
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWinning the fight against biofilms: the first six-month study showing no biofilm formation on zwitterionic polyurethanes†
Huifeng
Wang
,
Daniel Edward
Christiansen
,
Shafigh
Mehraeen
and
Gang
Cheng
 *
*
Department of Chemical Engineering, The University of Illinois at Chicago, Chicago, IL 60607, USA. E-mail: gancheng@uic.edu; Web: http://https://gancheng.people.uic.edu
First published on 25th April 2020
Abstract
Biofilms have been a long-standing challenge for healthcare, water transport, and many other industries. They lead to bacterial growth and infections in animals, food products, and humans, cause premature removal of the implanted materials or devices from patients, and facilitate fouling and corrosion of metals. Despite some published and patented methods on minimizing the effects of biofilms for a short period (less than two weeks), there exists no successful means to mitigate or prevent the long-term formation of biofilms. It is even more challenging to integrate critical anti-fouling properties with other needed physical and chemical properties for a range of applications. In this study, we developed a novel approach for combining incompatible, highly polar anti-fouling groups with less polar, mechanically modifying groups into one material. A multifunctional carboxybetaine precursor was designed and introduced into polyurethane. The carboxybetaine precursors undergo rapid, self-catalyzed hydrolysis at the water/material interface and provide critical anti-fouling properties that lead to undetectable bacterial attachment and zero biofilm formation after six months of constant exposure to Pseudomonas aeruginosa and Staphylococcus epidermidis under the static condition in a nutrient-rich medium. This zwitterionic polyurethane is the first material to demonstrate both critical anti-biofilm properties and tunable mechanical properties and directly validates the unproven anti-fouling strategy and hypothesis for biofilm formation prevention. This approach of designing ‘multitasking materials’ will be useful for the development of next generation anti-fouling materials for a variety of applications.
Introduction
Biofilms consist of clusters of bacterial cells, which are surrounded by extracellular polymeric substances and nucleic acids, allowing them to stick to and grow on nearly all existing materials. They are difficult to treat once formed, and humankind has suffered from biofilm-associated infection for all of history. Since the industrial revolution, biofouling has posed an even more severe challenge to a broad range of industries due to increased marine travel and trade and use of the polymeric materials in healthcare and general-use products. Unfortunately, we have not been able to adequately control biofilms and tackle these problems, leading to life-threating infections,1–3 marine fouling,4 and biocorrosion of pipelines5 and other surfaces.6 Treatment, lost-productivity, and other effects caused by biofilms in healthcare and marine industries alone cost the United States an estimated 55 billion USD per annum.7,8 Apart from economic savings, improving the quality of life and protecting the environment by not using toxic biocides are enough motivations to make the fight against biofilms a global priority.As of yet, there have been no methods or tools for effectively resisting biofilm formation for more than two weeks. Traditional preemptive approaches employ biocides and antibiotics to mitigate biofilms; however, studies have shown that these methods are ineffective in preventing formation of biofilms and treating them9 and may trigger initial biofilm formation,10 and/or generate resistance to antimicrobial agents.11 Also, the effectiveness of antimicrobial-impregnated materials that release antimicrobial agents remains unproven.12 A third approach utilizes chemical/mechanical kill-on-contact materials;13,14 however, these materials decline in efficacy from use over time, promote resistance, and/or leave behind carcasses of killed microorganisms on the surface, providing new substrates for biofilms to adhere and grow on.15,16 Due to the intrinsic limitations of the aforementioned strategies, a fourth, the anti-fouling/non-stick approach, wherein a substrate resists any bacterial adhesion leading to no biofilm formation, has been studied extensively in the past twenty years.17,18 The central hypothesis of this tactic is that a hydration layer atop a material would prevent the adsorption of biomacromolecules and function as a conditioning layer and mediate cell adhesion and biofilm formation.17,18 By preventing non-specific adsorption of biomacromolecules, there can be no cell debris or decrease in effectiveness from “wear and tear,”; thus, the material will not lose its anti-fouling ability. Given the simple method of preventing biofouling and no apparent negative side-effects, the anti-fouling approach seems to be the most practical strategy for preventing biofilm formation.2,13 However, while there have been considerable advancements in the abilities of these materials over the past few decades, so far, none of them have exhibited successful anti-biofilm properties beyond two weeks in complex nutrient-rich systems.
One of the current most widely used anti-fouling materials is poly(ethylene glycol) (PEG) due to its commercial availability, high hydrophilicity, and compatibility with a broad range of solvents and standard building blocks for polymer synthesis.19 Although PEG has been explored as a material-component in nearly all anti-fouling applications, none of the long-term biofouling challenges, such as marine fouling,4 healthcare-associated infections,20 foreign body responses (FBRs),21 and medical device-induced thrombosis,22 have been adequately addressed. Recent studies have observed that PEG cannot effectively resist protein adsorption from blood23 and triggers FBRs24,25 and immune responses,26,27 suggesting that PEG does not carry the critical anti-fouling properties (<5 ng cm−2 of fibrinogen adsorbed on a surface) necessary for long-term applications in complex biological systems.28
Recently, zwitterionic polymers such as poly(phosphorylcholine),29 poly(carboxybetaine),30 and poly(sulfobetaine)31,32 have been investigated for their outstanding anti-fouling properties.33 Unlike PEG, zwitterionic polymers form a highly dense hydration layer via ionic hydration.34 In 2007, for the first time, Dr Jiang et al. showed that poly(carboxybetaine) polymer brushes could resist biofilm formation for up to 10 days with less than 6% bacteria compared to unmodified glass at room temperature.17 This study showed the potential of zwitterionic polymers in resisting biofilms; however, no transformative progress has been made since then due to several factors: first, zwitterionic homopolymers cannot be used alone due to their high water solubility. Second, incompatibility between highly polar zwitterionic materials and other nonpolar building blocks leads to phase segregation that compromises the mechanical properties (ductility, durability, tensile, and compressive strength) of the material. Third, existing zwitterionic copolymers exhibit higher melting temperatures,35 which make them challenging to use with common commercial polymer processing methods, such as extrusion and injection molding. Thus, zwitterionic materials have primarily been studied as interfacial materials by being grafted from or grafted to the surface of supporting materials or as porous and soft hydrogels. To address this challenge, Dr Jiang et al. developed the first coating-free zwitterionic polymeric elastomer,36 but its anti-biofilm properties were not investigated. Among materials that form a supporting structure, polyurethane (PU) is particularly attractive due to its mechanical properties, chemical stability, and, most importantly, tunability, i.e., controllable material strength and thermal stability.37,38 PU is widely utilized in the medical industry for a variety of applications, such as catheters,39–41 tissue scaffolding,42–44 drug delivery vehicles,45,46 and various other implanted biomedical devices.47,48 Tunability of PU is achieved by varying the structure of the backbone through altering the ratios of reactants.49,50 PU consists of diisocyanates with a chain extender or cross-linker as the hard segment and a polyol as the soft segment, whereby the mechanical strength of a PU corresponds to the abundance and structure of the hard segment, while the soft segments dictate elastic behavior.51 There have been some studies on the development of zwitterionic PUs via surface modification52,53 or backbone conjugation of PU.54 However, none of these materials have demonstrated long-term resistance to biofilms in protein-rich media. It has been an enormous challenge to create a stable, consistent, and effective anti-fouling surface using the surface modification approach by a time and cost-saving process for real-world applications.
Due to the above limitations, anti-fouling polymers have not yet demonstrated their full potential in mitigating biofilms. There is an urgent need for a material platform that can validate the widely-used anti-fouling hypothesis for biofilm formation prevention, achieve “zero” bacterial cell attachment and biofilm formation, and be readily adapted for a broad range of applications.
For the abovementioned reasons, in this study, our objective is to create a material platform with tunable properties to mitigate biofilms, develop a simple and effective design strategy that can be adapted for the development of multifunctional materials, and validate the widely-used anti-fouling hypothesis for the management of long-term biofouling challenges. To achieve our goal, we designed a multifunctional carboxybetaine precursor (diethylamine ethyl acetate (DEAEA)) and introduced DEAEA diol into the backbone of PU via the same reaction mechanism used in PU synthesis. The chemical and physical properties of zwitterionic polyurethane were then tuned by altering the feed ratios of DEAEA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :glycerol:hexamethylene diisocyanate (HDI). The ability of the material to resist protein adsorption and cell attachment was investigated. A six-month anti-biofilm study was conducted against two challenging biofilm-forming strains, Pseudomonas aeruginosa PAO1 and Staphylococcus epidermidis.
:glycerol:hexamethylene diisocyanate (HDI). The ability of the material to resist protein adsorption and cell attachment was investigated. A six-month anti-biofilm study was conducted against two challenging biofilm-forming strains, Pseudomonas aeruginosa PAO1 and Staphylococcus epidermidis.
Results and discussion
Synthesis of PCBGUs
To avoid the drawbacks of the surface modification approach (such as time and cost-consuming processes, unsatisfactory long-term stability, etc.), anti-fouling groups have to be directly incorporated into PU during polymer synthesis. However, existing zwitterionic functional groups contain both anions and cations that form highly polar inner salts, which are incompatible with nonpolar solvents and reactants, such as diisocyanate and other polyols. For this reason, we designed a multifunctional DEAEA diol as both a chain extender of the PU backbone and an anti-fouling precursor. DEAEA contains an uncharged tertiary amine and a carboxylate ester. DEAEA exhibits similar solubility and polarity to HDI and polyols in commonly-used organic solvents. It can be efficiently synthesized via the Michael type reaction55 of diethanolamine and ethyl acrylate, usually producing more than 95% yield. PUs with different monomer ratios were synthesized via a one-pot reaction using glycerol as a crosslinker and hard segment (Fig. 1). We designed five polymers with different stoichiometric ratios of hydroxyl groups in DEAEA:hydroxyl groups of glycerol:isocyanate groups in HDI (100:0:100, 75:25:100, 50:50:100, 25:75:100 and 0:100:100) named PCBGU-100, PCBGU-75, PCBGU-50, PCBGU-25, and PGU, respectively. DEAEA plays two primary roles in the material: first, when DEAEA on the surface of the material is exposed to water, it undergoes fast and self-catalyzed hydrolysis to generate zwitterionic carboxybetaine functional groups, which provide critical anti-fouling properties. Second, DEAEA also functions as the soft segment in PCBGU, allowing for tunability of the mechanical, swelling, and anti-fouling properties by adjusting the ratio of soft and hard domains, as demonstrated in the following sections. For a control, PEG-based polyurethane (PEGCU-50) with a stoichiometric ratio of 50:50:100 (hydroxyl groups in PEG 2K:hydroxyl groups of glycerol:isocyanate groups in HDI) was synthesized and used for anti-fouling studies.
 | ||
| Fig. 1 (a) Synthetic route of PCBGU and its hydrolytic reaction in water. (b) Schematic diagram of zwitterionic polyurethane formation after hydrolysis. | ||
Fourier transform infrared spectroscopy (FT-IR)
FT-IR spectra of PCBGUs are shown in Fig. 2(a). The absence of absorbance at 2250–2270 cm−1, which is caused by –NCO stretching, indicates that the reaction proceeded to completion, and all isocyanate groups were completely depleted. The absorption bands of N–H stretching vibrations were observed at 3321 cm−1, C–H stretching at 2922 cm−1, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching at 1698 cm−1, and C–O stretching at 1140 cm−1. N–H deformation bands were found in the range of 1500–1600 cm−1. The sharp peaks ranging from 1690–1700 cm−1 indicate CO stretching vibrations of urethane and the carboxyl group on the DEAEA side chain. An aliphatic C–H stretching mode of 2880–2929 cm−1 and a carboxylic stretching absorption band at 3735–3770 cm−1 were also observed. Moreover, IR spectra demonstrate the characteristic peaks for C–O–C stretching of the hard segment at 1140 cm−1. These signature stretching bands indicate the formation of PU and the depletion of the reactants.
O stretching at 1698 cm−1, and C–O stretching at 1140 cm−1. N–H deformation bands were found in the range of 1500–1600 cm−1. The sharp peaks ranging from 1690–1700 cm−1 indicate CO stretching vibrations of urethane and the carboxyl group on the DEAEA side chain. An aliphatic C–H stretching mode of 2880–2929 cm−1 and a carboxylic stretching absorption band at 3735–3770 cm−1 were also observed. Moreover, IR spectra demonstrate the characteristic peaks for C–O–C stretching of the hard segment at 1140 cm−1. These signature stretching bands indicate the formation of PU and the depletion of the reactants.
 | ||
| Fig. 2 Fourier-transform infrared spectra and thermal properties of PCBGUs with varying component ratios. (a) FT-IR spectra, (b) TGA, (c) DTG, and (d) DSC profiles of PCBGU samples. | ||
Thermal stability and transition
The thermal stability and transition behavior of polymeric materials, which affect their melting point, decomposition, and glass transition temperature, play significant roles in the manufacturing processes of most polymeric materials. Thermal stabilities were determined by thermogravimetric analysis (TGA). At temperatures greater than 200 °C, decomposition of typical PUs occurs in three stages.56 In the first stage, alcohol and isocyanate groups are formed from urethane bond decomposition. Subsequently, dimerization of isocyanates to form carbodiimides prevents the complete volatilization of the resulting chain fragments. Then hydroxyl groups of the alcohols react with carbodiimides and form relatively stable N-substituted ureas. In the final stage, at higher temperatures, the N-substituted ureas degrade to volatile products and yield a small amount of carbon char. The TGA and differential-TGA (DTG) profiles (Fig. 2(b)) of PCBGUs show the three aforementioned steps of thermal decomposition. The first peak is attributed to the decomposition of urethane bonds, and the second and third peaks are related to the decomposition of N-substituted ureas. PCBGU-100, PCBGU-75, PCBGU-50, and PCBGU-25 show an average 43% mass loss between 200 and 300 °C in the first stage and around 42–48% combined mass loss in the second and third stages from 300 to 500 °C. PGU, which has the highest crosslinking density, shows a higher decomposition temperature between 250 and 350 °C with 46% mass loss in the first stage, and exhibit 44% mass loss from 370 to 500 °C in the second and third stages. Finally, all materials left a small amount of residue (7.3–15.8 wt%) above 500 °C. The TGA study indicates that PCBGUs have a similar and high thermal stability to commercially-available PUs, which is highly desired.57Thermal transition (i.e., melting and glass transition) behaviors of PCBGUs were studied by differential scanning calorimetry (DSC), as shown in Fig. 2(c). The glass transition temperature and melting temperature of PCBGUs are shown in Table 1. The glass transition temperatures of PCBGU-100, PCBGU-75, PCBGU-50, PCBGU-25, and PGU were 39.7 °C, 12.0 °C, 15.3 °C, 38.3 °C, and 45.3 °C, respectively. Except for PCBGU-100, the glass transition temperatures of all other materials increased with increasing cross-linking density. PCBGU-100 consists of only DEAEA and HDI and forms an alternating linear copolymer; thus, the polymer chains were more oriented, leading to a higher transition temperature of 40 °C. For all samples, as the hard segment content increased, the melting temperature rose due to a higher cross-link density. Future rheology studies can be used to explore how the mechanical properties of the material will change over such a small temperature range. The DSC study shows that we can control the glass transition temperature and melting temperature of PCBGU by adjusting its composition.
| Sample | T g (°C) | T m (°C) |
|---|---|---|
| PCBGU-100 | 39.7 | 203.6 |
| PCBGU-75 | 12.0 | 221.7 |
| PCBGU-50 | 15.3 | 227.0 |
| PCBGU-25 | 38.3 | 227.8 |
| PGU | 45.3 | 247.3 |
Compression and rheological properties
To evaluate the mechanical tunability of PCBGUs, compression testing was performed. As shown in Fig. 4(a) and Table 2, before hydrolysis, PCBGU-100, PCBGU-50 and PGU exhibited good mechanical properties with both high compressive modulus (76.2, 105.3 and 96.9 MPa) and high breaking strain (>98.9%, >104.7%, and >101.6%). The materials remain intact under a load of 5000 N, which is the upper limit of our load cell. These results suggest a good interfacial strength between soft and hard domains. Surprisingly, PCBGU-75 and PCBGU-25 demonstrated lower compressive moduli (3.2 MPa and 36.9 MPa) and lower breaking strains (60.2% and 91.3%) than other materials. This phenomenon may be caused by the less organized microscopic structures of PCBGU-75 and PCBGU-25. As Fig. 3(a) shows, after equilibration in water, PCBGU-100, PCBGU-75, PCBGU-50, PCBGU-25, and PGU exhibit lower compressive moduli (5.6 MPa, 2.0 MPa, 7.8 MPa, 2.3 MPa, and 22.5 MPa, respectively) because the absorbed water molecules reduce the inter/intramolecular interactions of polymer chains. Although all PCBGUs after equilibration in water showed lower compressive moduli compared to those before hydrolysis, their compressive moduli are still significantly higher than that of other zwitterionic polymers that have lower compressive moduli up to a few hundred kPa but similar high breaking strains (>50%).58,59 An earlier study reported that a highly chemically-crosslinked zwitterionic polymer showed a high modulus up to 100 MPa,60 but its breaking strain was not reported.60 In addition, the moduli of PCBGUs are in the upper-level range of polyurethane elastomers, which have been used in medical applications, such as tubing, packaging (e.g., IV solution bags), coatings, and soft-touch components.61 It is worth noting that typical linear zwitterionic polymers cannot carry any load in water because they are highly soluble in water. In contrast to the existing zwitterionic polymers, linear PCBGU-100 after hydrolysis still shows a high compressive modulus of 5.5 MPa and a moderately high breaking strain of 45.8%. The high modulus and breaking strain of linear PCBGU-100 are generated by a high density of the hydrogen bond-forming urethane group in the polymer. Among all materials, PGU displayed the highest compressive modulus, which can be attributed to PGU's highest crosslinking density among all samples. Interestingly, although PGU shows a high breaking strain (≫101.6%) before equilibration in water, its breaking strain after equilibration in water reduced to 32.7%, which is much lower than that of all PCBGUs.| Before hydrolysis | After hydrolysis | ||||
|---|---|---|---|---|---|
| Compressive modulus (MPa) | Breaking strain (%) | Compressive modulus (MPa) | Breaking strain (%) | Swelling ratio (%) | |
| PCBGU-100 | 76.2 | ≫98.9 | 5.5 | 45.8 | 35.3 |
| PCBGU-75 | 3.2 | 60.2 | 2.0 | 51.7 | 10.1 |
| PCBGU-50 | 105.3 | ≫104.7 | 7.8 | 74.2 | 9.4 |
| PCBGU-25 | 36.9 | 91.3 | 2.3 | 52.7 | 8.4 |
| PGU | 96.9 | ≫101.6 | 22.5 | 32.7 | 9.8 |
 | ||
| Fig. 3 Mechanical and rheological properties of PCBGU. Compression stress–strain curves of PCBGU (a) before and (b) after hydrolysis. Rheological behavior of (c) PCBGU-100, (d) PCBGU-75, (e) PCBGU-50, and (f) PCBGU-25 at 100 °C. | ||
The rheological study showed that PCBGU-50 exhibits the highest strain rate of all materials tested, indicating better elasticity and higher interfacial strength between soft and hard domains. The frequency-sweep rheological studies at 100 °C showed that all samples were solid-like and still held high shear strength. PCBGU-25 and PCBGU-50 demonstrate higher moduli than PCBGU-75 and PCBGU-100 due to higher crosslinking densities. Both compression and rheological studies clearly show that the soft segment, DEAEA, can be used to tune the mechanical properties over an extensive range. If it is needed, the mechanical properties can be tuned by adding a third polyol, such as 1,4-butanediol. We expect that the addition of 1,4-butanediol will increase the modulus and reduce swelling. Because PCBGU-25 that contains only 25 mol% DEAEA as polyol shows sufficient anti-fouling properties, there is a large window for us to combine DEAEA with other polyols to tune the mechanical properties of this material platform.
Swelling and hydrolysis of PCBGUs
Controllable swelling of polymeric materials is a critical parameter for many commercial and industrial applications. The swelling ratios of all materials were characterized, and the results are shown in Table 2. The swelling ratios of PCBGUs vary with the feed ratio between DEAEA and glycerol. The swelling and water uptake of a material are determined by its solubility and crosslinking density. In general, higher solvent solubility of a material leads to higher swelling; however, the crosslinking density, including both chemical and physical crosslinking, adversely affects swelling. Thus, it was expected that the lower crosslinking density in a PCBGU with a lower glycerol/DEAEA ratio would lead to higher swelling. Surprisingly, all other PCBGUs, except for PCBGU-100 that contains no glycerol, show a low swelling ratio of 8–10% and are similar to PGU that does not contain DEAEA. Although linear PCBGU-100 has the highest swelling ratio (35.3%) among all materials due to lack of glycerol as a crosslinker and the higher CB content, it remains an intact solid after equilibration in water, unlike current linear zwitterionic polymers that dissolve entirely in water. The hydrolysis of DEAEA produces hydrophilic carboxybetaine groups that increase the water uptake of the material after equilibration in water. Although PCBGUs and PGU have different swelling ratios in water, their dimensions did not change at all. We believe the physical crosslinking network formed by the strong inter- and intra-molecular hydrogen bonds among urethane groups significantly limited water penetration and swelling. In contrast to PCBGUs, the dimension of PEGCU-50, our control material, increases over 300% after equilibration in water. It should be noted that low deformation properties of PCBGUs are beneficial for applications, such as coatings for stents, heart valves, and catheters.To resist nonspecific adsorption of biomacromolecules and biofilm formation, a surface needs to carry effective and sufficient anti-fouling groups. PCBGUs can produce highly active anti-fouling carboxybetaine groups via hydrolysis of the ethyl ester of DEAEA. It should be noted that the beta-amino ester of DEAEA undergoes much faster hydrolysis due to two reasons, the higher acidity of the carboxylate group and the self-catalyzed hydrolysis properties, which will be expanded upon in the following sentences. To understand the hydrolysis process of PCBGUs, the pH values of DI water containing each sample were recorded during hydrolysis, shown in Fig. 4(c). All solutions were basic initially, and the pH values decreased rapidly for the first 24 hours, eventually reaching equilibrium within a few days. The self-catalyzed hydrolysis properties, as evidenced by the high initial pH in the solution, are caused by the immediate protonation of the tertiary amines of DEAEA components at the interface after the materials were submerged in water. The resulting hydroxide ions then catalyze the hydrolysis of the ester bond of DEAEA and produce carboxylate groups. The carboxylate group will form inner salts with the protonated tertiary amine. Because the hydrolysis of ester bonds consumes hydroxide ions, the higher pH value at the initial stage leads to faster hydrolysis and a higher pH change rate. Eventually, the pH value of the solution slowly approaches the neutral state when the material reaches equilibrium. Compared to PCBGU-75 and PCBGU-100, PCBGU-25 and PCBGU-50 had slightly higher final pH values in the solution. Because the pKa of the carboxylate is higher than the pKb of the tertiary amine in CB,62 lower CB contents in PCBGU-25 and PCBGU-50 than other PCBGUs might cause slightly higher pH values after materials reach equilibrium. The higher DEAEA ratio and lower crosslinking in PCBGU-100 and PCBGU-75 cause a slightly higher swelling ratio and result in the exposure of more internal DEAEA components to water; thus, faster water penetration leads to a higher local pH value and promotes the materials to complete hydrolysis and reach equilibrium more quickly.
 | ||
| Fig. 4 The hydrolytic process of PCBGUs monitored by pH change and surface protein adsorption and cell attachment. Protein adsorption on PCBGU surfaces after submersion in (a) water and (b) pH 8.5 Tris buffer for different lengths of time. (c) pH change of PCBGU solution as a function of time. (d) Cell attachment of PCBGU after hydrolysis. The cell densities on PEGCU-50 and PGU hydrogel surfaces were (15.0 ± 1.9) × 104 cells per cm2 and (62.2 ± 2.6) × 104 cells per cm2, correspondingly. For the PCBGU-100, PCBGU-75, PCBGU-50, and PCBGU-25 surface, there was no observable cell attachment. | ||
Although it took a few days for the whole material to reach equilibrium, the surface needs much less time to do so because of the higher pH at the initial stage. As it was mentioned in the previous section, although the moduli of PCBGUs decrease after hydrolysis, these materials still behave like an elastomer with a high modulus in the MPa range and a high breaking strain (>50%). In addition, all PCBGUs show no dimensional change after the equilibration in water. It will be up to the type of application to decide whether the whole material needs to be hydrolyzed before use. If it is needed, PCBGUs can be pretreated to reach equilibrium. Self-catalyzed hydrolysis is a great advantage for production because it avoids the addition and removal of acids or bases during post-processing treatments (if any). If a shorter processing time is preferred, PCBGUs can reach equilibrium in a moderately basic solution in hours.
Protein adsorption
It has been reported that adsorbed biomacromolecules on a material surface serve as a conditioning layer to trigger biofouling, including biofilms, fibrosis, foreign body responses, and thrombosis.2,24,63,64 Among biomacromolecules, fibrinogen (Fg) has been widely used as a standard in vitro screening tool to assess the anti-fouling properties of materials due to its ability to adsorb to a variety of surfaces.65 Therefore, we used fluorescein isothiocyanate (FITC)-labeled Fg to assess the anti-biomolecule adsorption properties of PCBGUs and the commercially-available medical-grade API-PU as the control material. The adsorption of FITC-labeled Fg on all surfaces was normalized to that of API-PU at a given time. As shown in Fig. 4(a and b), API-PU shows the highest protein adsorption among all materials. Before hydrolysis, PCBGUs show moderate Fg adsorption ranging from 30% to 65% compared to API-PU. As hydrolysis proceeds, the adsorption of Fg on all PCBGU surfaces in DI water (Fig. 4(a)) and pH 8.5 buffer (Fig. 4(b)) drastically decreases. After two hours in pH 8.5 buffer, all PCBGUs quickly switch to an anti-fouling state, and Fg adsorption (Table 3) on PCBGU surfaces is less than 4% of that on API-PU, indicating that the surface can effectively resist non-specific protein adsorption. In water, self-catalyzed hydrolysis requires a longer time for complete hydrolysis because the hydrolysis of carboxybetaine reduces the hydroxide concentration that drives hydrolysis. Within 48 hours, all PCBGUs switch to the anti-fouling state with less than 3% Fg adsorption relative to API-PU (Table 3). In this study, we synthesized PEGCU-50, which contains 50 mol% PEG as the polyol and used it as the positive anti-fouling control, given the consideration of PEG as the gold standard among anti-fouling materials. It is believed that a higher ratio of the anti-fouling moiety improves the anti-fouling performance of the material; however, a high PEG content often causes significant swelling of the material, making it very fragile and difficult to handle. PEGCU-50 shows a much higher swelling than all PCBGUs with over 300% dimension change and, despite the high PEG content, showed 47% Fg adsorption before equilibration in water. After equilibration in both DI water and pH 8.5 buffer, PEGCU-50 still shows over 25% Fg adsorption compared to the API-PU control surface.| Fg adsorption (%) | |||||
|---|---|---|---|---|---|
| PCBGU-100 | PCBGU-75 | PCBGU-50 | PCBGU-25 | PEGCU-50 | |
| After 2 days in DI water | 0.4 | 2.9 | 2.0 | 1.6 | 25.3 |
| After 2 hours in pH 8.5 buffer | 0.6 | 2.7 | 3.6 | 2.6 | 29.0 |
Previous studies observed that the higher water content and hydrophilicity of a subject material led to improved anti-fouling properties.66,67 Simultaneously, it is generally valid that hydrophilic materials provide better short-term anti-fouling properties compared to hydrophobic materials.68 However, many studies, including our own, show that the chemical structure of the material determines the critical anti-fouling properties for long-term applications. For example, our previous study found that the carboxybetaine-functionalized dextran hydrogel is much more resistant to protein adsorption and cell attachment than unmodified dextran hydrogel with a similar water content.69 It should be noted that dextran is commonly used as an anti-fouling material to reduce protein adsorption and cell attachment.60,70,71 Another previous study of ours showed that zwitterionic carboxybetaine polymers highly resist protein adsorption in both the high-density polymer brushes form and the porous hydrogel form.58 Although the higher water content of these materials is in favor of their anti-fouling properties, PEGCU-50, with a much higher water content than all PCBGUs, shows much worse anti-fouling properties than PCBGUs. Another concern was that proteins may be trapped in the polymer network materials. However, previous studies demonstrated that the anti-fouling properties of porous zwitterionic materials are independent of crosslinking density and water content.60,70–72 All PCBGUs, regardless of swelling and crosslinking density, showed strong resistance to protein adsorption and long-term biofilm formation. Fg is a fairly large molecule (∼45 nm in length) with a molecular weight of 340 kDa. PCBGU-50 has the same crosslinking density as PEGCU-50, but shows much better resistance to Fg. Furthermore, PGU, which has the highest crosslinking density and a similar swelling to PCBGU, shows the worst anti-fouling properties. Our ongoing study has also seen uncrosslinked zwitterionic PUs with sufficient carboxybetaine contents highly resist protein adsorption (data not shown). It should be noted that each DEAEA unit contains one anti-fouling carboxybetaine group, but one PEG2000 molecule contains 45 ethylene glycol anti-fouling units. So, this study clearly shows that carboxybetaine is a much more efficient anti-fouling group than PEG and further confirms that the chemical structure of the material instead of swelling determines the critical anti-fouling properties.
Twenty years ago, Dr Horbett et al. observed that surfaces coated with PEG with less than 5 ng cm−2 Fg adsorption could reduce platelet activation and delay blood coagulation.73 Due to the complexity of the study, platelet activation and blood coagulation experiments are often conducted in vitro for a few hours. Materials with moderately reduced protein adsorption, such as PEG, have demonstrated transient positive results for short-term (a few days) applications in less challenging environments, but our recent studies28 unveiled that the critical anti-fouling properties, significantly lower than 5 ng cm−2 Fg adsorption, are needed for long-term biofouling challenges. Among all tested materials in this study, PCBGU-100 showed the lowest protein adsorption, 0.4% after two days of hydrolysis in water and 0.6% after two hours of hydrolysis in pH 8.5 Tris buffer. Because the critical anti-fouling threshold is at such a low level, it is challenging to measure the non-specific protein adsorption of the surfaces of bulk materials. Surface plasmon resonance (SPR) and quartz crystal microbalance (QCM), which have been used in our previous studies, are more sensitive than the fluorescence method that was used in this study, but these techniques are limited to thin films, or polymer brushes less than a few hundred nanometers. Although further study is needed to develop a more suitable and sensitive method to quantify the non-specific adsorption of biomacromolecules at extremely low levels, our method confirms PCBGU's excellent anti-fouling capability.
Mammalian cell attachment
In addition to biofilm-associated infection, medical devices and biomaterials also suffer from FBR and material-induced thrombosis, which are initiated by protein adsorption from blood and body fluids, followed by the attachment of macrophages, platelets, and fibroblasts. The adsorbed proteins function as the conditioning layer to facilitate the attachment of the fibroblast or other types of cells. We used NIH-3T3 fibroblast cells to further evaluate the anti-fouling properties of PCBGUs in resisting mammalian cell attachment. To better mimic the in vivo environment in which the materials are consistently exposed to blood or body fluids, PCBGU and control samples were first immersed in 100% fetal bovine serum (FBS) for two days to allow the adsorption of serum proteins and then the samples were incubated with NIH-3T3 fibroblast cells at 37 °C for 24 hours. After 24 hours, the cell attachment on all surfaces was measured by bright-field microscopy. The results (Fig. 4(d)) show that the cell densities on PEGCU-50 and PGU hydrogel surfaces were (15.0 ± 1.9) × 104 cells per cm2 and (62.2 ± 2.6) × 104 cells per cm2, respectively, with no attached cell observed on all PCBGU surfaces. Our method for the cell attachment study is more relevant to the in vivo environment and is more challenging than the commonly-used in vitro procedures, in which the material without pretreatment with 100% FBS is incubated with cell suspension in 10% FBS for 24–48 hours. Our results demonstrate that zwitterionic CB moieties are capable of resisting mammalian cell adhesion in the complex protein-rich medium. It is worth mentioning that PEG-based PU74 has been widely explored for anti-fouling purposes in both academia and industry. However, this study and previous studies23,28 clearly show that the anti-fouling properties of PEG are insufficient in complex solutions or in vivo environments for long-period blood-contacting applications.Bacterial attachment and biofilm formation
Previous studies have shown that hydrophilic polymers can resist bacterial attachment for a few hours, and PEG exhibited delayed biofilm formation for up to two days.75 Meanwhile, well-defined PCB brushes have also demonstrated less than 6% biofilm formation of P. aeruginosa PAO1 up to ten days.17 However, no anti-fouling material has shown zero biofilm formation for over two weeks in the complex systems, which contain a high concentration of biomacromolecules and are more relevant to healthcare and industrial enviroments. In this study, we selected two active biofilm-forming strains, P. aeruginosa PAO1 and S. epidermidis (both initially isolated from infected patients) to evaluate the real-world utility of PCBGU. P. aeruginosa is the most common pathogen causing infections of indwelling catheters and foreign body implants because of its ability to form biofilms on various materials and resist many commonly-used antibiotics. In this work, the biofilm formation of both strains was conducted under the challenging static condition in the complex protein-rich medium. The tested surfaces were placed in solution and challenged continuously by both biomacromolecules and bacteria. Unlike the flow condition, in which the bacteria can be washed off by shear stress, the static conditions allow the bacteria to either replicate on the surface or attach to the surface from the solution. To ensure that nutrients would not be a limiting factor for biofilm development, we used a full growth medium containing a sufficiently high concentration of biomacromolecules from tryptone (10 g L−1) and yeast extract (5 g L−1) and each day, 50% of the culture was replaced with fresh medium. Under this static condition, biofilms quickly formed on the Petri dish and control surfaces. Within three days, 85.4% of the surface of API-PU was covered by P. aeruginosa PAO1. PEG-based PEGCU-50 showed delayed biofilm formation at the initial stage, with 28.5% surface coverage by P. aeruginosa PAO1 after three days. After one week, the commercially available API-PU samples were fully covered by a biofilm, and the PEGCU-50 surface showed significant biofilm formation with 45.9% surface coverage (Fig. 5). After two weeks, both API-PU and PEGCU-50 surfaces were fully covered by P. aeruginosa PAO1. Meanwhile, all PCBGU surfaces not only show no biofilm formation but have barely any attached cells on them throughout the six-month study. | ||
| Fig. 5 Fluorescence images for the biofilm formation of P. aeruginosa PAO1 on PCBGU-100, PCBGU-75, PCBGU-50, and PCBGU-25 hydrogels. Commercially available API-PU and PEGCU-50 were used as control materials. The scale bar is 20 μm. | ||
It has been reported that two Staphylococci strains, S. aureus and S. epidermidis, account for about two-thirds of infections.76 Also stated in that report is that S. epidermidis is a predominant cause of implant-associated diseases, a key theme in our studies. After three days, the surface coverage of S. epidermidis on the API-PU surface reached 91.8%, while PEGCU-50 showed 11.8% surface coverage. After a week of continuous exposure, the PEGCU-50 was nearly 100% covered by a S. epidermidis biofilm (Fig. 6). For the remainder of the six-month study, API-PU and PEGCU-50 remained fully covered by S. epidermidis biofilm. Meanwhile, all PCBGU surfaces showed nearly zero bacterial attachment and biofilm growth after six months. To the best of our knowledge, this is the first successful demonstration that the anti-fouling approach can effectively resist biofilm formation at nearly a “zero” level for over six months. Although the experiment can be extended, we expect similar results under the same conditions. Our study confirmed that carboxybetaine is highly effective in resisting biofilm formation, and 25% of CB diols in PCBGU is sufficient to provide critical anti-biofilm properties. We believe that a lower CB content might work, but the threshold CB content ratio has to be determined in the future. This study clearly shows that commercial and industrial materials with moderate anti-fouling properties, such as PEG, cannot provide meaningful anti-fouling properties for long-term applications in complex biological systems, which are rich in biomacromolecules. The capability of PCBGU to resist long-term biofilm formation makes PCBGU a strong candidate for biomedical applications.
 | ||
| Fig. 6 Fluorescence images for biofilm formation of S. epidermidis on PCBGU-100, PCBGU-75, PCBGU-50, and PCBGU-25 hydrogels. Commercially available API-PU and PEGCU-50 were used as control materials. The scale bar is 20 μm. | ||
Although many studies, including some of our own, use flow systems to study biofilms for up to a few days,17 the anti-fouling approach/material is applicable for both static and flow conditions. For biomedical applications, the majority of implants, except for blood-contacting implants, operate under static or semi-static conditions, which further validates our use of the static system. P. aeruginosa can form a biofilm under both aerobic and anaerobic conditions, though it was observed that the structure and composition of extracellular polymeric substances of biofilms are different under aerobic and anaerobi-c growth conditions.77 In this first long-term study, we did not control the dissolved oxygen level, and both strains formed dense biofilms on the surfaces of all containers and control materials. In contrast, all PCBGU surfaces showed no biofilm formation and undetectable individual cell attachment. It is also incredibly challenging to maintain a flow system using a rich medium for weeks and months. Biofilms will often clog the tubing, grow along the inside of the tubing, and contaminate the fresh medium reservoir. Because of the large surface area of the system, biofilms on the surface of the tubing deplete oxygen and nutrients from the fresh medium before it reaches any biofilm on the sample. There are some studies in the literature that demonstrate that some bacteria adhere better at the initial adhesion stage under flow than in static conditions.78–80 For example, Escherichia coli attaches better on the surface of mammalian erythrocytes79 and the monomannose-coated surface.78 The shear stress helps the initial attachment of P. aeruginosa on glass and polydimethylsiloxane surfaces.80 It was also observed by Thomen et al. that mechanical force affects the adhesion but does not directly influence the growth rate of biofilms.81 Our static study cannot rule out that some bacteria may adhere better on the anti-fouling surfaces under flow conditions, so, in the future, it is worth studying the biofilm formation on PCBGU surfaces under all conditions. However, we are optimistic about PCBGU's performance under the static condition presented currently.
Conclusion
In this work, we developed a simple but novel approach to introduce high potency anti-fouling carboxybetaine groups into polyurethanes without significantly changing the synthesis and processing conditions. All produced PCBGUs had three key features: an ability to resist most protein adsorption over an extensive hydrolysis study, complete resistance to NIH-3T3 mammalian cells over twenty four hours, and absolute biofilm formation prevention up to six months. Simultaneously, by adjusting CB ratios, the compression strength and swelling ratio could be controlled. Also, for the first time, we validated the widely-used but unproven anti-fouling hypothesis for long-term biofouling management and demonstrated the effectiveness of this approach. This study also contributes to a better understanding of the structure–function relationships for zwitterionic polymers, allowing researchers to create customized materials and develop formulations that may perform even better. The PCBGU platform with tunable mechanical properties and unmatched anti-fouling properties has promising potential in a broad range of industries.Methods
Materials
Diethanolamine, ethyl acrylate, and fluorescein isothiocyanate isomer 1 were purchased from Alfa Aesar (Haverhill, MA). Glycerol and Fibrinogen from human plasma (Fg) were purchased from Millipore Sigma (St. Louis, MO, USA). 1,6-Diisocyanatohexane (HDI) was purchased from Acros Organics (Pittsburg, PA, USA). Dimethylformamide was purchased from EMD Millipore (Burlington, MA, USA). The viability/cytotoxicity assay kit for live bacteria and dead cells was purchased from Biotium (Fremont, CA, USA).Synthesis of diethanolamine ethyl acetate (DEAEA)
Diethanolamine (50 g, 0.476 mol) was added into ethyl acrylate (48 g, 0.48 mol) dropwise while nitrogen was purged, and the resulting solution was stirred overnight at 35 °C. The reaction mixture was concentrated in a Randolph rotary evaporator then purified by flash chromatography using a dichloromethane and methanol mixture (4:1) as the mobile phase to yield DEAEA as a colorless oil with 95% yield. 1H NMR (400 MHz, CDCl3, ppm): 3.46 (t, 4H), 2.74 (t, 2H), 2.51 (t, 4H), 2.36 (t, 2H), 1.14 (t, 3H), 4.01 (m, 2H).
Synthesis of poly(carboxybetaine-glycerol-urethanes) (PCBGUs)
Five formulations of PCBGU were made using the following ratios of hydroxyl groups in DEAEA:hydroxyl groups of glycerol:isocyanate groups in HDI (100:0:100, 75:25:100, 50:50:100, 25:75:100 and 0:100:100). The PU-prepolymer (DEAEA with HDI) was synthesized in a three-necked round bottom flask equipped with a mechanical stirrer, temperature controller, and nitrogen inlet. Before synthesis, DEAEA was placed into a vacuum oven at 110 °C for 2 h to remove moisture. Once the DEAEA was placed in the flask, HDI was added dropwise. Anhydrous DMF was added once the viscosity increased, approximately 0.5 h later. This solution was stirred at 400 rpm for 2 h at 80 °C before glycerol was added dropwise. After another 30 minutes of stirring at the same rate and temperature, the mixed solution was poured into PTEF dishes and stored in the oven at 100 °C for 12 h without a vacuum. The resulting polyurethane film was dried at 100 °C in the oven under vacuum for another 12 h to remove residual solvent. After drying, the films were peeled off and cut into discs with a biophysical punch (8 mm diameter, 2 mm thickness). Excess material was used for thermogravimetric analysis, differential scanning calorimetry, and hydrolysis studies described below. The PEGCU-50 (96 wt% of PEG-2000 and 4 wt% of glycerol) control material was also synthesized by the same procedure.
Fourier transform infrared spectroscopy (FT-IR)
Fourier transform infrared spectroscopy (FT-IR) analysis was carried out on a Nexus 870 spectrometer (Thermo Nicolet, USA) in attenuated total reflection (ATR) mode. Each sample was scanned 64 times in the range of 400–4000 cm−1 with a spectral resolution of 2 cm−1.Thermogravimetric analysis (TGA)
Thermogravimetric analysis (TGA/SDTA 851e, Mettler Toledo, USA) was used to study the thermal stability of the PU materials. Approximately 4 mg of a specimen from the excess material for each sample was taken for measurements and the temperature was increased from 50 to 700 °C at a rate of 10 °C min−1 with a continuous N2 flow of 50 mL min−1.Differential scanning calorimetry (DSC)
DSC was performed using a thermal analyzer DSC 822e (Mettler Toledo, USA) for all materials to measure the phase transition temperature. About 7 mg of excess from each sample was taken for the measurements, and a heating rate of 10 °C min−1 from −40 to 250 °C was employed with a continuous N2 flow of 50 mL min−1.Hydrolysis kinetics
One 0.5 g sample of each ratio was submerged into 5 mL of DI water. The pH value was recorded every 5 min for the first 30 min, then every hour for the first day and then once a day for the next nine days.Swelling ratio
To study the swelling behavior, one disc of each ratio was submerged in 10 mL ultrapure water until pH equilibrium was reached after two weeks. Afterwards, each sample was weighed before being placed in a freeze-dryer to be lyophilized. Once all water is removed, the sample is weighed again. The swelling ratio, a comparison of these weights indicating cross-link density, was calculated using,where Q is the swelling ratio, MS is the mass after swelling, and MD is the mass after lyophilization.
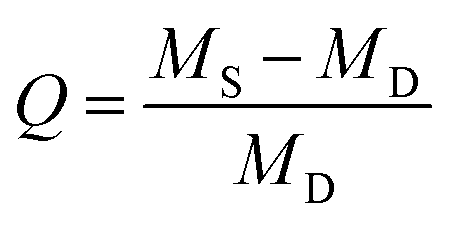
Compression strength
The stress–strain curves of polyurethane samples were evaluated using a Shimadzu EZ-Test Compact Bench Testing Machine (Shimadzu Corporation, Nakagyo-Ku, Kyoto, Japan). A disc of each ratio and the samples from the hydrolysis study were compressed to failure at a rate of 1 mm min−1 with a 5000 N load cell.Rheological study
A stress controller rotation rheometer (ARES-G2) equipped with 25 mm parallel plates was used to conduct rheological studies. To confirm that the experiments were conducted in the linear viscoelastic regime, strain sweep measurements were performed from 0.1% to 100% with a frequency of 10 rad s−1. Further frequency sweep measurements were conducted from 0.1 to 100 rad s−1 with a strain of 1% at 100 °C.Protein adsorption
The adsorption of proteins on PU hydrogels was determined by a fluorescence method previously described by Cao et al.82 Discs of the PCBGU hydrogel, API-PU membrane (positive control), and PEGCU-50 (negative control) were submerged into DI water and pH-8.5 Tris buffer for varying lengths of time (0, 1, 4, 24 and 48 h for DI water: 15, 30, 60, 90 and 120 min for pH-8.5 Tris buffer). Subsequently, the samples were transferred into a sterile 24-well plate, and 1 mL of FITC-labelled human fibrinogen (FITC-Fg) solution (1 mg mL−1) was added to each well. All samples were immersed in the solution for 30 minutes to equilibrate and allow proteins to adsorb. Afterward, samples were rinsed with phosphate buffer saline (PBS) three times to remove loosely adsorbed proteins. Protein adsorption on surfaces was visualized with an Olympus IX81 fluorescent microscope (Olympus, Japan) with a 20× objective lens through a FITC filter at a fixed exposure time (200 ms) for all samples. ImageJ software was used to quantify the fluorescence intensity of each sample.Mammalian cell attachment
After fresh hydrogels of each ratio were equilibrated in water and placed in a 24-well plate, NIH-3T3 cells were seeded at 105 cells per well concentration in a serum medium consisting of DMEM, 10% fetal bovine serum (FBS), and 1% penicillin–streptomycin and kept in an incubator with 5% CO2 at 37 °C for 24 hours. Surface cell coverage was visualized with the same fluorescence microscope as in protein adsorption studies with a 10× objective lens.Biofilm formation assay
Before being immersed in the bacterial solution, all samples were equilibrated in water. Pseudomonas aeruginosa PAO1 and Staphylococcus epidermidis cells were washed three times with sterile PBS and subsequently resuspended in sterile LB media at a concentration of 106 cells per mL. 50% of the solution was replaced with fresh sterile LB media each day of the study, and the temperature of the system was maintained at 25 °C. After varying lengths of time (1, 2, and 3 days, and 1, 2, 4, 8, and 10 weeks, and then 3, 4, 5, and 6 months), the accumulated bacteria were stained with DMAO dyes and recorded in situ using the same microscope as before with a 40× objective lens through a FITC filter.Statistical analysis
All experiments were repeated three times. Data were demonstrated as mean ± standard deviation. Statistical significance (p < 0.05) was evaluated using the one-way analysis of variance (ANOVA) followed by Bonferroni's post hoc test.Data availability
All data needed to evaluate the conclusions in this paper are present in the paper and/or the ESI.† Additional data related to this paper may be provided from the authors by request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the US National Science Foundation (DMR-1454837 and DMR-1741935). Parts of this work were conducted using the Soft Matter Characterization Facility of the University of Chicago. The authors wish to thank Philip Griffin for his aid with some experiments and data analysis.References
- D. M. Siddiq and R. O. Darouiche, Nat. Rev. Urol., 2012, 9, 305–314 CrossRef CAS PubMed.
- Z. K. Zander and M. L. Becker, ACS Macro Lett., 2018, 7, 16–25 CrossRef CAS.
- P. W. Stone, Expert Rev. Pharmacoecon. Outcomes Res., 2009, 9, 417–422 CrossRef PubMed.
- J. A. Callow and M. E. Callow, Nat. Commun., 2011, 2, 244 CrossRef PubMed.
- N. Kip and J. A. van Veen, ISME J., 2015, 9, 542–551 CrossRef CAS PubMed.
- C. C. C. R. de Carvalho, Front. Mar. Sci., 2018, 5, 126 CrossRef.
- J. D. Bryers, Biotechnol. Bioeng., 2008, 100, 1–18 CrossRef CAS PubMed.
- A. M. Rouhi, Chem. Eng. News, 1998, 76, 41–42 CrossRef.
- D. Davies, Nat. Rev. Drug Discovery, 2003, 2, 114–122 CrossRef CAS PubMed.
- L. R. Hoffman, D. A. D'Argenio, M. J. MacCoss, Z. Y. Zhang, R. A. Jones and S. I. Miller, Nature, 2005, 436, 1171–1175 CrossRef CAS PubMed.
- T. F. C. Mah and G. A. O'Toole, Trends Microbiol., 2001, 9, 34–39 CrossRef CAS PubMed.
- C. R. Arciola, F. I. Alvi, Y. H. An, D. Campoccia and L. Montanaro, Int. J. Artif. Organs, 2005, 28, 1119–1125 CrossRef CAS PubMed.
- M. Chen, Q. Yu and H. Sun, Int. J. Mol. Sci., 2013, 14, 18488–18501 CrossRef PubMed.
- S. R. Shah, A. M. Tatara, R. N. D'Souza, A. G. Mikos and F. K. Kasper, Mater. Today, 2013, 16, 177–182 CrossRef CAS.
- D. Ausbacher, L. Lorenz, B. Pitts, P. S. Stewart and D. M. Goeres, Lett. Appl. Microbiol., 2018, 66, 231–237 CrossRef CAS PubMed.
- G. Cheng, H. Xite, Z. Zhang, S. F. Chen and S. Y. Jiang, Angew. Chem., Int. Ed., 2008, 47, 8831–8834 CrossRef CAS PubMed.
- G. Cheng, G. Z. Li, H. Xue, S. F. Chen, J. D. Bryers and S. Y. Jiang, Biomaterials, 2009, 30, 5234–5240 CrossRef CAS PubMed.
- Q. Shao and S. Y. Jiang, Adv. Mater., 2015, 27, 15–26 CrossRef CAS PubMed.
- S. Lowe, N. M. O'Brien-Simpson and L. A. Connal, Polym. Chem., 2015, 6, 198–212 RSC.
- R. O. Darouiche, Clin. Infect. Dis., 2001, 33, 1567–1572 CrossRef CAS PubMed.
- J. M. Anderson, A. Rodriguez and D. T. Chang, Semin. Immunol., 2008, 20, 86–100 CrossRef CAS PubMed.
- I. H. Jaffer, J. C. Fredenburgh, J. Hirsh and J. I. Weitz, J. Thromb. Haemostasis, 2015, 13, S72–S81 CrossRef PubMed.
- J. Ladd, Z. Zhang, S. Chen, J. C. Hower and S. Jiang, Biomacromolecules, 2008, 9, 1357–1361 CrossRef CAS PubMed.
- M. D. Swartzlander, C. A. Barnes, A. K. Blakney, J. L. Kaar, T. R. Kyriakides and S. J. Bryant, Biomaterials, 2015, 41, 26–36 CrossRef CAS PubMed.
- A. D. Lynn, T. R. Kyriakides and S. J. Bryant, J. Biomed. Mater. Res., Part A, 2010, 93, 941–953 Search PubMed.
- X. Y. Wang, T. Ishida and H. Kiwada, J. Controlled Release, 2007, 119, 236–244 CrossRef CAS PubMed.
- Y. C. Hsieh, H. E. Wang, W. W. Lin, S. R. Roffler, T. C. Cheng, Y. C. Su, J. J. Li, C. C. Chen, C. H. Huang, B. M. Chen, J. Y. Wang, T. L. Cheng and F. M. Chen, Theranostics, 2018, 8, 3164–3175 CrossRef CAS PubMed.
- H. Wang, Y. Hu, D. Lynch, M. Young, S. Li, H. Cong, F.-J. Xu and G. Cheng, ACS Appl. Mater. Interfaces, 2018, 10, 37609–37617 CrossRef CAS PubMed.
- J. Cao, N. Chen, Y. Chen and X. Luo, Int. J. Mol. Sci., 2010, 11, 1870–1877 CrossRef CAS PubMed.
- P. N. Coneski and J. H. Wynne, ACS Appl. Mater. Interfaces, 2012, 4, 4465–4469 CrossRef CAS PubMed.
- C. F. Ma, H. Zhou, B. Wu and G. Z. Zhang, ACS Appl. Mater. Interfaces, 2011, 3, 455–461 CrossRef CAS PubMed.
- C. H. Wang, C. F. Ma, C. D. Mu and W. Lin, RSC Adv., 2017, 7, 27522–27529 RSC.
- Z. G. Estephan, P. S. Schlenoff and J. B. Schlenoff, Langmuir, 2011, 27, 6794–6800 CrossRef CAS PubMed.
- M. He, K. Gao, L. Zhou, Z. Jiao, M. Wu, J. Cao, X. You, Z. Cai, Y. Su and Z. Jiang, Acta Biomater., 2016, 40, 142–152 CrossRef CAS PubMed.
- S. H. Ye, Y. Hong, H. Sakaguchi, V. Shankarraman, S. K. Luketich, A. D'Amore and W. R. Wagner, ACS Appl. Mater. Interfaces, 2014, 6, 22796–22806 CrossRef CAS PubMed.
- H.-C. Hung, P. Jain, P. Zhang, F. Sun, A. Sinclair, T. Bai, B. Li, K. Wu, C. Tsao, E. J. Liu, H. S. Sundaram, X. Lin, P. Farahani, T. Fujihara and S. Jiang, Adv. Mater., 2017, 29, 1700617 CrossRef PubMed.
- A. Burke and N. Hasirci, Adv. Exp. Med. Biol., 2004, 553, 83–101 CrossRef CAS PubMed.
- R. J. Zdrahala and I. J. Zdrahala, J. Biomater. Appl., 1999, 14, 67–90 CrossRef CAS PubMed.
- P. Volkow, C. Vazquez, O. Tellez, C. Aguilar, L. Barrera, E. Rodrgiuez, D. Vilar-Compte, J. Zinser, E. Calderon, J. R. Perez-Padilla and A. Mohar, Am. J. Infect. Control, 2003, 31, 392–396 CrossRef PubMed.
- P. J. Nowatzki, R. R. Koepsel, P. Stoodley, K. Min, A. Harper, H. Murata, J. Donfack, E. R. Hortelano, G. D. Ehrlich and A. J. Russell, Acta Biomater., 2012, 8, 1869–1880 CrossRef CAS PubMed.
- S. Suresh and R. A. Black, J. Biomater. Appl., 2015, 29, 1028–1038 CrossRef PubMed.
- S. A. Guelcher, Tissue Eng., Part B, 2008, 14, 3–17 CrossRef CAS PubMed.
- T. W. Chuang and K. S. Masters, Biomaterials, 2009, 30, 5341–5351 CrossRef CAS PubMed.
- C. Alperin, P. W. Zandstra and K. A. Woodhouse, Biomaterials, 2005, 26, 7377–7386 CrossRef CAS PubMed.
- H. Chen, Y. Li, Y. Liu, T. Gong, L. Wang and S. Zhou, Polym. Chem., 2014, 5, 5168–5174 RSC.
- G. Morral-Ruiz, P. Melgar-Lesmes, C. Solans and M. J. Garcia-Celma, J. Controlled Release, 2013, 171, 163–171 CrossRef CAS PubMed.
- J. P. Santerre, K. Woodhouse, G. Laroche and R. S. Labow, Biomaterials, 2005, 26, 7457–7470 CrossRef CAS PubMed.
- Y. Hong, S. H. Ye, A. Nieponice, L. Soletti, D. A. Vorp and W. R. Wagner, Biomaterials, 2009, 30, 2457–2467 CrossRef CAS PubMed.
- T. Choi, J. Weksler, A. Padsalgikar and J. Runt, J. Biomater. Sci., Polym. Ed., 2011, 22, 973–980 CrossRef CAS PubMed.
- Z. Liu, X. Wu, X. Yang, D. Liu, C. Jun, R. Sun, X. Liu and F. Li, Biomacromolecules, 2005, 6, 1713–1721 CrossRef CAS PubMed.
- S. Gogolewski, Colloid Polym. Sci., 1989, 267, 29 CrossRef.
- C. Wang, C. Ma, C. Mu and W. Lin, RSC Adv., 2017, 7, 27522–27529 RSC.
- R. S. Smith, Z. Zhang, M. Bouchard, J. Li, H. S. Lapp, G. R. Brotske, D. L. Lucchino, D. Weaver, L. A. Roth, A. Coury, J. Biggerstaff, S. Sukavaneshvar, R. Langer and C. Loose, Sci. Transl. Med., 2012, 4, 153ra132 Search PubMed.
- P. N. Coneski and J. H. Wynne, ACS Appl. Mater. Interfaces, 2012, 4, 4465–4469 CrossRef CAS PubMed.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2013, 26, 724–744 CrossRef.
- M. Berta, C. Lindsay, G. Pans and G. Camino, Polym. Degrad. Stab., 2006, 91, 1179–1191 CrossRef CAS.
- D. K. Chattopadhyay and D. C. Webster, Prog. Polym. Sci., 2009, 34, 1068–1133 CrossRef CAS.
- B. Cao, L. L. Li, Q. Tang and G. Cheng, Biomaterials, 2013, 34, 7592–7600 CrossRef CAS PubMed.
- W. Q. Zhao, Y. N. Zhu, J. M. Zhang, T. Xu, Q. S. Li, H. S. Guo, J. W. Zhang, C. G. Lin and L. Zhang, J. Mater. Sci., 2018, 53, 13813–13825 CrossRef CAS.
- L. R. Carr, Y. B. Zhou, J. E. Krause, H. Xue and S. Y. Jiang, Biomaterials, 2011, 32, 6893–6899 CrossRef CAS PubMed.
- V. Kanyanta and A. Ivankovic, J. Mech. Behav. Biomed. Mater., 2010, 3, 51–62 CrossRef PubMed.
- J. C. Lee, H. Y. Wu, Q. Tang, B. Cao, H. F. Wang, H. B. Cong, J. Zhe, F. J. Xu and G. Cheng, Langmuir, 2015, 31, 9965–9972 CrossRef PubMed.
- N. Park, B. Kwon, I. Kim and J. Cho, J. Membr. Sci., 2005, 258, 43–54 CrossRef CAS.
- G. D. Bixler and B. Bhushan, Philos. Trans. R. Soc., A, 2012, 370, 2381–2417 CrossRef CAS PubMed.
- T. A. Horbett, J. Biomed. Mater. Res., Part A, 2018, 106, 2777–2788 CrossRef CAS PubMed.
- R. G. Chapman, E. Ostuni, M. N. Liang, G. Meluleni, E. Kim, L. Yan, G. Pier, H. S. Warren and G. M. Whitesides, Langmuir, 2001, 17, 1225–1233 CrossRef CAS.
- E. Ostuni, R. G. Chapman, R. E. Holmlin, S. Takayama and G. M. Whitesides, Langmuir, 2001, 17, 5605–5620 CrossRef CAS.
- G. Cheng, Z. Zhang, S. F. Chen, J. D. Bryers and S. Y. Jiang, Biomaterials, 2007, 28, 4192–4199 CrossRef CAS PubMed.
- B. Cao, L. L. Li, H. Y. Wu, Q. Tang, B. B. Sun, H. Dong, J. Zhe and G. Cheng, Chem. Commun., 2014, 50, 3234–3237 RSC.
- L. R. Carr, J. E. Krause, J. R. Ella-Menye and S. Y. Jiang, Biomaterials, 2011, 32, 8456–8461 CrossRef CAS PubMed.
- L. R. Carr, H. Xue and S. Y. Jiang, Biomaterials, 2011, 32, 961–968 CrossRef CAS PubMed.
- H. Wang, Y. Hu, D. Lynch, M. Young, S. Li, H. Cong, F. Xu and G. Cheng, ACS Appl. Mater. Interfaces, 2018, 10, 37609–37617 CrossRef CAS PubMed.
- W. B. Tsai, J. M. Grunkemeier and T. A. Horbett, J. Biomed. Mater. Res., 1999, 44, 130–139 CrossRef CAS PubMed.
- Z. Karahaliloğlu, Fibers Polym., 2017, 18, 2135–2145 CrossRef.
- A. Roosjen, H. C. van der Mei, H. J. Busscher and W. Norde, Langmuir, 2004, 20, 10949–10955 CrossRef CAS PubMed.
- I. Meskin, Pediatr. Rev., 1998, 19, 105–106 CrossRef CAS PubMed.
- S. S. Yoon, R. F. Hennigan, G. M. Hilliard, U. A. Ochsner, K. Parvatiyar, M. C. Kamani, H. L. Allen, T. R. DeKievit, P. R. Gardner, U. Schwab, J. J. Rowe, B. H. Iglewski, T. R. McDermott, R. P. Mason, D. J. Wozniak, R. E. W. Hancock, M. R. Parsek, T. L. Noah, R. C. Boucher and D. J. Hassett, Dev. Cell, 2002, 3, 593–603 CrossRef CAS PubMed.
- W. E. Thomas, L. M. Nilsson, M. Forero, E. V. Sokurenko and V. Vogel, Mol. Microbiol., 2004, 53, 1545–1557 CrossRef CAS PubMed.
- W. E. Thomas, E. Trintchina, M. Forero, V. Vogel and E. V. Sokurenko, Cell, 2002, 109, 913–923 CrossRef CAS PubMed.
- S. Lecuyer, R. Rusconi, Y. Shen, A. Forsyth, H. Vlamakis, R. Kolter and H. A. Stone, Biophys. J., 2011, 100, 341–350 CrossRef CAS PubMed.
- P. Thomen, J. Robert, A. Monmeyran, A. F. Bitbol, C. Douarche and N. Henry, PLoS One, 2017, 12, e0175197 CrossRef PubMed.
- B. Cao, L. Li, H. Wu, Q. Tang, B. Sun, H. Dong, J. Zhe and G. Cheng, Chem. Commun., 2014, 50, 3234–3237 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc06155j |
| This journal is © The Royal Society of Chemistry 2020 |