
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTripyrrin-armed isosmaragdyrins: synthesis, heterodinuclear coordination, and protonation-triggered helical inversion†
Chengjie
Li‡
 a,
Kai
Zhang‡
a,
Masatoshi
Ishida
b,
Qizhao
Li
a,
Keito
Shimomura
b,
Glib
Baryshnikov
c,
Xin
Li
c,
Mathew
Savage
d,
Xin-Yan
Wu
a,
Sihai
Yang
d,
Hiroyuki
Furuta
*b and
Yongshu
Xie
*a
a,
Kai
Zhang‡
a,
Masatoshi
Ishida
b,
Qizhao
Li
a,
Keito
Shimomura
b,
Glib
Baryshnikov
c,
Xin
Li
c,
Mathew
Savage
d,
Xin-Yan
Wu
a,
Sihai
Yang
d,
Hiroyuki
Furuta
*b and
Yongshu
Xie
*a
aKey Laboratory for Advanced Materials, Joint International Research Laboratory of Precision Chemistry and Molecular Engineering, Feringa Nobel Prize Scientist Joint Research Center, Shanghai Key Laboratory of Functional Materials Chemistry, School of Chemistry and Molecular Engineering, East China University of Science & Technology, Shanghai 200237, China. E-mail: yshxie@ecust.edu.cn
bDepartment of Chemistry and Biochemistry, Graduate School of Engineering, Center for Molecular Systems, Kyushu University, Fukuoka 819-0395, Japan. E-mail: hfuruta@cstf.kyushu-u.ac.jp
cSchool of Biotechnology, KTH Royal Institute of Technology, SE-10691 Stockholm, Sweden
dSchool of Chemistry, University of Manchester, Manchester M13 9PL, UK
First published on 4th February 2020
Abstract
Oxidative ring closure of linear oligopyrroles is one of the synthetic approaches to novel porphyrinoids with dinuclear coordination sites and helical chirality. The spatial arrangement of the pyrrolic groups of octapyrrole (P8) affected the position of the intramolecular oxidative coupling of the pyrrolic units; tripyrrin-armed isosmaragdyrin analogue (1) containing a β,β-linked bipyrrole moiety was synthesized regioselectively in a high yield by using FeCl3. NiII-coordination at the armed tripyrrin site of 1 allowed the formation of diastereomeric helical twisted complexes (2A and 2B) and succeeding CuII-coordination at the macrocyclic core afforded heterodinuclear NiII/CuII-complexes (3A and 3B). Each of them comprised a pair of separable enantiomers, exhibiting P- and M-helices, respectively. Notably, diastereomeric interconversion from 2A to 2B was quantitatively achieved as a consequence of helical transformation under acidic conditions.
Introduction
Recently, increasing attention has been focused on expanded porphyrin analogues bearing large π-conjugated oligo-pyrrolic scaffolds because of their diverse structural properties.1 The expanded porphyrins can serve as unique ligands for multi-metal coordination and adopt tunable three-dimensional conformations (e.g., figure-eight, triangle, spiral twisted, etc.) with aromaticity-dependent near-infrared optical properties.2 In some cases, topologically twisted metallo-porphyrinoids serve as chiral sensing components.3 The metalation-induced switchable function of expanded porphyrinoids (such as changes in stereoisomeric structures) should be fascinating for applications in environmentally-responsive molecular machines.4To build such large π-conjugated porphyrinoids, various approaches have been developed, mostly by acid-catalyzed condensation and organometallic cross-coupling reactions.5 Among them, oxidative ring closure of linear oligopyrroles could produce bipyrrole-containing porphyrinoids directly linked at the two terminal pyrroles in the α–α′ or α-N modes (Fig. 1a).6 However, less investigation has been carried out on oxidative coupling between β,β′-pyrrolic C-Hs using a rational approach, which may be due to the intrinsic low reactivity of pyrrolic β-CHs compared to α-CHs and the lower acidity of CHs than typical N-Hs.7 Moreover, the oligopyrranes commonly used in the oxidative ring closure reactions are usually too flexible to regulate the targeted reactive sites close to each other.
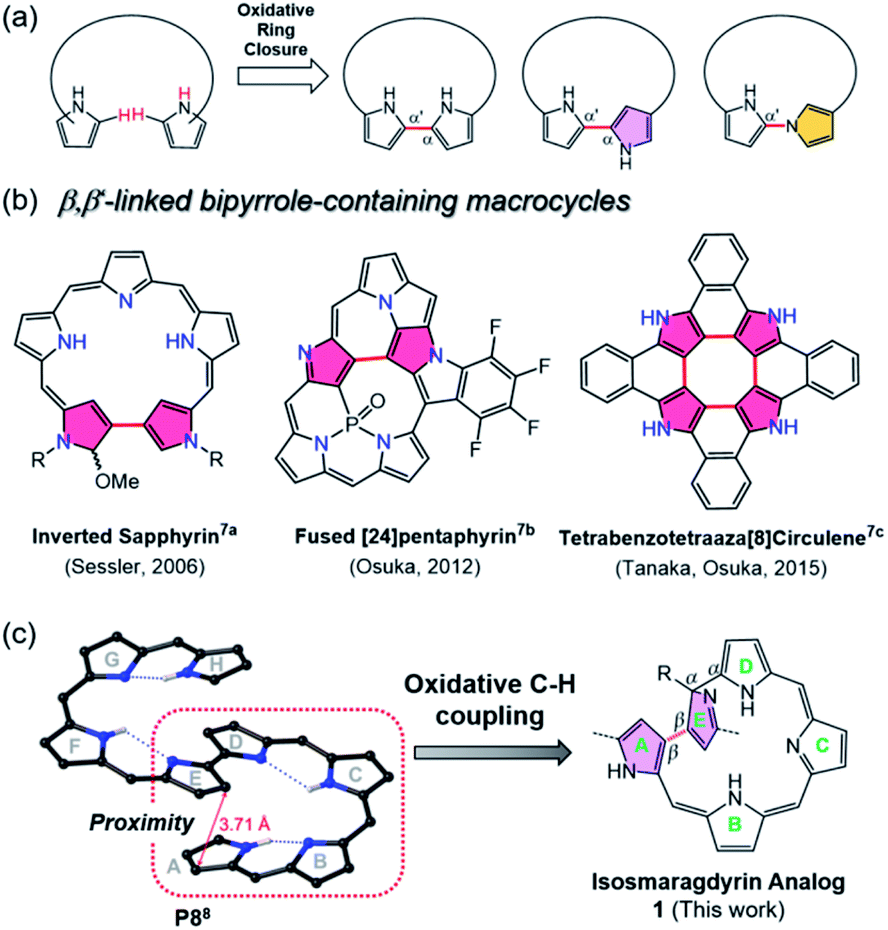 | ||
| Fig. 1 (a) Typical synthetic approaches for constructing bipyrrole-containing porphyrinoids linked in different modes through oxidative coupling of oligopyrroles. (b) Representative macrocyclic compounds containing β,β′-linked (red) bipyrrole moieties. (c) The oxidative coupling reaction of a helical oligopyrrole, P8, where the appropriate pyrrole rings are present spatially in close proximity, giving a new isosmaragdyrin analogue 1. Meso-aryl rings are omitted for clarity. | ||
Inspired by the oxidative coupling reactions of tetraaza[8]circulenes (depicted in Fig. 1b)7c and triply fused porphyrin tapes7d where the reaction sites (i.e., β-pyrrole CHs) reside spatially in close proximity, we here report the synthesis of a novel β,β-linked isosmaragdyrin-based macrocycle (1) from a fully conjugated linear octapyrrole (P8)8 with terminal α-acyl substituents by oxidative aromatic coupling using FeCl3. The octapyrrin P8 adopts a spiral conformation through its intrinsic hydrogen-bonding network, which resulted in positioning the specific CHs of pyrroles in close proximity. Indeed, two pyrrolic β-carbon atoms in P8 on rings A and E are close to each other with a short distance of 3.7 Å in the crystal (Fig. 1 and S24†), which enables a regioselective internal ring closure through the oxidative C–H coupling to afford a tripyrrin-armed pentapyrrolic macrocycle containing a β,β-linked bipyrrole moiety in high yield (Fig. 1c). Notably, the appended tripyrrin moiety and one of the outward-pointing pyrrolic nitrogen atoms of the macrocycle constitute a peripheral N4 coordination site and the isosmaragdyrin core serves as an internal N3 donor site, hence 1 provides hetero-bismetal chelation spheres. Taking advantage of this unique structure, metal-dependent complexation (with NiII and CuII ions) at both the peripheral and the isosmaragdyrin cores successfully afforded helical diastereomeric mononuclear NiII complexes (2A and 2B) and heterodinuclear NiIICuII complexes (3A and 3B). Each of these complexes contains a pair of enantiomers exhibiting P- and M-helices, where all four optically pure isomers (i.e., M,R-x, P,R-x, P,S-x, and M,S-x; x = 2 or 3) have been successfully separated. Unprecedentedly, diastereomeric interconversion from 2A to 2B was quantitatively achieved by the addition of trifluoroacetic acid (TFA), thus enabling transformation between the P- and M-helices. These results provide a practical approach for developing novel porphyrinoids through oxidative ring closure at the spatially adjacent β-positions of elaborately designed conjugated oligopyrroles.
Results and discussion
The oxidative cyclization of linear octapyrrin P8 was performed using 10 equivalents of anhydrous FeCl3 in CH2Cl2/MeOH for 24 h at room temperature. The β-positions of the spatially adjacent pyrrole units were regioselectively linked to afford 1 in a high yield of 88% (Scheme 1). The resulting macrocycle can be regarded as a novel isosmaragdyrin analogue (Fig. 1c).9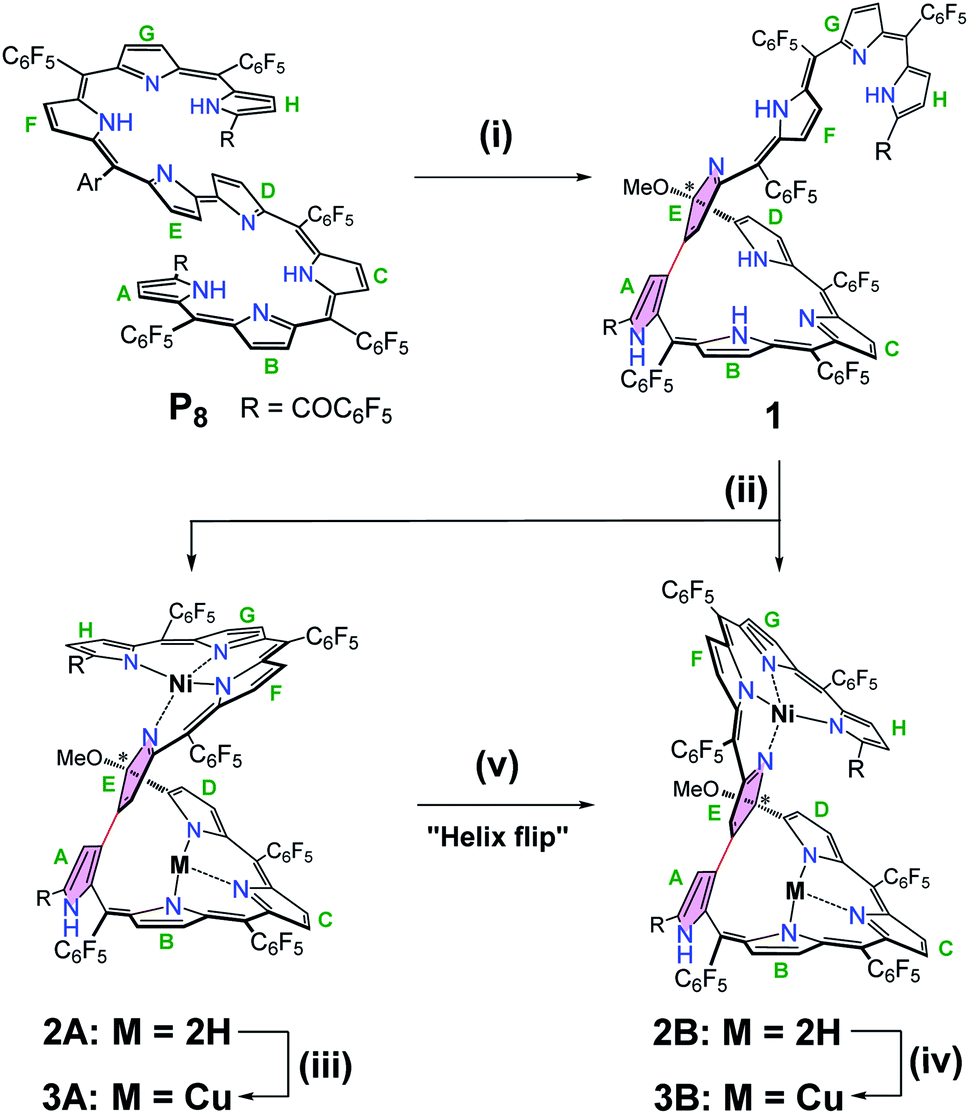 | ||
Scheme 1 Stepwise syntheses of pentaphyrin 1 and metal complexes 2 and 3. The labels, A–H, are defined to identify each pyrrole ring. Conditions: (i) FeCl3, CH2Cl2/MeOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50), 88%; (ii) Ni(OAc)2·4H2O, CH2Cl2/MeOH, 63% for 2A, 19% for 2B; (iii) Cu(OAc)2·H2O, MeOH, 81%; (iv) Cu(OAc)2·H2O, MeOH, 91%; (v) TFA, CH2Cl2. :50), 88%; (ii) Ni(OAc)2·4H2O, CH2Cl2/MeOH, 63% for 2A, 19% for 2B; (iii) Cu(OAc)2·H2O, MeOH, 81%; (iv) Cu(OAc)2·H2O, MeOH, 91%; (v) TFA, CH2Cl2. | ||
In the macrocycle 1, π-conjugation was disrupted, as evident from the blue-shifted absorption spectrum (λmax = 1073 and 638 nm for P8 and 1, respectively) (Fig. S11†). The high resolution mass spectrum (HRMS) of 1 showed a molecular ion peak at m/z = 2010.1192 (M+), consistent with a molecular formula of 1: C89H22F40N8O3 (indicating the formal addition of a methoxy group to P8, i.e., loss of two hydrogens by oxidation with FeCl3, followed by the addition of MeOH). The 1H NMR spectrum of 1 in CDCl3 indicates the unsymmetrical structure and shows two sets of signal patterns in the entire region, suggesting the presence of two conformational isomers in solution (Fig. S1 and S4†).10 For example, the signal of the OCH3 group appeared at δ = 3.19 and 3.09 ppm with a ratio of 0.85:1 (calculated from the integral) for the two isomers. On the basis of the preliminary crystal structure of 1, a tripyrrin appended pentaphyrin structure consisting of a direct β,β-linkage between the terminal ring A and the middle ring E was verified (Fig. S25†).11 The unusual β,β-bonding mode tends to exhibit a significant distortion of the pentapyrrolic core similar to that observed in the tetrapyrrolic corrorin.6d Notably, a methoxy group was found at the α-position of one of the directly linked pyrroles (ring E), thus generating an sp3 hybridized carbon linkage. The isosmaragdyrin macrocycle of 1 can be thus regarded as a nonaromatic derivative of modified isosmaragdyrin.
Owing to the intrinsic pentaphyrin cavity and flexible oligopyrrin arm, isosmaragdyrin derivative 1 may bind two metal ions and exhibit unexpected properties, such as external-stimuli induced helical transfer.12 Actually, 1 allows the coordination of metal cations at the tripyrrin side chain (Scheme 1). When a methanol solution of 1 and 50 equiv. of Ni(OAc)2·4H2O was heated at reflux in the presence of NaOAc under anaerobic conditions, two greenish products, that is, isomeric mononuclear NiII complexes 2A and 2B were obtained in 63% and 19% yields, respectively (Scheme 1). The HRMS of 2A and 2B showed essentially identical values of m/z = 2066.0459 and 2066.0432 (Fig. S18–S19†), respectively, indicating that the molecular formula is C89H20F40N8O3Ni, which is consistent with the mononuclear NiII chelate. Later the X-ray crystallographic analysis disclosed the structures of 2A and 2B with helical twists (Fig. 2a–d).13 The NiII cation is wrapped with three N atoms from the tripyrrin unit (rings F–H) and the fourth N atom from the confused pyrrole (ring E), while the inner cavity of the macrocycle remains uncoordinated. The directly linked pyrrole rings A and E are located almost coplanar, and the rings C and H are at opposite sides of the mean plane defined by rings A and E in 2A. However, the rings C and H in 2B are arranged on the same side of the plane. Thus, 2A and 2B can be regarded as pseudo-trans and pseudo-cis conformations, respectively. The methoxy-attached pyrrolic α-carbon (i.e., C20) for 2A is sp3 hybridized, which can be evidenced by the C20–C21, C20–C19, C20–O, and C20–N distances of 1.495(8), 1.536(7), 1.410(6), and 1.498(7) Å, respectively, typical values for C–C, C–O, and C–N single bonds.13 Similar bond lengths around C20 were also observed in 2B and the rough crystal structure of 1 (Fig. S25†). The pseudo-tetrahedral geometry of the sp3 carbon center can be further elucidated by the bond angles at C20 lying in the range from 103.7(4) to 111.6(4)°, which are close to 109.5°, a typical value for Td carbon atoms. Thus, C20 is a chiral center. Meanwhile, the helical structure of the tetrapyrrin (biliverdin-like) NiII-complex leads to another source of molecular chirality. As a result, there are four possible chiral isomers, namely, M,R-2, P,R-2, M,S-2, and P,S-2, where R and S represent the chirality of C20, and M (left-handed) and P (right-handed)14 denote the handedness of the biliverdin-like unit. Indeed, a pair of enantiomers, M,R-2 and P,S-2, was present in the crystals of the racemic compound 2A. Likewise, the racemic crystal 2B comprises enantiomers, P,R-2 and M,S-2. Hence, compounds 2A and 2B can be regarded as diastereomeric isomers.
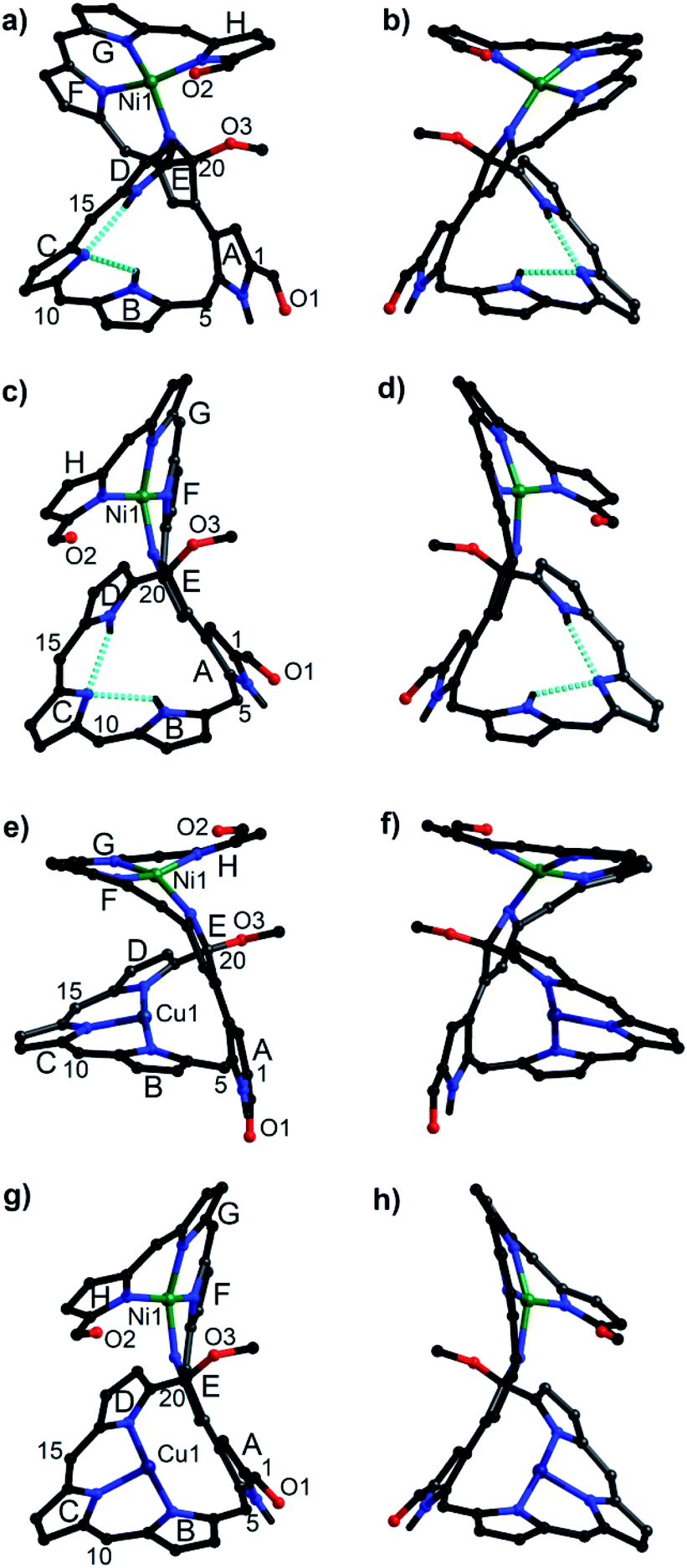 | ||
| Fig. 2 Molecular structures of 2A (a and b), 2B (c and d), 3A (e and f), and 3B (g and h). (a) and (b) A pair of enantiomers in the racemic crystals of 2A, specifically M,R-2 for (a) and P,S-2 for (b).16 Similarly, P,R-2 for (c), M,S-2 for (d), M,R-3 for (e), P,S-3 for (f), P,R-3 for (g), and M,S-3 for (h). C6F5 groups and the hydrogen atoms attached to carbon atoms are omitted for clarity. | ||
Further metalation of 2A and 2B with NiII ions did not proceed under the above conditions. Instead, the reaction of 2A and 2B with 10 equiv. of Cu(OAc)2 in a mixture of CH2Cl2/MeOH (1:1) at reflux for 2 h afforded hetero-dinuclear NiII/CuII complexes, 3A and 3B, in the yields of 81% and 91%, respectively (Scheme 1). The molecular ion peaks at m/z = 2126.9590 and 2126.9608, respectively, agree with the dinuclear NiIICuII coordination structures (Fig. S20 and S21†). The broad active EPR spectra of complexes, 3A and 3B, are consistent with the coordination of the paramagnetic CuII ion (Fig. S22 and S23†).15 Incorporation of a CuII ion into the inner N atoms of the macrocyclic core was also identified by X-ray crystallographic analysis (Fig. 2e–h).11 Notably, the CuII cations in 3A and 3B coordinate to the nitrogen atoms of pyrrole rings B, C, and D in a T-shaped manner and the vacant site of the square-planar CuII ion seems to be occupied by neighboring sp2 carbons (Cu–C: 2.380–2.748 Å) presumably through CuII-arene interactions (Fig. S27 and S28†).16 From the viewpoint of molecular chirality, complex 3A has the same chirality as 2A, consisting of enantiomers M,R-3 and P,S-3 (Fig. 2e and f). Likewise, the crystals of 3B comprise enantiomers P,R-3 and M,S-3 (Fig. 2g and h).
The aforementioned addition of a methoxy group introduces a chiral center into the macrocycle of 1. To understand the chiroptical properties of the isosmaragdyrin derivatives, enantiomeric separation was conducted using a chiral HPLC column with a mixture of n-hexane/2-propanol as the eluent. A pair of enantiomers of 1 (S-1 and R-1, respectively) was thus separated roughly in a 1:1 ratio (Fig. S2a†). The first fraction 1-I and the second 1-II displayed opposite Cotton effects in the circular dichroism (CD) spectra; they can be assigned as R-1 and S-1, respectively, judging from the data obtained experimentally and theoretically using time-dependent density functional theory (TD-DFT) calculations (Fig. S2b†). The absolute configuration of C20 remains intact during metalation; that is, coordination of R-1 with NiII ions afforded a diastereomeric mixture of M,R-2 and P,R-2, while the corresponding reaction of S-1 likewise afforded P,S-2 and M,S-2. These diastereomers can be separated on conventional silica gel columns. Along this way, subsequent complexation of a CuII ion produced a pair of pure chiral isomers (M,R-3, P,R-3) and (P,S-3, M,S-3), respectively. According to the CD spectra of the resulting enantiomers 2 and 3 (Fig. S34–S37†), the intense Cotton effects were observed in the series of M,R-x and P,S-x (Δε ≈ 400 M−1 cm−1) compared to those of M,S-x and P,R-x (Δε ≈ 100 M−1 cm−1). These stereostructure-dependent spectral features could be rationalized by theoretical simulations (Fig. S39–S42†). The different strengths of Δε may reflect the extent of magnetic dipole transition moments for each electronic transition of the complexes.17
Unprecedentedly, it was found that the isomer 2A could be quantitatively converted to 2B upon treating with 1 equiv. of TFA in CH2Cl2 for 12 h (Scheme 1). TFA-induced stereo-transformation was further analyzed by CD, absorption and 1H NMR spectroscopy. Upon treatment with TFA, the CD spectrum of M,R-2 in CH2Cl2 was spontaneously changed to the same spectra obtained from the acidic solution of P,R-2 while retaining the stereochemistry (Fig. 3). Absorption spectroscopy also showed the clear conversion from 2A to 2B in the presence of TFA in CH2Cl2 or MeOH (Fig. S12, S13, and S15†). Whereas, the profile of 2B remained intact under the same conditions. The 1H NMR spectra also assisted in tracking the helix interconversion processes between 2A and 2B. When the solution of 2A in CD2Cl2 was treated with TFA, the signals of 2A gradually disappeared and subsequently, a new set of signals appeared identical to what is seen for 2B under acidic conditions (Fig. S8†). In CDCl3 or CD2Cl2, broadening of a specific NH peak for 2B was observed upon addition of TFA (Fig. S7 and S9†).
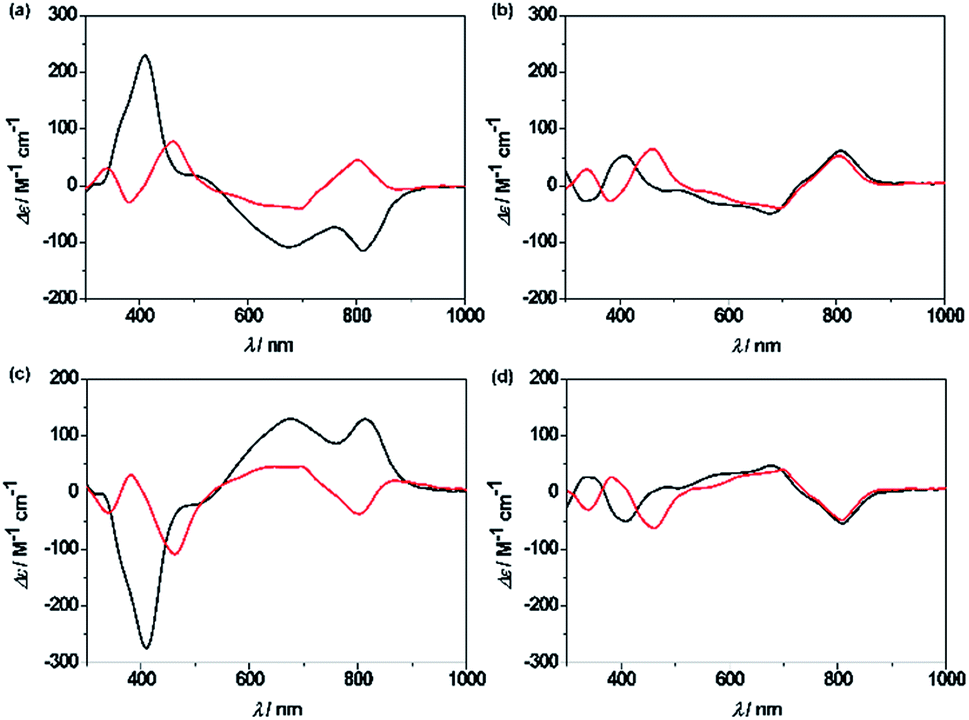 | ||
| Fig. 3 CD spectra of the complexes in CH2Cl2 in the absence (black line) and presence (red line) of TFA, (a) M,R-2, (b) P,R-2, (c) P,S-2, and (d) M,S-2. | ||
Although the mechanism for the helical inversion is yet unknown, TFA-assisted protonation at the most probable imino nitrogen site of the isosmaragdyrin core of 2A and 2B could occur, which affected the hydrogen bonding network in the core and caused a conformational change, as inferred from the broadening of the NH signals of the resulting species in the 1H NMR spectrum (Scheme S1†). Intervening of the associated counter anion in this helical inversion sterically or through metal coordination could not be excluded.18 The solution of 2A or 2B in CD3CN reached an equilibrium state (the molar ratio 2A:2B ≈ 3:2 from the integrals of the signals) by treatment with TFA (Fig. S10†). The solvent basicity may affect the protonation/deprotonation dynamics of the above system. In the case of CuII/NiII complexes, 3A and 3B reached equilibria in various media in the presence of acid. Demetallation of CuII ions of 3A and 3B seems to have occurred, judging from the resulting absorption spectra, especially in CH3CN (Fig. S14–S16†).
The calculated energies of all conformers enabled the interpretation of the relative stability of the isomers.19 In particular, the pseudo-trans conformation of 2A has a favorable van der Waals interaction, whereas the pseudo-cis conformation of 2B has better coplanarity between the coordinating and non-coordinating moieties. As a result, the pseudo-cis conformation for 2B is more stable than the pseudo-trans one for 2A (Fig. 4), which is in contrast to the observation that the yield of 2A is much higher than that of 2B during the nickel complexation of 1. The nickel complex 2A could thus be a kinetically favorable product, while 2B is a thermodynamically favorable one. The conformation of the helical structure of 2 is unaffected by further metal coordination. For complexes 3, 3B is also more stable than 3A, but the net stabilization effect for 2 is more remarkable than that for 3 (ΔE = 13 kJ mol−1vs. 4.2 kJ mol−1), which agrees with the ease of transformation for 2. The activation energy for this helical inversion process should be rather high, judging from the unidirectional helical changes, which might be relevant to the unusually high rotation barriers of the 3,3′-bipyrrole system.20 Consistently, 2A/2B and 3A/3B were not observed to interconvert while using temperature-dependent CD measurements. On the other hand, we can see from Fig. 4c and d that the protonation of the free-base compound 2 and corresponding Cu complex 3 does not cause visible changes in the conformational structures of the initial non-protonated species, but the energy differences between P,S-2/3H and M,S-2/3H become smaller relative to the non-protonated cases (Fig. 4a and b). Hence, we can assume that protonation might also decrease the activation barrier for the helical inversion processes leading to easier equilibria between the protonated 2A/2B and 3A/3B species.
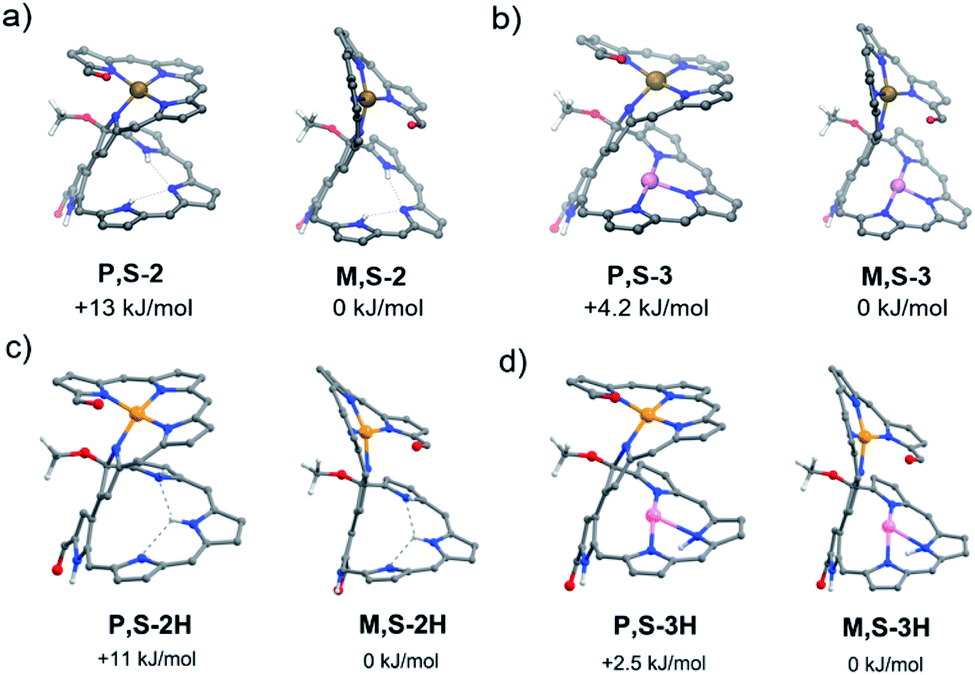 | ||
| Fig. 4 Relative energies between two conformers of (a) 2A and 2B, (b) 3A and 3B, (c) 2AH and 2BH, and (d) 3AH and 3BH. The energies of P,S-2, P,S-2H, P,S-3, and P,S-3H (corresponding to 2A, 2AH, 3A, and 3AH) were calculated relative to those of M,S-2, M,S-2H, M,S-3, and M,S-3H (corresponding to 2B, 2BH, 3B, and 3BH), respectively. | ||
Conclusions
In summary, the β-positions of two pyrrole rings close to each other in the pre-organized framework of conjugated octapyrrin P8 were linked selectively and efficiently by the FeCl3-promoted oxidative cyclization reaction. As a result, a tripyrrin appended doubly N-confused isosmaragdyrin analogue 1 has been obtained with concurrent addition of a methoxy group to one of the α-positions of the bipyrrole moiety. Stepwise coordination of 1 with NiII and CuII led to the formation of diastereomeric mononuclear NiII complexes 2A/2B and hetero-dinuclear NiIICuII complexes 3A/3B. X-ray crystal structures clearly revealed the unique helical oligopyrrin–Ni complex appended pentaphyrin structures of 2 and 3. The optical resolution of 1 followed by stepwise coordination afforded all four separable isomers, M,R-x, P,R-x, P,S-x, and M,S-x (x = 2 or 3). Intriguingly, the diastereomeric transformation between P- and M-twists from 2A to 2B was achieved upon simple treatment with acid (e.g., TFA). The combination of an open-chain oligopyrrin and a chiral porphyrinoid backbone has been demonstrated to be effective for achieving tunable chirality with external stimuli, and may be applied as a component of molecular machines or motors.21 In brief, oxidation of conjugated oligopyrroles paves a new way to the synthesis of a diversity of porphyrinoids, particularly through unique β,β-linkages.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the financial support for the work at ECUST by Shanghai Municipal Science and Technology Major Project (Grant No. 2018SHZDZX03) and the international cooperation program of Shanghai Science and Technology Committee (17520750100), NSFC (21971063, 21772041, 21702062, and 21811530005), the Fundamental Research Funds for the Central Universities (WK1616004, 222201717003, and 222201714013), and Program of Introducing Talents of Discipline to Universities (B160170). Part of the work in Kyushu was supported by Grants-in-Aid (JP19K05439 and JP19H04586) from the Japan Society for the Promotion of Science (JSPS) and Tokuyama Science Foundation. The authors thank Research Center of Analysis and Test of East China University of Science and Technology for help with the characterization.Notes and references
- (a) S. Saito and A. Osuka, Angew. Chem., Int. Ed., 2011, 50, 4342–4373 CrossRef CAS PubMed; (b) M. Stępień, N. Sprutta and L. Latos-Grażyński, Angew. Chem., Int. Ed., 2011, 50, 4288–4340 CrossRef PubMed; (c) V. V. Roznyatovskiy, C. H. Lee and J. L. Sessler, Chem. Soc. Rev., 2013, 42, 1921–1933 RSC; (d) X. S. Ke, H. M. Zhao, X. R. Zou, Y. Y. Ning, X. Cheng, H. M. Su and J. L. Zhang, J. Am. Chem. Soc., 2015, 137, 10745–10752 CrossRef CAS PubMed; (e) Y. Y. Lu, Y. C. Cheng, C. J. Li, J. X. Luo, W. Q. Tang, S. L. Zhao, Q. Y. Liu and Y. S. Xie, Sci. China: Chem., 2019, 62, 994–1000 CrossRef CAS.
- (a) T. D. Lash, Angew. Chem., Int. Ed., 2000, 39, 1763–1767 CrossRef CAS; (b) W. Miao, Z. Y. Zhu, Z. X. Li, E. Hao and L. J. Jiao, Chin. Chem. Lett., 2019, 30, 1895–1902 CrossRef CAS; (c) T. K. Chandrashekar and S. Venkatraman, Acc. Chem. Res., 2003, 36, 676–691 CrossRef CAS PubMed; (d) A. Ghosh, Angew. Chem., Int. Ed., 2004, 43, 1918–1931 CrossRef CAS PubMed; (e) J. Y. Shin, K. S. Kim, M. C. Yoon, J. M. Lim, Z. S. Yoon, A. Osuka and D. Kim, Chem. Soc. Rev., 2010, 39, 2751–2767 RSC; (f) Q. Z. Li, C. J. Li, J. Kim, M. Ishida, X. Li, T. T. Gu, X. Liang, W. H. Zhu, H. Ågren, D. Kim, H. Furuta and Y. S. Xie, J. Am. Chem. Soc., 2019, 141, 5294–5302 CrossRef CAS PubMed.
- (a) G. I. Vargas-Zúñiga and J. L. Sessler, Adv. Inorg. Chem., 2018, 71, 327–377 CrossRef; (b) J. M. Lintuluoto, K. Nakayama and J. Setsune, Chem. Commun., 2006, 3492–3494 RSC; (c) H. Ruffin, G. N. M. B. Boussambe, T. Roisnel, V. D. B. Boitrel and S. Le Gac, J. Am. Chem. Soc., 2017, 139, 13847–13857 CrossRef CAS PubMed.
- B. L. Feringa, Angew. Chem., Int. Ed., 2017, 56, 11060–11078 CrossRef CAS PubMed.
- (a) K. Mitsuno, T. Yoshino, I. Gupta, S. Mori, S. Karasawa, M. Ishida and H. Furuta, Angew. Chem., Int. Ed., 2017, 56, 14252–14256 CrossRef CAS PubMed; (b) R. Mysliborski, K. Hurej, M. Pawlicki and L. Latos-Grażyński, Angew. Chem., Int. Ed., 2018, 57, 16866–16870 CrossRef CAS PubMed; (c) T. Soya, H. Mori and A. Osuka, Angew. Chem., Int. Ed., 2018, 57, 15882–15886 CrossRef CAS PubMed; (d) X. S. Ke, T. Kim, Q. He, V. M. Lynch, D. Kim and J. L. Sessler, J. Am. Chem. Soc., 2018, 140, 16455–16459 CrossRef CAS PubMed; (e) D. G. Xie, Y. Liu, Y. T. Rao, G. Kim, M. B. Zhou, D. H. Yu, L. Xu, B. S. Yin, S. B. Liu, T. Tanaka, N. Aratani, A. Osuka, Q. Y. Liu, D. Kim and J. X. Song, J. Am. Chem. Soc., 2018, 140, 16553–16559 CrossRef CAS PubMed; (f) Y. K. Maurya, K. Noda, K. Yamasumi, S. Mori, T. Uchiyama, K. Kamitani, T. Hirai, K. Ninomiya, M. Nishibori, Y. Hori, Y. Shiota, K. Yoshizawa, M. Ishida and H. Furuta, J. Am. Chem. Soc., 2018, 140, 6883–6892 CrossRef CAS PubMed; (g) Q. Z. Li, M. Ishida, H. Kai, T. T. Gu, C. J. Li, X. Li, G. Baryshnikov, X. Liang, W. H. Zhu, H. Ågren, H. Furuta and Y. S. Xie, Angew. Chem., Int. Ed., 2019, 58, 5925–5929 CrossRef CAS PubMed.
- (a) J. Setsune, Chem. Rev., 2017, 117, 3044–3101 CrossRef CAS PubMed; (b) Y. S. Xie, P. C. Wei, X. Li, T. Hong, K. Zhang and H. Furuta, J. Am. Chem. Soc., 2013, 135, 19119–19122 CrossRef CAS PubMed; (c) P. C. Wei, K. Zhang, X. Li, D. Y. Meng, H. Ågren, Z. P. Ou, S. Ng, H. Furuta and Y. S. Xie, Angew. Chem., Int. Ed., 2014, 53, 14069–14073 CrossRef CAS PubMed; (d) K. Fujino, Y. Hirata, Y. Kawabe, T. Morimoto, M. T. Srinivasan, Y. Miseki, A. Kudo and H. Furuta, Angew. Chem., Int. Ed., 2011, 50, 6855–6859 CrossRef CAS PubMed; (e) H. Furuta, H. Maeda and A. Osuka, J. Am. Chem. Soc., 2001, 123, 6435–6436 CrossRef CAS PubMed.
- (a) J. L. Sessler, D. G. Cho, M. Stępień, V. Lynch, J. Waluk, Z. S. Yoon and D. Kim, J. Am. Chem. Soc., 2006, 128, 12640–12641 CrossRef CAS PubMed; (b) T. Higashino and A. Osuka, Chem. Sci., 2012, 3, 103–107 RSC; (c) F. Chen, Y. S. Hong, S. Shimizu, D. Kim, T. Tanaka and A. Osuka, Angew. Chem., Int. Ed., 2015, 54, 10639–10642 CrossRef CAS PubMed; (d) T. Tanaka and A. Osuka, Chem. Soc. Rev., 2015, 44, 943–969 RSC.
- K. Zhang, M. Savage, X. Li, Y. Jiang, M. Ishida, K. Mitsuno, S. Karasawa, T. Kato, W. H. Zhu, S. H. Yang, H. Furuta and Y. S. Xie, Chem. Commun., 2016, 52, 5148–5151 RSC.
- (a) Y. Pareek, M. Ravikanth and T. K. Chandrashekar, Acc. Chem. Res., 2012, 45, 1801–1816 CrossRef CAS PubMed; (b) J. L. Sessler, J. M. Davis and V. Lynch, J. Org. Chem., 1998, 63, 7062–7065 CrossRef CAS PubMed.
- HPLC chart of 1 suggests the presence of two independent isomers in the solution state at room temperature (Fig. S1†), which is consistent with the observed two sets of NMR signals. These two isomers tend to interconvert quickly within the laboratory timescale.
- CCDC 1509772 (1), 1430596 (2A), 1430597 (2B), 1430598 (3A), and 1430599 (3B) contain the supplementary crystallographic data for this paper.
- (a) T. Mizutani, S. Yagi, A. Honmaru and H. Ogoshi, J. Am. Chem. Soc., 1996, 118, 5318–5319 CrossRef CAS; (b) T. Mizutani, N. Sakai, S. Yagi, T. Takagishi, S. Kitagawa and H. Ogoshi, J. Am. Chem. Soc., 2000, 122, 748–749 CrossRef CAS.
- (a) B. Liu, X. F. Li, M. Stępień and P. J. Chmielewski, Chem. - Eur. J., 2015, 21, 7790–7797 CrossRef CAS PubMed; (b) K. Zhang, J. D. Zhang, X. Li, R. Guo, H. Ågren, Z. P. Ou, M. Ishida, H. Furuta and Y. S. Xie, Org. Lett., 2015, 17, 4806–4809 CrossRef CAS PubMed.
- (a) D. Sakamaki, D. Kumano, E. Yashima and S. Seki, Angew. Chem., Int. Ed., 2015, 54, 5404–5407 CrossRef CAS PubMed; (b) J. I. Setsune, A. Tsukajima, N. Okazaki, J. M. Lintuluoto and M. Lintuluoto, Angew. Chem., Int. Ed., 2009, 48, 771–775 CrossRef CAS PubMed.
- (a) N. Grzegorzek, M. Pawlicki, L. Szterenberg and L. Latos-Grażyński, J. Am. Chem. Soc., 2009, 131, 7224–7225 CrossRef CAS PubMed; (b) P. J. Chmielewski, Angew. Chem., Int. Ed., 2010, 49, 1359–1361 CrossRef CAS PubMed.
- The copper(II)–arene (cation-π) interactions have been occasionally observed. See the examples; (a) S. Saito, K. Furukawa and A. Osuka, Angew. Chem., Int. Ed., 2009, 48, 8086–8089 CrossRef CAS PubMed; (b) P. J. Chmielewski, L. Latos-Grażyński and I. Schmidt, Inorg. Chem., 2000, 39, 5475–5482 CrossRef CAS PubMed; (c) N. Grzegorzek, E. Nojman, L. Szterenberg and L. Latos-Grażyński, Inorg. Chem., 2013, 52, 2599–2606 CrossRef CAS PubMed.
- The HOMO–LUMO energy gaps were obtained from DFT calculations and were in general agreement with the electrochemical data. See Fig. S29–S33, S43 and Table S1.†.
- (a) P. C. Knipe, S. Thompson and A. D. Hamilton, Chem. Sci., 2015, 6, 1630–1639 RSC; (b) A. Gerus, K. Ślepokura and J. Lisowski, Inorg. Chem., 2013, 52, 12450–12460 CrossRef CAS PubMed.
- All the calculations were achieved with Gaussian09 program package at the B3LYP/6-31G* level. See Supporting Information for calculation details.†.
- S. Chatterjee, G. L. Butterfoss, M. Mandal, B. Paul, S. Gupta, R. Bonneau and P. Jaisankar, RSC Adv., 2016, 6, 71245–71249 RSC.
- J. F. Stoddart, Chem. Soc. Rev., 2009, 38, 1802–1820 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1509772, 1430596–1430599. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc06197e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |