
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of ABn-type colloidal molecules by polymerization-induced particle-assembly (PIPA)†‡
Dan
Li§
a,
Xi
Chen§
a,
Min
Zeng
a,
Jinzhao
Ji
a,
Yun
Wang
a,
Zhenzhong
Yang
 *b and
Jinying
Yuan
*a
*b and
Jinying
Yuan
*a
aKey Laboratory of Organic Optoelectronics and Molecular Engineering, Department of Chemistry, Tsinghua University, Beijing 100084, China. E-mail: yuanjy@mail.tsinghua.edu.cn
bDepartment of Chemical Engineering, Tsinghua University, Beijing 100084, China. E-mail: yangzhenzhong@tsinghua.edu.cn
First published on 28th January 2020
Abstract
Conventional synthesis of colloidal molecules (CMs) mainly depends on particle-based self-assembly of patchy building blocks. However, direct access to CMs by the self-assembly of isotropic colloidal subunits remains challenging. Here, we report the mass production of ABn-type CMs by polymerization-induced particle-assembly (PIPA), using a linear ABC triblock terpolymer system. Starting from diblock copolymer spheres, the association of spheres takes place in situ during the polymerization of the third block. The third blocks aggregate into attractive domains, which connect spheres into CMs. The stability of CMs is ensured, as long as the conversions are limited to ca. 50%, and the pH is low. The valence of ABn-type CMs (n = 2–6) is determined by the volume ratio of the polymer blocks. By tuning the volume ratio, 78.5% linear AB2-type CMs are yielded. We demonstrate that polymerization-induced particle-assembly is successful for the scalable fabrication of ABn-type CMs (50 g L−1), and can be easily extended to vastly different triblock terpolymers, for a wide range of applications.
Introduction
Colloidal molecules (CMs) are well-defined colloidal clusters with precise symmetry of molecular structures and have drawn extensive interest because of their unique properties1,2 and potential as building blocks for low-coordination open structures.3,4 With compartmental functional domains and directional interaction, CMs are promising in biological and medical applications,5,6 self-assembly,7,8 physical chemistry,9–11 and materials science and technology.12–14 Through particle-based self-assembly, CMs with molecule-like symmetries are enabled using monodisperse patchy building blocks with a suitable shape or interaction anisotropy. There are continuing efforts to develop methods toward patchy building blocks,15,16 including colloidal crystal templating,17 seeded emulsion polymerization,18 assembly of oppositely charged particles,19–21 DNA origami,22–24 self-assembly of block copolymers,25–28 and emulsion solvent-evaporation methods.29–31 However, these methods struggle in fabricating controllable patches with desired size (10–100 nm). In this size regime, the synthesis of monodisperse building block is difficult, and irregularity causes defects which amplify throughout the hierarchical self-assembly. Therefore, efficient access to CMs remains challenging.Over the past few years, self-assembly of block copolymers (BCPs) has emerged as an elegant bottom-up technique to produce soft nanoparticles with controllable structures, sizes, and functions.32–34 The polymerization-induced self-assembly (PISA) technique has experienced great success in the mass production of self-assembled BCPs, where the synthesis and self-assembly of BCPs are achieved simultaneously.35–41 Inspired by PISA, we envision that the synthesis of well-defined CMs might also be realized by chemical synthesis and self-assembly in one-step, without the fabrication and purification of patchy colloidal building blocks. Using isotropic diblock copolymer spheres as seeds, the polymerization of third blocks produces attractive patches on spheres, which combine colloids into anisotropic CMs in situ. Therefore, CMs are achieved directly via seeded polymerization, without the preparation and purification of patchy intermediates. We address this strategy as polymerization-induced particle-assembly (PIPA).
Herein, we demonstrate a direct approach for the mass production of BCP CMs based on dispersion RAFT polymerization. We perform the polymerization using spherical seeds of poly(N,N-dimethylaminoethyl methacrylate)-b-poly(benzyl methacrylate) (PDMA-b-PBzMA). The polymerization of 2-hydroxypropyl methacrylate (HPMA) produces a linear PDMA-b-PBzMA-b-PHPMA triblock terpolymer. Thus, PHPMA domains function as attractive patches and connect colloid atoms (CAs) into ABn-type CMs in situ. The stability of CMs is ensured by limiting conversions to ca. 50%, and the pH is ∼ 2. Without post-purification, PDMA70-b-PBzMA96-b-PHPMA70 yields 78.5% AB2 CMs as prepared, in concentrated solution (50 g L−1). The polymer-based AB2-type CMs are intrinsically self-assembled, soft and programmable, offering the possibility to act as subunits for higher-level structures. We have realized the large-scale preparation of ABn-type CMs via polymerization-induced self-assembly of spherical colloids, which is an efficient strategy achieving CMs and other hierarchical nanoparticles (HNPs) by synthesis and self-assembly in one step. This strategy can be readily applied to vastly different block copolymer systems, which would largely enrich the functional complexity of CMs and broaden their applications.
Results and discussion
Scheme 1 shows the synthesis of AB2-type CMs by aqueous seeded RAFT polymerization. Spherical PDMA70-b-PBzMA96 seeds are produced by dispersion RAFT polymerization of BzMA in ethanol, using a PDMA macro-chain transfer agent. The PDMA70-b-PBzMA96 seeds are then transferred into water by dialysis, and a desired amount of aqueous HCl is added to adjust pH to ∼ 2. The protonation of the PDMA stabilizer provided electrostatic repulsion for the stabilization of CMs.42,43 The aqueous seeded polymerization of HPMA generated a PDMA70-b-PBzMA96-b-PHPMA70 triblock terpolymer, which self-assembles hierarchically into AB2-type CMs.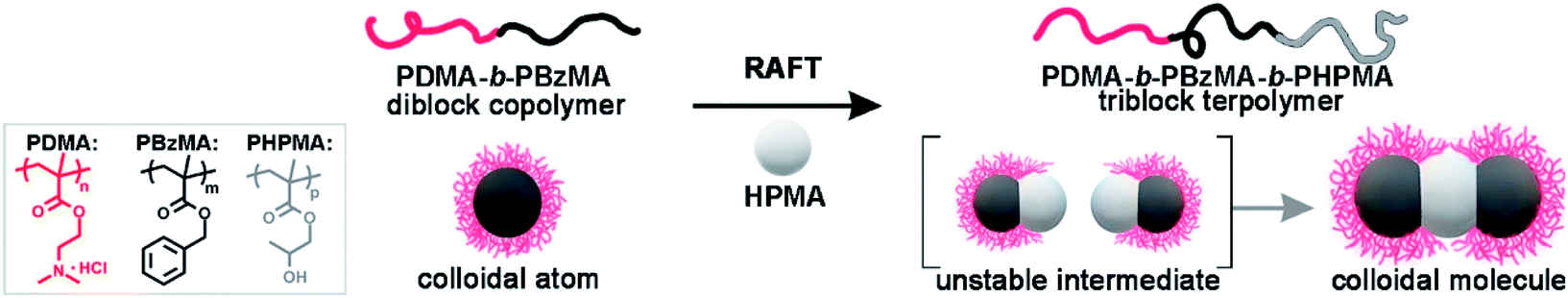 | ||
| Scheme 1 The synthesis of AB2-type CMs through polymerization-induced particle-assembly (PIPA). | ||
Fig. 1a shows the PDMA70-b-PBzMA96 spheres with a core diameter of 31 nm. The polymerization of HPMA produces a linear PDMA70-b-PBzMA96-b-PHPMA70 triblock terpolymer (Đ = 1.59), as studied by 1H NMR and SEC (Fig. S3 and S4‡). A relatively high dispersity is due to the relatively low blocking efficiency caused by the embedding of CTA in PBzMA cores44 and the crosslinking of PHPMA.45 The growth of PHPMA blocks leads to the shift of particle size from 31 nm to 52 nm, indicating the formation of nanoparticle clusters (Fig. 1b). The cryo-TEM image (Fig. 1e) shows well-defined three-segment clusters (aka AB2-CMs), resulting from seeded polymerization. We can find two dark grey CAs on each end as peripheral CAs, with the same size of PDMA70-b-PBzMA96 CAs (31 nm). The central CAs are much brighter in observation, suggesting a different chemical composition of middle CAs from cap CAs.
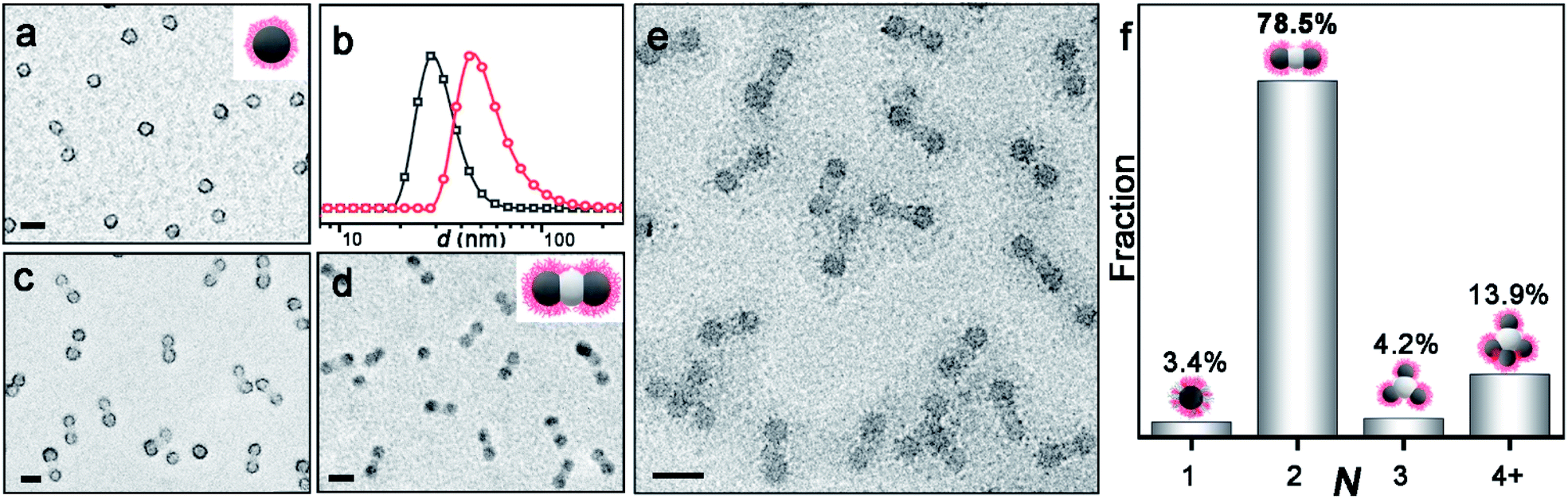 | ||
| Fig. 1 (a) TEM image of PDMA70-b-PBzMA96 CAs after PTA-staining of the PDMA corona with the inset schematic structure. (b) DLS profiles of PDMA70-b-PBzMA96 CAs (black) and PDMA70-b-PBzMA96-b-PHPMA70 CMs (red). (c and d) TEM image of PDMA70-b-PBzMA96-b-PHPMA70 CMs after PTA-staining and RuO4-staining, respectively; inset shows the schematic structure of AB2-type CM. (e) Cryo-TEM image of CMs. (f) Distribution of ABn-type CMs with varied valence numbers (N) obtained by statistically analysing 735 random particles by cryo-TEM. The valence number (N): 1, 2, 3, and 4+ correspond to AB1, AB2, AB3, and ABn (n ≥ 4), respectively. All the scale bars are 50 nm. | ||
The composition of AB2 CMs is further confirmed by TEM characterization of samples stained with RuO4 and phosphotungstic acid (PTA), respectively. Fig. 1c shows that PDMA is located at the surface of peripheral CAs (PTA staining: PDMA black, PBzMA grey, and PHPMA not visible). And Fig. 1d reveals that satellite CAs are comprised of PBzMA (RuO4 staining: PBzMA black, and PDMA and PHPMA not observable). Thus, we confirmed that central CAs were composed of newly formed PHPMA blocks. As described above, we have achieved PDMA70-b-PBzMA96-b-PHPMA70 AB2 CMs in situ during the polymerization, and the bonding angle of B-A-B is essentially 180° (Fig. S5‡), in analogy to the linear molecule of CO2.
Notably, PDMA70-b-PBzMA96-b-PHPMA70 forms the major product of AB2-type CMs (78.5%), with relative distribution shown in Fig. 1f. The sample was collected and analyzed as prepared, without multi-step purification. And the aqueous dispersion is more concentrated (50 g L−1) than previously reported selective solvents (5 g L−1).25,46 The above experimental results demonstrate that this approach is robust and reliable for the mass preparation of AB2-type CMs, which is especially beneficial for scalable applications.
The polymerization conditions for CM synthesis were optimized after kinetic study. Targeting PDMA70-b-PBzMA96-b-PHPMA200 with a 10 wt% solid content, the polymerization reached 71.1% monomer conversion at 1.5 h (Fig. S7‡). And at 78.3% conversion (2 h), a mixture of AB4 and large clusters was obtained. When the polymerization reached 90.7% conversion (4 h), large clusters were obtained possibly due to inter-cluster association and crosslinking between large PHPMA patches. To avoid clustering, we synthesize CM samples with the control of monomer conversion at ca. 50%.
A series of PDMA-b-PBzMA-b-PHPMA triblock terpolymer samples showed the morphology evolution of the ABn-type CMs. PDMA70-b-PBzMA96-b-PHPMA20 remains as symmetric spheres (Fig. 2a). This is understandable because PHPMA is too short to form visible domains by phase separation. For PDMA70-b-PBzMA96-b-PHPMA46 with a longer PHPMA chain, 37.8% AB2 CMs coexist with 58.3% spherical colloids (Fig. 2b). This is because, as the PHPMA domain exceed a critical size during continuing polymerization, the PDMA chains that stabilize the colloids are no longer able to cover them. As a result, the colloids become unstable due to largely increased surface energy (as will be discussed in detail later) and tend to bind together and form CMs to minimize the surface. Only a small number of Janus intermediates are captured in samples with a larger molecular weight (Fig. S6‡). Then AB2 dominates PDMA70-b-PBzMA96-b-PHPMA70 with a purity of 78.5% (Fig. 2c). Further growth of PHPMA leads to ABn-type CMs with n larger than two. PDMA70-b-PBzMA96-b-PHPMA109 generates a mixture of 50.2% AB2, 15.7% AB3, 22.9% AB4 (Fig. 2d). The hydrodynamic diameter of clusters grows significantly with the growth of PHPMA (Fig. 2e).
 | ||
Fig. 2 (a–d) TEM images of PDMA70-b-PBzMA96-b-PHPMAz with varied z (20, 46, 70, and 109) after PTA-staining (PDMA black, PBzMA grey, and PHPMA invisible). Scale bars are 50 nm. (e) DLS profiles of the colloid samples. PDMA70-b-PBzMA96-b-PHPMAz is abbreviated to D70B96Hz. (f) Distribution of CMs with varied valence numbers (N) determined by statistically analyzing the TEM images with the inset VH/VB ratios. (g) Number average valence (Nn) of PDMA70-b-PBzMA96-b-PHPMAz CMs against VH/VB. Error bars represent standard deviation. 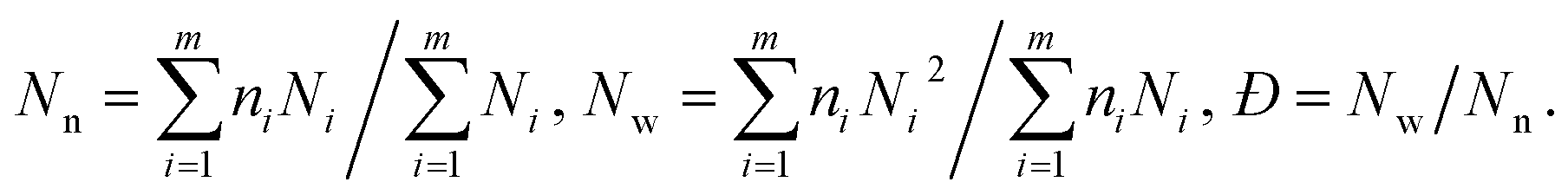 | ||
The increasing DP of PHPMA leads to the increase of the PHPMA/PBzMA volume ratio (VH/VB), which plays a key role in determining the size and valence of CMs. Fig. 2f shows the distribution of valence shifts towards a higher value, as VH/VB increases. As VH/VB increases from 0.34 to 1.87, the number average of valence (Nn) gradually increases from 1 to 2.73 (Fig. 2g). For PDMA70-b-PBzMA96-b-PHPMA70, the valence distribution is relatively narrow for a large-scale fabrication of CMs (Nn = 2.31, Đ = 1.11). Overall, we have generated a diversity of ABn-type CMs with mean valence increasing with VH/VB.
When VH/VB reached 5.48 (PDMA70-b-PBzMA96-b-PHPMA319), we have observed CMs of different valences (n = 3–6) with molecular precision (Fig. 3a), i.e. linear AB2, triangular AB3, tetrahedral AB4, trigonal bipyramidal AB5, square pyramidal AB5 and octahedral AB6, which are colloidal analogues of molecules like beryllium chloride (BeCl2), boron trifluoride (BF3), carbon tetrachloride (CCl4), antimonic chloride (SbCl5), iodine pentafluoride (IF5) and sulfur hexafluoride (SF6), respectively. In contrast, using larger CAs targeting VH/VB ≤ 1, linear colloidal polymers (PDMA70-b-PBzMA321-b-PHPMA169) are obtained, by the polymerization of “hamburger” intermediates (Fig. 3b).
 | ||
| Fig. 3 (a) Self-assembly of the PDMA70-b-PBzMA96-b-PHPMA319 triblock terpolymer toward CMs with VH/VB > 1; inset shows the cryo-TEM image of the unstable Janus intermediate. TEM images of varied PDMA-b-PBzMA-b-PHPMA CMs after PTA-staining; inset shows schematic structures and molecules. (b) Illustrative self-assembly of PDMA-b-PBzMA-b-PHPMA at VH/VB ≤ 1. Cryo-TEM (bottom, left) and TEM images (bottom, right) after PTA-staining of PDMA70-b-PBzMA321-b-PHPMA169 colloidal chains via the “Hamburger” (insert) structure. Scale bars are 50 nm. | ||
We therefore propose the formation mechanism of ABn-type CMs by polymerization-induced particle-assembly (PIPA): (1) PDMA-b-PBzMA forms spherical core–shell CAs with PBzMA blocks as rubbery cores (Tg = 56.8 °C),47 and PDMA chains as soluble stabilizers locating on the surface. (2) The polymerization produces PHPMA blocks which phase separate with PBzMA blocks, and aggregate into PHPMA micro-domains. (3) When the attractive PHPMA domains reach a critical size, they combine into PHPMA central CAs and unify PDMA-b-PBzMA spheres into colloidal clusters. (4) The structure of colloidal clusters is determined by VH/VB. When VH/VB > 1, the assembly of unstable monovalent Janus intermediates produces ABn-type CMs, with n regulated by VH/VB. When VH/VB ≤ 1, divalent hamburger-shape intermediates form colloidal chains.
This is in agreement with the principle of solvent-based fabrication of multicompartment micelles (MCMs), reported by Müller and coworkers.25,26 They have systematically studied the self-assembly of an ABC triblock terpolymer system (A as the attractive corona, B as the core, and C as the repulsive corona). The strong phase separation between A and B generates a patchy structure based on VA/VB. VA/VB > 1 yields monovalent Janus intermediates, with one attractive A patch and one repulsive C patch, which generate cluster-like MCMs. Oppositely, VA/VB ≤ 1 leads to divalent “hamburger” subunits with two attractive A patches and one repulsive C patch, which then assemble into chain-like MCMs. This concept has been validated by vastly different linear triblock terpolymer systems for the fabrication of both spherical MCMs and linear MCMs.
The formation of ABn-like CMs requires PHPMA domains protruding from PBzMA cores, not growing inside PBzMA cores. The interfacial energy is the critical parameter determining the CM formation during polymerization. Compared to the entropy cost of stretching chains, and the repulsive interactions among stabilizer chains, interfacial energies are dominant in the formation of ABn-type CMs. After neglecting free energies contributed by core-stretching and corona repulsion, the whole free energy change is written as
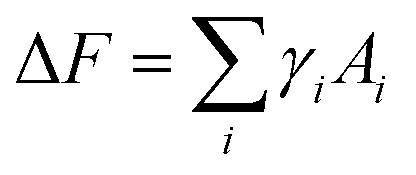
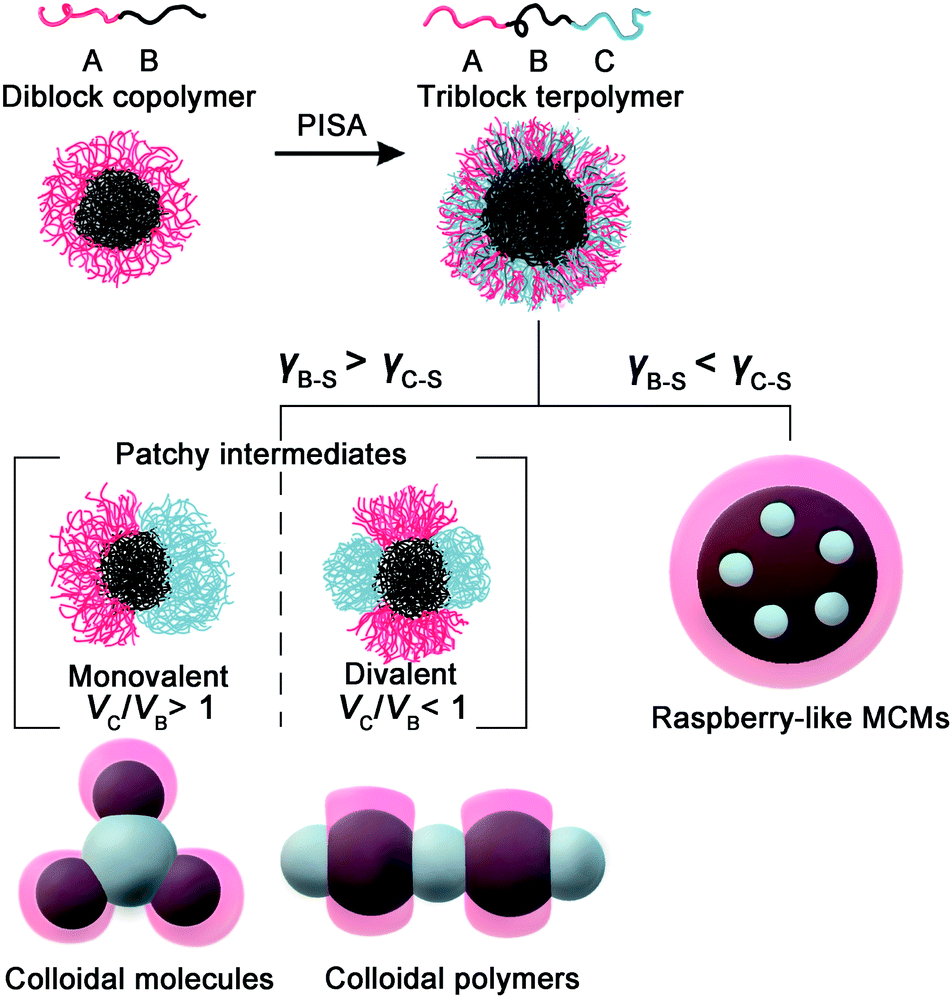 | ||
| Scheme 2 Illustrations of PIPA targeting colloidal molecules, colloidal polymers, and raspberry-like MCMs, respectively, by tuning the interfacial tensions (γB–S and γC–S) and the volume ratio (VC/VB). | ||
The PIPA strategy is an extension of PISA for the rational design and mass production of BCP nanoparticles with structural hierarchies. The design of the ABC triblock terpolymer requires (1) strong phase separation between B and C blocks and (2) monomer C suitable for PISA. And the morphology of HNPs is tuned by the interfacial tension and the VC/VB of the ABC triblock terpolymer. Based on these requirements, PIPA could be applied to vastly different triblock systems. For example, we also studied the aqueous seeded polymerization of diacetone acrylamide (DAAm), using PDMA70-b-PBzMA96 spheres as seeds. PDMA70-b-PBzMA96-b-PDAAm95 (Đ = 1.38) assembled into a mixture of AB2 and AB4 CMs (Fig. S21‡), with PDAAm as an attractive domain. As the DP of PDAAm increased, PDMA70-b-PBzMA96-b-PDAAm114 (Đ = 1.52) produced majorly AB4 CMs (Fig. S22‡). Our strategy could be readily applied to different triblock terpolymer systems for the fabrication of HNPs with different hierarchies and functionalities. Moreover, the discussed considerations of directing the process may well serve as an inspiration for other hierarchical self-assembly systems.
Conclusions
In summary, we have developed a straightforward way to fabricate BCP ABn-type CMs by seeded polymerization. A variety of well-defined CMs have been fabricated, including linear AB2, triangular AB3, tetrahedral AB4, trigonal bipyramidal AB5, square pyramidal AB5 and octahedral AB6. At a low volume ratio, colloidal chains were achieved by assembly of “hamburger” intermediates. Polymerization-induced particle-assembly allows the large scale production of the CMs, e.g. AB2-CMs (78.5% purity, more than 1017 particles, 50 g L−1). The CMs can serve as new building blocks for more complex structures such as colloidal chains and colloidal networks. The PIPA approach could be readily applied to vastly different triblock terpolymer systems for the fabrication of functional HNPs and has unique advantages for further applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Danyang Li and Xiaofeng Hu at the National Center for Protein Science (Beijing) Tsinghua Cryo-EM Facility for the technical support, and Prof. Xiaosong Wang at the University of Waterloo for helpful discussion. This work was supported by the National Natural Science Foundation of China (21871162 and 51573086).Notes and references
- Q. Chen, J. K. Whitmer, S. Jiang, S. C. Bae, E. Luijten and S. Granick, Science, 2011, 331, 199–202 CrossRef CAS PubMed.
- C. Zeng, Y. Chen, K. Kirschbaum, K. J. Lambright and R. Jin, Science, 2016, 354, 1580–1584 CrossRef CAS PubMed.
- P.-E. Rouet, C. Chomette, E. Duguet and S. Ravaine, Angew. Chem., Int. Ed., 2018, 57, 15754–15757 CrossRef CAS PubMed.
- Z. Wang, Z. Wang, J. Li, S. T. H. Cheung, C. Tian, S.-H. Kim, G.-R. Yi, E. Ducrot and Y. Wang, J. Am. Chem. Soc., 2019, 141, 14853–14863 CrossRef CAS PubMed.
- D. J. Lunn, J. R. Finnegan and I. Manners, Chem. Sci., 2015, 6, 3663–3673 RSC.
- L. Yang, L. Meng, J. Song, Y. Xiao, R. Wang, H. Kang and D. Han, Chem. Sci., 2019, 10, 7466–7471 RSC.
- K. Miszta, J. de Graaf, G. Bertoni, D. Dorfs, R. Brescia, S. Marras, L. Ceseracciu, R. Cingolani, R. van Roij, M. Dijkstra and L. Manna, Nat. Mater., 2011, 10, 872–876 CrossRef CAS PubMed.
- Z. Yang, J. Wei, Y. I. Sobolev and B. A. Grzybowski, Nature, 2018, 553, 313–318 CrossRef CAS PubMed.
- A. Walther and A. H. E. Müller, Chem. Rev., 2013, 113, 5194–5261 CrossRef CAS PubMed.
- Q. Yan, J. Yuan, Z. Cai, Y. Xin, Y. Kang and Y. Yin, J. Am. Chem. Soc., 2010, 132, 9268–9270 CrossRef CAS PubMed.
- R. Schreiber, J. Do, E.-M. Roller, T. Zhang, V. J. Schüller, P. C. Nickels, J. Feldmann and T. Liedl, Nat. Nanotechnol., 2013, 9, 74–78 CrossRef PubMed.
- B. Ruzicka, E. Zaccarelli, L. Zulian, R. Angelini, M. Sztucki, A. Moussaïd, T. Narayanan and F. Sciortino, Nat. Mater., 2010, 10, 56–60 CrossRef PubMed.
- W. Liu, M. Tagawa, H. L. Xin, T. Wang, H. Emamy, H. Li, K. G. Yager, F. W. Starr, A. V. Tkachenko and O. Gang, Science, 2016, 351, 582–586 CrossRef CAS PubMed.
- Z. Huang, J. Gong and Z. Nie, Acc. Chem. Res., 2019, 52, 1125–1133 CrossRef CAS PubMed.
- F. Li, D. P. Josephson and A. Stein, Angew. Chem., Int. Ed., 2011, 50, 360–388 CrossRef CAS PubMed.
- E. Duguet, A. Désert, A. Perro and S. Ravaine, Chem. Soc. Rev., 2011, 40, 941–960 RSC.
- L. Yao, Q. Li, Y. Guan, X. X. Zhu and Y. Zhang, ACS Macro Lett., 2018, 7, 80–84 CrossRef CAS.
- A. Perro, E. Duguet, O. Lambert, J.-C. Taveau, E. Bourgeat-Lami and S. Ravaine, Angew. Chem., Int. Ed., 2009, 48, 361–365 CrossRef CAS PubMed.
- N. B. Schade, M. C. Holmes-Cerfon, E. R. Chen, D. Aronzon, J. W. Collins, J. A. Fan, F. Capasso and V. N. Manoharan, Phys. Rev. Lett., 2013, 110, 148303 CrossRef PubMed.
- C. Yi, Y. Yang and Z. Nie, J. Am. Chem. Soc., 2019, 141, 7917–7925 CrossRef CAS PubMed.
- Z. Gong, T. Hueckel, G.-R. Yi and S. Sacanna, Nature, 2017, 550, 234–238 CrossRef PubMed.
- Y. Tian, T. Wang, W. Liu, H. L. Xin, H. Li, Y. Ke, W. M. Shih and O. Gang, Nat. Nanotechnol., 2015, 10, 637–644 CrossRef CAS PubMed.
- M. Y. Ben Zion, X. He, C. C. Maass, R. Sha, N. C. Seeman and P. M. Chaikin, Science, 2017, 358, 633–636 CrossRef CAS PubMed.
- W. Ren, S. Wen, S. A. Tawfik, Q. P. Su, G. Lin, L. A. Ju, M. J. Ford, H. Ghodke, A. M. van Oijen and D. Jin, Chem. Sci., 2018, 9, 4352–4358 RSC.
- A. H. Gröschel, F. H. Schacher, H. Schmalz, O. V. Borisov, E. B. Zhulina, A. Walther and A. H. E. Müller, Nat. Commun., 2012, 3, 710 CrossRef PubMed.
- A. H. Gröschel, A. Walther, T. I. Löbling, F. H. Schacher, H. Schmalz and A. H. E. Müller, Nature, 2013, 503, 247 CrossRef PubMed.
- Z. Zhang, C. Zhou, H. Dong and D. Chen, Angew. Chem., Int. Ed., 2016, 55, 6182–6186 CrossRef CAS PubMed.
- Z. Zhang, H. Li, X. Huang and D. Chen, ACS Macro Lett., 2017, 6, 580–585 CrossRef CAS.
- R. Deng, H. Li, F. Liang, J. Zhu, B. Li, X. Xie and Z. Yang, Macromolecules, 2015, 48, 5855–5860 CrossRef CAS.
- É. Ducrot, M. He, G.-R. Yi and D. J. Pine, Nat. Mater., 2017, 16, 652–657 CrossRef PubMed.
- J. S. Oh, S. Lee, S. C. Glotzer, G.-R. Yi and D. J. Pine, Nat. Commun., 2019, 10, 3936 CrossRef PubMed.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- X. Wang, G. Guerin, H. Wang, Y. Wang, I. Manners and M. A. Winnik, Science, 2007, 317, 644 CrossRef CAS PubMed.
- H. Sun, D. Liu and J. Du, Chem. Sci., 2019, 10, 657–664 RSC.
- X. Chen, L. Liu, M. Huo, M. Zeng, L. Peng, A. Feng, X. Wang and J. Yuan, Angew. Chem., Int. Ed., 2017, 56, 16541–16545 CrossRef CAS PubMed.
- X.-F. Xu, C.-Y. Pan, W.-J. Zhang and C.-Y. Hong, Macromolecules, 2019, 52, 1965–1975 CrossRef CAS.
- J. C. Foster, S. Varlas, B. Couturaud, Z. Coe and R. K. O'Reilly, J. Am. Chem. Soc., 2019, 141, 2742–2753 CrossRef CAS PubMed.
- P. Yang, Y. Ning, T. J. Neal, E. R. Jones, B. R. Parker and S. P. Armes, Chem. Sci., 2019, 10, 4200–4208 RSC.
- X. Wang, S. Man, J. Zheng and Z. An, ACS Macro Lett., 2018, 7, 1461–1467 CrossRef CAS.
- S. Guan, Z. Deng, T. Huang, W. Wen, Y. Zhao and A. Chen, ACS Macro Lett., 2019, 8, 460–465 CrossRef CAS.
- S. Li, H. Nie, S. Gu, Z. Han, G. Han and W. Zhang, ACS Macro Lett., 2019, 8, 783–788 CrossRef CAS.
- M. Williams, N. J. W. Penfold and S. P. Armes, Polym. Chem., 2016, 7, 384–393 RSC.
- D. Zhou, S. Dong, R. P. Kuchel, S. Perrier and P. B. Zetterlund, Polym. Chem., 2017, 8, 3082–3089 RSC.
- Y. Zhang, L. Yu, X. Dai, L. Zhang and J. Tan, ACS Macro Lett., 2019, 8, 1102–1109 CrossRef CAS.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS.
- A. H. Gröschel, A. Walther, T. I. Löbling, J. Schmelz, A. Hanisch, H. Schmalz and A. H. E. Müller, J. Am. Chem. Soc., 2012, 134, 13850–13860 CrossRef PubMed.
- M. Huo, M. Zeng, D. Li, L. Liu, Y. Wei and J. Yuan, Macromolecules, 2017, 50, 8212–8220 CrossRef CAS.
- F. S. Bates, Science, 1991, 251, 898–905 CrossRef CAS.
- P. Chambon, A. Blanazs, G. Battaglia and S. P. Armes, Macromolecules, 2012, 45, 5081–5090 CrossRef CAS.
- W. Kong, W. Jiang, Y. Zhu and B. Li, Langmuir, 2012, 28, 11714–11724 CrossRef CAS PubMed.
- D. K. Owens and R. C. Wendt, J. Appl. Polym. Sci., 1969, 13, 1741–1747 CrossRef CAS.
- D. H. Kaelble, J. Adhes., 1970, 2, 66–81 CrossRef CAS.
Footnotes |
| † This work is dedicated to Professor Caiyuan Pan, University of Science and Technology of China, for his 80th birthday in May, 2020. |
| ‡ Electronic supplementary information (ESI) available: Experimental details and figures. See DOI: 10.1039/d0sc00219d |
| § These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |