
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthetic biohybrid peptidoglycan oligomers enable pan-bacteria-specific labeling and imaging: in vitro and in vivo†
Jing-Xi
He
ab,
Kim
Le Mai Hoang
a,
Shu Hui
Kho
ac,
Zhong
Guo
a,
Wenbin
Zhong
b,
Kishore Reddy
Venkata Thappeta
b,
Rubí
Zamudio-Vázquez
b,
Sin Ni
Hoo
a,
Qirong
Xiong
b,
Hongwei
Duan
b,
Liang
Yang
d,
Mary B.
Chan-Park
 *b and
Xue-Wei
Liu
*a
*b and
Xue-Wei
Liu
*a
aSchool of Physical and Mathematical Sciences, Nanyang Technological University, 21 Nanyang Link, Singapore 637371, Singapore. E-mail: xuewei@ntu.edu.sg
bSchool of Chemical and Biomedical Engineering, Nanyang Technological University, 62 Nanyang Drive, Singapore 637459, Singapore
cNTU Institute for Health Technologies, Nanyang Technological University, Singapore
dSchool of Medicine, Southern University of Science and Technology, Shenzhen, 518055, China
First published on 26th February 2020
Abstract
Peptidoglycan is the core component of the bacterial cell wall, which makes it an attractive target for the development of bacterial targeting agents. Intercepting its enzymatic assembly with synthetic substrates allows for labeling and engineering of live bacterial cells. Over the past two decades, small-molecule-based labeling agents, such as antibiotics, D-amino acids or monosaccharides have been developed for probing biological processes in bacteria. Herein, peptidoglycan oligomers, substrates for transglycosylation, are prepared for the first time using a top-down approach, which starts from chitosan as a cheap feedstock. A high efficiency of labeling has been observed in all bacterial strains tested using micromolar substrates. In contrast, uptake into mammalian cells was barely observable. Additional mechanistic studies support a hypothesis of bacteria-specific metabolic labeling rather than non-specific binding to the bacterial surface. Eventually, its practicality in bacterial targeting capability is demonstrated in resistant strain detection and in vivo infection models.
Introduction
The rise of multidrug-resistant bacterial pathogens is becoming a global threat to public health, thus driving the relentless search for new and more effective antibiotics.1 The biosynthesis pathway for bacterial cell walls remains an attractive family of drug targets, due to its ubiquitous presence across all bacterial phyla and its complete absence in human cells.2 In fact, many of the most successful broad-spectrum antibiotics such as vancomycin, penicillin, and the associated beta-lactam families specifically disrupt various stages of bacterial cell wall biogenesis.3 Further, this feature may also be the target of bacteria-specific targeting agents underpinning various biosensing agents.The major constituent of bacterial cell walls, in both Gram-positive and Gram-negative bacteria, is the network of peptidoglycan (PG). The PG network forms a resilient structure that protects bacteria against varying osmotic pressures, and provides an anchoring platform for extracytosolic proteins such as Braun's lipoprotein.4 At the molecular level, PG contains repeating units of N-acetyl-glucosamine (NAG) linked N-acetyl-muramic acid (NAM), with the latter covalently linked to a pendant pentapeptide. Different bacterial strains demonstrate variance in the exact structure of the pentapeptides, which are all cross-linked to the same NAG-NAM oligosaccharide chains to generate the three-dimensional network of PG that is characteristic of all bacterial cell walls.
NAG-NAM oligosaccharide elongation and peptide cross-linking are thus shared processes in all bacteria, mediated by a variety of peptidoglycan glycosyltransferases (PGTs) and transpeptidases (TPs) respectively (Fig. 1). TPs, in particular, have been the target for several mechanistic or application studies, using antibiotics or derivatives of D-amino acids developed in recent years.5 However, little is known about the PGTs and their enzymatic properties, in part due to the limited availability of both relevant substrates, which have no shortcuts like D-amino acids for TPs, and appropriate in vitro assays. Towards this end, some encouraging efforts have been devoted to synthesizing the lipid II–VIII compounds as PGT substrates, through chemical or enzymatic pathways.6 Through these substrates, researchers have gathered invaluable information on PGT enzymatic properties, and made early attempts to chemically engineer the substrates into potential inhibitors for mechanistic studies, structure–activity relationship studies, and novel antibiotic designs.7 However, one of the most major obstacles inhibiting further progress was the difficulty in obtaining substrate derivatives, which have to be prepared through carefully designed orthogonal chemical protections. An overwhelming number of steps are required to obtain just the tetrasaccharide unit, reported at nearly 63 steps starting from monosaccharides,8 even though the tetrasaccharide is still suboptimal. Longer N-saccharide oligomers are likely to be more biologically relevant substrates for PGTs and TPs (Fig. 1).6b,9 Here we present an efficient 9-step approach for synthesizing biohybrid peptidoglycan oligomers (PGOs) from chitosan, a plentiful biopolymer readily available from all crustacean wastes. We also demonstrated that our biohybrid PGOs are successfully incorporated into the cell walls of various bacterial strains. Our PGOs can be customized for various needs in mechanistic studies, bacterial bioimaging and novel drug designs.
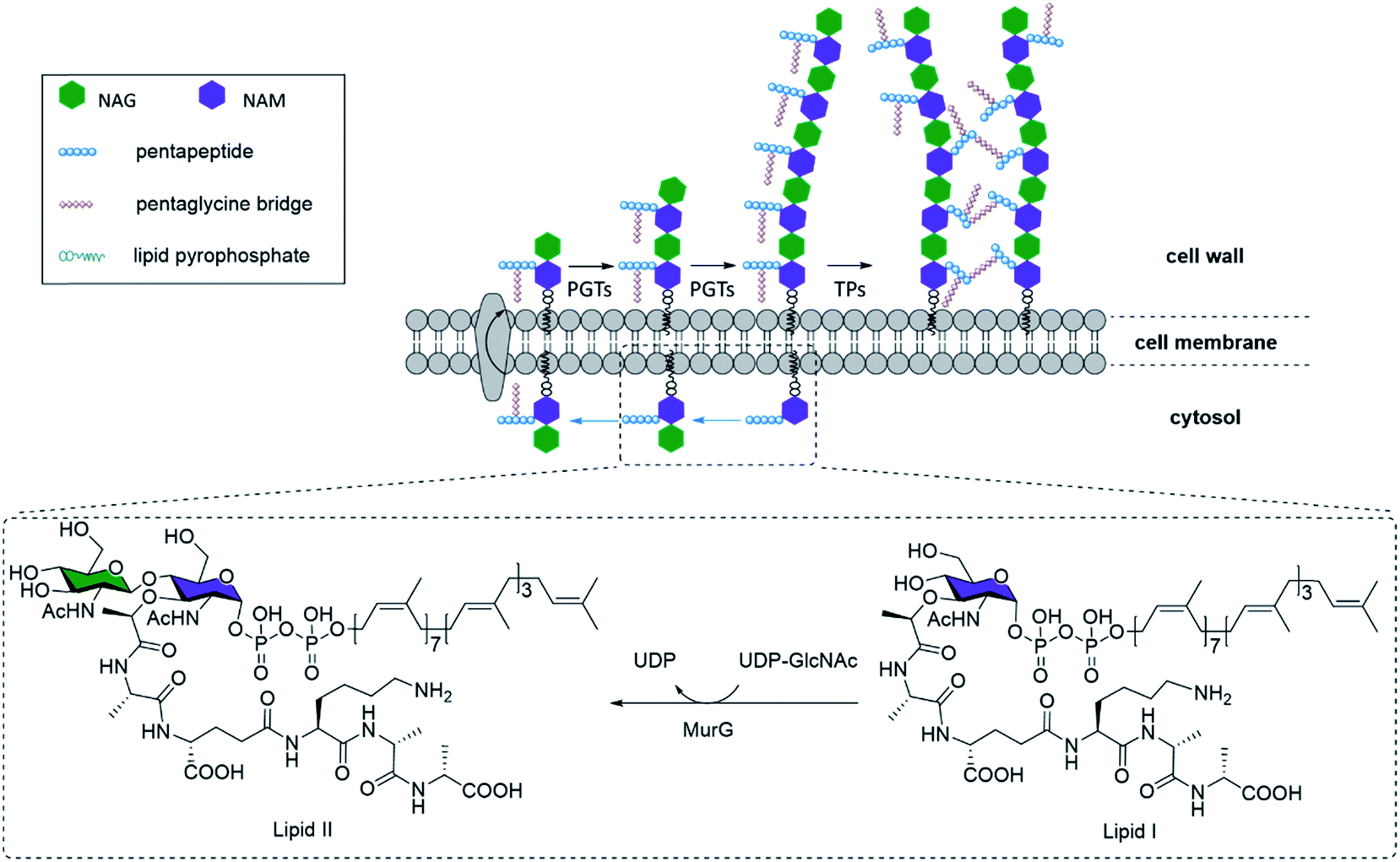 | ||
| Fig. 1 Peptidoglycan biosynthesis in bacteria. MurG is an enzyme that catalyzes the glycosylation reaction between UDP-GlcNAC, which contains NAG (green), and the lipid I substrate, which contains NAM (purple) and a pendant pentapeptide, to form the lipid-linked NAG-NAM β-(1,4) disaccharide known as lipid II. After a pentaglycine bridge is added to lipid II, it is translocated to the exterior surface of the cell membrane and incorporated into the peptidoglycan network by peptidoglycan glycosyltransferases (PGTs) to form peptidoglycan oligomers. Cross-linking by transpeptidases (TPs) completes peptidoglycan biosynthesis. Some TPs bind to penicillin, and are thus known as penicillin-binding proteins (PBPs). | ||
Results and discussion
Synthesis of PGOs
Starting from low molecular weight chitosan 1, the phthaloyl group was first used to protect the free amino group at C-2 from subsequent transformation (Fig. 2). The bulky triisopropylsilyl group was delivered to the primary alcohol and anomeric hydroxyl group at the reducing terminal successfully. C-3 hydroxy groups on chitosan are relatively inert towards this reaction unless harsher conditions are applied. This results in the formation of substrate 3. Next, 2-bromopropanoate was introduced at C-3 by SN2 chemistry, with a molar ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for the glucosamine unit: 2-bromopropanoate, approximating the alternating pattern of the peptidoglycan repeat motif, to give us the desired substrate 4. The remaining hydroxyl groups were then capped by acetyl esterification. We noted the improvement in organic solvent solubility along the protection scheme, from substrate 2 forming a dispersion in DMF to substrate 5 dissolving in CH2Cl2 as well as other commonly used organic solvents.
:1 for the glucosamine unit: 2-bromopropanoate, approximating the alternating pattern of the peptidoglycan repeat motif, to give us the desired substrate 4. The remaining hydroxyl groups were then capped by acetyl esterification. We noted the improvement in organic solvent solubility along the protection scheme, from substrate 2 forming a dispersion in DMF to substrate 5 dissolving in CH2Cl2 as well as other commonly used organic solvents.
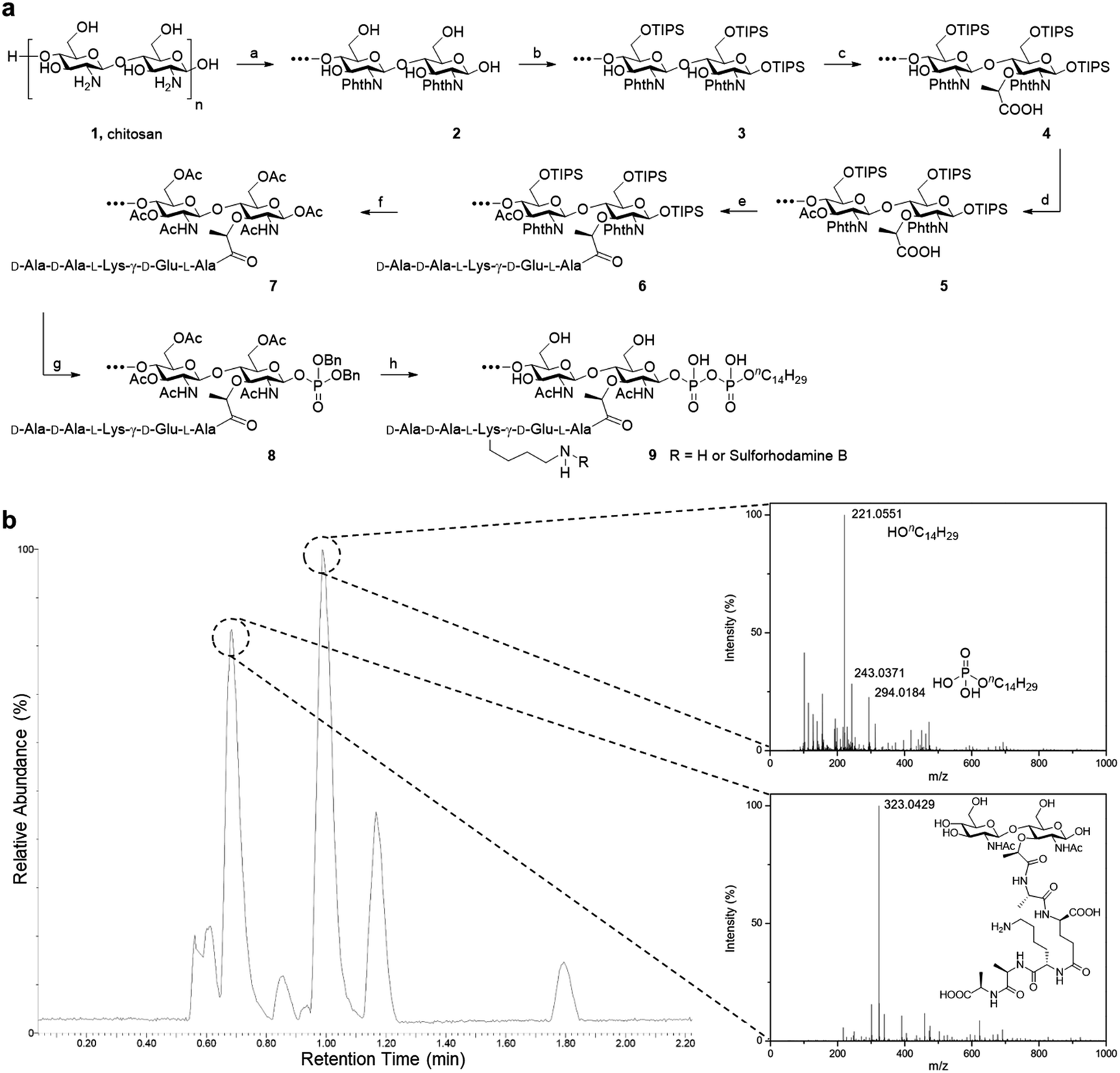 | ||
| Fig. 2 Synthesis of PGO. (a) Total synthesis of PGO 9 and rhodamine-labeled PGO 9 from low molecular weight chitosan 1. Reactions and conditions: (a) phthalic anhydride (3 equiv.), AcOH/H2O; (b) imidazole, TIPSCl (7 equiv.), DMF; (c) NaH, 2-bromopropanoic acid (0.5 equiv.), DMF; (d) acetic anhydride (5 equiv.), pyridine; (e) peptide 13 (1 equiv.), HATU, HOAt, DIEA, DMF; (f) TBAF, hydrazine, AcOH, MeOH, Ac2O; (g) (1) MeNH2, (2) 1H-tetrazole, dibenzyl N,N-diisopropylphosphoramidite (2 equiv.), (3) tert-butyl hydroperoxide, CH2Cl2; (h) (1) H2, Pd/C, MeOH, (2) nC14H29OPO(OH)2 (2 equiv.), DMF, (3) LiOH, MeOH/H2O (v/v, 1:1). Protections for the peptide are omitted in this scheme, and synthesis details are provided in the ESI.† (b) LC-ESI-TOF of metabolites from the lysozyme degradation assay. Two major resultant peaks at t = 0.69 min and t = 0.99 min correspond to the NAG-NAM subunit and the phospholipid respectively. The remaining peaks were from metabolites of the enzyme and the buffer used in the lysozyme degradation assay (Fig. S15†). | ||
The pentapeptide was prepared based on established literature, following the sequence found in S. aureus and E. faecalis.5a The protecting groups on the side chain and terminal functionalities were selected to be Fmoc for amine and benzyl or methyl ester for carboxylic acids, so that global deprotection can be realized at the end of synthesis with lithium hydroxide. The amide linkage between the pentapeptide and substrate 5 was achieved under HATU/HOAt/DIEA coupling conditions. Following partial cleavage and global protection with acetic anhydride, dibenzyl monophosphate can be introduced at the reducing terminus. Hydrogenation was used to liberate the phosphate so that it can be coupled with a second lipid-linked monophosphate, to yield the desired oligosaccharide pyrophosphate 9 (Fig. 2a). We opted to employ the tetradecanyl lipid linker in place of the natural polyprenyl lipid to simplify preparation routes, taking into consideration that the tetradecanyl linker has been found to display a better binding affinity to MurG, one of the key PGTs.7b The crude mixture was dialyzed with deionized water, filtered and lyophilized to furnish the pure oligomeric final product as a beige solid.
As it is a polysaccharide derivative, we collected its NMR and GPC profiles to analyse the molecular structure and molecular weight. Furthermore, the product's purity was substantiated with reversed-phase HPLC. As peak broadening was observed in the GPC chromatograms and the elution time was shorter than expected, we inferred the existence of self-assemblies with an increased hydrodynamic radius. This was confirmed by DLS of PGOs in solution, which showed nanoparticles of around 80 nm in diameter (Fig. S14b†). Comparison of integration values from sugar, peptide and lipid moieties in the NMR spectrum suggested a statistical ratio of 1:0.5:0.1, which totaled around 5 kDa molecular weight on average. This result agreed well with the theoretical outcome, as our synthesis started from chitosan of <3 kDa molecular weight and finished with a design where half of the sugar repeating units were grafted by the peptide. Finally, the finer structure of PGOs was evaluated with the lysozyme degradation assay. Lysozyme binds to tetrasaccharides, or longer motifs, in peptidoglycan and cleaves the glycosidic linkages in the middle. LC-ESI-TOF MS analysis revealed the presence of an m/z fragment consisting of a NAG-NAM with a pendant pentapeptide (Fig. 2b). The presence of this fragment, but not NAG-NAG or NAM-NAM fragments (Fig. S15†), supported our hypothesis that the oligomeric product 9 has mainly an alternating NAG-NAM pattern, as expected for a PGO.
There could exist a small amount of NAG-NAG or NAM-NAM moieties due to the nature of the polymer being a mixture of similar compounds, but the majority of our product comprised NAG-NAM moieties as the only observed peak in mass spectra.
Specific labelling of bacterial cell walls with PGOs
To label and visualize bacteria using PGOs, sulforhodamine B acid chloride (rhodamine) was covalently bound to the free amine on the side chain of lysine moieties.10 The lysine residue has been extensively modified without significant influence on substrates' enzymatic activities or the viability of bacteria in turn.7d,11 Furthermore, the dye of choice, rhodamine, can be easily coupled to the lysine amine and has a wavelength suitable for the stimulated emission depletion (STED) technique in super-resolution microscopy studies. We cultured bacterial strains in the presence of PGOs–rhodamine. Nutrient-rich media such as MHB and BHI were introduced, and the PGOs–rhodamine was incubated at varying concentrations and durations. The optimal incubation time was found to be an hour long, with 100 μg mL−1 of PGOs–rhodamine. A total of 6 bacterial strains were evaluated for this study, of which 4 were Gram-positive and 2 were Gram-negative (Fig. 3a). Among these, methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant Enterococcus faecalis and Pseudomonas aeruginosa are clinically relevant strains that represent multi-drug resistant (MDR) bacteria. The membrane dye FM 1-43fx was used as an indicator dye for bacterial cell surface localization (Fig. 3a). Based on our super-resolution STED microscopy studies, PGOs–rhodamine was colocalized with FM 1-43fx in all of the strains tested, suggesting PGOs were successfully incorporated into the bacterial cell walls of all 6 tested strains (Fig. 3a). In addition, we confirmed that the PGOs–rhodamine does not get into mammalian 3T3 cells (Fig. 3c), demonstrating the utility of PGOs–rhodamine as a bacteria-specific indicator.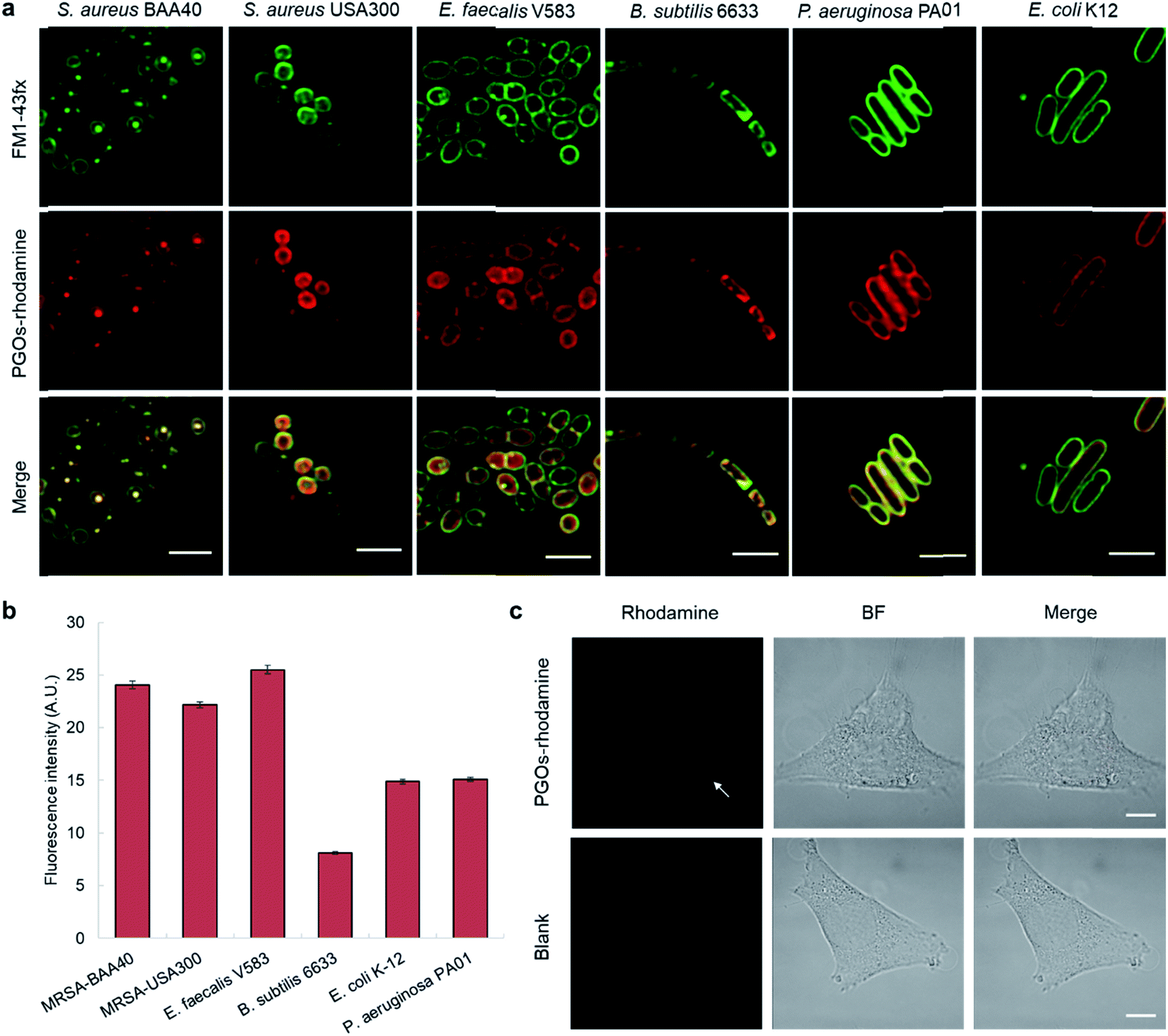 | ||
| Fig. 3 PGOs are successfully incorporated into both Gram-positive and -negative bacterial cell walls. (a) Fluorescence STED confocal studies of 6 different bacterial strains. Green color marks fluorescence from the membrane dye FM 1-43fx, red color marks fluorescence from the PGOs–rhodamine, and yellow color indicates colocalization of the two fluorophores in bacterial cells. Scale bar = 2 μm. (b) Relative fluorescence intensity on bacterial surfaces after incorporation of PGOs. A total of above one hundred cells from each strain were used for measurements (Tables S1–S6†), and the average PGOs–rhodamine fluorescence signal per cell was measured for comparison. (c), Confocal microscopy images of 3T3 cells incubated with (top row) and without (bottom row) PGOs–rhodamine. Incubation was done at 100 μg mL−1 substrate concentration for 1 hour, which was the same as the condition adopted for bacteria. No significant fluorescence signals were detected in treated cells, with trace amounts (white arrow) hypothetically from non-specific adsorption and endocytosis. Scale bar = 10 μm. | ||
After confirming the specificity of PGOs towards bacteria, we went on to investigate how the two interacted with each other. Quantification of fluorescence signals, following the method reported by Burgess et al., demonstrated that both Gram-negative and Gram-positive species showed significant uptake and labeling by our PGOs (Fig. 3b).12 The broad spectrum of efficient uptake and incorporation exhibited by PGOs makes them potentially useful substrates for further design of a new class of bacterial bioimaging reagents.
To confirm and verify which component of the PGOs led to robust uptake and incorporation, we used the rhodamine fluorophore to label the entire PGO, the peptide conjugated-chitosan, and the pentapeptide. The rhodamine-labeled substrates, and rhodamine alone, were incubated together with E. faecalis, and incorporation was measured by quantifying bacterial cell surface fluorescence intensity. The total intensity was normalized by the area of fluorescence.
Of all the substrates tested, only bacteria incubated with the rhodamine-labeled PGOs showed high fluorescence intensities (Fig. 4). These results are consistent with a previous crystal structure report which suggested that every moiety, including the oligosaccharide chain and the pentapeptide of PGOs, is required for effective binding to the active site of PGT enzymes.13 Thus our results proved that the accumulation of PGO–rhodamine was due to the biological incorporation of PGOs into the bacterial cell surface, and not due to trivial accumulation of any of the components or breakdown products of PGOs–rhodamine.
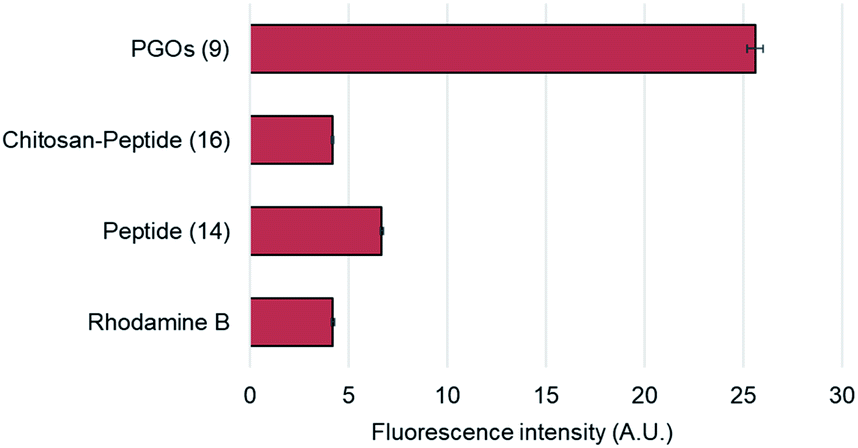 | ||
| Fig. 4 Cell surface relative fluorescence intensity of E. faecalis after incubation with different substrates. A total of above one hundred cells from each strain were used for measurements (refer to ESI†), and the average fluorescence signal per cell was measured for comparison. | ||
To characterize the modified bacterial cell wall with PGOs incorporated, we took images of higher resolution with STED confocal microscopy. Cells of S. aureus and E. faecalis showed thick cell walls as reflected by signals of PGOs–rhodamine (Fig. 5a). Upon closer observation, we noticed that PGOs–rhodamine resided on the inner part of the cell surface compared to FM 1-43fx, which was adsorbed directly onto the bacterial cell surface. This incomplete colocalization of FM1-43fx and PGO–rhodamine further ruled out the possibility that the PGOs–rhodamine had been only physically adsorbed onto the exterior of bacterial cells, and indicated that it was specifically transported into the bacterial cell surface layer. This was also supported by cryo-electron microscopic studies of the PGOs–rhodamine labeled bacterial cell wall, which showed a slightly thicker layer indicative of the incorporation of rhodamine into the PG network (Fig. S18†).
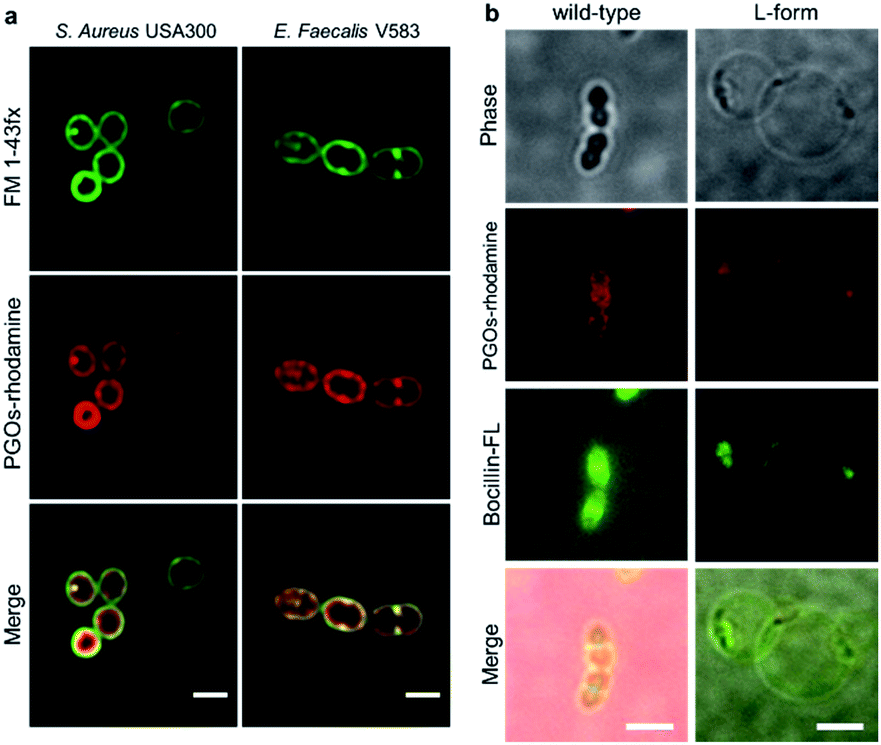 | ||
| Fig. 5 Further proof of PGO incorporation into bacterial cell walls. (a) Magnified STED confocal microscopy images of S. aureus and E. faecalis, scale bar = 1 μm. (b) Localization of PGOs–rhodamine and Boc-FL in wild-type and L-form E. faecalis OG1RF imaged with TIRF microscopy. Scale bar = 1 μm for wild-type and 10 μm for L-form respectively. PGOs–rhodamine (red) and Boc-FL (green) are colocalized in the septa of wild-type cells, and the punctae of L-form cells. | ||
We also used the L-form of the Gram-positive Enterococcus bacteria to further study the biological role of PGO incorporation into bacterial cell walls. L-form bacteria are deficient in cell wall components/fragments on their surface.14 Here we used BOCILLIN FL (Boc-FL), a fluorescent penicillin derivative, to label PBPs and cell walls specifically. Incubation of PGOs–rhodamine (red) and Boc-FL (green) with the wild type cells resulted in an intense fluorescence due to PGO and penicillin respectively in the septum region and also at the circumferential cell surface (Fig. 5b, left row), whereas L-form cell surfaces showed only a few distinct fluorescent punctae that are overlapping from both PGOs–rhodamine and Boc-FL (Fig. 5b, right row). The images always showed colocalization of PGOs and penicillin derivatives, whether it is the division septa of the wild type strain or the distinctive punctae in the L-form strain's cell surfaces. We also attempted to compare the localization of PGOs to the membrane dye polymyxin B-BODIPY FL. Strong uniform membranous staining by the polymyxin B derivative was observed in the L-form cells, including the region with a dense membrane material as indicated (Fig. S16a-bottom row†). In comparison, the PGOs localized in discrete punctae on the L-form cell surfaces. Hence, we can conclude that PGOs' localization patterns within bacterial cell walls closely resemble those of native PG substrates.
Furthermore, we used isothermal titration calorimetry (ITC) to check the binding interactions between PGOs and PGT proteins (Fig. S26†). We used E. coli K12 PBP1a, since we have shown that E. coli K12 avidly incorporates PGOs, and E. coli K12 PBP1a is known to recognize large PG substrates.6b,9 Interestingly, the interaction between PGOs and PBP1a had an exceptionally high exothermic enthalpy (ΔH) of −33 kcal mol−1 which defied a perfect fit into binding models. As an earlier study by Wong and Cheng showed a similar trend in the raw heat graph (without curve fitting and calculation) of lipid II's interaction with PGT, we deemed this result plausible, probably due to the extensive hydrogen bonding interaction present in multivalent binding of the oligomers.7d We found that the binding energy for PGOs and E. coli K12 PBP1a had ΔG = −9.8 ± 0.2 kcal mol−1 and Kd of 83 nM, which are higher than literature reports, being −4 to −6 kcal mol−1 and around 10 μM respectively.
With all the mechanistic studies conducted, the mode of action for PGO incorporation was proposed to be that PGOs and their metabolites intercept the PG biosynthesis pathway (Fig. 1) and thread into parts of the PG strands after binding with PBPs. The efficient metabolic labelling of the bacterial cell wall has been pioneered by Kahne and VanNieuwenhze using derivatives of D-amino acids.5a,15 As small molecules, functionalized D-amino acids could be readily synthesized on a practical scale. However, the small size of the D-amino acid molecule implies that any modification (e.g. with a fluorescent dye) would significantly alter the recognition and incorporation of D-amino acid by bacteria. Hence, the metabolic labeling process might need relatively high concentrations to enable cellular uptake, as it was noted that the minimal concentration to reach an acceptable signal to noise ratio of 3 was around 0.5 mM of D-amino acid with at least 3 washes.16 Furthermore, with a small binding pocket for the interaction between TPs and D-amino acids, bacteria could readily elude such labeling via mutation, as demonstrated in the case of MRSA, which acquired resistance from a small change in the TPs' binding pocket.17 In contrast, the smallest substrate of PGTs known is lipid II, and there has been no report on the compound's derivative being successfully used for metabolic labeling of bacteria, possibly due to the high synthetic challenges arising from conventional bottom-up assembly. The synthetic PGOs presented in this work may offer an alternative strategy to label bacteria with excellent specificity and efficiency.
Diagnostic potential of PGOs
We have further explored the potential of the synthetic PGOs for application as a diagnostic tool. Such a tool has to be sensitive to low bacterial concentration and to generate results fast enough to be sufficiently practical. Hence, varying bacterial concentrations have been incubated with PGOs–rhodamine for 1 hour, and then subjected to quantification with a fluorospectrometer (Fig. 6a).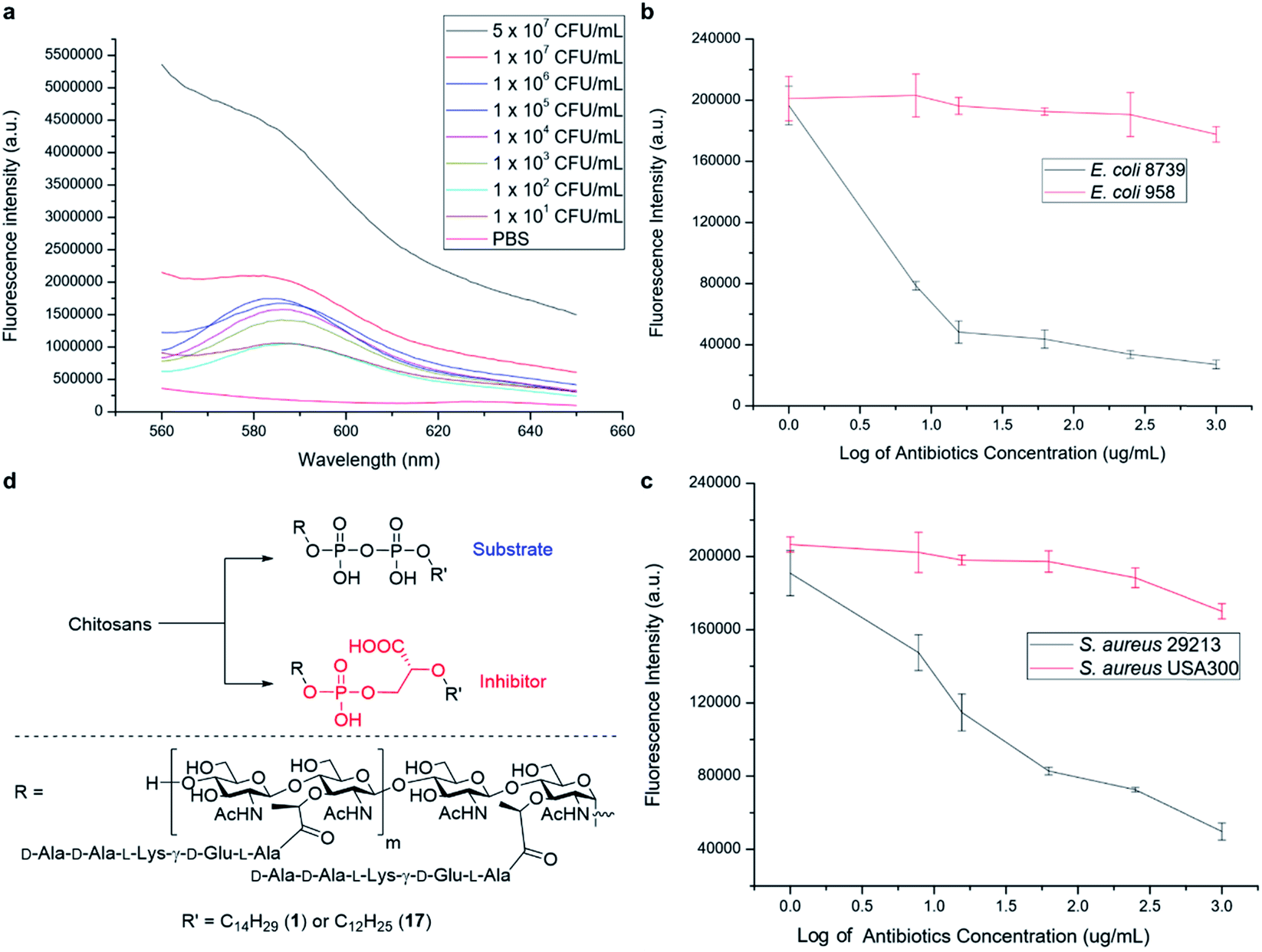 | ||
| Fig. 6 Capability of PGOs as a bacteria detection or killing tool. (a) Fluorescence intensity of the E. coli EC958 suspension labelled with PGOs–rhodamine; all suspensions (including PBS control) were incubated with PGOs–rhodamine at 200 μg mL−1 for 1 hour and washed with PBS before measurement of fluorescence with a fluorospectrometer. (b and c) Fluorescence intensity comparison of resistant and susceptible E. coli and S. aureus respectively at 106 CFU mL−1. The samples were incubated with 0–1000 μg mL−1 penicillin G sodium salt for 2 hours, washed, and then incubated with 50 μg mL−1 PGOs–rhodamine for 1 hour consecutively. Eventually, the bacterial samples were washed and resuspended in 1 mL PBS for analysis with a fluorospectrometer. (d) Preparation of an antibacterial agent with minimal modifications to the PGO synthetic route. The key difference is highlighted as pyrophosphate being converted into a phosphoglycerate. | ||
Significant fluorescence was observed in bacteria with concentration even as low as 101 CFU mL−1 when compared to a PBS control, and the intensity aligned well with bacterial concentration.
Knowing that detection of bacteria with PGOs–rhodamine had a low limit, we then moved on to address a concern closely pertaining to real life circumstances involving antibiotic resistant strains. Infections caused by such bacteria required far more careful treatment due to their complexity, and high lethality in turn. Therefore, enormous investment is being dedicated to the development of methods to timely diagnose bacterial infections involving antibiotic resistance. The broad spectrum of bacteria recognizing PGOs as substrates was demonstrated in earlier experiments, and thus we conceived it was possible to identify the presence of resistant strains in a rapid manner using PGOs. As the effective uptake of the synthetic PG substrate relied heavily on cell growth and metabolism, bacterial samples were treated with antibiotics first to inhibit the growth of susceptible cells. Subsequently, PGOs–rhodamine was added for fluorescence analysis. Notably, a significant and consistent difference in fluorescence intensity was observed between resistant and susceptible strains for the two bacterial species tested, namely E. coli and S. aureus (Fig. 6b and c). The selective labelling of resistant bacterial strains, E. coli 958 and S. aureus USA300, in combination with PGOs' excellent selectivity for bacterial cells in contrast to mammalian cells, would allow us to identify the presence of antibiotic resistant bacteria from samples of infection in a facile and practical manner, without the need of culturing potentially hazardous samples of infection.
In addition, we briefly explored the therapeutic potential of PGOs with minimal modifications. A few antibiotics have been marketed as PGT inhibitors, with the most well studied being the moenomycin family. The compounds have excellent activity and there is no report of induced resistance, yet they are not optimized for human treatment due to their poor pharmacokinetic properties.18 Intrigued by an earlier study which demonstrated antibacterial activities using moenomycin analogues on lipid I/II scaffolds,19 we sought to test the potential of PGOs through the same modification, which was the conversion of the pyrophosphate aglycone to a phosphoglycerate (Fig. 6d). The new product had promising MIC values compared with literature reports, having a value of 32 μg mL−1 against both antibiotic susceptible S. aureus ATCC29213 and a resistant strain MRSA USA300. In contrast, the cytotoxicity against mammalian cells was found to be very low, with an IC50 value of 2048 μg mL−1. This contrast illustrates the potential of PGOs as a platform to build on for the development of novel antibacterial agents.
Based on the exploration of PGOs' diagnostic and therapeutic potential in vitro, we advanced to in vivo studies to test their capability of detecting bacteria. Mice were infected with bacteria via the intraperitoneal injection of bacteria, followed by intravenous injection of PGOs tagged by an NIR dye Cy7.5. As shown in Fig. 7a, PGOs–Cy7.5 was cleared faster in non-infected mice (left) compared to infected ones (right). The liver and kidney are two major organs infected in the sepsis model, and we can clearly see that the clearance of PGOs was retarded due to bacterial infection, as demonstrated by the strong fluorescence in the liver area for infected mice in comparison with insignificant fluorescence for non-infected ones (Fig. 7a). To confirm the correlation between the observed fluorescence intensity and bacterial CFU count, we dissected the mice 8 hours after injection of PGO–Cy7.5. Stronger fluorescence, along with a higher bacterial count was found in the kidney, liver and muscle (leg) of infected mice compared to non-infected controls, as shown in Fig. 7b and c. The results of comparison well demonstrated the in vivo bacterial targeting effect, and in turn the applicability, as a diagnostic tool, of our PGOs with minor modification with Cy7.5 dye.
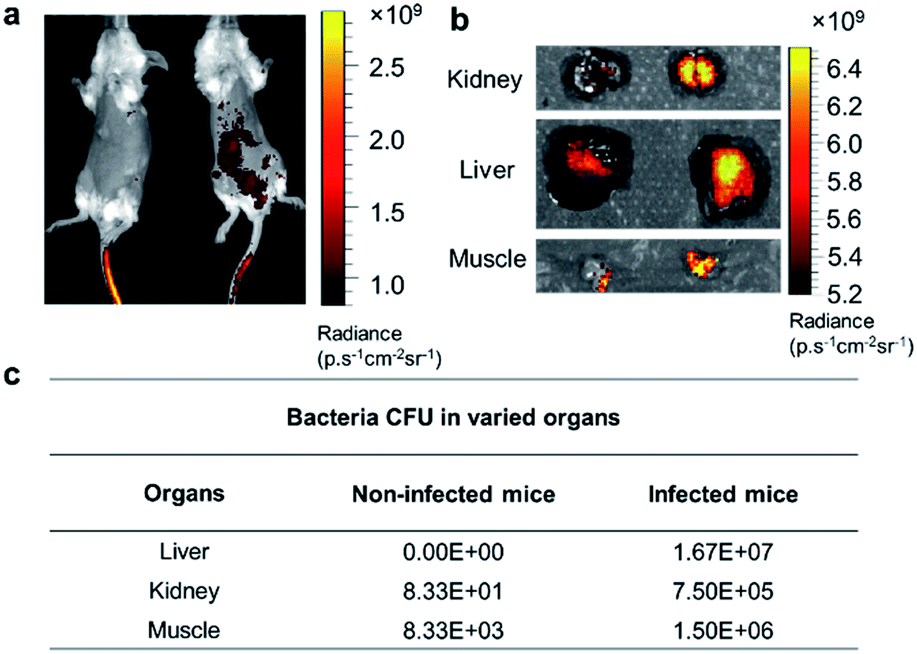 | ||
| Fig. 7 Imaging of PGOs–Cy7.5 in vivo. (a) Representative images of non-infected mice (left) and infected mice (right), 8 hours after receiving intravenous injection of PGOs–Cy7.5. (b and c) Fluorescence and CFU count respectively from the excised kidney, liver and muscle (leg) of non-infected mice (left column) and infected mice (right). | ||
Real time in vivo imaging provides a practical tool to gather information about a lesion in an accurate and timely manner, so doctors would be able to advise on treatments more efficiently, yet it is more developed for anticancer studies.20 More constraints, however, are seen when the target of interest is bacterial infection. Antibiotics have been adopted for targeting bacteria through specific binding,5c although they are always haunted by the concern of AMR, which could be both the cause of a false negative result and the outcome of using such agents per se. On the other hand, monoclonal antibodies or antimicrobial peptides have also been commonly used for bacteria targeting, but they suffer from high cost or a short duration of contact before metabolized or cleared. More recently, metabolic labelling has been realized in vivo, but it required high concentration and topical application, presumably due to a similarly short duration to take effect within the body.21 In contrast, PGOs have demonstrated high sensitivity and selectivity to bacterial infection, while demanding a low dosage and short time to take effect.
Conclusion
In summary, we report the top-down facile transformation of inexpensive chitosan into biohybrid PGOs that can be successfully incorporated into the cell walls of different bacterial strains. The polymeric substrate could be suitably modified for bioimaging applications as demonstrated above, with a hypothesized metabolic interaction pathway. Our bio-synthetic hybrid PGOs could constitute a versatile platform to facilitate further mechanistic studies of the biosynthesis process of bacterial cell walls, or to be developed, after minor modifications, into diagnostic tools. Further enzymatic assays are ongoing for in-depth investigation of the interaction mechanisms.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was performed in strict accordance with the guidelines for the care and use of laboratory animals from Nanyang Technological University (NTU) and was approved by the Institutional Animal Care and Use Committee (IACUC) of NTU (Singapore). Financial support from the Ministry of Education, Singapore (MOE 2013-T3-1-002) and Nanyang Technological University (RG120/18) is gratefully acknowledged. The authors acknowledge the facilities, and the scientific and technical assistance of the Advanced Bioimaging Core at The Academia, Singapore Health Services. Dr Jian Shi at Centre for BioImaging Sciences, National University of Singapore is acknowledged for cryo-TEM analysis. L-form enterococcus was received as a gift from Prof. Kimberly Kline's laboratory in Singapore Centre for Environmental Life Sciences Engineering, where the protocol was optimized.Notes and references
- (a) R. Hakenbeck, T. Grebe, D. Zähner and J. B. Stock, Mol. Microbiol., 1999, 33, 673–678 CrossRef CAS PubMed; (b) I. G. Boneca and G. Chiosis, Expert Opin. Ther. Targets, 2003, 7, 311–328 CrossRef CAS PubMed; (c) R. Hakenbeck, R. Brückner, D. Denapaite and P. Maurer, Future Microbiol., 2012, 7, 395–410 CrossRef CAS PubMed.
- (a) D. J. Waxman and J. L. Strominger, Annu. Rev. Biochem., 1983, 52, 825–869 CrossRef CAS PubMed; (b) J. V. Holtje, Microbiol. Mol. Biol. Rev., 1998, 62, 181–203 CrossRef CAS PubMed; (c) K. K. Wong and D. L. Pompliano, Adv. Exp. Med. Biol., 1998, 456, 197–217 CrossRef CAS PubMed; (d) A. Typas, M. Banzhaf, C. A. Gross and W. Vollmer, Nat. Rev. Microbiol., 2012, 10, 123–136 CrossRef CAS PubMed.
- (a) T. Schneider, T. Kruse, R. Wimmer, I. Wiedemann, V. Sass, U. Pag, A. Jansen, A. K. Nielsen, P. H. Mygind, D. S. Raventós, S. Neve, B. Ravn, A. M. J. J. Bonvin, L. De Maria, A. S. Andersen, L. K. Gammelgaard, H.-G. Sahl and H.-H. Kristensen, Science, 2010, 328, 1168–1172 CrossRef CAS PubMed; (b) L. L. Ling, T. Schneider, A. J. Peoples, A. L. Spoering, I. Engels, B. P. Conlon, A. Mueller, T. F. Schaberle, D. E. Hughes, S. Epstein, M. Jones, L. Lazarides, V. A. Steadman, D. R. Cohen, C. R. Felix, K. A. Fetterman, W. P. Millett, A. G. Nitti, A. M. Zullo, C. Chen and K. Lewis, Nature, 2015, 517, 455–459 CrossRef CAS PubMed.
- W. Vollmer, D. Blanot and M. A. de Pedro, FEMS Microbiol. Rev., 2008, 32, 149–167 CrossRef CAS PubMed.
- (a) T. J. Lupoli, H. Tsukamoto, E. H. Doud, T.-S. A. Wang, S. Walker and D. Kahne, J. Am. Chem. Soc., 2011, 133, 10748–10751 CrossRef CAS PubMed; (b) E. Kuru, H. V. Hughes, P. J. Brown, E. Hall, S. Tekkam, F. Cava, M. A. de Pedro, Y. V. Brun and M. S. VanNieuwenhze, Angew. Chem., Int. Ed., 2012, 51, 12519–12523 CrossRef CAS PubMed; (c) M. van Oosten, T. Schäfer, J. A. C. Gazendam, K. Ohlsen, E. Tsompanidou, M. C. de Goffau, H. J. M. Harmsen, L. M. A. Crane, E. Lim, K. P. Francis, L. Cheung, M. Olive, V. Ntziachristos, J. M. van Dijl and G. M. van Dam, Nat. Commun., 2013, 4, 2584 CrossRef PubMed; (d) S. E. Pidgeon, J. M. Fura, W. Leon, M. Birabaharan, D. Vezenov and M. M. Pires, Angew. Chem., Int. Ed., 2015, 54, 6158–6162 CrossRef CAS PubMed.
- (a) M. Lee, D. Hesek and S. Mobashery, in Chemical Glycobiology, American Chemical Society, 2008, vol. 990, pp. 54–78, ch. 3 Search PubMed; (b) H.-W. Shih, K.-T. Chen, T.-J. R. Cheng, C.-H. Wong and W.-C. Cheng, Org. Lett., 2011, 13, 4600–4603 CrossRef CAS PubMed; (c) L.-Y. Huang, S.-H. Huang, Y.-C. Chang, W.-C. Cheng, T.-J. R. Cheng and C.-H. Wong, Angew. Chem., Int. Ed., 2014, 53, 8060–8065 CrossRef CAS PubMed; (d) Y. Qiao, V. Srisuknimit, F. Rubino, K. Schaefer, N. Ruiz, S. Walker and D. Kahne, Nat. Chem. Biol., 2017, 13, 793 CrossRef CAS PubMed; (e) N. Wang, H. Hasegawa, C.-y. Huang, K. Fukase and Y. Fujimoto, Chem.–Asian J., 2017, 12, 27–30 CrossRef CAS PubMed.
- (a) X.-Y. Ye, M.-C. Lo, L. Brunner, D. Walker, D. Kahne and S. Walker, J. Am. Chem. Soc., 2001, 123, 3155–3156 CrossRef CAS PubMed; (b) H. Liu, T. K. Ritter, R. Sadamoto, P. S. Sears, M. Wu and C.-H. Wong, ChemBioChem, 2003, 4, 603–609 CrossRef CAS PubMed; (c) S. Cho, Q. Wang, C. P. Swaminathan, D. Hesek, M. Lee, G.-J. Boons, S. Mobashery and R. A. Mariuzza, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8761–8766 CrossRef CAS PubMed; (d) H.-W. Shih, Y.-F. Chang, W.-J. Li, F.-C. Meng, C.-Y. Huang, C. Ma, T.-J. R. Cheng, C.-H. Wong and W.-C. Cheng, Angew. Chem., Int. Ed., 2012, 51, 10123–10126 CrossRef CAS PubMed.
- M. Lee, D. Hesek, D. A. Dik, J. Fishovitz, E. Lastochkin, B. Boggess, J. F. Fisher and S. Mobashery, Angew. Chem., Int. Ed., 2017, 56, 2735–2739 CrossRef CAS PubMed.
- (a) Y. Zhang, E. J. Fechter, T.-S. A. Wang, D. Barrett, S. Walker and D. E. Kahne, J. Am. Chem. Soc., 2007, 129, 3080–3081 CrossRef CAS PubMed; (b) T.-S. A. Wang, T. J. Lupoli, Y. Sumida, H. Tsukamoto, Y. Wu, Y. Rebets, D. E. Kahne and S. Walker, J. Am. Chem. Soc., 2011, 133, 8528–8530 CrossRef CAS PubMed.
- G. T. Hermanson, in Bioconjugate Techniques, Academic Press, Boston, 3rd edn, 2013, pp. 395–463, DOI:10.1016/B978-0-12-382239-0.00010-8.
- R. Sadamoto, K. Niikura, P. S. Sears, H. Liu, C.-H. Wong, A. Suksomcheep, F. Tomita, K. Monde and S.-I. Nishimura, J. Am. Chem. Soc., 2002, 124, 9018–9019 CrossRef CAS PubMed.
- (a) A. Burgess, S. Vigneron, E. Brioudes, J.-C. Labbé, T. Lorca and A. Castro, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 12564–12569 CrossRef CAS PubMed; (b) R. A. McCloy, S. Rogers, C. E. Caldon, T. Lorca, A. Castro and A. Burgess, Cell Cycle, 2014, 13, 1400–1412 CrossRef CAS PubMed.
- C.-Y. Huang, H.-W. Shih, L.-Y. Lin, Y.-W. Tien, T.-J. R. Cheng, W.-C. Cheng, C.-H. Wong and C. Ma, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 6496–6501 CrossRef CAS PubMed.
- S. Chang and S. N. Cohen, Mol. Gen. Genet., 1979, 168, 111–115 CrossRef CAS PubMed.
- E. Kuru, H. V. Hughes, P. J. Brown, E. Hall, S. Tekkam, F. Cava, M. A. de Pedro, Y. V. Brun and M. S. VanNieuwenhze, Angew. Chem., Int. Ed., 2012, 51, 12519–12523 CrossRef CAS PubMed.
- E. Kuru, S. Tekkam, E. Hall, Y. V. Brun and M. S. Van Nieuwenhze, Nat. Protoc., 2014, 10, 33 CrossRef PubMed.
- (a) K. T. Bæk, A. Gründling, R. G. Mogensen, L. Thøgersen, A. Petersen, W. Paulander and D. Frees, Antimicrob. Agents Chemother., 2014, 58, 4593–4603 CrossRef PubMed; (b) R. J. Fair and Y. Tor, Perspect. Med. Chem., 2014, 6, 25–64 Search PubMed.
- B. Ostash and S. Walker, Nat. Prod. Rep., 2010, 27, 1594–1617 RSC.
- S. Dumbre, A. Derouaux, E. Lescrinier, A. Piette, B. Joris, M. Terrak and P. Herdewijn, J. Am. Chem. Soc., 2012, 134, 9343–9351 CrossRef CAS PubMed.
- H. Wang, R. Wang, K. Cai, H. He, Y. Liu, J. Yen, Z. Wang, M. Xu, Y. Sun, X. Zhou, Q. Yin, L. Tang, I. T. Dobrucki, L. W. Dobrucki, E. J. Chaney, S. A. Boppart, T. M. Fan, S. Lezmi, X. Chen, L. Yin and J. Cheng, Nat. Chem. Biol., 2017, 13, 415 CrossRef CAS PubMed.
- (a) D. Mao, F. Hu, Kenry, S. Ji, W. Wu, D. Ding, D. Kong and B. Liu, Adv. Mater., 2018, 30, 1706831 CrossRef PubMed; (b) W. Wang, L. Lin, Y. Du, Y. Song, X. Peng, X. Chen and C. J. Yang, Nat. Commun., 2019, 10, 1317 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc06345e |
| This journal is © The Royal Society of Chemistry 2020 |