
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhanced CH3OH selectivity in CO2 hydrogenation using Cu-based catalysts generated via SOMC from GaIII single-sites†
Erwin
Lam
a,
Gina
Noh
a,
Ka Wing
Chan
 a,
Kim
Larmier
a,
Dmitry
Lebedev
a,
Keith
Searles
a,
Patrick
Wolf
a,
Olga V.
Safonova
b and
Christophe
Copéret
*a
a,
Kim
Larmier
a,
Dmitry
Lebedev
a,
Keith
Searles
a,
Patrick
Wolf
a,
Olga V.
Safonova
b and
Christophe
Copéret
*a
aDepartment of Chemistry and Applied Biosciences, ETH Zurich, Vladimir Prelog Weg 2, CH-8093 Zurich, Switzerland. E-mail: ccoperet@ethz.ch
bPaul Scherrer Institute, CH-5232 Villigen, Switzerland
First published on 26th February 2020
Abstract
Small and narrowly distributed nanoparticles of copper alloyed with gallium supported on silica containing residual GaIII sites can be obtained via surface organometallic chemistry in a two-step process: (i) formation of isolated GaIII surface sites on SiO2 and (ii) subsequent grafting of a CuI precursor, [Cu(OtBu)]4, followed by a treatment under H2 to generate CuGax alloys. This material is highly active and selective for CO2 hydrogenation to CH3OH. In situ X-ray absorption spectroscopy shows that gallium is oxidized under reaction conditions while copper remains as Cu0. This CuGa material only stabilizes methoxy surface species while no formate is detected according to ex situ infrared and solid-state nuclear magnetic resonance spectroscopy.
Introduction
The transformation of CO2 into value-added products is a promising strategy to mitigate the further increase of CO2 in the earth's atmosphere that is connected to environmental changes.1–4 In that context, the hydrogenation of CO2 yields CH3OH that can be used as a base chemical for the production of fuels or as a fuel itself, thus generating a closed carbon-fuel-cycle, provided that efficient CO2 capture and storage, and efficient H2 production from renewable energy sources are available.5 Major issues associated with the hydrogenation of CO2 into CH3OH are the selectivity of the process and the long-term stability of catalysts. Regarding selectivity, the hydrogenation of CO2 can also yield CO via, for instance, the reverse water gas shift (RWGS) reaction or the decomposition of methyl formate, that can be formed in situ via secondary reactions.6,7 Heterogeneous catalysts that are able to form CH3OH with good selectivity and activity mainly consist of copper-based catalysts together with zinc oxide (e.g. Cu/ZnO or Cu/ZnO/Al2O3), gallium oxide (e.g. Cu/ZnO/Ga2O3) or zirconium oxide (Cu/ZrO2).8–23 In the case of ZrO2, it has been shown that the Lewis acidic surface ZrIV sites stabilize reaction intermediates (CO2, formates and methoxy) and improve the activity and methanol selectivity. The origin of the promotional effect of zinc or gallium oxide is, however, not fully understood.24–29 For instance, the formation of highly active CuZn surface alloys or interfacial sites between Cu and ZnO have both been proposed.24–29 The promotional effect of gallium oxide has been far less studied.27,30Recently, it has been shown that surface organometallic chemistry (SOMC) combined with thermolytic molecular precursors (TMP) constitutes a powerful synthetic strategy to generate supported metal nanoparticles surrounded by well-defined isolated promoter sites that allows investigating the role of interfacial sites via in situ spectroscopic methods.31,32 This approach can also yield supported alloyed nanoparticles, depending on the selection of metal and promoters (vide infra). The SOMC/TMP approach can be summarized as follows: in a first step, the support, SiO2, is dehydroxylated at 700 °C to obtain isolated surface silanol (Si–OH) groups (1 OH, nm−2) that are used as anchoring groups to graft the TMP.33 Post-thermal treatment at high temperature generates isolated low-coordinated metal surface sites, that are free of organic ligands, and restores ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Si–OH groups (Scheme 1a), onto which a second molecular precursor is grafted. Subsequent post-treatment under reducing conditions (H2) generates metal nanoparticles interfacing low-coordinated metal surface sites (Scheme 1b). This approach has allowed the generation of highly active and selective CO2 hydrogenation catalysts by supporting Cu nanoparticles on SiO2 containing isolated ZrIV and TiIV sites.34,35 These catalysts show high activity and CH3OH selectivity, but also suffer from the decrease of CH3OH selectivity at high conversion as observed for other CO2 hydrogenation catalysts.34 The outstanding activity and CH3OH selectivity of copper supported on silica containing TiIV isolated sites is particularly noteworthy, considering that Cu/TiO2 performs very poorly in CH3OH synthesis by favoring CO formation.36–39 This difference of catalyst performance has been ascribed to the site isolation of TiIV and the use of a non-reducible support, SiO2, thus allowing TiIV to play exclusively the role of a Lewis acid, that stabilizes reaction intermediates at the interface with Cu particles.35 Using a similar approach, i.e., the treatment under H2 of a grafted platinum(II) molecular precursor on isolated GaIII sites generates small and narrowly distributed PtGax nanoparticles stabilized by remaining GaIII sites that show high activity, selectivity and stability for propane dehydrogenation.40
Si–OH groups (Scheme 1a), onto which a second molecular precursor is grafted. Subsequent post-treatment under reducing conditions (H2) generates metal nanoparticles interfacing low-coordinated metal surface sites (Scheme 1b). This approach has allowed the generation of highly active and selective CO2 hydrogenation catalysts by supporting Cu nanoparticles on SiO2 containing isolated ZrIV and TiIV sites.34,35 These catalysts show high activity and CH3OH selectivity, but also suffer from the decrease of CH3OH selectivity at high conversion as observed for other CO2 hydrogenation catalysts.34 The outstanding activity and CH3OH selectivity of copper supported on silica containing TiIV isolated sites is particularly noteworthy, considering that Cu/TiO2 performs very poorly in CH3OH synthesis by favoring CO formation.36–39 This difference of catalyst performance has been ascribed to the site isolation of TiIV and the use of a non-reducible support, SiO2, thus allowing TiIV to play exclusively the role of a Lewis acid, that stabilizes reaction intermediates at the interface with Cu particles.35 Using a similar approach, i.e., the treatment under H2 of a grafted platinum(II) molecular precursor on isolated GaIII sites generates small and narrowly distributed PtGax nanoparticles stabilized by remaining GaIII sites that show high activity, selectivity and stability for propane dehydrogenation.40
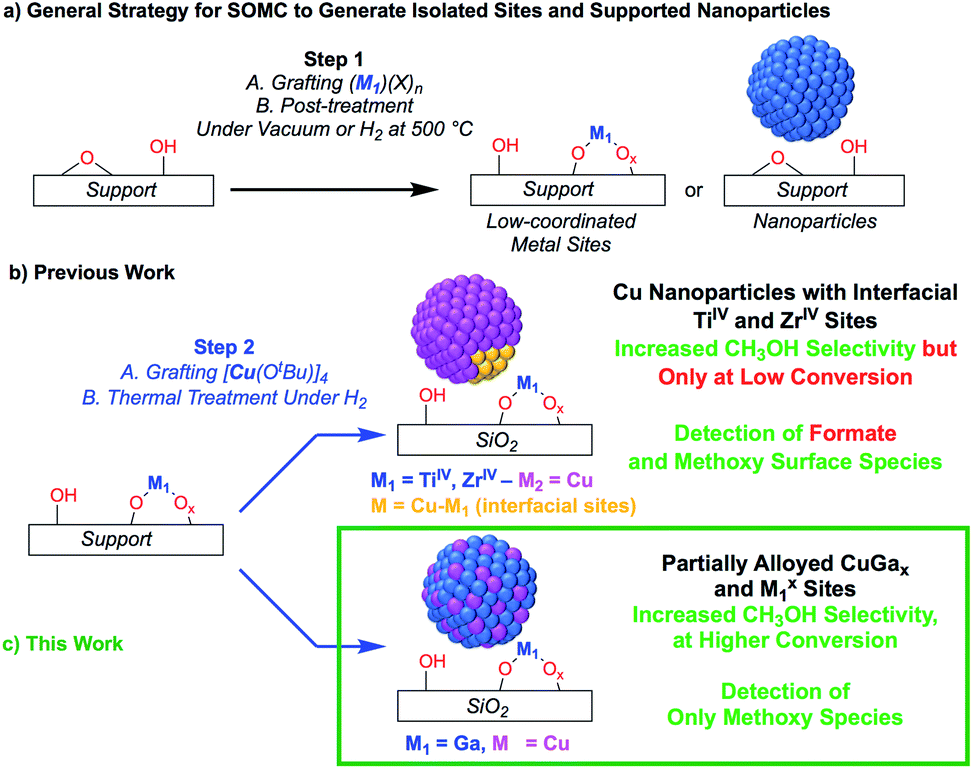 | ||
| Scheme 1 (a) General SOMC strategy to generate isolated sites or supported nanoparticles. (b and c) Utilizing isolated metal site supports generated from SOMC for metal and metal–alloy nanoparticle formation. | ||
Here, we reasoned that the SOMC/TMP approach would be a useful tool to investigate the behavior of gallium promoters in CO2 hydrogenation catalysts (Scheme 1c). We thus prepared Cu-based nanoparticles by grafting [Cu(OtBu)]4 on a silica support containing GaIII single-sites, followed by a treatment under H2. This approach yields nanoparticles consisting of CuGax alloys that evolved under CO2 hydrogenation conditions into Cu0 and GaIII according to in situ X-ray absorption spectroscopy (XAS). Such Cu-Ga/SiO2 catalysts display enhanced activity and selectivity at higher conversions in the hydrogenation of CO2 into CH3OH by comparison to other Cu-based catalysts. These improved performances are attributed to Cu0/GaIII interfaces that only stabilize methoxy and not formate intermediates, according to infrared (IR) and solid state nuclear magnetic resonance (NMR) spectroscopy.
Results and discussion
Catalyst synthesis and characterization
We first prepared well-defined GaIII sites on SiO2 with ca. 1.0 GaIII nm−2 by grafting and thermolysis of [Ga(OSi(OtBu)3)3(THF)]:41,42GaIII@SiO2 (ref. 42) (Mx@SiO2 denoted as isolated M surface sites in its x oxidation state) via an SOMC/TMP approach and then grafted [Cu(OtBu)]4 on residual surface silanols present in GaIII@SiO2. The IR spectra show the consumption of Si–OH groups (3747 cm−1) and the appearance of C–H stretching (2700–3000 cm−1) and bending (1300–1500 cm−1) bands, consistent with grafting of [Cu(OtBu)]4via protonolysis of the Si–OH group (Fig. S1†). Next, reduction under H2 at 500 °C removes all the organics and regenerate the Si–OH groups, as shown by IR spectroscopy, yielding Cu-Ga/SiO2 (Fig. 1a). Based on inductively coupled plasma optical emission spectroscopy (ICP-OES), a metal loading of 1.61 wt% gallium and 3.88 wt% copper is obtained for Cu-Ga/SiO2 (corresponding to a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 Cu:Ga atomic ratio).
:2 Cu:Ga atomic ratio).
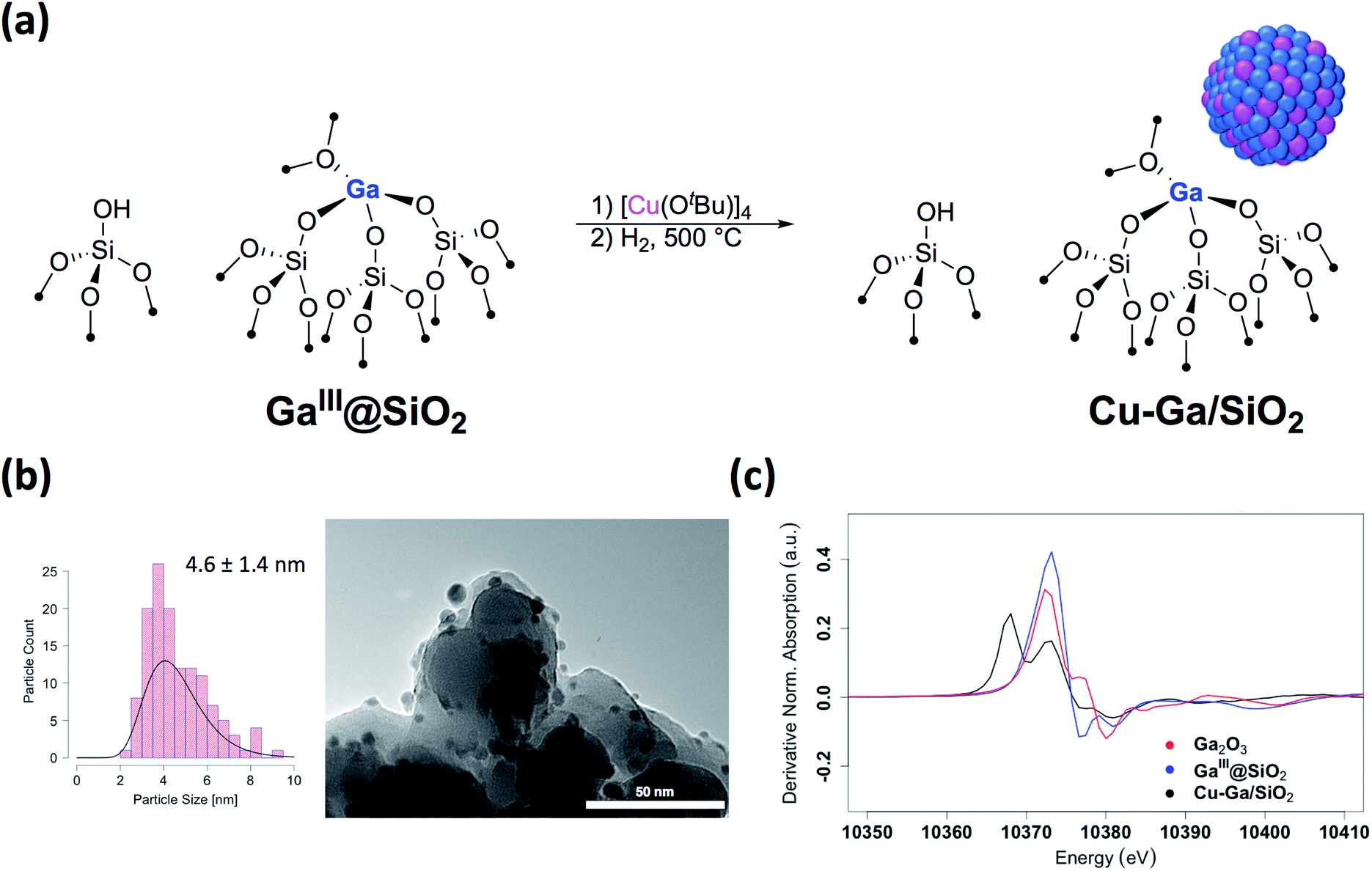 | ||
| Fig. 1 (a) Schematic procedure for grafting of [Cu(OtBu)]4 on GaIII@SiO2 followed by reduction under H2 at 500 °C. (b) Particle size distribution and TEM images of Cu-Ga/SiO2. (c) First derivative of the XANES spectra at the Ga K-edge of Cu-Ga/SiO2 and reference samples. | ||
A specific surface area of ca. 200 m2 g−1 – determined by N2 physisorption isotherms and Brunauer–Emmett–Teller43 (BET) analysis (Table S1†) – is obtained, similar to this of the initial material. Transmission electron microscopy (TEM) studies show the formation of small and narrowly distributed nanoparticles on SiO2 (4.6 ± 1.4 nm) (Fig. 1b), which corresponds to Cu–Ga alloy nanoparticles (vide infra). These particles are slightly larger than what was found for the corresponding Cu/SiO2 (2.9 ± 1.3 nm) prepared via a similar approach.34 Energy-dispersive X-ray (EDX) mapping of as-prepared Cu-Ga/SiO2, introduced to the microscope without exposure to air using a vacuum transfer TEM sample holder, shows that gallium is found in the same region as the copper nanoparticles, but it is also dispersed throughout the support (Fig. S2†). In addition, the particle size distribution of Cu-Ga/SiO2 from TEM of samples with and without exposure to air does not change, indicating the absence of copper and gallium redispersion under oxidizing conditions (Fig. S3†). N2O titration at 90 °C, that selectively titrates the surface of Cu nanoparticles, was performed, resulting in around 55 μmol gcat−1 surface sites for Cu-Ga/SiO2 (assuming a 1:2 stoichiometry between N2O and the Cu surface site) (Table S1†), which is similar for what is obtained for Cu/SiO2 (50 μmol gcat−1),34 despite the larger particle sizes for Cu-Ga/SiO2. It is possible that N2O reacts with reduced gallium sites leading to a higher N2O uptake similar to what is observed with zinc in case of Cu/ZnO/Al2O3.24 Chemisorption experiments using H2 at 40 °C indicate around 60 μmol gcat−1 surface sites (assuming a 1:2 stoichiometry between H2 and the surface site) for Cu-Ga/SiO2, consistent with the number obtained via N2O titration (Table S1 and Fig. S4†). No crystalline phases are observed by powder X-ray diffraction, as expected from the amorphous nature of the SiO2 support and the presence of small metal nanoparticles (Fig. S5†). The presence of residual Lewis acidic gallium sites is supported by pyridine adsorption and IR spectroscopy,44 by the presence of ring vibrational bands of pyridine at 1621 cm−1, characteristic for Lewis acidic surface sites, likely associated with GaIII sites (Fig. S6†). Upon adsorption of pyridine, it persists even at 500 °C under high-vacuum (10−5 mbar) indicating strongly bound pyridine (for details see ESI†). IR spectroscopy (Fig. S7†) of the catalysts upon adsorption of CO (90 mbar) at room temperature shows stretching bands at 2102 cm−1 for Cu-Ga/SiO2 corresponding to CO bonded to the metal nanoparticle which are slightly red-shifted with respect to what is observed for pure Cu/SiO2 at 2106 cm−1.
Further information regarding the oxidation states and structural environment of copper and gallium in Cu-Ga/SiO2 is obtained by the XAS spectra at the copper and gallium K-edges for the as-prepared catalysts stored under inert conditions (Fig. 1c). The Cu K-edge X-ray absorption near-edge structure (XANES) spectrum of Cu-Ga/SiO2 shows an edge energy at 8979 eV consistent with reduced Cu0 but the near edge features of Cu-Ga/SiO2 are different from the Cu/SiO2 or Cu foil spectrum (Fig. S8†). Fitting of the extended X-ray absorption fine structure (EXAFS) shows the presence of gallium (N = 4 ± 3 at 2.57 Å) and copper (N = 7 ± 2 at 2.54 Å) scattering paths (Table 1 and Fig. S9†), suggesting the formation of a CuGax alloy phase. The XANES spectrum of Cu-Ga/SiO2 at the gallium K-edge shows an edge energy of 10368 eV, which is 5 eV lower than the edge energy of GaIII@SiO2 or Ga2O3, as reference samples (Fig. 1c and S10†). The shift in the edge energy indicates the presence of reduced gallium species, while the feature observed in GaIII@SiO2 or Ga2O3 at 10373 eV is still present, albeit lower in intensity, in Cu-Ga/SiO2 as shown in the first derivative of the XANES spectrum (Fig. 1c). Further analysis and fitting of the EXAFS (Table 1) at the Ga K-edge reveals the presence of Cu neighbors (N = 8 ± 2 at 2.57 Å) consistent with the EXAFS fitting result at the Cu K-edge with part of the gallium forming a metal alloy phase with copper. In addition, there are oxygen neighbors (N = 2 ± 1 at 1.81 Å), which are attributed to remaining isolated GaIII sites (Fig. S11†), consistent with pyridine adsorption and IR spectroscopy (vide supra). Considering the number of oxygen neighbors (N = 4) found in GaIII@SiO2,42 we estimated that approx. 50% of Ga is in the form of GaIII sites in Cu-Ga/SiO2. This indicates formation of CuGax alloy with x ≈ 0.2 based on respective copper (3.88 wt%) and gallium (1.61 wt%) loadings and taking into account that ca. 50% of gallium sites are present in its reduced form.
Overall, the XAS spectra show that reduction of the samples after Cu grafting (500 °C under H2) leads to a partial reduction of GaIII with the formation of CuGax alloys along with remaining GaIII sites. These finding contrast with what was observed for Cu-Ti/SiO2 (ref. 35) and Cu-Zr/SiO2 (ref. 34) prepared in a similar fashion from TiIV and ZrIV single-sites that remained isolated upon Cu nanoparticle formation.
Catalytic performance in CO2 hydrogenation
The catalytic performance of Cu-Ga/SiO2 in CO2 hydrogenation was evaluated at 230 °C under a total pressure of 25 bar and 3:1 H2:CO2. Following exposure to air, the material is reduced at 300 °C under H2 in the reactor prior to catalysis. The effect of contact time on the catalytic activity/selectivity by varying the gas flowrate and the intrinsic formation rates extrapolated to zero contact time are evaluated and compared with Cu/SiO2 and Cu-Zr/SiO2 benchmark materials (Fig. S12 and S13†). Catalytic tests are carried out at conversions below 7%, which are far from thermodynamic equilibrium (15% with a CH3OH selectivity of 43%)6 under the given reaction condition. The intrinsic formation rate for CH3OH is 1.3 g h−1 gCu−1 for Cu-Ga/SiO2 that is 4 times higher than Cu/SiO2 and also slightly higher than Cu-Zr/SiO2 (Fig. 2a). Note that the product formation rates on the support itself (GaIII@SiO2) are below detection limits. The intrinsic CO formation rate of Cu-Ga/SiO2 (0.1 g h−1 gCu−1) is 3 times lower as compared to Cu/SiO2 or Cu-Zr/SiO2 (0.3 g h−1 gCu−1), making Cu-Ga/SiO2 a better catalysts with an improved selectivity to CH3OH products of 93% (CH3OH/DME = 30) with only 7% of CO. The formation of DME likely arise from the subsequent dehydration of CH3OH, indicating a significant Lewis acidity for this support (vide supra).45 Remarkably, Cu-Ga/SiO2 shows a higher CH3OH selectivity than unpromoted Cu/SiO2 (48%) or even Cu-Zr/SiO2 (77%).34 At longer contact times (Fig. S14†), both CH3OH and CO formation rates decrease (with a slightly larger decrease for CH3OH formation rates (Fig. S14†)), suggesting product inhibition for both processes for Cu-Ga/SiO2. The decrease of activity with contact time is a key limiting factor for a high productivity of CH3OH for the Cu-Ga/SiO2 catalyst. The product inhibition is likely associated with the blocking of Lewis acidic GaIII sites by H2O/CH3OH that in turn reduces CH3OH and CO formation rates. Note that in case of Cu/SiO2, where no Lewis acidic (interfacial) sites are present to assist the conversion of CO2, CH3OH and CO formation rates remain independent of contact time. Product inhibition was also observed for the related Cu-Ti/SiO2 and Cu-Zr/SiO2 catalysts. However, the major difference between Cu-Ti/SiO2 or Cu-Zr/SiO2 and Cu-Ga/SiO2 is that in the former cases, the contact time affects the CH3OH formation rates more than CO formation rates, leading to a decrease of CH3OH selectivity. This dramatic decrease of selectivity is not observed for Cu-Ga/SiO2, also indicating that CO likely forms via different mechanisms. A high selectivity toward CH3OH is maintained for Cu-Ga/SiO2 (>89%; taking into account DME that is initially formed from CH3OH) at ca. 3% conversions vs. 71% and 60% selectivity for Cu-Ti/SiO2 and Cu-Zr/SiO2, respectively at the same conversion (Fig. 2b and S15†). After 30 hours of reaction, Cu-Ga/SiO2 deactivates for both CH3OH and CO formation by 75% and 80%, respectively. Analysis of the spent catalyst shows a similar particle size distribution by TEM for Cu-Ga/SiO2 of 4.9 ± 1.6 nm compared to the fresh catalyst (Fig. S16†). The absence of any crystalline phases by powder X-ray diffraction (Fig. S3†) further indicates the absence of significant sintering throughout the catalytic testing. The deactivation of the catalyst could originate from a slightly decreased amount of accessible metal sites as shown by N2O titration of the fresh/spent catalyst (Table S1†).
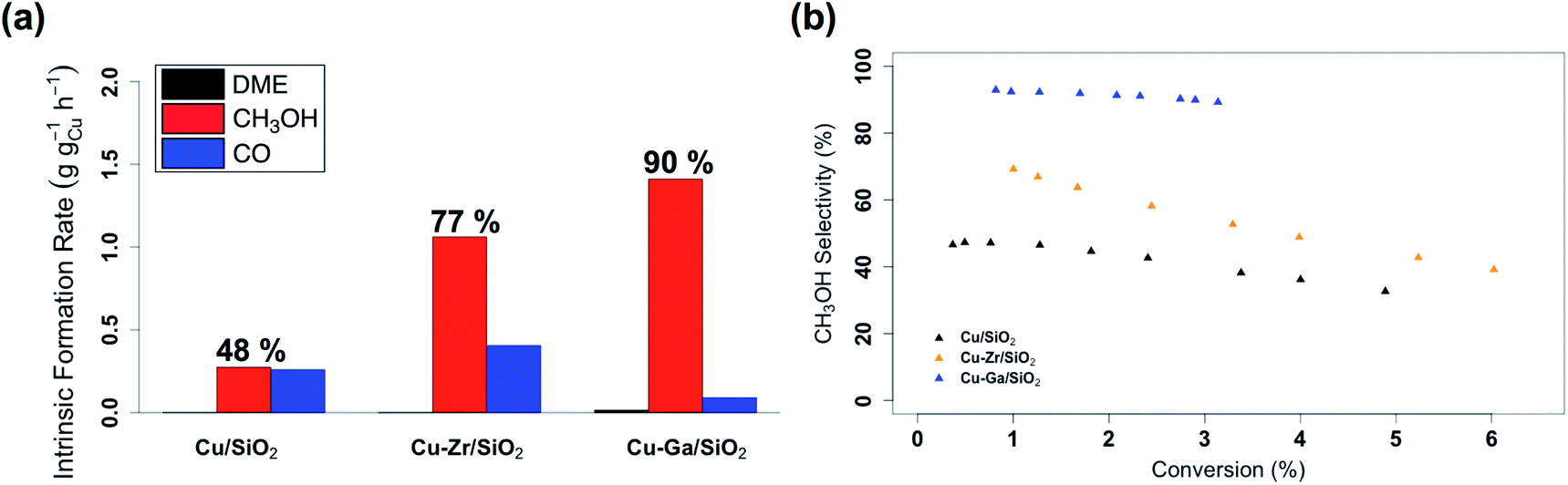 | ||
| Fig. 2 (a) Intrinsic formation rates for CO, CH3OH and DME and (b) overall CH3OH selectivity vs. conversion for Cu/SiO2, Cu-Zr/SiO2 and Cu-Ga/SiO2. | ||
In situ X-ray absorption spectroscopy
The role of gallium, especially the effect of metal alloy formation and its consequence in promoting the selective formation of CH3OH, were further investigated by in situ XAS at the copper K-edge and the gallium K-edges for Cu-Ga/SiO2 (Fig. 3). The X-ray absorption spectra are first recorded after oxidation of the catalyst in air, followed by reduction at 300 °C under H2. The reduced catalyst was then cooled down to 230 °C and the reaction gas consisting of CO2 and H2 (1:3) was introduced at 1 bar and then pressurized to 5 bar.
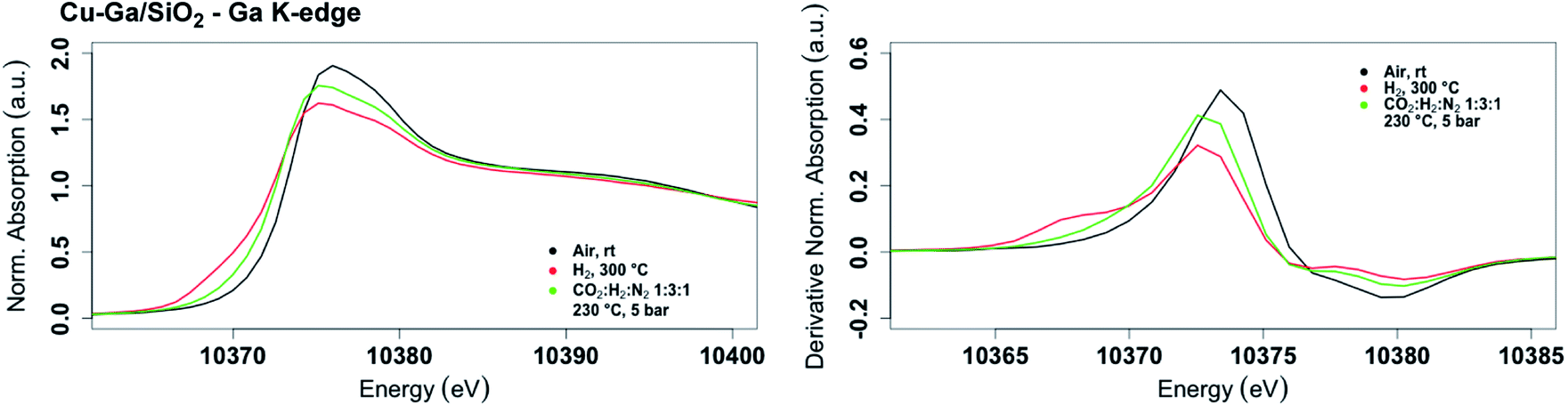 | ||
| Fig. 3
In situ XANES spectrum (left) and its first derivative (right) at the gallium K-edge for Cu-Ga/SiO2 under air at room temperature, reduced at 300 °C under H2 and under reaction conditions at 5 bar with CO2:H2:N2 (1:3:1) at 230 °C. | ||
The XANES spectrum of Cu-Ga/SiO2 at the gallium K-edge after reduction under H2 shows a decreased white line intensity and the appearance of a feature at lower energy (10368 eV) indicative of reduced gallium sites.42 The XANES spectrum has a lower intensity of the feature at that energy (10368 eV) compared to the as-prepared catalyst, indicating that the gallium sites are more difficult to reduce following exposure to air. The feature toward lower energy associated with reduced gallium sites only appears when copper is present and is not observed for GaIII@SiO2 treated under H2 (Fig. S17†). Changes in GaIII@SiO2 during in situ XAS is only due to changes in the oxygen coordination number upon heating at high temperature most likely due to removal of water (due to exposure to air) according to EXAFS fitting (Table S2 and Fig. S18–S20†). The absence of reduced gallium sites for the material without copper (GaIII@SiO2) suggests that copper, most likely in close interaction with gallium, is necessary to reduce GaIII to Ga0. Under reaction conditions at 5 bar (1:3 ratio of CO2:H2), the white line intensity increases and the feature toward lower energy disappears, indicating full oxidation of gallium sites (Fig. 4).
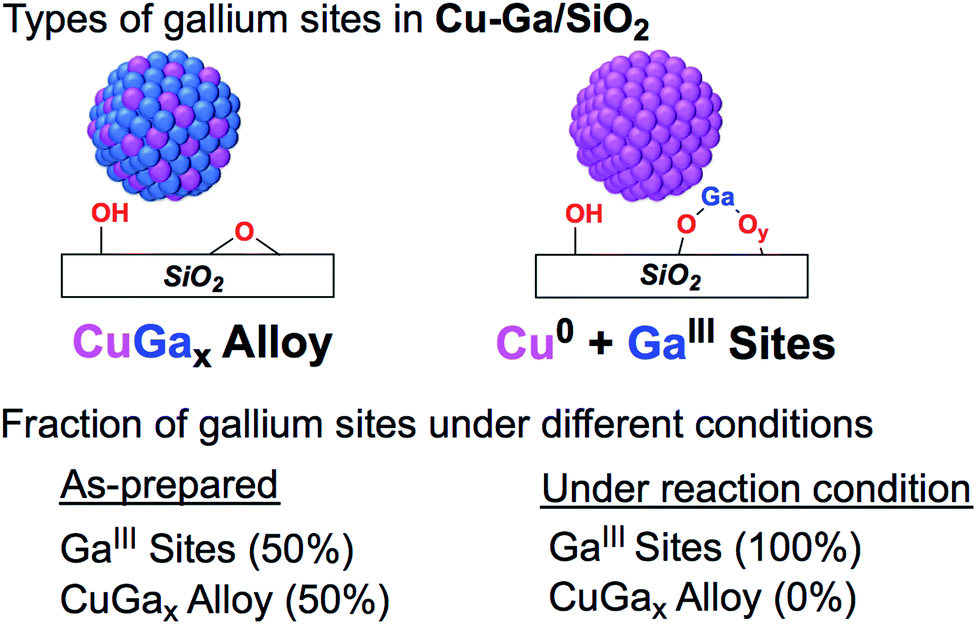 | ||
| Fig. 4 Fraction of gallium sites on Cu-Ga/SiO2 of the as prepared catalyst and under reaction condition according XAS. | ||
Copper is fully reduced upon reaction with H2 at 300 °C and under reaction condition according to XAS (Fig. S21†). Furthermore, Lewis acidic surface sites are present in the catalysts after exposure to air followed by reduction as shown by pyridine adsorption and IR spectroscopy (Fig. S22†). This data suggests that the Lewis acidic sites from GaIII would be responsible for the promotion of CH3OH synthesis, similarly to what is observed for Cu/ZrO2 or related Cu-Zr/SiO2.17,34
Ex situ solid state NMR spectroscopy
In order to determine possible intermediates on gallium under reaction conditions, 1H–13C HETCOR spectra of Cu-Ga/SiO2 (reduced after exposure to air) are recorded after reacting the catalyst with 5 bar of 1H2:13CO2 (3:1) for 12 hours at 230 °C followed by evacuating the gas phase under high vacuum (10−5 mbar) at room temperature. The NMR spectra of Cu-Ga/SiO2 shows a cross-peak at around 3/50 ppm (1H/13C), which is indicative of methoxy species (Fig. S23†) and the presence of dimethyl ether as evidenced by the additional cross-peak at 3/60 ppm (1H/13C) (Fig. S23†), consistent with the observed formation of dimethyl ether during catalysis. Notably, no formate species on Lewis acidic gallium sites (present in the case of Cu/ZrO2, Cu-Zr/SiO2, Cu-Ti/SiO2 or Cu/Al2O3 systems)6,17,34,35 are observed, indicating that GaIII Lewis acidic sites possibly favor the subsequent hydrogenation of formate into methoxy species and/or increase the thermodynamic stability of methoxy in comparison to formate species. This is also confirmed by the IR spectra of the ex situCu-Ga/SiO2 sample after reacting with 1H2/13CO2 (Fig. S24†) showing the 13C–H stretches at around 2954 and 2855 cm−1 indicating the presence of methoxy, while no band associated with formate species are observed. While formate species are also likely formed as reaction intermediates on Cu-Ga/SiO2 under reaction condition, the lower stability of formate compared to methoxy would be consistent with (and explains) the higher CH3OH selectivity of this material in contrasts to other systems. Indeed, stable formate species have been shown to be able to generate methyl formate that readily decomposes into CO.6 Further work is needed to investigate the formation (or not) of formate species as key intermediate in this Cu-Ga/SiO2 system.
Conclusions
The use of SOMC was explored in order to understand the promotional effect of gallium in Cu-based CO2 hydrogenation catalysts, starting from well-defined silica-supported GaIII sites as an initial support. This approach generates small and narrowly distributed silica-supported CuGax nanoparticles along with residual GaIII Lewis acidic sites. This is in contrast to previously studied well-defined isolated ZrIV and TiIV sites on SiO2 that yield Cu nanoparticles surrounded with isolated metal interfacial sites.34,35 These materials are readily oxidized to generate the corresponding CuO and GaIII sites upon exposure to air, but can be partially reduced back to CuGax alloys under H2. These CuGax systems display improved catalytic performances in the hydrogenation of CO2, allowing the increase in the overall CH3OH (CH3OH + DME) selectivity (up to ca. 90%) at higher conversion (3%) by comparison with the benchmark catalysts, Cu-Zr/SiO2 and Cu-Ti/SiO2. Under reaction conditions, the silica-supported CuGax de-alloys yielding Cu nanoparticles and GaIII sites indicating that the increased activity and selectivity is likely due to an increased interfacial area between Cu0 and GaIIIOx that would promote CH3OH formation. In fact, methoxy surface species are the only observed intermediates according to ex situ solid state NMR or IR. This study overall shows the subtle difference between promoters; it opens new ways to tailor CH3OH selective catalysts. We are currently exploring other promoters to understand their role and to design improved CO2 hydrogenation catalysts via a more rational design.Conflicts of interest
There are no conflicts to declare.Acknowledgements
E. L., G. N., K. L., D. L. and P. W. were supported by the SCCER Heat and Energy Storage program (InnoSuisse). We acknowledge PSI Super-XAS for beamtime (proposal #20180825). Scott R. Docherty (ETH Zürich) and Jan Alfke (ETH Zürich) are acknowledged for helpful discussions and assistance in the processing of the XAS data.References
-
G. A. Olah, A. Goeppert and G. K. Surya Prakash, Beyond Oil and Gas: The Methanol Economy, Wiley-VCH, Weinheim, Germany, 2nd edn, 2011 Search PubMed
.
- A. Goeppert, M. Czaun, J.-P. Jones, G. K. Surya Prakash and G. A. Olah, Chem. Soc. Rev., 2014, 43, 7995–8048 RSC
- A. Olah George, Angew. Chem., Int. Ed., 2012, 52, 104–107 CrossRef PubMed
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS
- A. Álvarez, A. Bansode, A. Urakawa, A. V. Bavykina, T. A. Wezendonk, M. Makkee, J. Gascon and F. Kapteijn, Chem. Rev., 2017, 117, 9804–9838 CrossRef
- E. Lam, J. J. Corral-Pérez, K. Larmier, G. Noh, P. Wolf, A. Comas-Vives, A. Urakawa and C. Copéret, Angew. Chem., Int. Ed., 2019, 58, 13989–13996 CrossRef CAS PubMed
- A. Y. Rozovskii and G. I. Lin, Top. Catal., 2003, 22, 137–150 CrossRef CAS
- K. T. Jung and A. T. Bell, Catal. Lett., 2002, 80, 63–68 CrossRef CAS
- A. Baiker, M. Kilo, M. Maciejewski, S. Menzi and A. Wokaun, Stud. Surf. Sci. Catal., 1993, 75, 1257–1272 CrossRef
- M. Behrens, F. Studt, I. Kasatkin, S. Kühl, M. Hävecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B.-L. Kniep, M. Tovar, R. W. Fischer, J. K. Nørskov and R. Schlögl, Science, 2012, 336, 893–897 CrossRef CAS
- J. Schumann, T. Lunkenbein, A. Tarasov, N. Thomas, R. Schlögl and M. Behrens, ChemCatChem, 2014, 6, 2889–2897 CrossRef CAS
- J. D. Grunwaldt, A. M. Molenbroek, N. Y. Topsøe, H. Topsøe and B. S. Clausen, J. Catal., 2000, 194, 452–460 CrossRef CAS
- S. Kuld, M. Thorhauge, H. Falsig, C. F. Elkjær, S. Helveg, I. Chorkendorff and J. Sehested, Science, 2016, 352, 969–974 CrossRef CAS
- T. Lunkenbein, J. Schumann, M. Behrens, R. Schlögl and M. G. Willinger, Angew. Chem., Int. Ed., 2015, 54, 4544–4548 CrossRef CAS PubMed
- J. Toyir, P. Ramírez de la Piscina, J. L. G. Fierro and N. s. Homs, Appl. Catal., B, 2001, 34, 255–266 CrossRef CAS
- J. Toyir, P. Ramírez de la Piscina, J. L. G. Fierro and N. s. Homs, Appl. Catal., B, 2001, 29, 207–215 CrossRef CAS
- K. Larmier, W. C. Liao, S. Tada, E. Lam, R. Verel, A. Bansode, A. Urakawa, A. Comas-Vives and C. Copéret, Angew. Chem., Int. Ed., 2017, 56, 2318–2323 CrossRef CAS
- K. Samson, M. Śliwa, R. P. Socha, K. Góra-Marek, D. Mucha, D. Rutkowska-Zbik, J. F. Paul, M. Ruggiero-Mikołajczyk, R. Grabowski and J. Słoczyński, ACS Catal., 2014, 4, 3730–3741 CrossRef CAS
- J. F. Edwards and G. L. Schrader, J. Phys. Chem., 1984, 88, 5620–5624 CrossRef CAS
- T. Fujitani, I. Nakamura, T. Uchijima and J. Nakamura, Surf. Sci., 1997, 383, 285–298 CrossRef CAS
- O. Martin, C. Mondelli, A. Cervellino, D. Ferri, D. Curulla-Ferré and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2016, 55, 11031–11036 CrossRef CAS PubMed
- F. Arena, G. Italiano, K. Barbera, S. Bordiga, G. Bonura, L. Spadaro and F. Frusteri, Appl. Catal., A, 2008, 350, 16–23 CrossRef CAS
- E. Lam, K. Larmier, S. Tada, P. Wolf, O. V. Safonova and C. Copéret, Chin. J. Catal., 2019, 40, 1741–1748 CrossRef
- S. Kuld, C. Conradsen, P. G. Moses, I. Chorkendorff and J. Sehested, Angew. Chem., Int. Ed., 2014, 53, 5941–5945 CrossRef CAS PubMed
- A. Le Valant, C. Comminges, C. Tisseraud, C. Canaff, L. Pinard and Y. Pouilloux, J. Catal., 2015, 324, 41–49 CrossRef CAS
- C. Tisseraud, C. Comminges, T. Belin, H. Ahouari, A. Soualah, Y. Pouilloux and A. Le Valant, J. Catal., 2015, 330, 533–544 CrossRef CAS
- J. Nakamura, Y. Choi and T. Fujitani, Top. Catal., 2003, 22, 277–285 CrossRef CAS
- P. C. K. Vesborg, I. Chorkendorff, I. Knudsen, O. Balmes, J. Nerlov, A. M. Molenbroek, B. S. Clausen and S. Helveg, J. Catal., 2009, 262, 65–72 CrossRef CAS
- P. L. Hansen, J. B. Wagner, S. Helveg, J. R. Rostrup-Nielsen, B. S. Clausen and H. Topsøe, Science, 2002, 295, 2053–2055 CrossRef CAS
- J. C. Medina, M. Figueroa, R. Manrique, J. Rodríguez Pereira, P. D. Srinivasan, J. J. Bravo-Suárez, V. G. Baldovino Medrano, R. Jiménez and A. Karelovic, Catal. Sci. Technol., 2017, 7, 3375–3387 RSC
- C. Copéret, Acc. Chem. Res., 2019, 52, 1697–1708 CrossRef
- C. Copéret, Nat. Energy, 2019, 4, 1018–1024 CrossRef
- C. Copéret, A. Comas-Vives, M. P. Conley, D. P. Estes, A. Fedorov, V. Mougel, H. Nagae, F. Núñez-Zarur and P. A. Zhizhko, Chem. Rev., 2016, 116, 323–421 CrossRef
- E. Lam, K. Larmier, P. Wolf, S. Tada, O. V. Safonova and C. Copéret, J. Am. Chem. Soc., 2018, 140, 10530–10535 CrossRef CAS PubMed
- G. Noh, E. Lam, J. L. Alfke, K. Larmier, K. Searles, P. Wolf and C. Copéret, ChemSusChem, 2019, 12, 968–972 CrossRef CAS PubMed
- K. H. Lee and J. S. Lee, Korean J. Chem. Eng., 1995, 12, 460–465 CrossRef CAS
- C. Liu, X. Guo, Q. Guo, D. Mao, J. Yu and G. Lu, J. Mol. Catal. A: Chem., 2016, 425, 86–93 CrossRef CAS
- S. Kattel, B. Yan, Y. Yang, J. G. Chen and P. Liu, J. Am. Chem. Soc., 2016, 138, 12440–12450 CrossRef CAS
- G. Noh, S. R. Docherty, E. Lam, X. Huang, D. Mance, J. L. Alfke and C. Copéret, J. Phys. Chem. C, 2019, 123, 31082–31093 CrossRef CAS
- K. Searles, K. W. Chan, J. A. Mendes Burak, D. Zemlyanov, O. Safonova and C. Copéret, J. Am. Chem. Soc., 2018, 140, 11674–11679 CrossRef CAS PubMed
- J. P. Dombrowski, G. R. Johnson, A. T. Bell and T. D. Tilley, Dalton Trans., 2016, 45, 11025–11034 RSC
- K. Searles, G. Siddiqi, O. V. Safonova and C. Copéret, Chem. Sci., 2017, 8, 2661–2666 RSC
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS
- E. P. Parry, J. Catal., 1963, 2, 371–379 CrossRef CAS
- R. S. Schiffino and R. P. Merrill, J. Phys. Chem., 1993, 97, 6425–6435 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc00465k |
| This journal is © The Royal Society of Chemistry 2020 |