
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNon-covalent allosteric regulation of capsule catalysis†
Vicente
Martí-Centelles
 ,
Rebecca L.
Spicer
and
Paul J.
Lusby
*
,
Rebecca L.
Spicer
and
Paul J.
Lusby
*
EaStCHEM School of Chemistry, University of Edinburgh, Joseph Black Building, David Brewster Road, Edinburgh, Scotland EH9 3FJ, UK. E-mail: Paul.Lusby@ed.ac.uk
First published on 2nd March 2020
Abstract
Allosteric regulation is an essential biological process that allows enzymes to modulate their active site properties by binding a control molecule at the protein exterior. Here we show the first example of capsule catalysis in which activity is changed by exotopic binding. This study utilizes a simple Pd2L4 capsule that can partition substrates and external effectors with high fidelity. We also present a detailed, quantitative understanding of how effector interactions alter both substrate and transition state binding. Unlike other allosteric host systems, perturbations are not a consequence of large mechanical changes, rather subtle electronic effects resulting from weak, non-covalent binding to the exterior surface. This investigation paves the way to more sophisticated allosteric systems.
Introduction
Compartmentalization is a common theme that underpins various areas from logic gates,1 trans-membrane transport2 through to molecular machines3 and allosteric regulation of catalysis.4 Recently, multicavity metallosupramolecular architectures5 able to allocate different guest molecules in localized cavities have been reported (Fig. 1, left).6 However, there are both challenges and unexplored opportunities with such systems. The challenges are most obviously complex ligand synthesis and difficult self-assembly reactions. While selective binding in different sites has been demonstrated,6a,b as has allosteric influence of one cavity by another,6a,c quantifying how binding at one site affects another is limited.7 Such allosteric regulation usually take place through steric factors,6a,c wherein the binding at one site produces a large mechanical re-arrangement of the global structure. To date, the influence that allosteric binding has on capsule catalysis has not been described.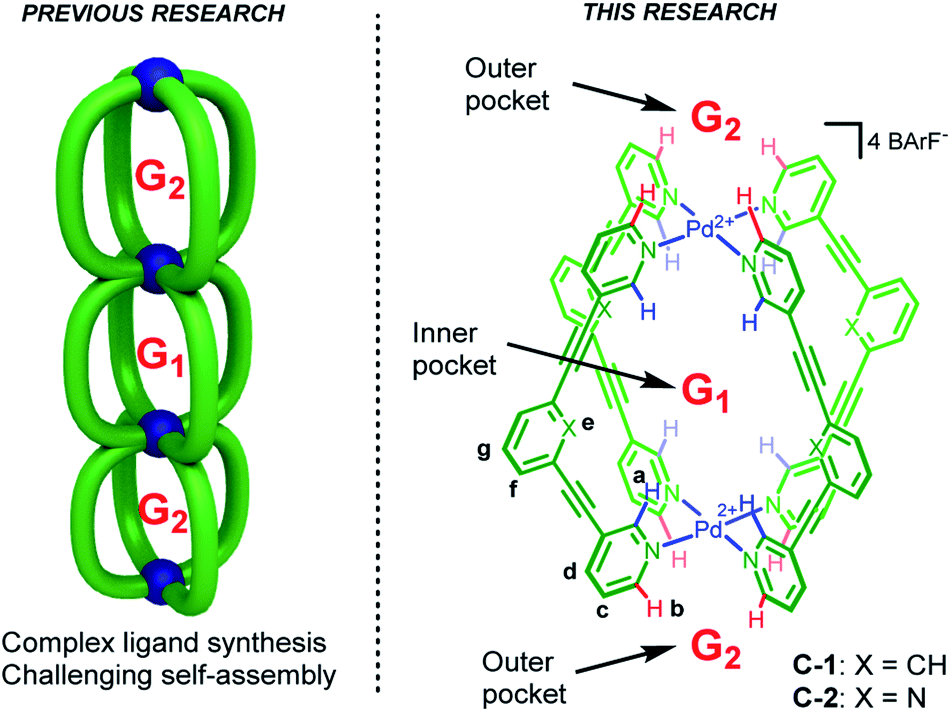 | ||
| Fig. 1 Compartmentalized guest binding using metallosupramolecular hosts. | ||
The multi-binding site approach we envisaged negates the need for complex architectures (Fig. 1, left), rather it uses a minimalist, mono-cavity system at which external effectors could interact (Fig. 1, right).7 The exterior of most coordination capsules are swamped by counter-ion interactions, leaving little scope for binding more specific or weaker effectors. Also, counter-ion binding is often poorly defined, a consequence of dominant non-directional coulombic effects. In contrast, cage compounds C-1 and C-2 (Fig. 1) only interact weakly with the associated BArF counteranions (BArF = B(3,5-(CF3)2C6H3)4−),8a moreover, they possess both inner and outer H-bond donor pockets (shown in blue and red, respectively) that can interact with complementary neutral molecules. Furthermore, the functional properties of these cages make them prime candidates to explore allosterically regulated binding and catalysis.8
Results and discussion
Experiments with C-1 and C-2 have shown that no observable exotopic binding occurs with quinone compounds.9 This emphasises that the synergistic interaction of both internal H-bond pockets is crucial for quinone encapsulation.8 It was therefore clear that any outside effector would need to be a very strong hydrogen bond acceptor and be size and/or shape mismatched for the cavity. We were thus pleased to see that the addition of excess Ph3PO (10 eq.) to a sample of C-1 induced significant change in the exotopic 1H NMR signals: a significant downfield shift (ca. 1 ppm) of the exterior H-bond donor atom (Hb, Fig. 2a and b) and smaller yet significant changes to neighbouring protons (Hc/d). Also, only small changes in the internal resonances (Ha and He) were observed. When an excess (10 eq.) of benzoquinone (Bq) was added to the Ph3PO containing sample (Fig. 2c), changes characteristic of guest encapsulation were observed: Ha moved downfield by 0.4 ppm and the inner equatorial protons (He) shift upfield due to shielding by the guest. At the same time, the peaks associated with the outside resonances change little, showing high fidelity binding of both molecules. Similar spectroscopic changes were observed for C-2 (see Fig. S1†).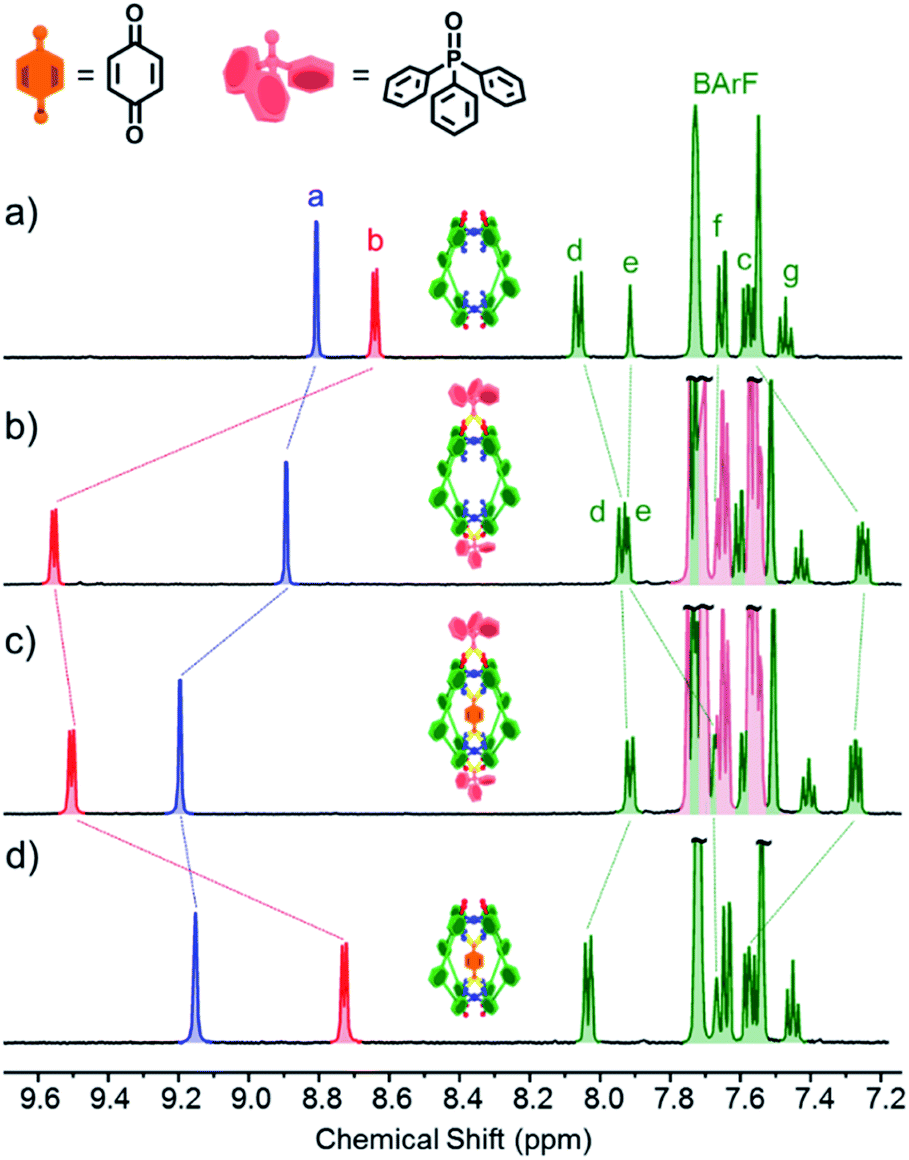 | ||
| Fig. 2 1H NMR (500 MHz, CD2Cl2, 300 K) of (a) cage C-1 (0.5 mM); (b) C-1 (0.5 mM) + Ph3PO (5 mM); (c) C-1 (0.5 mM) + Ph3PO (5 mM) + benzoquinone (5 mM); (d) cage C-1 (0.5 mM) + benzoquinone (5 mM). Exo and endotopic CH acidic protons coloured red and blue respectively. See Fig. 1 for assignments. | ||
Initially, the exotopic association constants (K11, K12; Table 1) were determined using 1H NMR titration. The data from these experiments fits the binding isotherm for a statistical 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 model (see Fig. S6 and S11†) and also a 1:1 model based on twice the concentration of host (Fig. S7 and S12†). The statistical model yielded association constants of K11 = 8200 M−1 and K12 = 2100 M−1 for both C-1 and C-2 with Ph3PO (Table 1, entries 1 and 6), using the assumptions that K11 = 4 × K12 and ΔδHG2 = 2 × ΔδHG.10 The lack of cooperativity between exotopic sites likely stems from the remoteness of the exterior recognition sites, and that binding requires little reorganization. These results also show that unlike guest encapsulation, the central phenyl (C-1) and pyridyl (C-2) rings have little effect on exterior binding.
:2 model (see Fig. S6 and S11†) and also a 1:1 model based on twice the concentration of host (Fig. S7 and S12†). The statistical model yielded association constants of K11 = 8200 M−1 and K12 = 2100 M−1 for both C-1 and C-2 with Ph3PO (Table 1, entries 1 and 6), using the assumptions that K11 = 4 × K12 and ΔδHG2 = 2 × ΔδHG.10 The lack of cooperativity between exotopic sites likely stems from the remoteness of the exterior recognition sites, and that binding requires little reorganization. These results also show that unlike guest encapsulation, the central phenyl (C-1) and pyridyl (C-2) rings have little effect on exterior binding.
|
|
||||
|---|---|---|---|---|
| Entry | Equilibrium | Cage | Guest | K A (M−1) |
| a Ref. 8a. b Ref. 8b. c Ref. 8c. | ||||
| 1 | K 11 | C-1 | — | 8200 |
| K 12 | 2100 | |||
| 2a | K Q | C-1 | Benzoquinone | 7900 |
| 3 | K Q2 | C-1 | Benzoquinone | 2800 |
| 4a | K Q | C-1 | Pentacenedione | 8 × 108 |
| 5 | K Q11 | C-1 | Pentacenedione | 2900 |
| K Q12 | 725 | |||
| 6 | K 11 | C-2 | — | 8200 |
| K 12 | 2100 | |||
| 7b | K Q | C-2 | Benzoquinone | 1100 |
| 8 | K Q2 | C-2 | Benzoquinone | 250 |
| 9c | K Q | C-2 | Pentacenedione | 8.9 × 105 |
| 10 | K Q2 | C-2 | Pentacenedione | 2.5 × 105 |
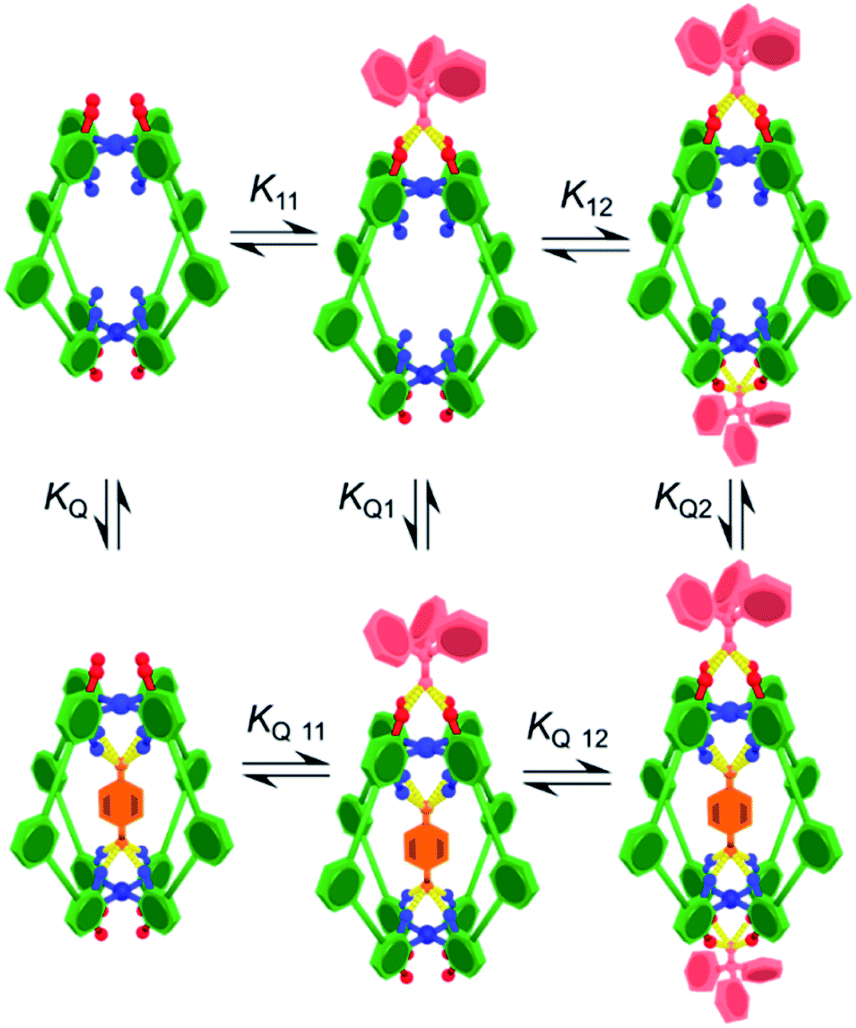
To determine how the binding of the guest is affected by the exotopic interactions, benzoquinone was titrated into cage solutions containing 10 equivalents of Ph3PO. Under these conditions the major species in solution are the 1:2 cage–(Ph3PO)2 complexes (89% of 1:2 complex and 11% of 1:1 complex and less than 1% free cage). These titration sets were analysed using a 1:1 binding isotherm (Fig. S17 and S19†), producing excellent fits that gave a KQ2 value of 2800 M−1 for C-1(Ph3PO)2 (Table 1, entry 3) and 250 M−1 for C-2(Ph3PO)2 (Table 1, entry 8). These values represent reductions of 65% and 77% with respect to the association constants of 7900 M−1 and 1100 M−1 in the absence of Ph3PO (Table 1, entries 2 and 7).8
In order to determine how the binding of the allosteric sites is affected by the internal guest, we reasoned that utilising a sub-stoichiometric ratio of strong binding, slow exchange quinone would expedite this process as it would facilitate simultaneous monitoring of both free and filled cages. As anticipated, titrating Ph3PO into C-1 in the presence of half an equivalent of pentacenedione (KA = 8 × 108 M−1; Table 1, entry 4) showed the characteristic downfield shift in the exotopic protons of both the empty and filled cages (Fig. 3a). It was also apparent that the changes in the filled cage were more gradual. Fitting this data (Fig. 3b and S9†) confirmed this empirical observation, with KQ11 = 2900 M−1 and KQ12 = 725 M−1 for pentacenedione⊂C-1 (Table 1, entry 5), a 65% reduction in exotopic Ph3PO binding compared to empty C-1. The low solubility of pentacenedione⊂C-2 supramolecular complex hampered a similar 1H NMR titration with Ph3PO.11 However, we were able to use a UV/Vis titration at much lower concentration to measure how Ph3PO exterior binding affects the association of the same strong guest pentacenedione with C-2 (Fig. 3c and d). This approach yielded a KQ2 value for C-2 with pentacenedione of 2.5 × 105 M−1 compared to KQ of 8.9 × 105 M−1 in the absence of Ph3PO (Table 1, entries 9 and 10) corresponding to a 72% reduction of the association constant. This indicates that exterior binding affects both weak and strong binding quinone guests similarly.
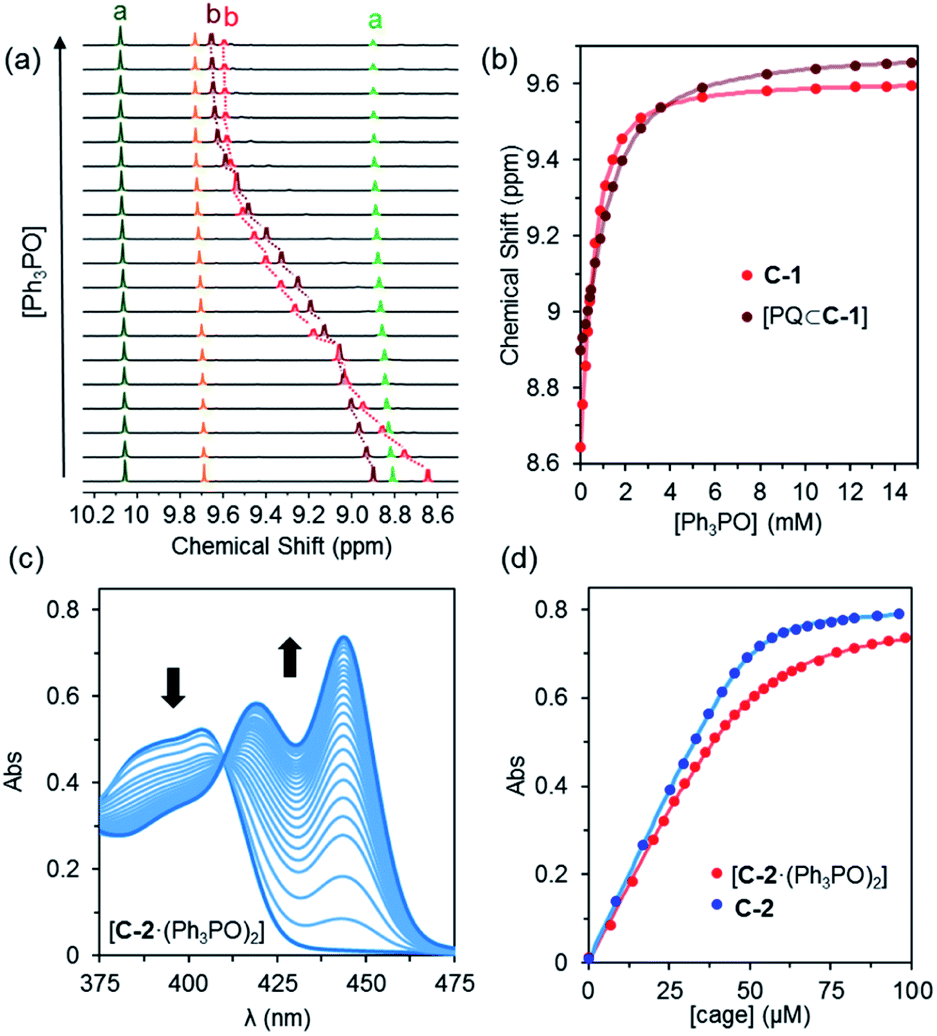 | ||
| Fig. 3 (a) Partial 1H NMR spectra (600 MHz, CD2Cl2, 300 K) for the titration of C-1 (0.48 mM) and pentacenequinone (0.25 mM) with Ph3PO. (b) Binding curves based on proton Hb for C-1 only (red) and pentacenequinone⊂C-1 (brown). The solid points are experimental data, the continuous brown line the fitted binding isotherm, and the continuous red line is the predicted binding isotherm using the previously determined association constants for cage only. (c) UV-Vis titration of pentacenedione (50 μM) with cage C-2 (0–2 equiv.) in the presence of Ph3PO (5 mM). (d) A comparison of binding curves for the encapsulation of pentecenedione with free C-2 and C-2(Ph3PO)2. Absorbance measured at 444 nm C-2(Ph3PO)2 and 447 nm (C-2). | ||
The destabilization of quinone binding by Ph3PO and vice versa is interesting, especially considering the direction of the 1H NMR chemical shifts. These spectra show that (a) Ph3PO binding to the exterior site causes a slight deshielding of the interior H-bond protons (Ha, Fig. 2a and b) and (b) quinone binding also causes slight deshielding of the exterior protons (Hb, Fig. 2a and d). The reduction in electron density of these binding site H-bond atoms should correspond to an increased H-bond donor capacity, which should translate to mutually stronger binding. We have also analysed the known X-ray data of C-1 and C-2 to gain insight into the structural changes that occur upon guest encapsulation (Fig. S42–S47†). These structures show little conformational change between “empty”, quinone and simple anion containing cages, suggesting there is no obvious mechanical effect that could explain the reduction in binding. Considering these contradictory observations, we suggest that field effects12 contribute to binding, wherein the positively charged Pd2+ ions are attracted to the electron rich oxygen atoms of the guest and effector. Binding one species therefore partially “neutralizes” the charge on Pd2+, leading to the observed mutual destabilization.
Remotely regulating activity is a key aspect of biological catalysis. Considering the frequently-drawn parallels between enzyme and capsule catalysis,13 it is therefore surprising that external regulation of the latter has not been previously described, as far as we are aware. We were therefore interested to determine the consequence of effector binding on the previously described Diels–Alder activity of cages C-1 and C-2.8b Considering that free C-1 does not accelerate the reaction of benzoquinone and isoprene (or other small dienes), it is perhaps unsurprising that adding excess Ph3PO does not change this (Fig. 4). In contrast, when the effector was added to the C-2 catalyzed reaction, a marked reduction in the overall rate was observed (Fig. 4), from a kobs of 3.4 M−1 h−1 with C-2 only, to 1.4 M−1 h−1 in the presence of Ph3PO (Table 2). We note that Ph3PO does not affect the uncatalyzed reaction. We have also examined the effect of adding a small phosphine oxide, which can bind to both the inside and outside pockets. As expected, adding Et3PO has a much larger influence on catalysis, reducing the rate of product formation to not much above the background reaction (Fig. 4), indicating that this sterically un-encumbered hydrogen bond acceptor also competes with benzoquinone for the capsule's cavity.
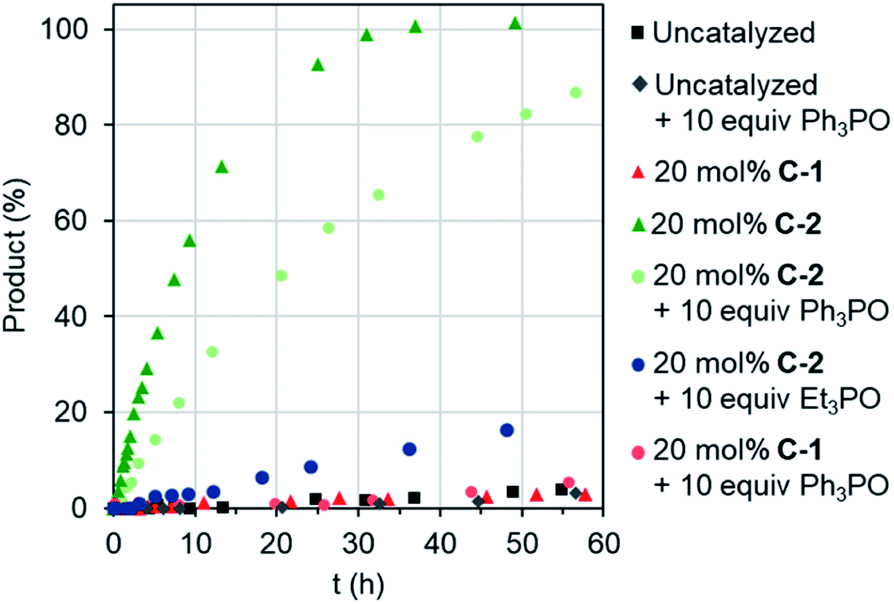 | ||
| Fig. 4 Allostreic regulation of capsule catalysis. Evolution of Diels–Alder product for the reaction of benzoquinone and isoprene. | ||
| Catalyst | Diene | k obs (M−1 h−1) | k cat (M−1 h−1) | k cat/kuncat | K Ass TS (M−1) |
|---|---|---|---|---|---|
| C-2 | Isoprene | 3.4 | 24 | 400 | 4.3 × 105 |
| C-2(Ph3PO)2 | 1.4 | 17 | 290 | 7.2 × 104 | |
| C-2 | 1,3-Cyclohexadiene | 6 | 61 | 300 | 3.3 × 105 |
| C-2(Ph3PO)2 | 2.5 | 37 | 190 | 4.6 × 104 |
We have sought to understand and rationalize the multiple factors that contribute to this reduction in activity. The kcat values, and by extension kcat/kuncat, are surprisingly close considering the nearly 3-fold reduction in kobs (Table 2). This can be understood in terms of relative stabilization of the substrate and the transition state (TS) energies (Fig. 5). While the effector lowers the TS affinity (KAss TS) by nearly an order magnitude compared to C-2 only, corresponding to a 1.1 kcal mol−1 energy difference, this is offset by a smaller lowering of the substrate energy. This combination leads to only 0.2 kcal mol−1 difference in the catalytic energy barriers. However, the lower binding affinity for benzoquinone in the presence of Ph3PO corresponds to a drop from 14% to 7% initially bound substrate. Considering all the factors, it is therefore apparent that the effector-induced decrease in catalytic activity stems principally from the lower catalyst–substrate concentration. The DA reaction between benzoquinone and 1,3-cyclohexadiene shows similar trends (Table 2).
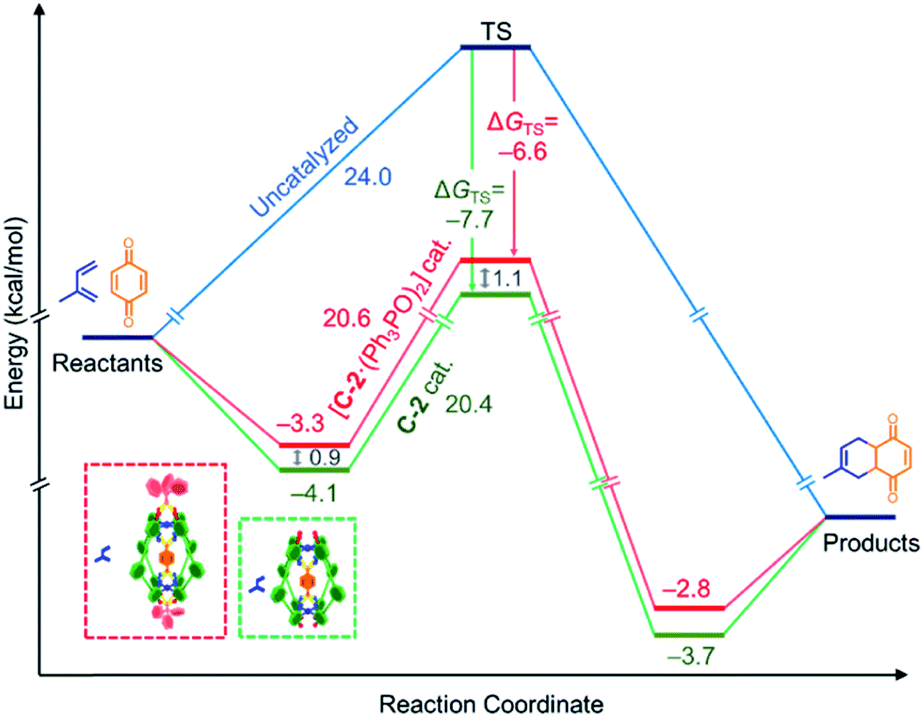 | ||
| Fig. 5 “Diels–Alderase” catalyst activity rationalized using substrate and TS stabilization effects: whereas the association constants are reduced by binding of Ph3PO to the allosteric pockets, the catalytic activity remains good. The uncatalyzed reaction is represented in blue, the C-2 catalyzed reaction in green, and the C-2(Ph3PO)2 in orange. | ||
Conclusions
We have shown how a simple, single cavity Pd2L4 capsules can recognize and partition multiple neutral molecules using endotopic and exotopic binding sites. Outer binding also modulates both guest binding and catalytic properties. The reduction in activity we observe also sheds further light on the non-covalent interactions involved in both substrate and TS recognition. We envisage that this greater understanding will pave the way to more sophisticated bio-mimetic catalyst systems.Experimental section
See ESI† for association constant determination by 1H NMR, UV-Vis and kinetic experiments.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Leverhulme Trust (RPG-2015-232). We acknowledge the EPSRC Critical Resource Catalysis Centre for Doctoral training (CRITICAT, EP/L016419/1) for a studentship for RLS.References
- (a) D. C. Magri, G. J. Brown, G. D. McClean and A. P. de Silva, J. Am. Chem. Soc., 2006, 128, 4950 CrossRef CAS PubMed; (b) S. Erbas-Cakmak, S. Kolemen, A. C. Sedgwick, T. Gunnlaugsson, T. D. James, J. Yoon and E. U. Akkaya, Chem. Soc. Rev., 2018, 47, 2228 RSC.
- (a) I. W. Park, J. Yoo, B. Kim, S. Adhikari, S. Kuk Kim, Y. Yeon, C. J. E. Haynes, J. L. Sutton, C. C. Tong, V. M. Lynch, J. L. Sessler, P. A. Gale and C. Lee, Chem.–Eur. J., 2012, 18, 2514–2523 CrossRef CAS PubMed; (b) P. A. Gale, J. T. Davis and R. Quesada, Chem. Soc. Rev., 2017, 46, 2497 RSC.
- (a) M. N. Chatterjee, E. R. Kay and D. A. Leigh, J. Am. Chem. Soc., 2006, 128, 4058 CrossRef CAS PubMed; (b) C. Cheng, P. R. McGonigal, J. F. Stoddart and R. D. Astumian, ACS Nano, 2015, 9, 8672 CrossRef CAS PubMed; (c) S. Kassem, T. van Leeuwen, A. S. Lubbe, M. R. Wilson, B. L. Feringa and D. A. Leigh, Chem. Soc. Rev., 2017, 46, 2592 RSC.
- (a) N. C. Gianneschi, P. A. Bertin, S. T. Nguyen, C. A. Mirkin, L. N. Zakharov and A. L. Rheingold, J. Am. Chem. Soc., 2003, 125, 10508 CrossRef CAS PubMed; (b) H. J. Yoon, J. Kuwabara, J. H. Kim and C. A. Mirkin, Science, 2010, 330, 66 CrossRef CAS PubMed.
- For reviews on coordination assemblies, see (a) M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418 CrossRef CAS PubMed; (b) R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810 CrossRef CAS PubMed; (c) M. D. Ward and P. R. Raithby, Chem. Soc. Rev., 2013, 42, 1619 RSC; (d) T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001 CrossRef CAS PubMed; (e) C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Chem. Rev., 2015, 115, 3012 CrossRef CAS PubMed; (f) S. Zarra, D. M. Wood, D. A. Roberts and J. R. Nitschke, Chem. Soc. Rev., 2015, 44, 419 RSC; (g) W. M. Bloch and G. H. Clever, Chem. Commun., 2017, 53, 8506 RSC.
- (a) S. Freye, J. Hey, A. Torras-Galán, D. Stalke, R. Herbst-Irmer, M. John and G. H. Clever, Angew. Chem., Int. Ed., 2012, 51, 2191 CrossRef CAS PubMed; (b) D. Preston, J. E. M. Lewis and J. D. Crowley, J. Am. Chem. Soc., 2017, 139, 2379 CrossRef CAS PubMed; (c) K. Yazaki, M. Akita, S. Prusty, D. K. Chand, T. Kikuchi, H. Sato and M. Yoshizawa, Nat. Commun., 2017, 8, 15914 CrossRef CAS PubMed.
- (a) J. Mendez-Arroyo, A. I. d'Aquino, A. B. Chinen, Y. D. Manraj and C. A. Mirkin, J. Am. Chem. Soc., 2017, 139, 1368 CrossRef CAS PubMed; (b) J. Nitschke, C. A. Schalley, B. S. Pilgrim, D. A. Roberts and L. K. S. von Krbek, Angew. Chem., Int. Ed., 2018, 57, 14121–14124 CrossRef PubMed.
- (a) D. P. August, G. S. Nichol and P. J. Lusby, Angew. Chem., Int. Ed., 2016, 55, 15022 CrossRef CAS PubMed; (b) V. Marti-Centelles, A. Lawrence and P. J. Lusby, J. Am. Chem. Soc., 2018, 140, 2862 CrossRef CAS PubMed; (c) T. A. Young, V. Martí-Centelles, J. Wang, P. J. Lusby and F. Duarte, J. Am. Chem. Soc., 2020, 142, 1300–1310 CrossRef PubMed.
- 2-tert-Butyl-1,4-benzoquinone is not encapsulated due to size, it also does not interact with the exotopic sites, see ref. 8a.
- G. Ercolani, C. Piguet, M. Borkovec and J. Hamacek, J. Phys. Chem. B, 2007, 111, 12195–12203 CrossRef CAS PubMed.
- Rapid formation of crystals of prevented a 1H NMR titration. See ESI† for the crystal structure of pentecenedione⊂C-2.
- (a) M. Gargiulo Holl, M. D. Struble, P. Singal, M. A. Siegler and T. Lectka, Angew. Chem., Int. Ed., 2016, 55, 8266 CrossRef PubMed; (b) L. Guan, M. Gargiulo Holl, C. R. Pitts, M. D. Struble, M. A. Siegler and T. Lectka, J. Am. Chem. Soc., 2017, 139, 14913 CrossRef CAS PubMed; (c) J. Simó Padial, J. Poater, T. Nguyen, P. Tinnemans, M. Bickelhaupt and J. Mecinović, J. Org. Chem., 2017, 82, 9418 CrossRef PubMed.
- (a) J. W. Michael, P. A. Ulmann and C. A. Mirkin, Angew. Chem., Int. Ed., 2011, 50, 114 CrossRef PubMed; (b) D. M. Kaphan, M. D. Levin, R. G. Bergman, K. N. Raymond and F. D. Toste, Science, 2015, 350, 1235 CrossRef CAS PubMed; (c) Q. Zhang and K. Tiefenbacher, Nat. Chem., 2015, 7, 197 CrossRef CAS PubMed; (d) Q.-Q. Wang, S. Gonell, S. H. A. M. Leenders, M. Dürr, I. Ivanović-Burmazović and J. N. H. Reek, Nat. Chem., 2016, 8, 225 CrossRef CAS PubMed; (e) W. Cullen, A. J. Metherell, A. B. Wragg, C. G. P. Taylor, N. H. Williams and M. D. Ward, J. Am. Chem. Soc., 2018, 140, 2821 CrossRef CAS PubMed; (f) L. R. Holloway, P. M. Bogie, Y. Lyon, C. Ngai, T. F. Miller, R. R. Julian and R. J. Hooley, J. Am. Chem. Soc., 2018, 140, 8078 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1978675. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc00341g |
| This journal is © The Royal Society of Chemistry 2020 |