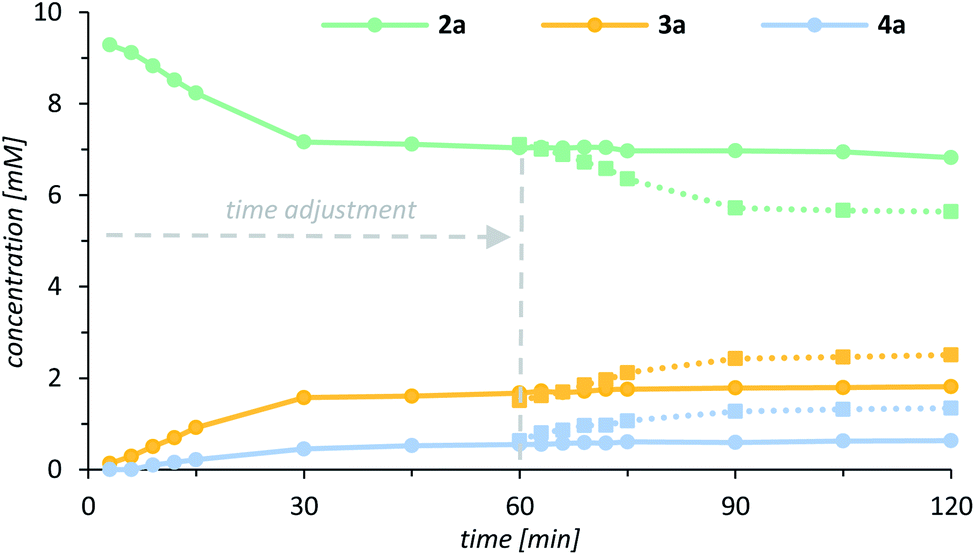 | ||
| Fig. 5 Rate profile for the enantioselective oxygenation of 2a (green) to alcohol 3a (yellow) and ketone 4a (blue) catalysed by the chiral manganese porphyrin complex 1 (solid lines). Overlay of the rate profile for the kinetic inhibition experiment following the “same excess” protocol30 with a time adjustment of 60 min (dashed lines). | ||
In fact, the experiment demonstrated, that there is no inhibition by neither of the products 3a or 4a suggesting that after 30 min the catalyst is eventually modified resulting in a significantly diminished activity. This observation was further corroborated by a third experiment, which was set up as usual with 1.0 equivalent of the quinolone substrate and 1.0 mol% of the catalyst 1. The reaction was initiated by addition of the oxidant (2.0 equiv.) and – as expected – ceased after 30 min at a conversion of 24%. Upon addition of a second portion of the catalyst 1 (1.0 mol% after 60 min) the reaction was resumed and another 19% of 2a were converted into 11% of the alcohol 3a and 8% of the ketone 4a conclusively matching the rate profile of the “same excess” experiment. It should be noted that the reaction was only relaunched by addition of catalyst, while addition of another portion iodosobenzene turned out to be inconsequential.
With respect to all rate profiles (see the ESI† for a complete set of data), it is conspicuous that the rapid decrease in catalytic activity correlated to the iodosobenzene concentration. At high concentration, with the quinolone as the limiting reagent, the manganese catalyst quickly lost its activity resulting in retarded conversion of the substrate to the alcohol 2a → 3a and of the alcohol to the ketone 3a → 4a (Fig. 3). In other words, the reaction slowed down but continued to show the typical rate profile of a consecutive reaction in which the alcohol is an intermediate en route to the ketone. The alcohol to ketone ratio 3a/4a decreased over time, irrespective of catalyst and oxidant concentration. For example, after prolonged reaction times the ratio 3a/4a after 1 h under the conditions of Scheme 1 (2.0 mol% 1, 1.0 equiv. of 2a, 2.0 equiv. PhIO) was 76/24 while it decreased to 38/62 after 24 h. In stark contrast, at a low iodosobenzene concentration with an excess of quinolone, the activity of the catalyst remained high until almost the entire iodosobenzene was consumed. The reaction is terminated due to the lack of oxidant before larger amounts of ketone can be formed. Along these lines, it is inevitable to keep the concentration of the quinolone substrate 2a high, if elevated turnover numbers (TONs) and reasonable yields for alcohol 3a are prioritized over the amount of 2a applied. Reducing the concentration of the oxidant to a minimum by portion-wise addition prevents catalyst degradation and can increase the lifetime of the catalyst. Under optimized reaction conditions, high TONs and excellent yields based on recovered starting material (up to 96%) were observed, while the enantioselectivity was maintained (up to 99% ee).
Conclusion
In summary, we have established that a manganese porphyrin complex linked to a chiral recognition site effectively catalyses a highly selective oxygen insertion into a variety of aliphatic and benzylic exocyclic methylene groups. A lactam recognition motif was employed which limits the scope of substrates to compounds with a matching hydrogen bonding site. In the present study, a comprehensive set of 3-substituted quinolones (27 examples, up to 64% yield) was successfully oxygenated adjacent to the 3-position of the heterocyclic carbon skeleton with outstanding site- and enantioselectivity (up to 99% ee). Catalyst 1 operates under the influence of two-point hydrogen bonding, whereupon the substrate is precisely oriented in a way that allows for no other than the pro-(S) hydrogen atom to be attacked. Kinetic experiments revealed that employing the oxidant as the limiting reagent can significantly increase the number of catalytic turnovers and improve the alcohol to ketone ratio thus providing excellent yields based on recovered starting material.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft (grant Ba 1372-17) is gratefully acknowledged. O. Ackermann and J. Kudermann are acknowledged for their help with HPLC and GLC analysis. We thank M. Stierle for synthetic assistance.Notes and references
- For reviews on C–H activation, see: (a) P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS PubMed; (b) C. G. Newton, S.-G. Wang, C. C. Oliveira and N. Cramer, Chem. Rev., 2017, 117, 8908–8976 CrossRef CAS PubMed; (c) J. F. Hartwig and M. A. Larsen, ACS Cent. Sci., 2016, 2, 281–292 CrossRef CAS PubMed; (d) H. M. L. Davies and D. Morton, J. Org. Chem., 2016, 81, 343–350 CrossRef CAS PubMed; (e) J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740–4761 RSC; (f) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885–1898 RSC; (g) K. Godula and D. Sames, Science, 2006, 312, 67–72 CrossRef CAS PubMed; (h) G. Dyker, Handbook of C–H transformations: applications in organic synthesis, Wiley-VCH, 2005 CrossRef; (i) J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507–514 CrossRef CAS PubMed.
- For reviews on site-selective C–H functionalization, see: (a) F. D. Toste, M. S. Sigman and S. J. Miller, Acc. Chem. Res., 2017, 50, 609–615 CrossRef CAS PubMed; (b) L. Ping, D. S. Chung, J. Bouffard and S.-g. Lee, Chem. Soc. Rev., 2017, 46, 4299–4328 RSC; (c) S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936–946 CrossRef CAS; (d) T. Brückl, R. D. Baxter, Y. Ishihara and P. S. Baran, Acc. Chem. Res., 2012, 45, 826–839 CrossRef PubMed; (e) W. R. Gutekunst and P. S. Baran, Chem. Soc. Rev., 2011, 40, 1976–1991 RSC.
- For reviews on the C–H oxygenation reactions, see: (a) G. B. Shul'pin, Catalysts, 2016, 6, 50 CrossRef; (b) E. P. Talsi and K. P. Bryliakov, Coord. Chem. Rev., 2012, 256, 1418–1434 CrossRef CAS; (c) T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362–3374 CrossRef CAS PubMed; (d) R. Curci, L. D'Accolti and C. Fusco, Acc. Chem. Res., 2006, 39, 1–9 CrossRef CAS PubMed; (e) L. Que and W. B. Tolman, Nature, 2008, 455, 333–340 CrossRef CAS PubMed; (f) M. Costas, M. P. Mehn, M. P. Jensen and L. Que, Chem. Rev., 2004, 104, 939–986 CrossRef CAS PubMed.
- For a recent example on a selective benzylic mono-oxygenation, see: L. Tanwar, J. Börgel and T. Ritter, J. Am. Chem. Soc., 2019, 141, 17983–17988 CrossRef CAS PubMed.
- For reviews on cytochrome P450 and bioengineered congeners, see: (a) T. L. Poulos, Chem. Rev., 2014, 114, 3919–3962 CrossRef CAS PubMed; (b) R. Fasan, ACS Catal., 2012, 2, 647–666 CrossRef CAS; (c) J. C. Lewis, P. S. Coelho and F. H. Arnold, Chem. Soc. Rev., 2011, 40, 2003–2021 RSC; (d) P. R. Ortiz de Montellano, Chem. Rev., 2010, 110, 932–948 CrossRef CAS PubMed; (e) P. R. Ortiz de Montellano, Cytochrome P450: structure, mechanism, and biochemistry, Springer Science & Business Media, 2005 CrossRef.
- For reviews on metal–salen complexes, see: (a) K. C. Gupta and A. K. Sutar, Coord. Chem. Rev., 2008, 252, 1420–1450 CrossRef CAS; (b) N. S. Venkataramanan, G. Kuppuraj and S. Rajagopal, Coord. Chem. Rev., 2005, 249, 1249–1268 CrossRef CAS; (c) T. Katsuki, Coord. Chem. Rev., 1995, 140, 189–214 CrossRef CAS.
- Selected examples: (a) K. Hamachi, R. Irie and T. Katsuki, Tetrahedron Lett., 1996, 37, 4979–4982 CrossRef CAS; (b) T. Hamada, R. Irie, J. Mihara, K. Hamachi and T. Katsuki, Tetrahedron, 1998, 54, 10017–10028 CrossRef CAS; (c) S.-I. Murahashi, S. Noji and N. Komiya, Adv. Synth. Catal., 2004, 346, 195–198 CrossRef CAS.
- For reviews on metalloporphyrins, see: (a) H. Lu and X. P. Zhang, Chem. Soc. Rev., 2011, 40, 1899–1909 RSC; (b) M. Costas, Coord. Chem. Rev., 2011, 255, 2912–2932 CrossRef CAS; (c) C.-M. Che, V. K.-Y. Lo, C.-Y. Zhou and J.-S. Huang, Chem. Soc. Rev., 2011, 40, 1950–1975 RSC; (d) M. Senge, Chem. Commun., 2011, 47, 1943–1960 RSC.
- Selected examples: (a) J. T. Groves and P. Viski, J. Am. Chem. Soc., 1989, 111, 8537–8538 CrossRef CAS; (b) J. T. Groves and P. Viski, J. Org. Chem., 1990, 55, 3628–3634 CrossRef CAS; (c) R. Zhang, W.-Y. Yu, T.-S. Lai and C.-M. Che, Chem. Commun., 1999, 1791–1792 RSC; (d) Z. Gross and S. Ini, Org. Lett., 1999, 1, 2077–2080 CrossRef CAS; (e) R. Zhang, W.-Y. Yu and C.-M. Che, Tetrahedron: Asymmetry, 2005, 16, 3520–3526 CrossRef CAS; (f) H. Srour, P. L. Maux and G. Simonneaux, Inorg. Chem., 2012, 51, 5850–5856 CrossRef CAS PubMed.
- For a recent overview of iron aminopyridine complexes, see: G. Olivo, O. Cussó and M. Costas, Chem.–Asian J., 2016, 11, 3148–3158 CrossRef CAS PubMed.
- Selected examples: (a) M. S. Chen and M. C. White, Science, 2007, 318, 783–787 CrossRef CAS PubMed; (b) P. E. Gormisky and M. C. White, J. Am. Chem. Soc., 2013, 135, 14052–14055 CrossRef CAS PubMed; (c) M. Milan, M. Bietti and M. Costas, ACS Cent. Sci., 2017, 3, 196–204 CrossRef CAS PubMed; (d) B. Qiu, D. Xu, Q. Sun, C. Miao, Y.-M. Lee, X.-X. Li, W. Nam and W. Sun, ACS Catal., 2018, 8, 2479–2487 CrossRef CAS; (e) M. C. White and J. Zhao, J. Am. Chem. Soc., 2018, 140, 13988–14009 CrossRef CAS PubMed; (f) R. V. Ottenbacher, E. P. Talsi, T. V. Rybalova and K. P. Bryliakov, ChemCatChem, 2018, 10, 5323–5330 CrossRef CAS; (g) J. Zhao, T. Nanjo, E. C. de Lucca and M. C. White, Nat. Chem., 2019, 11, 213–221 CrossRef CAS PubMed; (h) B. Qiu, D. Xu, Q. Sun, J. Lin and W. Sun, Org. Lett., 2019, 21, 618–622 CrossRef CAS PubMed.
- For reviews on catalysis based on non-covalent interactions, see: (a) H. J. Davis and R. J. Phipps, Chem. Sci., 2017, 8, 864–877 RSC; (b) A. J. Neel, M. J. Hilton, M. S. Sigman and F. D. Toste, Nature, 2017, 543, 637–646 CrossRef CAS PubMed; (c) M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1660–1733 RSC; (d) P. Dydio and J. N. H. Reek, Chem. Sci., 2014, 5, 2135–2145 RSC; (e) S. Carboni, C. Gennari, L. Pignataro and U. Piarulli, Dalton Trans., 2011, 40, 4355–4373 RSC; (f) E. Lindbäck, S. Dawaigher and K. Wärnmark, Chem.–Eur. J., 2014, 20, 13432–13481 CrossRef PubMed; (g) S. Das, G. W. Brudvig and R. H. Crabtree, Chem. Commun., 2008, 413–424 RSC.
- (a) R. Breslow, Acc. Chem. Res., 1980, 13, 170–177 CrossRef CAS; (b) R. Breslow, A. B. Brown, R. D. McCullough and P. W. White, J. Am. Chem. Soc., 1989, 111, 4517–4518 CrossRef CAS; (c) R. Breslow, X. Zhang, R. Xu, M. Maletic and R. Merger, J. Am. Chem. Soc., 1996, 118, 11678–11679 CrossRef CAS.
- (a) J. T. Groves and R. Neumann, J. Am. Chem. Soc., 1987, 109, 5045–5047 CrossRef CAS; (b) J. T. Groves and R. Neumann, J. Org. Chem., 1988, 53, 3891–3893 CrossRef CAS; (c) J. T. Groves and R. Neumann, J. Am. Chem. Soc., 1989, 111, 2900–2909 CrossRef CAS.
- (a) S. Das, C. D. Incarvito, R. H. Crabtree and G. W. Brudvig, Science, 2006, 312, 1941–1943 CrossRef CAS; (b) S. Das, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2008, 130, 1628–1637 CrossRef CAS PubMed.
- (a) D. Font, M. Canta, M. Milan, O. Cussó, X. Ribas, R. J. M. Klein Gebbink and M. Costas, Angew. Chem., Int. Ed., 2016, 55, 5776–5779 CrossRef CAS PubMed; (b) G. Olivo, G. Farinelli, A. Barbieri, O. Lanzalunga, S. Di Stefano and M. Costas, Angew. Chem., Int. Ed., 2017, 56, 16347–16351 CrossRef CAS PubMed; (c) G. Olivo, G. Capocasa, O. Lanzalunga, S. Di Stefano and M. Costas, Chem. Commun., 2019, 55, 917–920 RSC.
- D. S. Kemp and K. S. Petrakis, J. Org. Chem., 1981, 46, 5140–5143 CrossRef CAS.
- Selected examples on hydrogen bond-mediated molecular recognition induced by a similar structural motif: (a) J. Rebek, K. Williams, K. Parris, P. Ballester and K. S. Jeong, Angew. Chem., Int. Ed., 1987, 26, 1244–1245 CrossRef; (b) J. Rebek, B. Askew, P. Ballester, C. Buhr, S. Jones, D. Nemeth and K. Williams, J. Am. Chem. Soc., 1987, 109, 5033–5035 CrossRef CAS; (c) K. Williams, B. Askew, P. Ballester, C. Buhr, K. S. Jeong, S. Jones and J. Rebek, J. Am. Chem. Soc., 1989, 111, 1090–1094 CrossRef CAS; (d) R. K. Castellano, V. Gramlich and F. Diederich, Chem.–Eur. J., 2002, 8, 118–129 CrossRef CAS; (e) R. Faraoni, R. K. Castellano, V. Gramlich and F. Diederich, Chem. Commun., 2004, 370–371 RSC; (f) D. B. Varshney, X. Gao, T. Friščić and L. R. MacGillivray, Angew. Chem., Int. Ed., 2006, 45, 646–650 CrossRef CAS PubMed.
- For reviews on hydrogen bond-mediated catalysis, see: (a) N. R. Mote and S. H. Chikkali, Chem.–Asian J., 2018, 13, 3623–3646 CrossRef CAS PubMed; (b) X. Fang and C.-J. Wang, Chem. Commun., 2015, 51, 1185–1197 RSC; (c) X. Yu and W. Wang, Chem.–Asian J., 2008, 3, 516–532 CrossRef CAS PubMed; (d) A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713–5743 CrossRef CAS; (e) M. S. Taylor and E. N. Jacobsen, Angew. Chem., Int. Ed., 2006, 45, 1520–1543 CrossRef CAS PubMed; (f) P. R. Schreiner, Chem. Soc. Rev., 2003, 32, 289–296 RSC.
- For recent examples, see: (a) Y. Park and S. Chang, Nat. Catal., 2019, 2, 219–227 CrossRef CAS; (b) J.-P. Berndt, Y. Radchenko, J. Becker, C. Logemann, D. R. Bhandari, R. Hrdina and P. R. Schreiner, Chem. Sci., 2019, 10, 3324–3329 RSC; (c) X. Lu, Y. Yoshigoe, H. Ida, M. Nishi, M. Kanai and Y. Kuninobu, ACS Catal., 2019, 9, 1705–1709 CrossRef CAS; (d) S.-T. Bai, V. Sinha, A. M. Kluwer, P. R. Linnebank, Z. Abiri, P. Dydio, M. Lutz, B. de Bruin and J. N. H. Reek, Chem. Sci., 2019, 10, 7389–7398 RSC; (e) S. T. Bai, C. B. Bheeter and J. N. H. Reek, Angew. Chem., Int. Ed., 2019, 58, 13039–13043 CrossRef CAS PubMed; (f) W. Fang and B. Breit, Angew. Chem., Int. Ed., 2018, 57, 14817–14821 CrossRef CAS PubMed; (g) X. Li, C. You, Y. Yang, Y. Yang, P. Li, G. Gu, L. W. Chung, H. Lv and X. Zhang, Chem. Sci., 2018, 9, 1919–1924 RSC; (h) H. J. Davis, G. R. Genov and R. J. Phipps, Angew. Chem., Int. Ed., 2017, 56, 13351–13355 CrossRef CAS; (i) Z. Han, P. Li, Z. Zhang, C. Chen, Q. Wang, X.-Q. Dong and X. Zhang, ACS Catal., 2016, 6, 6214–6218 CrossRef CAS.
- For reviews on bioinspired oxygenation, see: (a) D. Vidal, G. Olivo and M. Costas, Chem.–Eur. J., 2018, 24, 5042–5054 CrossRef CAS; (b) M. Milan, M. Bietti and M. Costas, Chem. Commun., 2018, 54, 9559–9570 RSC.
- For seminal work on hydrogen bond-mediated molecular recognition utilizing a related lactam, see: (a) D. G. Lonergan, J. Riego and G. Deslongchamps, Tetrahedron Lett., 1996, 37, 6109–6112 CrossRef CAS; (b) D. G. Lonergan, J. Halse and G. Deslongchamps, Tetrahedron Lett., 1998, 39, 6865–6868 CrossRef CAS; (c) D. G. Lonergan and G. Deslongchamps, Tetrahedron, 1998, 54, 14041–14052 CrossRef CAS.
- For a recent perspective, see: F. Burg and T. Bach, J. Org. Chem., 2019, 84, 8815–8836 CrossRef CAS PubMed.
- (a) P. Fackler, C. Berthold, F. Voss and T. Bach, J. Am. Chem. Soc., 2010, 132, 15911–15913 CrossRef CAS PubMed; (b) P. Fackler, S. M. Huber and T. Bach, J. Am. Chem. Soc., 2012, 134, 12869–12878 CrossRef CAS PubMed; (c) F. Zhong and T. Bach, Chem.–Eur. J., 2014, 20, 13522–13526 CrossRef CAS PubMed.
- (a) T. Höke, E. Herdtweck and T. Bach, Chem. Commun., 2013, 49, 8009–8011 RSC; (b) J. R. Frost, S. M. Huber, S. Breitenlechner, C. Bannwarth and T. Bach, Angew. Chem., Int. Ed., 2015, 54, 691–695 CAS; (c) F. Burg, M. Gicquel, S. Breitenlechner, A. Pöthig and T. Bach, Angew. Chem., Int. Ed., 2018, 57, 2953–2957 CrossRef CAS PubMed.
- For mechanistic studies on the Mn-oxo species, see: (a) F. De Angelis, N. Jin, R. Car and J. T. Groves, Inorg. Chem., 2006, 45, 4268–4276 CrossRef CAS PubMed; (b) W. J. Song, M. S. Seo, S. DeBeer George, T. Ohta, R. Song, M.-J. Kang, T. Tosha, T. Kitagawa, E. I. Solomon and W. Nam, J. Am. Chem. Soc., 2007, 129, 1268–1277 CrossRef CAS PubMed; (c) N. Jin, M. Ibrahim, T. G. Spiro and J. T. Groves, J. Am. Chem. Soc., 2007, 129, 12416–12417 CrossRef CAS PubMed; (d) S. Fukuzumi, N. Fujioka, H. Kotani, K. Ohkubo, Y.-M. Lee and W. Nam, J. Am. Chem. Soc., 2009, 131, 17127–17134 CrossRef CAS PubMed; (e) X. Wu, M. S. Seo, K. M. Davis, Y.-M. Lee, J. Chen, K.-B. Cho, Y. N. Pushkar and W. Nam, J. Am. Chem. Soc., 2011, 133, 20088–20091 CrossRef CAS PubMed; (f) D. F. Leto, R. Ingram, V. W. Day and T. A. Jackson, Chem. Commun., 2013, 49, 5378–5380 RSC; (g) M. Guo, M. S. Seo, Y.-M. Lee, S. Fukuzumi and W. Nam, J. Am. Chem. Soc., 2019, 141, 12187–12191 CrossRef CAS PubMed.
- (a) M. Guo, T. Corona, K. Ray and W. Nam, ACS Cent. Sci., 2019, 5, 13–28 CrossRef CAS PubMed; (b) X. Huang and J. T. Groves, Chem. Rev., 2018, 118, 2491–2553 CrossRef CAS PubMed; (c) X. Huang and J. T. Groves, J. Biol. Inorg Chem., 2017, 22, 185–207 CrossRef CAS PubMed; (d) W. Liu, M.-J. Cheng, R. J. Nielsen, W. A. Goddard and J. T. Groves, ACS Catal., 2017, 7, 4182–4188 CrossRef CAS; (e) C. Arunkumar, Y.-M. Lee, J. Y. Lee, S. Fukuzumi and W. Nam, Chem.–Eur. J., 2009, 15, 11482–11489 CrossRef CAS PubMed; (f) B. Meunier, S. P. de Visser and S. Shaik, Chem. Rev., 2004, 104, 3947–3980 CrossRef CAS PubMed; (g) J. T. Groves and M. Van der Puy, J. Am. Chem. Soc., 1976, 98, 5290–5297 CrossRef CAS; (h) J. T. Groves and G. A. McClusky, J. Am. Chem. Soc., 1976, 98, 859–861 CrossRef CAS.
- (a) H. Kagan and J. Fiaud, Topics in Stereochemistry, John Wiley and Sons, Inc., New York, 1988 Search PubMed; (b) J. M. Keith, J. F. Larrow and E. N. Jacobsen, Adv. Synth. Catal., 2001, 343, 5–26 CrossRef CAS.
- For non-enzymatic examples and reviews on the kinetic resolution of racemic alcohols with high s-factors, see: (a) B. Tao, J. C. Ruble, D. A. Hoic and G. C. Fu, J. Am. Chem. Soc., 1999, 121, 5091–5092 CrossRef CAS; (b) S. Hashiguchi, A. Fujii, J. Takehara, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1995, 117, 7562–7563 CrossRef CAS; (c) S. Hashiguchi, A. Fujii, K. J. Haack, K. Matsumura, T. Ikariya and R. Noyori, Angew. Chem., Int. Ed., 1997, 36, 288–290 CrossRef CAS; (d) S. Y. Lee, J. M. Murphy, A. Ukai and G. C. Fu, J. Am. Chem. Soc., 2012, 134, 15149–15153 CrossRef CAS PubMed; (e) H. Pellissier, Adv. Synth. Catal., 2011, 353, 1613–1666 CrossRef CAS; (f) C. E. Müller and P. R. Schreiner, Angew. Chem., Int. Ed., 2011, 50, 6012–6042 CrossRef PubMed.
- (a) D. G. Blackmond, Angew. Chem., Int. Ed., 2005, 44, 4302–4320 CrossRef CAS PubMed; (b) J. S. Mathew, M. Klussmann, H. Iwamura, F. Valera, A. Futran, E. A. C. Emanuelsson and D. G. Blackmond, J. Org. Chem., 2006, 71, 4711–4722 CrossRef CAS PubMed; (c) D. G. Blackmond, J. Am. Chem. Soc., 2015, 137, 10852–10866 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures and full characterisation for all starting materials and products (2–10), spectroscopic data, NMR spectra, kinetic studies, etc. CCDC 1967795. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc06089h |
| This journal is © The Royal Society of Chemistry 2020 |