
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRoom temperature crystallization of amorphous polysiloxane using photodimerization†
Taylor
Wright
 a,
Yael
Petel
a,
Carson O.
Zellman
b,
Ethan R.
Sauvé
a,
Zachary M.
Hudson
a,
Carl A.
Michal
ac and
Michael O.
Wolf
*a
a,
Yael
Petel
a,
Carson O.
Zellman
b,
Ethan R.
Sauvé
a,
Zachary M.
Hudson
a,
Carl A.
Michal
ac and
Michael O.
Wolf
*a
aDepartment of Chemistry, University of British Columbia, Vancouver, BC, Canada V6T 1Z1. E-mail: mwolf@chem.ubc.ca
bDepartment of Chemistry, Simon Fraser University, 8888 University Drive, Burnaby, BC, Canada V5A 1S6
cDepartment of Physics and Astronomy, University of British Columbia, Vancouver, BC, Canada V6T 1Z1
First published on 28th February 2020
Abstract
Bulk crystallization in flexible polymeric systems is difficult to control due to the random orientation of the chains. Here we report a photo cross-linking strategy that results in simultaneous cross-linking and crystallization of polysiloxane chains into millimeter sized leaf-like polycrystalline structures. Polymers containing pendant anthracene groups are prepared and undergo [4+4] photocycloaddition under 365 nm irradiation at room temperature. The growth and morphology of the crystalline structures is studied using polarized optical microscopy (POM) and atomic force microscopy and is found to progress through three unique stages of nucleation, growth, and constriction. The mobility of the individual chains is probed using pulsed-field gradient (PFG) NMR to provide insights into the diffusion processes that may govern chain transport to the growing crystal fronts. The room temperature crystallization of this conventionally amorphous polymer system may allow for a new level of morphological control for silicone materials.
Introduction
Polymer crystallinity plays a major role in the optical, physical, and conductive properties of polymeric materials.1–3 In order to optimize polymers for modern applications such as flexible batteries and solar panels, crystallinity must be considered and controlled.4–6 Crystallization of linear polymers has been widely studied both experimentally and via computer simulations, but remains difficult to predict and control due to random entanglement of the chains.7–9 Highly crystalline polymers have been prepared through the use of rigid backbones and directing linkers,10 monomers featuring a high crystallization enthalpy,11 or the topological photopolymerization and modification of crystallized monomers to yield polymeric crystals,12,13 where pre-existing crystallinity is preserved after polymerization.Macroscopic crystallization of cross-linked polymers is much less common, owing partially to the low solubility of crosslinked materials making polymer crystal growth from dissolved solutions difficult. One method for the preparation of cross-linked polymer crystals is the concurrent polymerization and crystallization of small molecules into covalent-organic frameworks.14–16 This technique requires rigid linkers that promote regioregularity between cross-linking nodes. Others have explored the photochemical polymerization of anthracene-containing molecules to produce 2D polymer crystals, beginning with monomer single crystals or interfacial thin films that can be cross-linked into crystalline sheets.17–19
While crystallization in many commercial polymers has been extensively probed, poly(dimethylsiloxane) (PDMS) crystallization is much less reported. PDMS has been widely explored for use in microfluidics, flexible conductors, and biocompatible materials.20–22 Low cost, low toxicity, and elastomeric behaviour make PDMS an attractive material for soft-polymer applications. However, the high degree of flexibility and resulting low glass transition temperature (Tg) of linear PDMS requires temperatures below −70 °C for crystallization to occur and be stable.23,24 This temperature limitation severely limits the study of crystalline siloxane's properties. Cross-linking can raise the crystallization temperature, but gives materials with only semi-crystalline regions without discrete crystal formation.25,26 No room temperature stable polymer crystals of PDMS have been reported, likely owing to the highly amorphous nature of the flexible backbone.
Here, we use anthracene photodimerization to cross-link a linear PDMS polymer liquid, resulting in the crystallization of approximately 1 mm diameter polycrystalline structures in less than one hour of low intensity 365 nm irradiation. This is the first example of room temperature crystallization of highly amorphous PDMS to our knowledge. The use of photo-driven cross-linking allows for the growth of structures to be controlled by varying the duration or intensity of irradiation. The amount of anthracene on the polymer backbone has a large influence on the resulting crystal size and morphology, however in all cases the anthracene units represent less than 4% of the total number of repeat units in the flexible polymer backbone. This strategy of driving crystallization through rigid photo crosslinking nodes represents a significant departure from previously reported anthracene polymer crystals where numerous rigid repeat units in the backbone were employed to provide the order necessary for crystallization. This strategy may allow for new types of soft, elastomeric polymer crystals that can be studied and handled at room temperature.
Results and discussion
Polymer synthesis
The condensation of alkyl amines with acetoacetates is an efficient synthetic route for the preparation of dynamic polymer networks, requiring minimal solvent, purification, or reaction optimization.27–29 We employ the same strategy to covalently attach a photoactive moiety onto a linear silicone backbone.30 9-Anthracenemethanol was esterified to give 9-anthracene-methylacetoacetate (AnAc) in high yield (96%) in a single step. AnAc was condensed with a commercially available propylamine-containing PDMS copolymer (PDMS-NH2) to yield the anthracene-containing random co-polymers as viscous oils without the need for further purification (Fig. 1a). Polymers were prepared with 3, 7, and 25 molar equivalents of AnAc relative to the polymer backbone, named P3, P7, and P25 respectively. | ||
| Fig. 1 Polymer structure and crystallization after UV irradiation. (a) Structure of the anthracene-containing PDMS random co-polymer with varying amounts of anthracene. (b) [4+4] Photocycloaddition of anthracene and photo/thermal cycloreversion reaction, showing one of two possible dimer isomers. (c) Image of a thin film of P7 after irradiation. (d) Optical microscope image of P7 after irradiation. (e) Photograph of the leaves of a sequoia redwood tree. (f) Polarized optical microscope image of P7 under a cross-polarizer and 530 nm retardation plate showing a birefringent pattern. | ||
Characterization of photochemical dimerization
Photo-induced dimerization was carried out on drop-cast thin-films on glass substrates, irradiating from 3 cm above for one hour using a 4 W handheld UV lamp at 365 nm, without any co-crystallizing agent or solvent added to the system. The absorption spectrum of P7 was collected as a function of irradiation time (Fig. S3†). A decrease in the absorbance between 300 and 400 nm is consistent with [4+4] photocycloaddition of anthracene.31 No further changes in the absorption spectra were observed after 30 minutes of irradiation, with the percent of anthracene remaining after irradiation estimated at 25% from the residual peak intensity at 365 nm. Free-standing solid pieces of polymer (approximately 115 μm thick, Fig. S4†) could be removed from the substrate after irradiation using a blade and did not dissolve fully in dichloromethane (48% percent soluble fraction after 24 hours), supporting the photo cross-linking of the material from an oil to elastic solid. While anthracene dimerization at air interfaces can form anthracene endoperoxides, this process is non-competitive with dimerization for systems where the anthracene concentration is greater than 1 mM (AnAc concentration in P7 = 132 mM).32,33 Endoperoxide formation has been shown to inhibit the assembly and crystallization of anthracene molecules due to disruption of the planarity preventing close packing of the rings, and does not lead to cross-linking.34A simple viscosity experiment was performed on a bulk sample, inverting a sealed vial of the polymer before and after irradiation. A decrease in creep distance was observed as a function of irradiation time, resulting in a solid sample (no observable movement after inversion) after 120 minutes of irradiation (Fig. S5†). No changes were observed in the FT-IR spectrum of bulk samples or thin films after irradiation, indicating that the vinylogous urethane bonds were not disrupted (Fig. S6†). Vibrational changes in the anthracene moiety are masked by the position and intensity of the stretching frequencies of the PDMS backbone.
Microscopy of polycrystalline structures
Optical microscopy imaging of P7 after 15 minutes of irradiation shows dispersed polycrystalline spherical structures between 5–25 μm in diameter (Fig. S7†). Further irradiation of the same sample for up to 60 minutes of total irradiation time resulted in the outward growth of polycrystalline sheaf-like lamellar structures of a maximum diameter of approximately 1 mm (Fig. 1d). Image analysis of Fig. 1c shows approximately 23% of the film surface consists of crystalline structures, but this does not account for crystallinity below the surface. These structures closely resemble the lamellar growth of PLLA/PVP and PEO/PVP composites, which are formed from controlled cooling of linear polymer melts.35,36Each spherulitic structure is observed to nucleate from an individual spherical structure, with clearly delineated arms from which a fine structure grows. Images resemble the leaves of a redwood tree, and as such a seed and leaf model analogy is applied (Fig. 1e). Leaves are observed to grow into contact with neighbouring leaves but joining or overlap of two structures was not observed (Fig. S8†). Isotropic irradiation of bulk samples in a vial produced crystal structures of a similar maximum diameter but lacking the branching and leaf structure, indicating different crystal growth dynamics (Fig. S9†).
Differential scanning calorimetry of P7 films after irradiation did not show any significant thermal transitions from −80 to 150 °C, with no changes to endotherms on repeated runs (Fig. S10†). The Tg is expected to be <−120 °C, and Tm values for semi-crystalline cross-linked PDMS have been reported in the region of 25–100 °C.25,26 Crystalline spherulite formation of linear PDMS has been reported at temperatures below −90 °C but not for samples at room temperature.23,24 Heating samples to higher temperatures was not explored as the amine functionalities of PDMS-NH2 are not thermally stable above 150 °C. No visual change was observed for crystallized samples after four weeks at room temperature under ambient laboratory conditions. A crystallized film of P7 was brought to 100 °C for one hour, resulting in a change from the observed leaf structures to long needle-shaped crystals (Fig. S11 and S12†). PXRD measurements of the needle-containing film showed a new sharp peak at 11.7° (Fig. S13†).
Polymers were prepared with three different molar ratios of AnAc to PDMS-NH2 (Fig. 1a) and the compositional effect on crystal growth studied. P3 produced seed structures within 15 minutes of irradiation, and upon further irradiation stunted leaf structures were observed with a maximum cross-sectional size of approximately 110 μm (Fig. 2). P25 showed macroscopic buckling of the thin-film surface after ten minutes of irradiation, with no crystallization seen in the deformed regions (Fig. S14†). At the film edges where buckling was less pronounced, crystals of a maximum cross-sectional size of approximately 210 μm were observed (Fig. 2). These crystals exhibit a less spherical growth and lessened fine structure near the leaf tips compared with P7, and a much wider variance in average size.
 | ||
| Fig. 2 Compositional effect on crystalline growth. Optical microscope image of (top) P3 crystals after one hour of 365 nm irradiation and (bottom) P25 crystals after ten minutes of 365 nm irradiation focused on the outer edges. | ||
Irradiation of P25 beyond ten minutes did not result in further crystallization. The small growth of P3 is attributed to insufficient anthracene units per polymer, resulting in crystal surfaces not capable of nucleating further growth. Buckling in P25 suggests that cross-linking is too rapid for the growth of well-defined leaf structures to occur, attributed to the higher concentration of anthracene groups. Despite the large dependence of crystallization on anthracene content, for the three tested samples anthracene-containing subunits on the polymer represented only between 0.5–4% of the total number of repeat units per polymer chain. Polarized optical microscopy (POM) of P7 shows a distinct birefringence pattern that persists across the non-contiguous branches of the same leaf (Fig. 1f). These POM results are consistent with birefringence in PLLA/PVP composites that is proposed to arise from sudden discontinuous branching and not from a helical twist of the growing crystal.35 Growth in these cases proceeds until a defect or breakage in the crystalline structure results in new branches extending from the original portion. Analysis of the POM images of P7 shows a branching angle of 45° between the orange and blue regions along a single leaf direction (Fig. S15†). The seed structures do not exhibit the same crystallization pattern under the same imaging conditions (Fig. S16†). No polarization effects were observed in the P7 oil before irradiation.
X-ray studies of polymer crystals
Crystallinity in P7 was probed using X-ray diffraction. The WAXS spectrum shows a similar profile before and after irradiation (Fig. S18†), with only two diffuse peaks at q = 0.85 and 1.5 Å−1, attributed to the Si–O–Si and Si–CH3 components of the PDMS backbone respectively.37 The SAXS spectrum of irradiated P7 shows a broad peak at q = 0.15 Å−1 not present in the non-irradiated sample. A Guinier plot of the low q region of the SAXS spectrum of crystallized P7 films gives a calculated radius of gyration (Rg) of 9.4 Å after irradiation (Fig. S19†).38 This value is lower than Rg values for linear PDMS and globular proteins, indicating highly packed chains.39–41 Tight packing is further supported by the non-linearity of the Guinier plot at lower q values. The correlation distance, ξ, was calculated to be 61 Å from the Ornstein–Zernike plot of crystallized P7 and is interpreted as the distance between the cross-linking nodes (Fig. S20†).42,43PXRD measurements of P7 were performed before and after crystallization, as well as on PDMS-NH2. All samples show the same two broad diffraction peaks centered at 12° and 22° that align with the features in the WAXS spectrum (Fig. S21†). Liquid crystal mesogens containing PDMS portions have been reported, demonstrating the ability of PDMS chains to participate in self-assembly when guided by rigid, aromatic moieties.37,44 In our system, we propose that interactions between anthracene dimer crosslinking nodes produces ordering that is not present when crosslinking is done using flexible moieties, however the high weight percentage of amorphous PDMS prevents complete ordering here (Fig. S22†). For similar sheaf-like lamellar structures prepared using two-component polymer blends, X-ray diffraction peaks are more prominent for blends featuring a higher proportion of the crystallizing polymer versus the amorphous polymer.45–47 Blends featuring a high proportion of amorphous component have been reported that exhibit only diffuse diffraction while still assembling into macroscopic structures.48 Combined with the X-ray scattering measurements, this demonstrates that the birefringent structures are only weakly crystalline, exhibiting long-range ordering necessary for the observed POM results, but lacking extensive short-range crystalline order (Fig. S22†). We attribute the weak crystallinity to the high proportion of amorphous component (PDMS moieties far from an anthracene cross-linking point) that inhibits short-range ordering as well as dominating the PXRD spectra with the two diffuse peaks.
Polymer crystal growth mechanism
Laser scanning confocal microscopy was used to map the morphology of the leaves and their spatial relation to the seeds. Three distinct focal planes were observed, each spaced approximately 40 μm apart (Fig. 3a and ESI-Video 1†). The lowest plane contains seed structures from which no leaves grew, while the middle plane has a second layer of seeds. The third plane contains the leaf structures that are observed to grow from a seed structure in the middle focal plane. | ||
| Fig. 3 Confocal microscopy of seeds and leaves. (a) Images of crystallized P7 at different depths, showing three distinct focal planes, collected at 10× magnification. Inset numbers are distance from the lowest focal plane. (b) Schematic representation for the proposed stages of photo-induced crystallization proceeding through seed growth, crystal growth from an uppermost seed, and constriction of the bulk. | ||
Imaging only the edges of a leaf structure shows that the leaves grow up and out from the center, with a height difference of 11 μm for the observed region (Fig. S23†). This demonstrates that the leaves grow upwards towards the source of illumination from an embedded seed and suggests that only seeds near the middle of the film are capable of nucleating to produce the large leaf structures.
Anthracene dimerization is expected to stop once cross-linking sufficiently inhibits polymer motion, preventing anthracene molecules from adopting the correct orientation for photocycloaddition. Samples remain emissive after irradiation as approximately 25% of the anthracene is not dimerized (Fig. S24†). Fluorescence microscopy imaging to examine the distribution of non-dimerized anthracene in the samples (Fig. S25†), demonstrates that both seeds and leaves are clearly distinguishable from the background. This demonstrates the presence of non-dimerized anthracene molecules in the crystalline structures, possibly due to anthracene molecules becoming confined in the growing structure, preventing their photoreaction (Fig. S26†). A direct comparison between emission intensities in the bulk and crystalline regions is not possible due to the changes in absorbance and quantum yield between single crystal, powder, and dissolved anthracene samples.49
Confocal imaging suggests that the leaf structures grow upwards from seed structures embedded in the interior of the film. However, photoreaction is expected to occur first near the surface of the film where the light intensity is the greatest. Diffusion of polymer chains against a photo-induced cross-linking gradient has been previously reported for the preparation of heterogeneously swollen films.50,51 Diffusion in our samples was studied using pulsed field gradient (PFG) 1H NMR spectroscopy.52,53 The translational diffusion coefficient (Dt) and rotational time play important roles in crystallization events, governing the rates of mass transport and reorientation to the growing crystal facets respectively.54,55 Measurements were performed on liquid samples before irradiation and solid samples after irradiation as a function of anthracene content, probing the methyl protons on the PDMS backbone (Fig. 4, 5a and Table S2†). The relative rotational times were estimated from the T1/T2 ratio.56,57
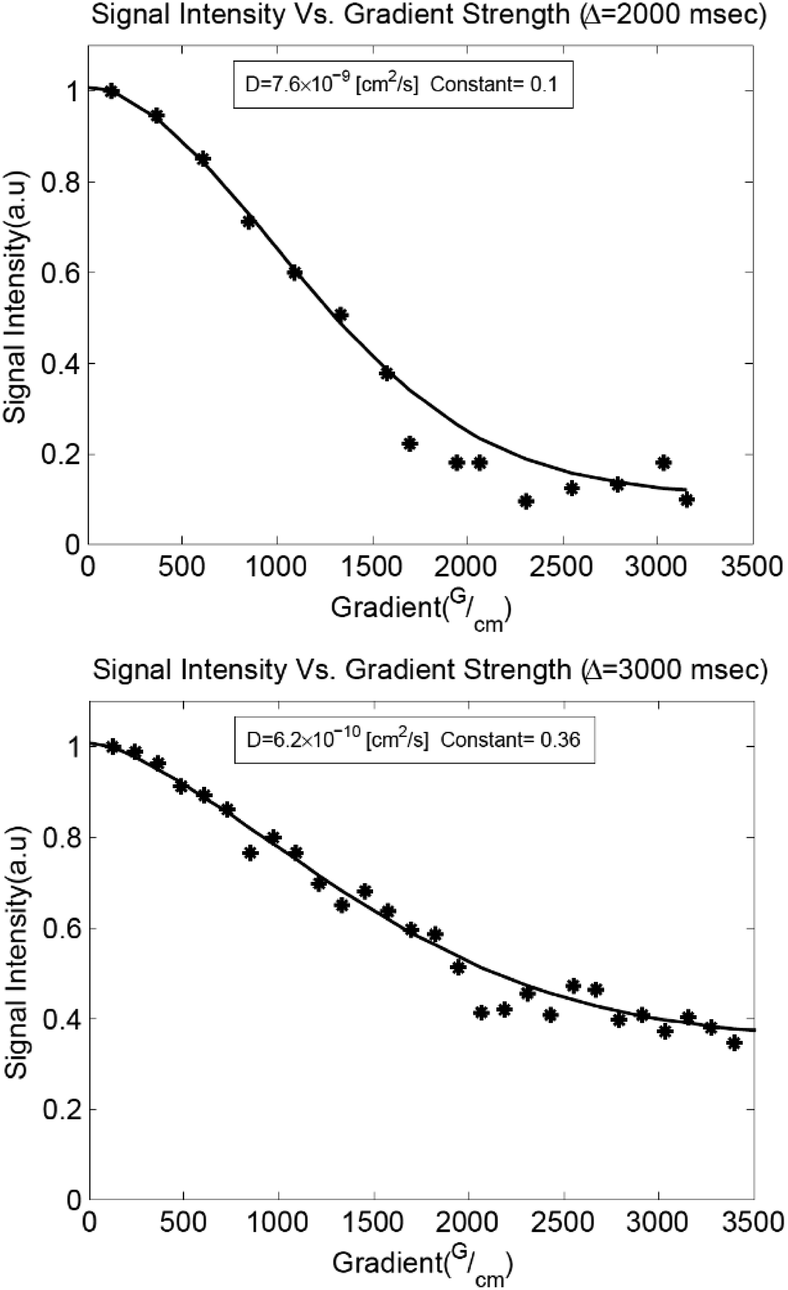 | ||
| Fig. 4 PFG 1H NMR diffusion measurements. Gaussian fits to the Stejskal–Tanner diffusion equation52 with an additive constant to account for slowly diffusing species are shown for liquid P7 (top) and irradiated solid P7 (bottom). | ||
 | ||
| Fig. 5 Calculated displacement for liquid polymer chains. (a) Translational diffusion coefficient (Dt) and relative rotational time (T1/T2) as a function of molar equivalents of anthracene, measured using PFG 1H NMR spectroscopy. (b) Maximum linear displacement distances for PDMS-NH2 and P7 as a function of time, as calculated from Dt. | ||
A decrease in Dt of nearly two orders of magnitude accompanied the addition of 3 equivalents of AnAc to the liquid polymer, with insignificant decreases upon addition of further molar equivalents (Fig. 5a). This suggests an aggregated state arising from the pendant anthracenes that is not impacted by further anthracene addition. The relative rotational times increase linearly with increasing AnAc equivalents. However, the increase in rotational times was less than an order of magnitude between P3 and P25. As such, diffusional limitations are not expected to play a major role in the larger degree of crystallization of P7 relative to P3 and P25. Given the morphology of the irradiated samples observed optically these findings are not surprising, as the mobility of the polymer likely depends on the proximity to crystallized structures. These measurements represent only the mobile components of the samples because the short T2 values of the crystalline components result in their suppression due to the filtering effects of the PFG pulse sequence.
The maximum translational diffusion lengths (assuming linear displacement) were plotted from Dt for PDMS-NH2 and P7 (before irradiation) as a function of time (Fig. 5b). P3 and P25 were omitted from this plot as the similar diffusion speeds to P7 results in a nearly identical profile. Crystallization of seeds in P7 is observed to begin within approximately 15 minutes of irradiation, while leaf growth began within 30 minutes.
From the data in Fig. 5b, it may be seen that an individual polymer chain is predicted to diffuse a maximum of 39 μm (15 minutes) and 55 μm (30 minutes). Confocal microscopy showed focal planes separated by approximately 40 μm between the layers of seeds and growing crystal leaf. As samples are irradiated from above, cross-linking and a reduction in motion should begin at the surface and propagate downward as irradiation continues. However, the leaves are observed to nucleate from seeds in the interior of the film and grow into a region that should already be cross-linked and thus unable to undergo reorientation for growth of the leaf structures. Based on the data in Fig. 5, it is reasonable to propose that diffusion from lower, non-cross-linked regions up to the growing crystalline front may be ongoing during irradiation, driven by the cross-linking gradient.
Atomic force microscopy (AFM) of crystallized P7 was performed in both non-contact mode and fast-force mapping contact mode to probe the surface topology and Young's modulus of the resulting films (Fig. 6a and b). The leaf structures are observable using AFM with feature heights of up to 1.3 μm relative to the surrounding flat film. Similarly shaped polymer lamellae typically exhibit a sheaf thickness on the order of tens of nanometers.58 Crystallization of the leaves must proceed prior to a bulk reduction in volume causing the film to constrict, forcing the tips of the crystal leaves to protrude from the film (Fig. 3b).
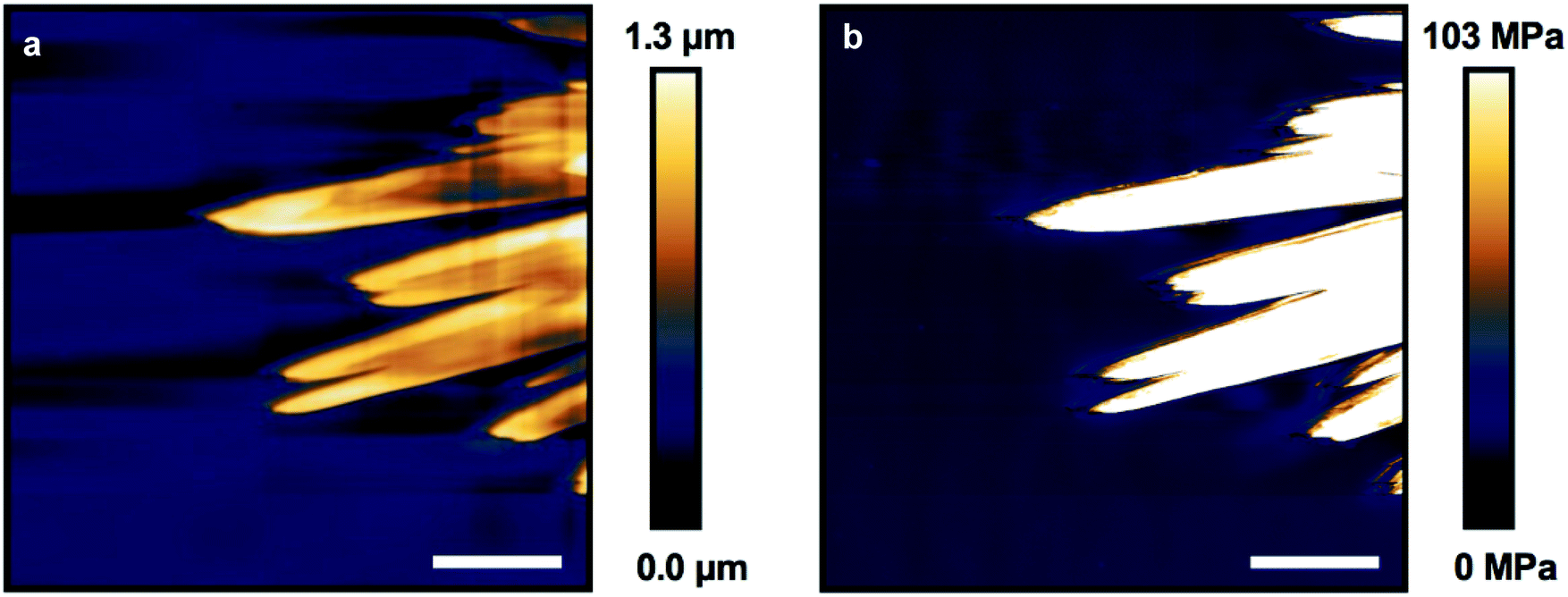 | ||
| Fig. 6 Atomic force microscopy imaging of crystalline leaves. (a) Non-contact mode showing feature height and (b) fast-force mapping contact mode showing Young's modulus of crystallized P7. Scale bar = 8 μm. | ||
Force mapping of the same exposed leaf structure shows a uniform Young's modulus of approximately 100 MPa across the crystals, an increase of nearly 10 times over the bulk material (Fig. S28†). This value is comparable to the reported modulus determined using AFM for ozone cross-linked PDMS, and shows a nearly 100 times increase over the modulus for bulk measurements of PDMS-NH2 cross-linked with flexible linkers.30,59 The modulus for organic crystals determined by AFM is typically between 5–10 GPa, orders of magnitude higher than those found for the leaf structures, supporting the incorporation of the PDMS units into the observed structures.60–62 The large contrast in bulk and crystal modulus may allow the leaves to act as structural reinforcement amongst the elastomeric bulk if this chemistry can be expanded to thicker, more mechanically robust samples.
Conclusions
In this work we report PDMS random co-polymers containing pendant anthracene groups suitable for [4+4] photocycloaddition. UV irradiation of thin films of these materials resulted in cross-linking accompanied by macroscopic organization into millimeter scale lamellar crystalline structures that are stable at room temperature. The final crystalline structure size and morphology was found to depend on the anthracene content in the system, with both high and low molar equivalents producing smaller structures than an intermediate amount. The crystallinity of these structures was studied using polarized optical microscopy and diffraction techniques and was found to be similar to lamellar crystalline phases prepared using more rigid polymer structures. However, the flexibility of the siloxane chain in our system represents a departure for lamellar crystalline formation as the rigid anthracene moieties represent less than four percent of the repeat units in the polymer chain. Diffusion NMR results coupled with insights from AFM imaging and confocal microscopy allowed for a growth model to be proposed that involves diffusion against a photo-induced cross-linking gradient resulting in the crystalline phases protruding up to a micron in height from the bulk film surface. The rigidity of the lamellar structures was probed using AFM indentation and found to be much greater than the surrounding bulk, which we envisage may allow for a new type of crystalline-reinforced silicone material.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by funding from the Natural Sciences and Engineering Research Council of Canada (NSERC) and the Canada Foundation for Innovation. We thank Dr Mark MacLachlan (UBC Chemistry) for discussions regarding POM and polymer crystallinity. Confocal microscopy measurements were performed by Dr Miki Fujita and Kevin Hodgson (UBC Bioimaging Facility). PXRD measurements were collected by Anita Lam (UBC Chemistry). DSC measurements were collected by David Ester (SFU 4D Labs). Fluorescence microscopy images were collected by Hyungki Kim (UBC Chemistry). E. R. S. thanks NSERC for a postgraduate scholarship, and Z. M. H. is grateful for support from the Canada Research Chairs program.References
- K. Okano, A. Shishido and T. Ikeda, Adv. Mater., 2006, 18, 523–527 CrossRef CAS.
- G. Perego, G. D. Cella and C. Bastioli, J. Appl. Polym. Sci., 2004, 59, 37–43 CrossRef.
- Z. Gadjourova, Y. G. Andreev, D. P. Tunstall and P. G. Bruce, Nature, 2001, 412, 520–523 CrossRef CAS PubMed.
- Z. He, B. Xiao, F. Liu, H. Wu, Y. Yang, S. Xiao, C. Wang, T. P. Russell and Y. Cao, Nat. Photonics, 2015, 9, 174–179 CrossRef CAS.
- S. Kwon, K. Yu, K. Kweon, G. Kim, J. Kim, H. Kim, Y. R. Jo, B. J. Kim, J. Kim, S. H. Lee and K. Lee, Nat. Commun., 2014, 6, 4183 CrossRef PubMed.
- D. Vonlanthen, P. Lazarev, K. A. See, F. Wudl and A. Heeger, J. Adv. Mater., 2014, 26, 5095–5100 CrossRef CAS PubMed.
- P. Welch and M. Muthukumar, Phys. Rev. Lett., 2001, 87, 218302 CrossRef CAS PubMed.
- M. Anwar and T. Schilling, Polymer, 2015, 76, 307–312 CrossRef CAS.
- R. Noriega, J. Rivnay, K. Vandewal, F. P. V. Koch, N. Stingelin, P. Smith, M. F. Toney and A. Salleo, Nat. Mater., 2013, 12, 1038–1044 CrossRef CAS PubMed.
- H. J. Lee, Y. R. Jo, S. Kumar, S. J. Yoo, J. G. Kim, Y. J. Kim, B. J. Kim and J. S. Lee, Nat. Commun., 2016, 7, 12803 CrossRef PubMed.
- X. Wang, G. Guerin, H. Wang, Y. Wang, I. Manners and M. A. Winnik, Science, 2007, 317, 644–647 CrossRef CAS PubMed.
- A. Matsumoto, K. Yokoi, S. Aoki, K. Tashiro, T. Kamae and M. Kobayashi, Macromolecules, 2002, 31, 2129–2136 CrossRef.
- A. Matsumoto, T. Odani, K. Sada, M. Miyata and K. Tashiro, Nature, 2000, 405, 328–330 CrossRef CAS PubMed.
- D. Beaudoin, T. Maris and J. D. Wuest, Nat. Chem., 2013, 5, 830–834 CrossRef CAS PubMed.
- Y. B. Zhang, J. Su, H. Furukawa, Y. Yun, F. Gándara, A. Duong, X. Zou and O. M. Yaghi, J. Am. Chem. Soc., 2013, 135, 16336–16339 CrossRef CAS PubMed.
- F. J. Uribe-Romo, J. R. Hunt, H. Furukawa, C. Klöck, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 4570–4571 CrossRef CAS PubMed.
- P. Kissel, D. J. Murray, W. J. Wulftange, V. J. Catalano and B. T. King, Nat. Chem., 2014, 6, 774–778 CrossRef CAS PubMed.
- M. J. Kory, M. Wörle, T. Weber, P. Payamyar, S. W. Van De Poll, J. Dshemuchadse, N. Trapp and A. D. Schlüter, Nat. Chem., 2014, 6, 779–784 CrossRef CAS PubMed.
- V. Müller, A. Hinaut, M. Moradi, M. Baljozovic, T. A. Jung, P. Shahgaldian, H. Möhwald, G. Hofer, M. Kröger, B. T. King, E. Meyer, T. Glatzel and A. D. Schlüter, Angew. Chem., Int. Ed., 2018, 57, 10584–10588 CrossRef PubMed.
- J. De Jong, R. G. H. Lammertink and M. Wessling, Lab Chip, 2006, 6, 1125–1139 RSC.
- X. Niu, S. Peng, L. Liu, W. Wen and P. Sheng, Adv. Mater., 2007, 19, 2682–2686 CrossRef CAS.
- J. H. Moon, D. H. Baek, Y. Y. Choi, K. H. Lee, H. C. Kim and S. H. Lee, J. Micromech. Microeng., 2010, 20, 025032 CrossRef.
- P. R. Sundararajan, G. K. Hamer and M. D. Croucher, Macromolecules, 1980, 13, 971–973 CrossRef CAS.
- P. R. Sundararajan, Polymer, 2002, 43, 1691–1693 CrossRef CAS.
- T. Dollase, H. W. Spiess, M. Gottlieb and R. Yerushalmi-Rozen, Europhys. Lett., 2002, 60, 390–396 CrossRef CAS.
- J. Van Damme, O. Van Den Berg, J. Brancart, L. Vlaminck, C. Huyck, G. Van Assche, B. Van Mele and F. Du Prez, Macromolecules, 2017, 50, 1930–1938 CrossRef CAS.
- W. Denissen, G. Rivero, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Adv. Funct. Mater., 2015, 25, 2451–2457 CrossRef CAS.
- T. Wright, T. Tomkovic, S. G. Hatzikiriakos and M. O. Wolf, Macromolecules, 2019, 52, 36–42 CrossRef CAS.
- W. Denissen, M. Droesbeke, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Nat. Commun., 2017, 8, 14857 CrossRef PubMed.
- T. Stukenbroeker, W. Wang, J. M. Winne, F. E. Du Prez, R. Nicolaÿ and L. Leibler, Polym. Chem., 2017, 8, 6590–6593 RSC.
- R. R. Islangulov and F. N. Castellano, Angew. Chem., Int. Ed., 2006, 45, 5957–5959 CrossRef CAS PubMed.
- H. Bouas-Laurent, A. Castellan, J. P. Desvergne and R. Lapouyade, Chem. Soc. Rev., 2001, 30, 248–263 RSC.
- P. Payamyar, K. Kaja, C. Ruiz-Vargas, A. Stemmer, D. J. Murray, C. J. Johnson, B. T. King, F. Schiffmann, J. Vandevondele, A. Renn, S. Götzinger, P. Ceroni, A. Schütz, L. T. Lee, Z. Zheng, J. Sakamoto and A. D. Schlüter, Adv. Mater., 2014, 26, 2052–2058 CrossRef CAS PubMed.
- W. Fudickar and T. Linker, Langmuir, 2010, 26, 4421–4428 CrossRef CAS PubMed.
- S. Nurkhamidah, E. M. Woo, Y. T. Yeh, F. Luo and V. Katiyar, Polymers, 2018, 10, 1–21 Search PubMed.
- G. Lugito, C. C. Su, Y. H. Wang and E. M. Woo, J. Polym. Res., 2017, 24, 166 CrossRef.
- K. Nickmans, J. N. Murphy, B. de Waal, P. Leclère, J. Doise, R. Gronheid, D. J. Broer and A. P. H. Schenning, J. Adv. Mater., 2016, 28, 10068–10072 CrossRef CAS PubMed.
- P. V. Konarev, V. V. Volkov, A. V. Sokolova, M. H. J. Koch and D. I. Svergun, J. Appl. Crystallogr., 2003, 36, 1277–1282 CrossRef CAS.
- J. S. Higgins, K. Dodgson and J. A. Semlyen, Polymer, 1979, 20, 553–558 CrossRef CAS.
- G. L. Jadav, V. K. Aswal, H. Bhatt, J. C. Chaudhari and P. S. Singh, J. Membr. Sci., 2012, 415–416, 624–634 CrossRef CAS.
- D. M. Smilgies and E. Folta-Stogniew, J. Appl. Crystallogr., 2015, 48, 1604–1606 CrossRef CAS PubMed.
- L. S. Ornstein and F. Zernike, Phys. Z., 1918, 19, 134 Search PubMed.
- S. Matsui, T. Kureha, Y. Nagase, K. Okeyoshi, R. Yoshida, T. Sato and D. Suzuki, Langmuir, 2015, 31, 7228–7237 CrossRef CAS PubMed.
- E. Ibarboure, J. Rodriguez-Hernández and E. Papon, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4668–4679 CrossRef CAS.
- T. P. Russell, H. Ito and G. D. Wignall, Macromolecules, 1988, 21, 1703–1709 CrossRef CAS.
- G. Crevecoeur and G. Groeninckx, Macromolecules, 1991, 24, 1190–1195 CrossRef CAS.
- S. Talibuddin, L. Wu, J. Runt and J. S. Lin, Macromolecules, 1996, 29, 7527–7535 CrossRef CAS.
- V. P. R. Silva, G. G. Silva, V. Caliman, J. Rieumont, C. O. B. de Miranda-Pinto, B. S. Archanjo and B. R. A. Neves, Eur. Polym. J., 2007, 43, 3283–3291 CrossRef CAS.
- R. Katoh, K. Suzuki, A. Furube, M. Kotani and K. Tokumaru, J. Phys. Chem. C, 2009, 113, 2961–2965 CrossRef CAS.
- R. Jellali, V. Bertrand, M. Alexandre, N. Rosière, M. Grauwels and C. Jérôme, Macromol. Biosci., 2017, 17, 1–12 CrossRef PubMed.
- R. Jellali, M. Alexandre and C. Jérôme, Polym. Chem., 2017, 8, 2499–2508 RSC.
- E. O. Stejskal and J. E. Tanner, J. Chem. Phys., 1965, 42, 288–292 CrossRef CAS.
- T. R. Saarinen and C. S. Johnson, J. Magn. Reson., 1988, 78, 257–270 Search PubMed.
- L. Gránásy, T. Pusztai, G. Tegze, J. A. Warren and J. F. Douglas, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2005, 72, 1–15 CrossRef PubMed.
- B. Crist and J. M. Schultz, Prog. Polym. Sci., 2016, 56, 1–63 CrossRef CAS.
- D. S. Korchuganov, I. E. Gagnidze, E. N. Tkach, A. A. Schulga, M. P. Kirpichnikov and A. S. Arseniev, J. Biomol. NMR, 2004, 30, 431–442 CrossRef CAS PubMed.
- C. Li, Y. Wang and G. J. Pielak, J. Phys. Chem. B, 2009, 113, 13390–13392 CrossRef CAS PubMed.
- W. Wang, X.-M. Zhai, B.-L. He, X.-J. Wen, Z.-P. Ma, F. Yuan and X.-F. Tang, Macromolecules, 2005, 38, 1717–1722 CrossRef.
- J. Song, D. Tranchida and G. J. Vancso, Macromolecules, 2008, 41, 6757–6762 CrossRef CAS.
- J. K. Sun, W. Li, C. Chen, C. X. Ren, D. M. Pan and J. Zhang, Angew. Chem., Int. Ed., 2013, 52, 6653–6657 CrossRef CAS PubMed.
- F. M. Capaldi, M. C. Boyce and G. C. Rutledge, J. Chem. Phys., 2006, 124, 214709 CrossRef PubMed.
- M. S. R. N. Kiran, S. Varughese, C. M. Reddy, U. Ramamurty and G. R. Desiraju, Cryst. Growth Des., 2010, 10, 4650–4655 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc06235a |
| This journal is © The Royal Society of Chemistry 2020 |