
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal–ligand cooperativity in the soluble hydrogenase-1 from Pyrococcus furiosus†
Gregory E.
Vansuch
a,
Chang-Hao
Wu
bc,
Dominik K.
Haja
b,
Soshawn A.
Blair
d,
Bryant
Chica
ae,
Michael K.
Johnson
d,
Michael W. W.
Adams
bd and
R. Brian
Dyer
 *a
*a
aDepartment of Chemistry, Emory University, Atlanta, Georgia 30222, USA. E-mail: briandyer@emory.edu
bDepartment of Biochemistry & Molecular Biology, University of Georgia, Athens, Georgia 30602, USA
cAskGene Pharma Inc., Camarillo, CA 93012, USA
dDepartment of Chemistry, University of Georgia, Athens, Georgia 30602, USA
eBiosciences Center, National Renewable Energy Laboratory, Golden, Colorado 80401, USA
First published on 30th July 2020
Abstract
Metal–ligand cooperativity is an essential feature of bioinorganic catalysis. The design principles of such cooperativity in metalloenzymes are underexplored, but are critical to understand for developing efficient catalysts designed with earth abundant metals for small molecule activation. The simple substrate requirements of reversible proton reduction by the [NiFe]-hydrogenases make them a model bioinorganic system. A highly conserved arginine residue (R355) directly above the exogenous ligand binding position of the [NiFe]-catalytic core is known to be essential for optimal function because mutation to a lysine results in lower catalytic rates. To expand on our studies of soluble hydrogenase-1 from Pyrococcus furiosus (Pf SH1), we investigated the role of R355 by site-directed-mutagenesis to a lysine (R355K) using infrared and electron paramagnetic resonance spectroscopic probes sensitive to active site redox and protonation events. It was found the mutation resulted in an altered ligand binding environment at the [NiFe] centre. A key observation was destabilization of the Nia3+–C state, which contains a bridging hydride. Instead, the tautomeric Nia+–L states were observed. Overall, the results provided insight into complex metal–ligand cooperativity between the active site and protein scaffold that modulates the bridging hydride stability and the proton inventory, which should prove valuable to design principles for efficient bioinspired catalysts.
Introduction
Metalloenzymes activate stable small molecules (H2, N2, CO2) for rapid, specific, and efficient redox chemistry,1,2 characteristics that are critical to establish in bioinspired systems.3–6 Consequently, searching for design principles of metalloenzymes has been an area of significant interest. It has long been apparent that cooperative interactions between the metal centre and protein scaffold are critical to enzyme function,2,7–9 and a well-established approach to probe structure–function relationships is to make mutations to the protein scaffold and relate changes of enzymatic activity to structural perturbations near the active site. Complementary spectroscopic probes of the active site can reveal changes of electronic structure and dynamics related to the enzyme activity. Our laboratory has used these approaches to study soluble hydrogenase 1 from Pyrococcus furiosus (Pf SH1),10 a model hydrogenase (H2ase) enzyme that catalyses the reversible oxidation of molecular hydrogen: H2 ↔ 2H+ + 2e−.Pf SH1 belongs the class of H2ases with the [NiFe] active site depicted in Fig. 1a,11,12 and more specifically to the group 3b cytoplasmic [NiFe]-H2ases that use NADP(H) as an external electron donor/acceptor during cellular metabolism.11,13,14 Although various [NiFe]-H2ases have adopted distinct features for optimal function in particular ecological and cellular environments,15,16 it is believed they utilize a similar mechanistic framework that includes (1) H2 diffusion along hydrophobic gas channels between the buried active site and protein surface, (2) electron transfer along a chain of iron–sulphur clusters between the active site and external donors/acceptors, and (3) proton transfer along a series of water molecules and amino acid residues between the active site and external environment.17 The [NiFe]-H2ases also share a common set of intermediate states that constitute the catalytic cycle,16,18 and a minimal sketch of turnover is depicted in Fig. 1b.19–26
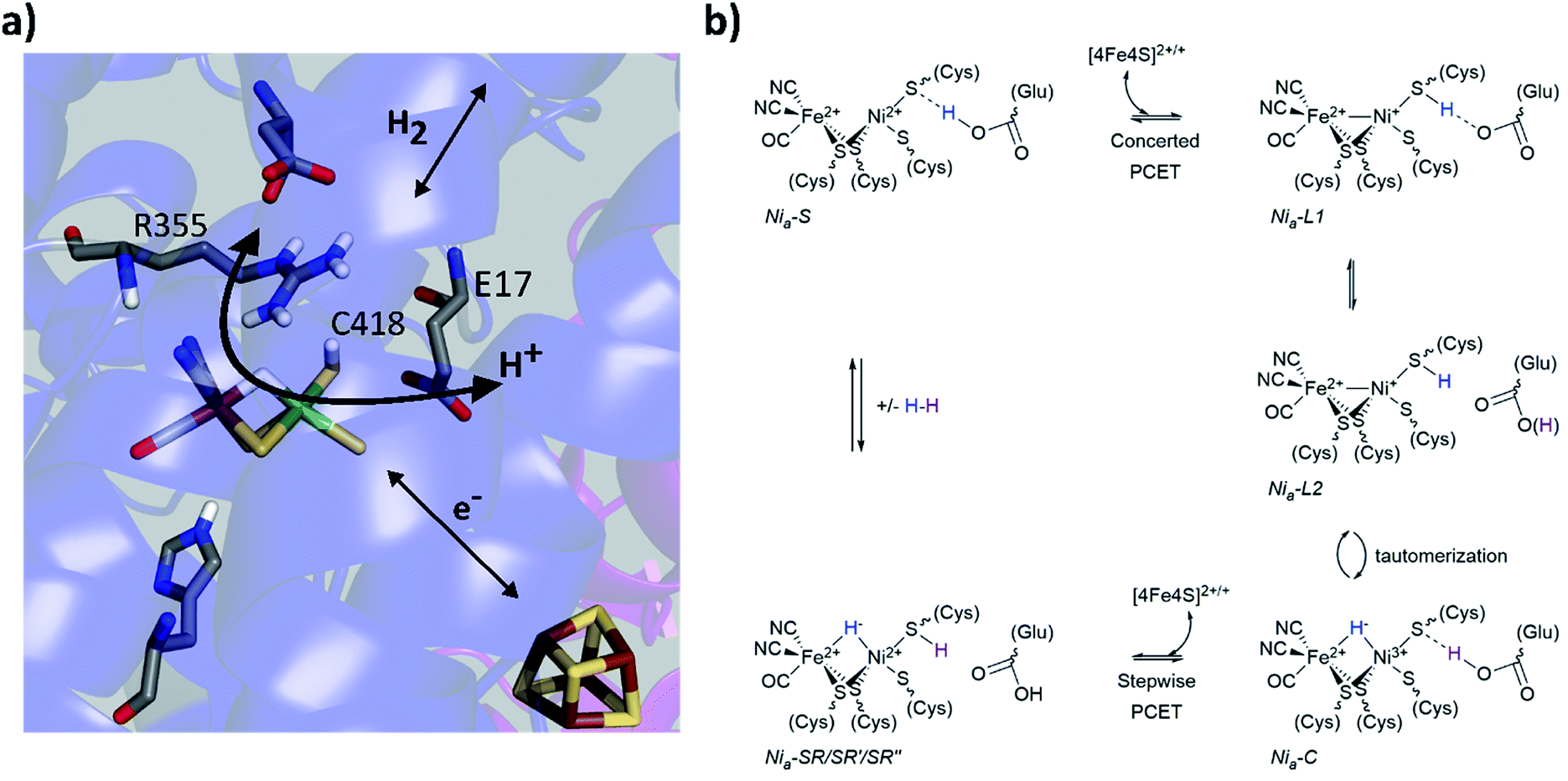 | ||
| Fig. 1 (a) The [NiFe]-active site based on the Nia2+–SR crystal structure from Desulfovibrio vulgaris Miyazaki F (PDB code 4U9I). Select conserved amino acid residues in the second coordination sphere are displayed, as well as the iron sulphur cluster proximal to the active site. Colour code: carbon (grey); nitrogen (blue); oxygen (red); hydrogen (white); sulphur (yellow), nickel (green) and iron (brown). (b) one proposed elementary proton-coupled electron transfer (PCET) mechanism at the [NiFe]-hydrogenase active site that is largely based on mechanistic investigations of Pf SH1. For simplicity, the proton limited Nia2+–SR substrates are listed with Nia2+–SR. | ||
Various aspects of the primary and secondary coordination sphere fine tune the electronic structure25,27–31 and further aid catalysis by stabilizing redox/protonation states necessary for turnover32 and facilitating electrostatic and dynamic processes for efficient substrate processing.10,17,33–35 Directly relevant to protons and hydrides, the primary sphere stores one proton and the two electrons of H2 as a bridging hydride in the Nia3+–C and Nia2+–SR states.26,36,37 The Nia3+–C state can adopt tautomeric forms that are broadly termed Nia+–L (Fig. 1b), and cryogenic FTIR spectroscopy with the Desulfovibrio vulgaris Miyazaki (Dv MF) enzyme demonstrated C418‡ is protonated in the Nia+–L2 state.23 An electron transfer from Nia+–L1 to an iron–sulphur cluster and a proton transfer to the secondary sphere forms Nia2+–S.16,19,21 The closest ionizable residue to C418 is the highly conserved glutamic acid E17 (Fig. 1a), which is crucial for the Nia2+–S ↔ Nia+–L1 transition because mutation to the non-ionizable amino acid glutamine (E17Q) shuts off the transition in Pf SH1 (ref. 10) and Escherichia coli Hyd-1 (Ec Hyd-1).38 A large H/D kinetic isotope effect observed for the Nia2+–S → Nia3+–C transition in Pf SH1,19 which requires a Nia+–L intermediate, indicated proton tunnelling through a hydrogen bond, meaning that protonated C418 hydrogen bonds to E17 in the Nia+–L1 state, and protonated E17 hydrogen-bonds to C418 in the Nia2+–S state.21,23 Thus, C418 and E17 act as a proton donor/acceptor pair during the Nia2+–S ↔ Nia+–L1 transition as illustrated in Fig. 1b.
Another residue known to be critical for substrate processing is the highly conserved arginine located above the active site exogenous ligand binding position (R355, Fig. 1a). Mutation of this residue to lysine (R355K) in Ec Hyd1 strongly attenuated enzyme activity,34,39 but the exact function(s) of R355 remain unclear17,34,35,40,41 and have yet to be investigated in Pf SH1. Here, we present steady state kinetic and equilibrium spectroscopic studies of the R355K variant in Pf SH1. Our results provided insight into possible roles of R355, and also allowed us to study a redox state under equilibrium conditions that is not accessible in native SH1. Importantly, H/D exchange kinetics indicated that reduced H2 oxidation activity in R355 is not solely a consequence of the initial step of H2 cleavage (Nia2+–S ↔ Nia2+–SR interconversion), and EPR and FTIR signatures demonstrated this mutation influences exogenous ligand binding at the active site. A key finding was the effect of the mutation on the Nia3+–C ↔ Nia+–L tautomeric equilibrium. Minimal Nia3+–C spectroscopic signatures were detected in R355K, which indicated the mutation alters the thermodynamic landscape and affects hydride reactivity. Observation of Nia+–L1 and Nia+–L2 signatures over a wide pH range provided a unique opportunity to further explore the communication between C418 and E17 when the nickel formally adopts a +1 redox state.
Results
Steady state kinetics
Fig. 2a shows the steady state H2 oxidation and H+ reduction rates of R355K compared to native and E17Q Pf SH1 at pH = 8.4 (standard pH values for when measuring Pf SH1 activity are 8.0–8.4 (ref. 13, 14 and 42)) and T = 80 °C (near optimal turnover conditions42). E17Q was utilized as a “control” variant because spectroscopic studies have indicated E → Q minimally perturbs active site electronics,10,24,38 making it a useful “baseline” mutation. The activity of E17Q was 20–30% of native SH1, which was consistent with a previous report.10 The H2 oxidation and H+ reduction activity of R355K was ∼2% and 16% relative to native SH1, respectively. Native and E17Q SH1 had a clear bias for H2 oxidation, with the bias defined as the ratio of the H2 oxidation rate versus the H+ reduction rate (kH2,Ox/kH+,red) at pH = 8.4. R355K had no obvious catalytic bias.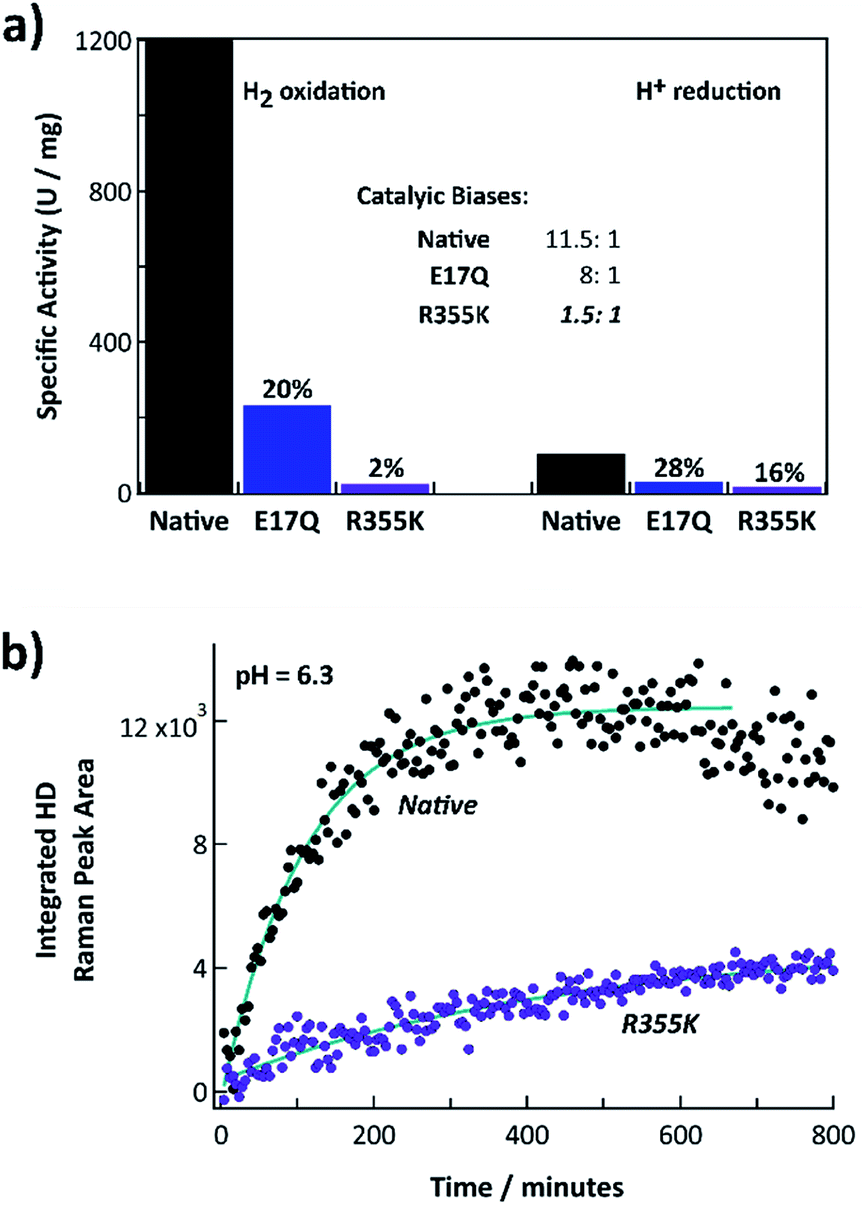 | ||
| Fig. 2 (a) H2 oxidation and H+ reduction activities of native, E17Q, and R355K SH1 at T = 80 °C, pH = 8.4 (100 mM HEPPS buffer). The inset indicates the corresponding catalytic biases of WT, E17Q, and R355K SH1, defined as the ratio of the rate of hydrogen oxidation versus hydrogen production. (b) Time course of H/D formation for native and R355K SH1 in 50 mM KPI and 1.5 mM dithionite at pH = 6.3. Raman spectra for select time points are provided in Fig. S3.† The R355K SH1 plot has been scaled to account for concentration differences (separate plots of native SH1 and R355K H/D exchange without scaling are provided in Fig. S4 and S5†). The wavenumber calibration and an example workup at a specific time point are provided in Fig. S1 and S2.† | ||
We also measured the H/D exchange rate of D2 substrate with native and R355K SH1 in H2O at pH = 6.3 (where it is known Pf SH1 has a large H/D exchange rate43) and T = 20 °C. Raman spectra of the gas headspace was used to follow the conversion of the D2 substrate to HD.43,44 The H/D exchange process is independent of electron transfer because it only requires the Nia2+–S ↔ Nia2+–SR interconversion (Scheme S1†), provided the proton transport pathway is in equilibrium with bulk solution. Exponential fits of the kinetics indicated 13% of the exchange rate was retained for R355K (Fig. 2b).
Electron paramagnetic resonance (EPR) spectroscopy
We utilized EPR spectroscopy to identify paramagnetic intermediates in native and R355K Pf SH1. The [NiFe] active site is known to display rhombic features corresponding to low symmetry S = ½Ni3+ or Ni+ electronic configurations.25,31 Because the γ-subunit of Pf SH1 harbours a [2Fe2S] cluster that exhibits a strong axial signal that overlaps with and masks the typical [NiFe]-hydrogenase gz features, the values of gz were estimated and fixed for the spectral fits,45 which were generated with the MATLAB EasySpin toolbox.46The main EPR feature in native enzyme was a signal with gy ∼ 2.13 and gx ∼ 2.22 for both pH = 6.7 and 9.3 (Fig. 3). We assigned these to Nia3+–C because the g-values closely corresponded to the Nia3+–C feature reported in Pf SH1 by Silva and co-workers,45 and because the difference between gy and gx was not consistent with oxidized/inactive paramagnetic states.31,45 A summary of Pf SH1 EPR work based on results from Silva and co-workers and our results is provided in Table S7.†
 | ||
| Fig. 3 X-Band EPR spectra of native and R355K SH1 (microwave power = 10 mW, temperature = 70 K except for the pH = 6.7 R355K sample, which was run at 50 K in order to detect signal). The gz region is not shown because of an overlapping signal due to a [2Fe2S] cluster (or other cofactor) as described in the text. Because of the stronger native SH1 signals and the sample dependent background in the g < 2.05 region, the maximum value of gy was normalized to 1 for each data set for visual clarity. Fits are located below the data and were scaled by the same factor as the respective spectrum. Individual plots of processed spectra and fits are provided in Fig. S6–S9.† The g-values are listed in Tables S3–S6.† The protein was isolated under anaerobic conditions and stored under 3–4% hydrogen at least one week. The samples were prepared in 25 mM MOPS (pH = 6.7) or 25 mM glycine (pH = 9.3) buffer. | ||
The R355K spectra were different (Fig. 3). The pH = 6.7 sample appeared to have three obvious features, and the pH = 9.3 sample one obvious feature. The pH = 9.3 sample feature, gy ∼ 2.11(6) and gx ∼ 2.36, was consistent with Nia+–L, which is clear by computing Δgy and Δgx between Nia3+–C and Nia+–L in various [NiFe]-hydrogenases (see Table S8†) and because the gy, gx set is not consistent with inactive paramagnetic states.31,45 The pH = 6.7 displayed three clear features. One signal assigned as Ni3+–X was similar to an oxidized inactive feature reported by Silva and co-workers.45 The other two features exhibited a gy value between what is expected for Nia3+–C and Nia+–L (gy ∼ 2.12), and corresponding gx values that more readily distinguish the two features as Nia3+–C (gx ∼ 2.215) and Nia+–L (gx ∼ 2.38, though this signal is overlapped with Ni3+–X and less clear than in the pH = 9.3 sample). Verification of the Nia+–L species was also clear from infrared spectroscopy (discussed below).
Fourier transform infrared (FTIR) spectroscopy
Intermediate states accessible under equilibrium conditions, including diamagnetic ones, were identified using infrared spectroscopy, which is a technique sensitive to the carbon monoxide and two cyanide vibrations of the catalytic core. The IR-detected stretching frequencies shift when the delocalized electron density on and nearby the core is perturbed due to π-backbonding between Fe and the CO and CN ligands. Thus, a specific νCO and νCN symmetric/antisymmetric pair report on a distinct active site electron count and extent of protonation.47 Multiple states caused the νCN region to be highly congested; thus, we relied on the typical νCO region of Pf SH1 (1900–1980 cm−1)19,21,48 to identify distinct states.The FTIR spectrum of native SH1 at pH = 7.2 is shown in Fig. 4a (dashed black line). Similar to what we have observed before under a nitrogen or ∼4% H2 atmosphere after anaerobic purification, the FTIR spectrum is dominated by a feature near 1967 cm−1, corresponding to Nia3+–C.19,21 Minor features corresponded to Nia2+–S (∼1950 cm−1), and Nia2+–SR and subforms18,49 Nia2+–SR′ and Nia2+–SR′′ (∼1954, 1940, and 1931 cm−1). Nia2+–SR′′ probably overlaps with the inactive Nir2+–S state, so both features may be present near 1931 cm−1.19,21,50
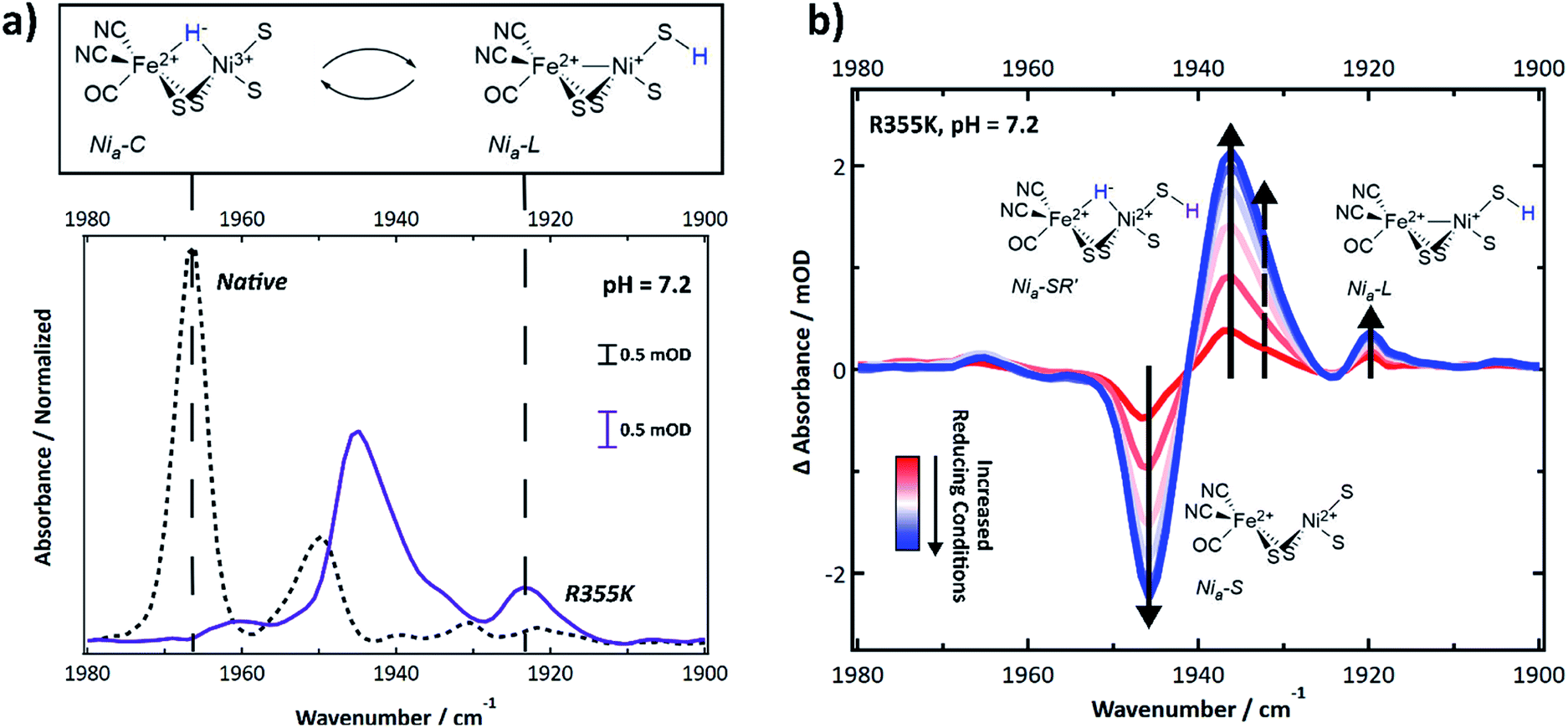 | ||
| Fig. 4 (a) FTIR spectrum of native and R355K SH1 at pH 7.2, prepared after anaerobic purification of the enzyme and storage under 3–4% H2. Native SH1 displays a dominant peak that corresponds to the Nia3+–C state. R355K displays a different distribution of states; unlike native SH1, the Nia3+–C state absorbance is not apparent. Instead, the tautomeric Nia+–L states are observed. The presented structure of Nia+–L assumes identical cysteine based protonation to what has been experimentally demonstrated in Dv MF (see ref. 23). (b) Difference spectra (light–dark) following an equilibrium photochemical reduction of R355K with CdSe/CdS core/shell nanocrystals (OD ∼ 0.15–0.3) and 30 mM 1,1′-trimethyene-2,2′-dipyridium dibromide as a redox mediator (E0 ∼ −550 mV vs. NHE) in 30 mM phosphate/50 mM MPA buffer (pH = 7.2). The illumination wavelength was 405 nm (∼4 mW). Abbreviated first coordination sphere structures are pictured with the corresponding induced absorbance or bleach. The dashed arrow corresponds to Nia-SR′′. A weak induced absorbance signal near 1965 cm−1 may correspond to Nia3+–C. Second derivative (νCO and νCN region) and absorbance spectra (νCO region) are provided in Fig. S10 and S11.† The νCN region of the difference spectra is shown in Fig. S12.† | ||
The R355K spectrum at pH = 7.2 is drastically different (Fig. 4a, solid purple line). The 1967 cm−1 feature due to Nia3+–C was missing. Instead, the tautomeric Nia+–L states were observed near 1920 cm−1,10,19,21 and observation of these states supports the assignment of the R355K Nia+–L EPR feature(s). Significant spectral overlap in the region > 1930 cm−1 complicated the assignment of other states by direct comparison to native SH1. To aid in the assignments, we generated redox difference spectra using an equilibrium photochemical reduction.50 Photoexcitation of CdSe/CdS dot-in-rod nanocrystals in the solution rapidly reduced a redox mediator (1,1′-trimethyene-2,2′-dipyridium dibromide, E0 = −550 mV vs NHE (ref. 51)), which caused the solution potential to decrease.52 The enzyme then equilibrated with the more reducing conditions. The spectral changes due to reduction are shown in the (light–dark) difference spectra (Fig. 4b). A strong bleach near 1946 cm−1 was assigned to depletion of the enzyme resting state, Nia2+–S. The strong induced absorbance near 1936 cm−1 was assigned to an increased population of the fully reduced state, Nia2+–SR′. There was also a small induced absorbance near 1920 cm−1 corresponding to Nia+–L. We attributed the very weak induced absorbance near 1965 cm−1 to Nia3+–C.
FTIR signatures of R355K were further investigated from pH 6.7 to 9.3. The normalized spectra and corresponding multicomponent Voigt fits are shown in Fig. 5a. It is clear that the 1944.8 ± 0.8 cm−1 feature (Nia2+–S, red) decreases in relative amplitude with increasing pH, whereas the 1939.0 ± 1.42 cm−1§ feature (Nia2+–SR′, light blue) increased in relative amplitude with increasing pH. The summed areas of these two features had no obvious pH dependence and after scaling for oscillator strength differences,53 corresponded to 33–41% of the total peak area at full width half maximum (see Table S12†). Plotting the population of these two states versus the estimated H2/H+ couple at each pH value shows these features appear in a redox window expected of active states (Fig. 5b),20,31,54 which further supports their assignments as Nia2+–S and Nia2+–SR′. In conjunction with our ability to observe R355K turnover from steady state kinetics and even methyl viologen reduction under mild conditions (e.g. 5% H2 and heat, see Fig. S28 and S29†) it is clear active states are accessible between pH = 6.7 and 9.3 under the conditions used.
 | ||
| Fig. 5 (a) FTIR spectra of R355K SH1 from pH 6.7–9.3 samples prepared under ∼4% H2 after anaerobic purification of the enzyme and storage under a 3–4% H2 for at least one week. Samples were prepared in in 25 mM MOPS (pH 6.7), 50 mM Tris (pH 7.5), 50 mM HEPPS (pH 8.0 and 8.5), or 25 mM glycine (pH 9.3) buffer. The pH 7.2 sample is identical to that from Fig. 4a. Spectra were normalized such that the area under the curve between 1880–2000 cm−1 was equal to 1 (see ESI†). Non-normalized spectra are provided in Fig. S14,† and second derivative spectra are presented in Fig. S13.† Individual normalized spectra are presented in Fig. S15–S20.† The peak positions, peak areas at full-width half maximum, and oscillator-strength corrected peak areas from the multicomponent Voigt spectral fits are provided in Tables S10–S12.† Individual components of the resultant spectral fits are plotted and colour coded by state as indicated in the legend. (b) The pH dependent populations of the Nia2+–S, Nia2+–SR′, Nia+–L1, and Nia+–L2 states from the oscillator strength corrected peak areas from Table S12;† the top axis estimates the H2/H+ couple of the cell for a given pH assuming 4% H2 (see ESI†). (c) Zoomed in spectra that focuses on the Nia+–L1 and Nia+–L2 states. | ||
Nia2+–SR′′ and Nir2+–S (pink) were observed near 1933.5 ± 0.6 cm−1. Nia+–L1 (yellow) was observed at 1917.8 ± 1.17 cm−1 and Nia+–L2 (brown) was observed at 1923.4 ± 0.8 cm−1. The weak feature observed near 1952 cm−1 may correspond to Nia2+–SR (dark blue); however, it did not display an obvious induced absorbance during the photochemical reduction that would be expected for Nia2+–SR,50 so it is likely an inactive state. At least one state was consistently observed >1955 cm−1 (grey). Because the feature did not change significantly during the photochemical (Fig. 4b, S10 and S11†), it likely corresponds to an inactive state that does not reactivate under the experimental conditions.
The summed populations of Nia+–L1 and Nia+–L2 had no obvious pH dependence and were ∼5–9% of the total peak area (Table S12†). However, the equilibrium between Nia+–L1 and Nia+–L2 exhibited a clear pH dependence (Fig. 5b and c). One possible explanation for the pH dependence would be an acid/base equilibrium involving formation of a hydrogen bond between C418 and the deprotonated form of E17 (ref. 21) (CysSH + HOGlu ↔ CysSH–OGlu; see Fig. 1b. This assumes Pf SH1 exhibits the same type of terminal cysteine protonation observed in Dv MF23). However, the data do not appear to be a simple monoprotic acid–base equilibrium. This is not surprising since E17 probably interacts with other protein residues, which will affect its interaction with the active site. We further address this in the discussion section.
Sample oxidation was monitored by two methods, one at pH 8.5 and the other at pH 9.3. In the first method (pH 8.5), the as prepared sample under ∼4% H2 was subjected to a temperature ramp between 8–70 °C which results in enzyme auto-oxidation through enzyme consumption of the hydrogen and/or loss of H2 from the infrared cell. As shown in Fig. S21,† the feature we have assigned as Nia2+–SR′ decreased in intensity as the temperature increased, and the Nia2+–S feature increased in intensity, which is exactly what is expected during auto-oxidation. In the second method (pH 9.3), the sample cell was stored in the air, which allowed the enzyme to oxidize over time. FTIR spectra were measured periodically over the course of a month (Fig. S22 and S23†). There was a growth of two states between ∼1958–1970 cm−1 as the sample adopted more oxidizing conditions (and perhaps 1950 cm−1 as well, see Fig. S22, S23 and Table S14†). Such peaks are consistent with known inactive states of native Pf SH1 near 1948, 1960, and 1963 cm−1.50 Under highly oxidized conditions, a peak we are unable to assign appeared near 2012 cm−1 (and perhaps 1998 cm−1). Interestingly, an unassigned and unusually high frequency state was recently observed at 1993 cm−1 in a similar [NiFe]-hydrogenase, Hydrogenophilus thermoluteolus TH-1T, under oxidized (as isolated) conditions.55
Exogenous carbon monoxide binding to R355K was also compared to CO binding in native and E17Q Pf SH1 at pH 8.0 (Fig. 6). Exogenous CO is known to bind terminally to the nickel atom56,57 and exhibit a distinct νCO peak around ∼2050–2060 cm−1 depending on the exact enzyme.31,57–59 Both native and E17Q SH1 displayed exogenous νCO peaks near 2044 cm−1 with a corresponding endogenous νCO peak near 1946 cm−1. These peak positions are consistent with a minor redshift of endogenous νCO relative to Nia2+–S (∼1950 cm−1 in native SH1) and the location of exogenous νCO in [NiFe]-hydrogenases following CO binding.31,57–59
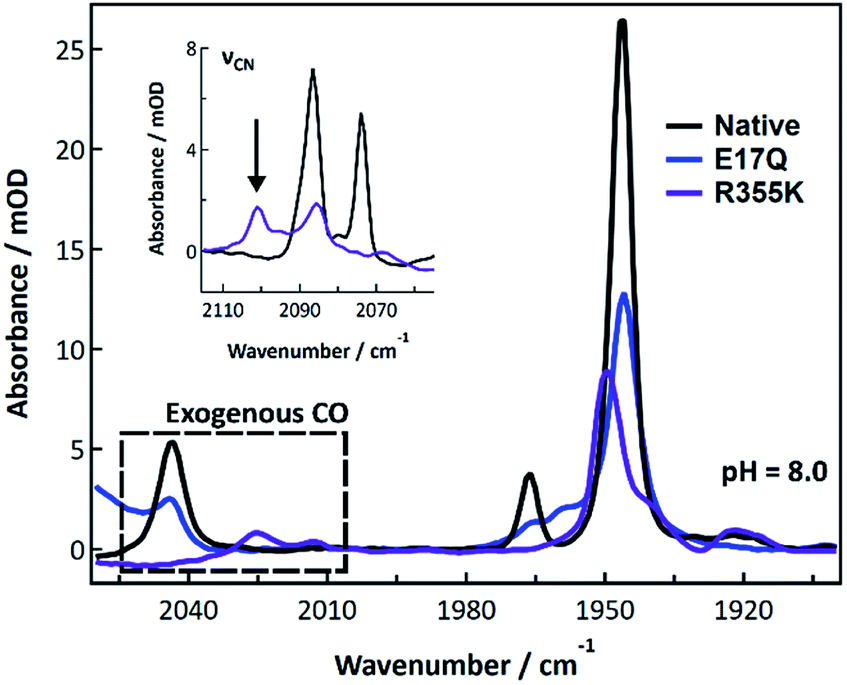 | ||
| Fig. 6 FTIR spectra of native, E17Q, and R355K SH1 at pH = 8.0 (50 mM HEPES or HEPPS buffer) after incubation with carbon monoxide. Both native and E17Q SH1 displayed similar endogenous and exogenous νCO peaks. The R355K spectrum displayed a broad distribution of endogenous νCO peaks, with a major peak located near 1950 cm−1. Two exogenous νCO peaks were located near 2013 and 2025 cm−1. Individual spectra, the corresponding second derivatives, and spectral fits are provided in Fig. S24–S27.† Peak positions are tabulated in Tables S15 and S16.† | ||
In contrast, the R355K spectrum showed a major endogenous νCO peak near 1949.5 cm−1, which is blue-shifted from the Nia2+–S peak of the variant (∼1946 cm−1). Surprisingly, two νCO features were observed near 2013 and 2025 cm−1, likely due to exogenous CO binding. The presence of Fe bound CO would be expected to give a significant trans effect, and the crystal structure of bound CO in the R355K variant from Ec Hyd-2 showed CO is terminally bound to the nickel atom.60 Therefore, it is likely these peaks are due to exogenous CO bound to nickel, although we cannot rule out a weak interaction with the iron. The νCN region also provides evidence for significant alteration of R355K SH1 behaviour in the presence of exogenous CO. A critical observation is shown in the inset of Fig. 6, where a feature near 2100 cm−1 is indicative of at least one cyanide frequency being blue-shifted compared to the Nia2+–S state, which is not seen in native SH1, nor E17Q (see Fig. S25–S27†). This is an unusually high frequency νCN feature for almost all known active, inactive, and CO bound enzyme states.10,19,21,31
Discussion
We have explored the role of the secondary sphere residue R355 in the soluble [NiFe]-hydrogenase SH1 from the hyperthermophilic organism Pyrococcus furiosus by mutating this residue to a lysine. Mutagenesis can sometimes cause dramatic changes to enzyme properties that are not obviously linked to changes in structure,61 making spectroscopic and kinetic studies necessary for mechanistic insight. Here, our steady state kinetic and equilibrium spectroscopic studies of R355K have provided new understanding of Pf SH1 catalysis.10,19,21 An important consideration is that despite extensive efforts over at least three decades, it has not been possible to crystallize Pf SH1; however, its similar spectroscopic behaviour compared to other [NiFe]-H2ases implies a similar structure to those studied by X-ray crystallography.18,19,48 Thus, we often support conclusions from our FTIR and EPR studies based on structure–function and electronic/computational studies of other [NiFe]-H2ases, especially the extensively characterized group 1 Dv MF and Ec Hyd-1 enzymes. However, it remains to be demonstrated that the specific mechanistic conclusions for Pf SH1 can be extended to other [NiFe] enzymes.Our kinetic assays showed R355K exhibits lower H2 oxidation and H+ reduction activity than native Pf SH1 (Fig. 2a). These results are similar to those for Ec Hyd-1,34,39 although the residual activity in SH1 is larger. Attenuated H/D exchange (Fig. 2b) demonstrated the mitigated activity is associated with active site properties. While it is somewhat surprising the H2 oxidation and H/D exchange rates of R355K are 2% and 13% of native enzyme respectively, the active site transformations of H/D exchange are more limited than what is necessary for H2 oxidation coupled with reduction of an external redox mediator. H2 oxidation involves H2 cleavage, product release, and fundamental active site redox chemistry via formation of Nia2+–S, Nia+–L, Nia3+–C, and Nia2+–SR/SR′/SR′′. In contrast, the H/D exchange process only involves Nia2+–S and Nia2+–SR (Scheme S1†), and simply demonstrates that R355K SH1 can cleave D2 and H2, exchange protons and deuterons with bulk solution, and release HD product or the double exchanged H2 product to solution. These differences make it difficult to compare the H/D exchange rates directly to the proton reduction/hydrogen oxidation rates. Nevertheless, both assays clearly demonstrated mitigated activity of R355K versus native Pf SH1; but they do not provide insight into how the R → K mutation affects the active site. Thus, we turn to the spectroscopic signatures that directly report on active site chemistry.
We first consider the FTIR spectra of external CO binding. We observed one exogenous νCO band in native and E17Q SH1, and the endogenous νCO and νCN bands were slightly redshifted relative to Nia2+–S, as observed for other [NiFe]-hydrogenases.31,57–59 In contrast, R355K exhibited exogenous νCO and νCN bands blueshifted relative to Nia2+–S. Furthermore, two exogenous νCO bands were observed at lower energy compared to the one exogenous band in native and E17Q SH1 (∼2013 and 2025 cm−1versus 2046 cm−1; Fig. 6). This shift is indicative of greater π-backbonding to the exogenous CO, and thus stronger binding of CO to the active site relative to native and E17Q SH1. Computational studies have indicated inclusion of R355 is critical for modelling exogenous CO ligand binding in [NiFe]-hydrogenases, which was attributed primarily to steric effects.59,62 When considering these former computational analyses together with the differences in the FTIR spectrum of exogenous CO bound R355K, it is clear R355 influences binding of the (albeit sterically demanding) external CO ligand.
The importance of R355 for ligand binding extends to the catalytically relevant Nia3+–C state and tautomeric Nia+–L states. Both here and in other reports, activated native SH1 has clear equilibrium IR and EPR detectable signatures diagnostic of Nia3+–C (Fig. 3 and 4a),19,21,45,48 with minimal to no Nia+–L, which is only observed in trace amounts at pH ≥ 8.5.21 In contrast, only a minor amount of Nia3+–C was observed for R355K in the EPR spectra and equilibrium photochemical reduction FTIR difference spectra (Fig. 3 and 4b). Although Nia2+–S and Nia2+–SR′ comprised the bulk of the IR detected spectral signatures, we also observed the Pf SH1 Nia3+–C tautomers, Nia+–L(1/2), at room temperature over a broad pH range (Fig. 5). The clear similarity of the IR signatures of Nia+–L in native19,21,50 and R355K highly suggests Nia+–C is destabilized relative to Nia+–L.
Nia+–L is not observed in most [NiFe]-hydrogenases under ambient conditions, and the same is true for native Pf SH1. This state is most commonly observed via photolysis of the Nia3+–C hydride under cryogenic conditions to trap the Nia+–L photoproducts.23,36,63 However, the Nia+–L tautomer has been readily observed under ambient conditions in some [NiFe]-hydrogenases,20,64 and the pKa and bond strength of the Nia3+–C hydride has been hypothesized to vary through subtle electronic and/or structural changes to partially explain such observations.18,65 A shift of tautomeric equilibrium to favour base protonation over metal-hydride formation has been demonstrated in a cationic [FeFe] mimic and iron–diphosphine complex via ion-pairing of the protonated base with anionic species such as BF4−.66,67 Furthermore, depending on electronic structure of a nickel atom, there is the possibility of the thermodynamic preference for thiolate protonation over nickel-hydride formation.68,69 The [NiFe] active site is electron rich given the nature of the coordinating ligands, and it is reasonable that R355 would act as a cationic species in an ion pairing like interaction with the hydride ligand in the Nia3+–C state, resulting in preferential hydride formation over cysteine base protonation. Thus, R355 modulates the Nia3+–C ↔ Nia+–L tautomeric equilibrium, and the mutation to lysine allows Nia+–L to be trapped under ambient conditions. Interestingly, the inverse situation is observed in [FeFe]-hydrogenases, in which unusual conditions, site-directed mutagenesis,70,71 or photochemistry at low temperatures72 is required for trapping the terminal iron-hydride.
The pH dependence of Nia+–L(1/2) (Fig. 5) is complex. The simplest model involves an acid/base equilibrium of E17, with a hydrogen bond forming between E17 and C418 when E17 is deprotonated (CysSH + HOGlu versus CysSH–OGlu; see also Fig. 1b).21 However, the rest of the protein scaffold must be considered because E17 is part of a proton transfer network. Thus, its acid base chemistry and conformation(s) are likely coupled to other components of the network which will affect its general properties and interaction with the active site, which would be similar to how the proton transfer pathway residues (and active site) are coupled in [FeFe]-hydrogenases.73–75 This indicates a complex cooperativity between the primary, secondary, and outer coordination sphere that affect active site properties of Pf SH1.
Lastly, another intriguing difference between native and R355K Pf SH1 is that only the proton limited fully reduced states of R355K, Nia2+–SR′ and a minor population of Nia2+–SR′′, were observed in the FTIR spectra (Fig. 4b and 5a). The proton limited Nia2+–SR′ state is observed to increase in population (and Nia2+–S to decrease) as the pH is raised or as the solution potential is decreased in the photochemical reduction experiment. No obvious IR band for the protonated fully reduced Nia2+–SR state was observed at any pH (although the weak inactive state band at 1952 cm−1 might mask the presence of a small population) and none was observed to accumulate in the photochemical reduction experiment. This behaviour is in contrast with native Pf SH1, for which we observe a strong Nia2+–SR band (1954 cm−1) that decreases as the pH is raised from 6.5 to 9.5 and increases as the solution potential is reduced.19,21,50 While exact differences between sub-forms of Nia2+–SR have yet to be determined,18,49,50 the >10 cm−1 redshift of νCO of Nia2+–SR′ compared to Nia2+–SR is consistent with deprotonation of an active site residue,76 with the most likely candidate being C418.26 Both Nia2+–SR and Nia2+–SR′ each has a bridging hydride that is likely destabilized in R355K (in the same manner as observed for the Nia3+–C state), consistent with the mitigated H/D exchange activity. We postulate that the destabilized hydride is readily protonated in the Nia2+–SR state by a nearby donor (possibly C418) to form H2, whereas this donor is deprotonated in Nia2+–SR′, allowing the hydride state to accumulate. Protonation of Nia2+–SR to form H2 would also reform the oxidized Nia2+–S state, which would explain the large amount of Nia2+–S observed at pH > 8 relative to what would be expected based on an estimated solution potential < −400 mV (Fig. 5a and b).54 This model is testable using time-resolved potential-jump experiments to observe the transient formation of Nia2+–SR prior to protonation of the hydride and formation of H2.
Conclusions
We have presented an electron paramagnetic and infrared spectroscopic study of the R355K variant in Pf SH1. The mutant enzyme revealed pieces of the complex cooperativity between the active site metal and protein scaffold. The arginine itself was found to be critical for controlling exogenous ligand binding; with regards to known intermediate states, it was demonstrated to control the Nia3+–C ↔ Nia+–L tautomeric equilibrium, which is relevant to proton/hydride management crucial for substrate processing. Further investigations of metal–ligand cooperativity in hydrogenases and other gas processing metalloenzymes via structure-functional studies should lead to a better understanding of design principles for sustainable bioinspired systems.Author contributions
GEV, BC, and MWWA conceived of the experiments. GEV performed the R355K infrared, CO inhibition infrared, and H/D exchange experiments. CW and DKH prepared and purified the enzyme. CW preformed H2 oxidation/production assays. SAB preformed the EPR experiments. BC performed the native enzyme infrared measurement and developed the specific photochemical reduction methodology used herein. GEV analysed and interpreted the data. RBD, MWWA, and MKJ supervised the experiments. GEV wrote the manuscript and RBD edited manuscript with input from all other authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
GEV thanks Monica Sanchez for the gift of the 1,1′-trimethylene-2,2′-dipyridium dibromide dye, Yuhgene Liu for the synthesis of NCS materials and Dr Ban-Seok Jeong for helpful advice and discussions. This research was funded by NSF grants DMR1808288 and CHE1807865 (awarded to RBD), grants to MKJ from NIH (R37GM062524) and NSF (MRI CHE1827968) and a grant (DE-FG05-95ER20175 to MWWA) from the Division of Chemical Sciences, Geosciences and Biosciences, Office of Basic Energy Sciences of the Department of Energy.References
- F. A. Armstrong and J. Hirst, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 14049–14054 CrossRef CAS
.
- D. C. Rees, Annu. Rev. Biochem., 2002, 71, 221–246 CrossRef CAS
- M. L. Reback, B. Ginovska, G. W. Buchko, A. Dutta, N. Priyadarshani, B. L. Kier, M. L. Helm, S. Raugei and W. J. Shaw, J. Coord. Chem., 2016, 69, 1730–1747 CrossRef CAS
- J. B. Geri, J. P. Shanahan and N. K. Szymczak, J. Am. Chem. Soc., 2017, 139, 5952–5956 CrossRef CAS
- E. M. Nichols, J. S. Derrick, S. K. Nistanaki, P. T. Smith and C. J. Chang, Chem. Sci., 2018, 9, 2952–2960 RSC
- J. A. Laureanti, M. O'Hagan and W. J. Shaw, Sustainable Energy Fuels, 2019, 3, 3260–3278 RSC
- B. L. Vallee and R. J. Williams, Proc. Natl. Acad. Sci. U. S. A., 1968, 59, 498–505 CrossRef CAS
- M. D. Wodrich and X. Hu, Nat. Rev. Chem., 2017, 2, 0099 CrossRef
- A. Hemschemeier and T. Happe, Nat. Rev. Chem., 2018, 2, 231–243 CrossRef CAS
- B. L. Greene, G. E. Vansuch, C.-H. Wu, M. W. W. Adams and R. B. Dyer, J. Am. Chem. Soc., 2016, 138, 13013–13021 CrossRef CAS
- J. W. Peters, G. J. Schut, E. S. Boyd, D. W. Mulder, E. M. Shepard, J. B. Broderick, P. W. King and M. W. W. Adams, Biochim. Biophys. Acta, Mol. Cell Res., 2015, 1853, 1350–1369 CrossRef CAS
-
The PyMOL Molecular Graphics System, version 1.3, Schrödinger, LLC: New York 2010 Search PubMed
- K. Ma, Z. H. Zhou and M. W. W. Adams, FEMS Microbiol. Lett., 1994, 122, 245–250 CrossRef CAS
- D. J. van Haaster, P. J. Silva, P.-L. Hagedoorn, J. A. Jongejan and W. R. Hagen, J. Bacteriol., 2008, 190, 1584–1587 CrossRef CAS
- H. S. Shafaat, O. Rüdiger, H. Ogata and W. Lubitz, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 986–1002 CrossRef CAS
- H. Tai, Y. Higuchi and S. Hirota, Dalton Trans., 2018, 47, 4408–4423 RSC
- J. C. Fontecilla-Camps, A. Volbeda, C. Cavazza and Y. Nicolet, Chem. Rev., 2007, 107, 4273–4303 CrossRef CAS
- P. A. Ash, R. Hidalgo and K. A. Vincent, ACS Catal., 2017, 7, 2471–2485 CrossRef CAS
- B. L. Greene, C. H. Wu, P. M. McTernan, M. W. Adams and R. B. Dyer, J. Am. Chem. Soc., 2015, 137, 4558–4566 CrossRef CAS
- R. Hidalgo, P. A. Ash, A. J. Healy and K. A. Vincent, Angew. Chem., Int. Ed., 2015, 54, 7110–7113 CrossRef CAS
- B. L. Greene, C. H. Wu, G. E. Vansuch, M. W. Adams and R. B. Dyer, Biochemistry, 2016, 55, 1813–1825 CrossRef CAS
- C. Bagyinka, J. P. Whitehead and M. J. Maroney, J. Am. Chem. Soc., 1993, 115, 3576–3585 CrossRef CAS
- H. Tai, K. Nishikawa, Y. Higuchi, Z.-w. Mao and S. Hirota, Angew. Chem., Int. Ed., 2019, 131, 13419–13424 CrossRef
- S. Dementin, B. Burlat, A. L. De Lacey, A. Pardo, G. Adryanczyk-Perrier, B. Guigliarelli, V. M. Fernandez and M. Rousset, J. Biol. Chem., 2004, 279, 10508–10513 CrossRef CAS
- M. Kampa, M.-E. Pandelia, W. Lubitz, M. van Gastel and F. Neese, J. Am. Chem. Soc., 2013, 135, 3915–3925 CrossRef CAS
- H. Ogata, K. Nishikawa and W. Lubitz, Nature, 2015, 520, 571–574 CrossRef
- A. L. De Lacey, V. M. Fernandez, M. Rousset, C. Cavazza and E. C. Hatchikian, J. Biol. Inorg Chem., 2003, 8, 129–134 CrossRef CAS
- A. Müller, I. Tscherny, R. Kappl, E. C. Hatchikian, J. Hüttermann and R. Cammack, J. Biol. Inorg Chem., 2002, 7, 177–194 CrossRef
- M. Kampa, W. Lubitz, M. van Gastel and F. Neese, J. Biol. Inorg Chem., 2012, 17, 1269–1281 CrossRef CAS
- S. Qiu, L. M. Azofra, D. R. MacFarlane and C. Sun, Phys. Chem. Chem. Phys., 2018, 20, 6735–6743 RSC
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS
- A. Abou-Hamdan, P. Ceccaldi, H. Lebrette, O. Gutierrez-Sanz, P. Richaud, L. Cournac, B. Guigliarelli, A. L. De Lacey, C. Léger, A. Volbeda, B. Burlat and S. Dementin, J. Biol. Chem., 2015, 290, 8550–8558 CrossRef CAS
- D. M. A. Smith, S. Raugei and T. C. Squier, Phys. Chem. Chem. Phys., 2014, 16, 24026–24033 RSC
- R. M. Evans, E. J. Brooke, S. A. Wehlin, E. Nomerotskaia, F. Sargent, S. B. Carr, S. E. Phillips and F. A. Armstrong, Nat. Chem. Biol., 2016, 12, 46–50 CrossRef CAS
- A. M. Escorcia and M. Stein, Front. Chem., 2018, 6, 164 CrossRef
- M. Brecht, M. van Gastel, T. Buhrke, F. Barbel and W. Lubitz, J. Am. Chem. Soc., 2003, 125, 13075–13083 CrossRef CAS
- S. Foerster, M. van Gastel, M. Brecht and W. Lubitz, J. Biol. Inorg Chem., 2005, 10, 51–62 CrossRef CAS
- R. M. Evans, P. A. Ash, S. E. Beaton, E. J. Brooke, K. A. Vincent, S. B. Carr and F. A. Armstrong, J. Am. Chem. Soc., 2018, 140, 10208–10220 CrossRef
- E. J. Brooke, R. M. Evans, S. T. Islam, G. M. Roberts, S. A. Wehlin, S. B. Carr, S. E. Phillips and F. A. Armstrong, Biochemistry, 2017, 56, 132–142 CrossRef CAS
- E. Szóri-Dorogházi, G. Maróti, M. Szóri, A. Nyilasi, G. Rákhely and K. L. Kovács, PLoS One, 2012, 7, e34666 CrossRef
- F. Oteri, M. Baaden, E. Lojou and S. Sacquin-Mora, J. Phys. Chem. B, 2014, 118, 13800–13811 CrossRef CAS
- F. O. Bryant and M. W. Adams, J. Biol. Chem., 1989, 264, 5070–5079 CAS
-
B. L. Greene, Ph.D. thesis, Emory University, 2015
- Y. Kawahara-Nakagawa, K. Nishikawa, S. Nakashima, S. Inoue, T. Ohta, T. Ogura, Y. Shigeta, K. Fukutani, T. Yagi and Y. Higuchi, Protein Sci., 2019, 28, 663–670 CrossRef CAS
- P. J. Silva, B. De Castro and W. R. Hagen, J. Biol. Inorg Chem., 1999, 4, 284–291 CrossRef CAS
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS
- M. Y. Darensbourg, E. J. Lyon and J. J. Smee, Coord. Chem. Rev., 2000, 206–207, 533–561 CrossRef CAS
- H. Wang, C. Y. Ralston, D. S. Patil, R. M. Jones, W. Gu, M. Verhagen, M. Adams, P. Ge, C. Riordan, C. A. Marganian, P. Mascharak, J. Kovacs, C. G. Miller, T. J. Collins, S. Brooker, P. D. Croucher, K. Wang, E. I. Steifel and S. P. Cramer, J. Am. Chem. Soc., 2000, 122, 10544–10552 CrossRef CAS
- B. Bleijlevens, F. A. Broekhuizen, A. L. De Lacey, W. Roseboom, V. M. Fernandez and S. P. J. Albracht, J. Biol. Inorg Chem., 2004, 9, 743–752 CrossRef CAS
-
B. C. Chica, Ph.D. thesis, Emory University, 2017
- R. F. Homer and T. E. Tomlinson, J. Chem. Soc., 1960, 2498–2503 RSC
- B. Chica, C.-H. Wu, Y. Liu, M. W. W. Adams, T. Lian and R. B. Dyer, Energy Environ. Sci., 2017, 10, 2245–2255 RSC
- J. O. Alben, P. P. Moh, F. G. Fiamingo and R. A. Altschuld, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 234–237 CrossRef CAS
- C. Fichtner, C. Laurich, E. Bothe and W. Lubitz, Biochemistry, 2006, 45, 9706–9716 CrossRef CAS
- J. Preissler, S. Wahlefeld, C. Lorent, C. Teutloff, M. Horch, L. Lauterbach, S. P. Cramer, I. Zebger and O. Lenz, Biochim. Biophys. Acta, Bioenerg., 2018, 1859, 8–18 CrossRef CAS
- H. Ogata, Y. Mizoguchi, N. Mizuno, K. Miki, S.-i. Adachi, N. Yasuoka, T. Yagi, O. Yamauchi, S. Hirota and Y. Higuchi, J. Am. Chem. Soc., 2002, 124, 11628–11635 CrossRef CAS
- Y. Ilina, C. Lorent, S. Katz, J.-H. Jeoung, S. Shima, M. Horch, I. Zebger and H. Dobbek, Angew. Chem., Int. Ed., 2019, 58, 18710–18714 CrossRef CAS
- K. A. Bagley, C. J. Van Garderen, M. Chen, W. H. Woodruff, E. C. Duin and S. P. J. Albracht, Biochemistry, 1994, 33, 9229–9236 CrossRef CAS
- A. L. De Lacey, C. Stadler, V. M. Fernandez, E. C. Hatchikian, H.-J. Fan, S. Li and M. B. Hall, J. Biol. Inorg Chem., 2002, 7, 318–326 CrossRef CAS
-
S. E. Beaton, Ph.D. thesis, University of Oxford, 2018
- E. Yikilmaz, J. Porta, L. E. Grove, A. Vahedi-Faridi, Y. Bronshteyn, T. C. Brunold, G. E. O. Borgstahl and A.-F. Miller, J. Am. Chem. Soc., 2007, 129, 9927–9940 CrossRef CAS
-
M. Kampa, Ph.D. thesis, Technische Universität Berlin, 2013
- J. W. Van der Zwaan, S. P. J. Albracht, R. D. Fontijn and E. C. Slater, FEBS Lett., 1985, 179, 271–277 CrossRef CAS
- M. Saggu, I. Zebger, M. Ludwig, O. Lenz, B. Friedrich, P. Hildebrandt and F. Lendzian, J. Biol. Chem., 2009, 284, 16264–16276 CrossRef CAS
- M.-E. Pandelia, P. Infossi, M. Stein, M.-T. Giudici-Orticoni and W. Lubitz, Chem. Commun., 2012, 48, 823–825 RSC
- M. E. Carroll, B. E. Barton, T. B. Rauchfuss and P. J. Carroll, J. Am. Chem. Soc., 2012, 134, 18843–18852 CrossRef CAS
- G. M. Chambers, S. I. Johnson, S. Raugei and R. M. Bullock, Chem. Sci., 2019, 10, 1410–1418 RSC
- W. Clegg and R. A. Henderson, Inorg. Chem., 2002, 41, 1128–1135 CrossRef CAS
- R. A. Henderson, J. Chem. Res., Synop., 2002, 407–411 CrossRef CAS
- M. Winkler, J. Duan, J. Esselborn, T. Happe, M. Senger, S. T. Stripp, F. Wittkamp, U.-P. Apfel and E. Hofmann, Nat. Commun., 2017, 8, 16115 CrossRef CAS
- D. W. Mulder, Y. Guo, M. W. Ratzloff and P. W. King, J. Am. Chem. Soc., 2017, 139, 83–86 CrossRef CAS
- C. Lorent, S. Katz, J. Duan, C. J. Kulka, G. Caserta, C. Teutloff, S. Yadav, U.-P. Apfel, M. Winkler, T. Happe, M. Horch and I. Zebger, J. Am. Chem. Soc., 2020, 142, 5493–5497 CrossRef CAS
- J. Duan, M. Senger, J. Esselborn, V. Engelbrecht, F. Wittkamp, U.-P. Apfel, E. Hofmann, S. T. Stripp, T. Happe and M. Winkler, Nat. Commun., 2018, 9, 4726 CrossRef
- M. Senger, V. Eichmann, K. Laun, J. Duan, F. Wittkamp, G. Knör, U.-P. Apfel, T. Happe, M. Winkler, J. Heberle and S. T. Stripp, J. Am. Chem. Soc., 2019, 141, 17394–17403 CrossRef CAS
- C. C. Pham, D. W. Mulder, V. Pelmenschikov, P. W. King, M. W. Ratzloff, H. Wang, N. Mishra, E. E. Alp, J. Zhao, M. Y. Hu, K. Tamasaku, Y. Yoda and S. P. Cramer, Angew. Chem., Int. Ed., 2018, 57, 10605–10609 CrossRef CAS
- A. Pardo, A. L. Lacey, V. M. Fernandez, H.-J. Fan, Y. Fan and M. B. Hall, J. Biol. Inorg. Chem., 2006, 11, 286–306 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: H/D exchange scheme; experimental details and data analysis; native and R355K EPR spectra; table of EPR signals; FTIR absorbance, photochemical difference and second derivative spectra; pH dependent FTIR spectra; tabulation of FTIR normalization values; peak positions; and relevant populations; temperature dependent pH = 8.5 FTIR spectra; pH = 9.3 oxidation FTIR spectra and tabulated peak positions; temperature dependent H2 oxidation of native and R355K SH1 with methyl viologen and 5% H2 UV-vis spectra; additional analyses and discussions. See DOI: 10.1039/d0sc00628a |
| ‡ Amino acid numberings correspond to soluble hydrogenase I from Pyrococcus furiosus (Pf SH1). |
| § Peak positions are presented as the average value and standard deviation of the tabulated positions in Table S7 between pH 6.7–9.3. |
| This journal is © The Royal Society of Chemistry 2020 |