
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTaking lanthanides out of isolation: tuning the optical properties of metal–organic frameworks†
Samantha L.
Anderson‡
 a,
Davide
Tiana‡
ab,
Christopher P.
Ireland‡
a,
Gloria
Capano
a,
Maria
Fumanal
a,
Andrzej
Gładysiak
a,
Stavroula
Kampouri
a,
Aiman
Rahmanudin
c,
Néstor
Guijarro
c,
Kevin
Sivula
c,
Kyriakos C.
Stylianou§
a and
Berend
Smit
*a
a,
Davide
Tiana‡
ab,
Christopher P.
Ireland‡
a,
Gloria
Capano
a,
Maria
Fumanal
a,
Andrzej
Gładysiak
a,
Stavroula
Kampouri
a,
Aiman
Rahmanudin
c,
Néstor
Guijarro
c,
Kevin
Sivula
c,
Kyriakos C.
Stylianou§
a and
Berend
Smit
*a
aLaboratory of Molecular Simulation (LSMO), Institut des Sciences et Ingénierie Chimiques, Valais Ecole Polytechnique Fédérale de Lausanne (EPFL), Rue de l’Industrie 17, CH-1951 Sion, Switzerland. E-mail: berend.smit@epfl.ch
bSchool of Chemistry, University College Cork, College Rd, Cork, Ireland
cLaboratory for Molecular Engineering of Optoelectronic Nanomaterials (LIMNO), Institut des Sciences et Ingénierie Chimiques (ISIC), Ecole Polytechnique Fédérale de Lausanne (EPFL), CH-1015 Lausanne, Switzerland
First published on 20th March 2020
Abstract
Metal organic frameworks (MOFs) are increasingly used in applications that rely on the optical and electronic properties of these materials. These applications require a fundamental understanding on how the structure of these materials, and in particular the electronic interactions of the metal node and organic linker, determines these properties. Herein, we report a combined experimental and computational study on two families of lanthanide-based MOFs: Ln-SION-1 and Ln-SION-2. Both comprise the same metal and ligand but with differing structural topologies. In the Ln-SION-2 series the optical absorption is dominated by the ligand and using different lanthanides has no impact on the absorption spectrum. The Ln-SION-1 series shows a completely different behavior in which the ligand and the metal node do interact electronically. By changing the lanthanide in Ln-SION-1, we were able to tune the optical absorption from the UV region to absorption that includes a large part of the visible region. For the early lanthanides we observe intraligand (electronic) transitions in the UV region, while for the late lanthanides a new band appears in the visible. DFT calculations showed that the new band in the visible originates in the spatial orbital overlap between the ligand and metal node. Our quantum calculations indicated that Ln-SION-1 with late lanthanides might be (photo)conductive. Experimentally, we confirm that these materials are weakly conductive and that with an appropriate co-catalysts they can generate hydrogen from a water solution using visible light. Our experimental and theoretical analysis provides fundamental insights for the rational design of Ln-MOFs with the desired optical and electronic properties.
Introduction
Metal–organic frameworks (MOFs) have now acquired a prominent position in the literature owing to their ability to form porous extended networks, with record breaking surface areas,1 low crystal density,2 and their potential in practical applications such as gas storage and separation.3–5 The inherent tunability of these versatile crystal structures make them an ideal playground to obtain a fundamental understanding of how systematic changes in the chemistry impact the performance or functionality of these materials. While a large body of research has been directed to gas separation and storage, less attention has focused on the optical and electronic properties of MOFs. Insights in these properties is of importance to rationally design MOFs with specific functionalities for emerging applications such as (photo)catalysis,6–8 sensing,9–13 non-linear optics,14,15 ferroelectricity,16 magnetism,17,18 and conductivity.19–24In this work, we focused on lanthanide MOFs (Ln-MOFs). Since the first observation of Ln(III) fluorescence induced by energy transfer from an optically active material,25 lanthanides opto-physical properties have been extensively studied for application in chemosensoring, optical fibers, and fluorescence microscopy.26–30 Their unique properties are a consequence of their electronic configuration. In the most common oxidation state, 3+, lanthanides can be considered as a closed-shell inert, isolated ion:26 the 4f valence orbitals are compact in space and interact little with the external environment. In the presence of optically active components (ligands or the matrix in which they are embedded), the excitation may be transferred via energy transfer to the empty f orbitals of Ln(III), from which the radiative decay to lower lying f orbitals will take place. This effect, called the antenna effect, is exploited in the most common Ln(III)-based materials applications. f–f transitions are allowed by the spectroscopic selection rules, with each lanthanide ion giving rise to a different and specific emission spectrum. This rooted the idea that the optical properties of Ln atoms can be understood in isolation. Indeed, given the interest for the Ln(III) luminescence, most studies focus on understanding the f–f transitions. Only a few studies focus on other excitations such as ligand-to-metal-charge-transfer (LMCT), metal-to-ligand-charge-transfer (MLCT), or metal-to-metal-charge-transfer (MMCT),27,31 but even these studies are discussed in relation with the f–f transitions (e.g., sensing).
The optical properties of lanthanide compounds are therefore usually rationalized based on the notion that the light absorption is fully localized on the ligands and the emission on the lanthanide metal. This is not ideal for all applications. For example, in photocatalysis, we aim to engineer a material that acts not just as an antenna, absorbing solar light, but that is also capable of transferring or localizing the charge (electron or hole) to an active catalytic site within the material. This will imply charge transfer from the optically active ligand to the Ln metal instead of energy transfer. In the present work, we report a study of two families of Ln(III) MOFs (hereafter named as Ln-SION-1 and Ln-SION-2) that are chemically equivalent but that have different structural topologies. We found that for the Ln-SION-2 series the traditional behaviour of ligand-centered absorption and isolated lanthanide ions holds. In sharp contrast, for the Ln-SION-1 series the opto-electronic properties can be tuned by changing the metal, making this system ideal to study the metal–ligand interactions.
We have previously reported the mechanism of the transformation of Tb-SION-2 into Tb-SION-1.32 In this work, we report the synthesis of the entire lanthanide SION-1 and SION-2 families together with a study of their optical absorption properties. In addition, we combine these experiments with a computational study to rationalize the observations. Our computational study demonstrates that by carefully selecting the correct topology and lanthanide/ligand combination we can engineer a material that absorbs light in a significant proportion of the visible spectrum, that promotes charge transfer from the ligand to the metal, and that is (weakly) photoconductive. The focus of this work is to obtain a better understanding of the metal–ligand interactions in Ln-MOFs, but we also show, as a proof of principle, that with an appropriate co-catalyst, this material is also weakly active for photo-catalytic generation of hydrogen from a water-based solution.
Results and discussion
Structure of Ln-SION-1 and Ln-SION-2
Ln-SION-1 and Ln-SION-2 are synthesised with the same Ln ions and ligand (2,5-dihydroxy-1,4-benzenedicarboxylic acid, H2DHBDC), but differ in their crystalline structures (see Fig. 1). The synthesis of the Tb version of this material was reported previously by our group;32 in the ESI† we described the synthesis method (Section 1.1, 1.2) and characterisation (Section 2, 3.1, 3.2) for the new lanthanides MOFs. | ||
| Fig. 1 Structures of Ln-SION-1 and Ln-SION-2 derived from the single crystal measurements: (A) the Ln-SION-1 structure, which shows a ladder-like chain of alternating lanthanide (green spheres) and oxygen atoms along the [111] direction (highlighted). (B) The coordination of the ligand and metal, showing the η1:η2:η2-bridging seen with the –OH groups on the parent H2DHBDC ligand, in addition to the carboxylate groups, involved in the bonding to the metal. (C) The Ln-SION-2 structure, which illustrates the contrast to the Ln-SION-1 structure, in that the metal atoms are linked as metal dimers (highlighted), and the ligand coordinates exclusively via the carboxylate oxygen atoms. (D) The coordination of the metal and ligand in Ln-SION-2, illustrating the binuclear Tb2(COO)6 cluster inter-connected by three distinct DHBDC ligands. | ||
Ln-SION-2 forms a 3-dimensional framework based on binuclear Ln2(COO)6 clusters interconnected by three DHBDC ligands in which the hydroxyl groups do not participate in metal coordination. In contrast, for Ln-SION-1 (Fig. 1C and D) we found alternating Ln(III) ions and O atoms, which form 21-symmetrical chains running along the [010] direction (Fig. 1C). The chain resembles a ladder, and within these ladder-like arrays, two types of ligands are present: the DHBDC ligands with protonated hydroxyl groups bound to the Ln(III) ion through a coordination mode identical with the η1:η1 bridging ligands in Ln-SION-2, and the fully de-protonated DOBDC with a η1:η2:η2 bridging mode making use of all O atoms to bind to the Ln(III). This highlights the primary difference between the structure of the two series. In Ln-SION-2 the hydroxyl groups of the DHBDC ligand do not participate in coordination while they do in Ln-SION-1. As a result, in Ln-SION-2 binuclear metal clusters separated by linkers are clearly distinguishable, whereas Ln-SION-1 forms a one-dimensional chain.
Optical properties
The differences in structure between Ln-SION-1 and Ln-SION-2 have a profound effect on the optical properties. Ln-SION-1 and Ln-SION-2 display different colours; Ln-SION-2 has a gold colour, while Ln-SION-1 is red. As both have the same lanthanide metal node and linker, their different colour suggests that both the lanthanide-based metal node and the H2DHBDC ligand interact with light.The ligand absorbs primarily in the UV-vis region, and is yellow in colour; its practical use in photocatalysis would be significantly enhanced if we could tune the light absorption more towards the visible part of the spectrum. To achieve this, one possibility is to promote electronic coupling between the ligand and the metal node orbitals. This will not only shift the absorption bands but also induce charge transfer upon excitation. The colour differences between the two series already indicate that such a coupling can occur in one of the series. One of the main motivations of this work was to obtain a deeper understanding of this coupling and how this understanding can be exploited to tune the optical properties of MOFs.
In Fig. 2 the diffuse reflectance spectra of the different Ln-SION-1 and Ln-SION-2 MOFs are compared with the free ligand H2DHBDC. The free ligand displays a strong absorption band in the UV to visible spectral region up to 470 nm (2.76 eV). The diffuse reflectance spectrum of Tb-SION-2 displays a comparable absorption band with that of the free H2DHBDC ligand; the slight blue shift seen is due to the ligand now being constrained in the MOF. In Fig. S11† the UV-vis results for the other lanthanides in the series are presented. These results show that changing the Ln(III) within the isostructural Ln-SION-2 family has very little effect on its absorption properties. This indicates that the absorption is fully localized in the ligand (for all Ln) and that there is no mixing between the metal node and the ligand states.
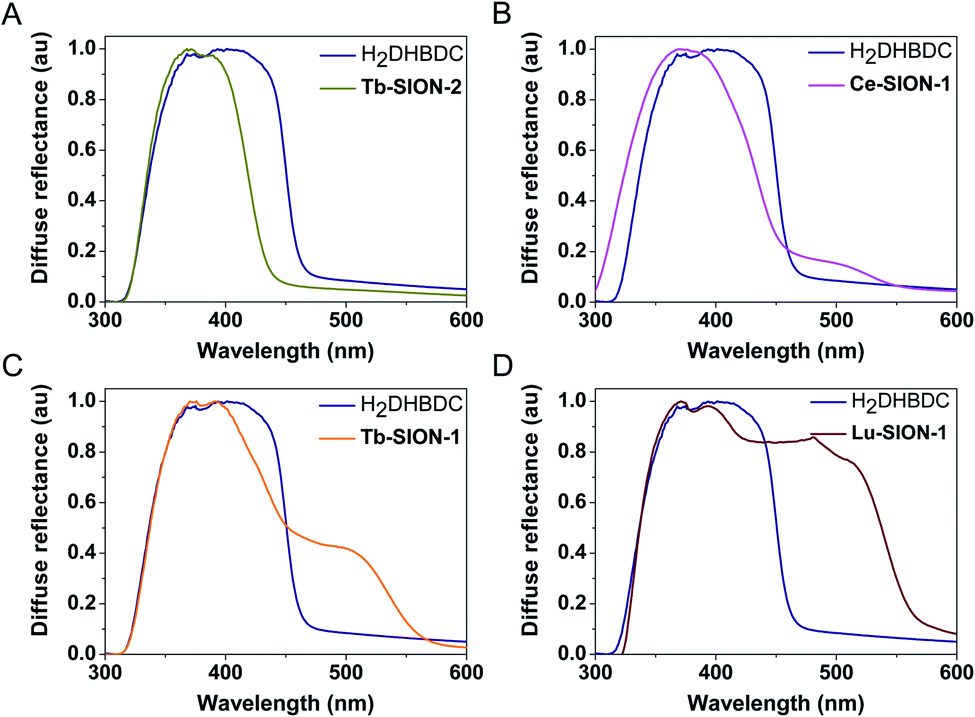 | ||
| Fig. 2 UV/vis spectra of Ln-SION-1, Ln-SION-2 and H2DHBDC. (A) Tb-SION-2 displays an absorption band comparable to the free H2DHBDC ligand. Evolution of the visible-light absorption band in (B) Ce-SION-1, (C) Tb-SION-1, and (D) Lu-SION-1. | ||
For the Ln-SION-1 family we observe very different optical absorption behavior. This family also displays the characteristic absorption band of the ligand at 470 nm, but in addition we observe an additional absorption band at lower energies depending on the lanthanide. Fig. 2 shows the appearance of a shoulder at ∼500 nm, and by changing the metal, Fig. S12 and S13† show that the intensity of this shoulder increases moving from early (Ce-Gd), Fig. S12,† to late (Tb-Lu) Ln's, Fig. S13.† These observations are the most important result of our work: we can exploit the MOFs flexibility for tuning the light absorption from mainly the UV region (e.g., Ln-SION-2 and Ce-SION-1), towards a significant portion of the visible light (e.g., Lu-SION-1 or Yb-SION-1), while promoting charge transfer states from the ligand to the metal nodes.
Molecular interpretation
To understand these differences in the optical properties of the Ln-SION-1, and Ln-SION-2 families, it is instructive to discuss the different scenarios of how the electronic structure of the ligand and metal can interact (see Fig. 3). Illumination of photo-active MOFs promotes an electron from the HOCOs (highest occupied crystal orbitals) to the LUCOs (lowest unoccupied crystal orbitals); or in band-structure terms, from the valence band to the conduction band. From the density of (electronic) states (DOS), we can obtain the energy gap between the HOCO and the LUCO. In some cases, this gap estimates the lowest wavelength in which the materials can absorb light. Fig. 3 shows the three different scenarios on how the metal node and the ligand can interact via orbital mixing. Two features need to be considered to achieve orbital mixing, namely spatial overlap (top figure) and appropriate energy matching (bottom figure). If there is appropriate energy matching but poor spatial overlap, the orbital mixing will still be minor and the orbital energies will remain the ones of the constituent units, namely the ligand and the metal node. In such a scenario the optical absorption is dominated by the ligand. This is what we observed for Ln-SION-2. The ligand is absorbing in the UV caused by a π → π* transition, and there is no influence of the lanthanide metal; so that the orbitals are too far apart either in energy or in space for overlapping. | ||
| Fig. 3 The different scenarios on how the metal node and linker can interact to change the optical properties. The bottom view gives illustrates the projected band structure and the top view the crystal orbitals. The main result of this work is that we need to fulfilled two conditions to tune the optical properties: we need that the orbitals are at the right energy and at the same time a sufficient overlap of these orbitals in space. | ||
To extend the absorption towards the visible light, we need to ensure orbital mixing between the ligand and the metal centre to stabilize those mixed electronic states and lower the band gap. For this to happen we need to ensure that the metal centre and ligand are in sufficient spatial proximity and energy matching. Fig. 3 shows the scenarios in which an increasing amount of spatial orbital overlap between the ligand and the metal orbitals occurs, as well as the 5d and π* orbital energies approach. The increased spatial overlapping is caused by the contraction of the lanthanide ion in the Ln-SION-1 series which allows the metal–ligand bond distance to decrease. In Ln-SION-1, the 4f orbitals are continually filled through the series, which shortens the Ln–O bond and brings the ligand and lanthanide metal centre in closer proximity, opening up the possibility for the 5d Ln metal and π* DOBDC orbitals to overlap. The shift of the absorption towards the visible light increases from the early Ln-SION-1, where a little spatial overlapping occurs, to an optimal overlap of the orbitals for the late Ln-SION-1.
To use such arguments for the design of MOFs it is important to quantify the scenarios shown in Fig. 3. For this reason, we have carried out detailed DFT calculations to determine the structure and the electronic properties of the Ln-SION-1 and Ln-SION-2 series.
DFT calculations
We have been able to successfully optimize both the Ln-SION-1 and Ln-SION-2 structures, with Tb-SION-1 and Tb-SION-2 results in good agreement with the single crystal experimental data (see ESI† Section 5 for computed cell parameters). The DFT calculations showed that the SION-1 structure has a lower energy than SION-2 for all lanthanides. This agrees with the experimental observation of SION-2 being a metastable structure that upon further heating, undergoes partial dissolution, and is then reassembled into the thermodynamically more stable SION-1 phase.32 The DFT calculations of the Ln-SION-1 structure confirmed that the bond length between the Ln(III) and oxido O atom of DOBDC shortens. The results of this optimization are summarized in Table S2 in the ESI.†
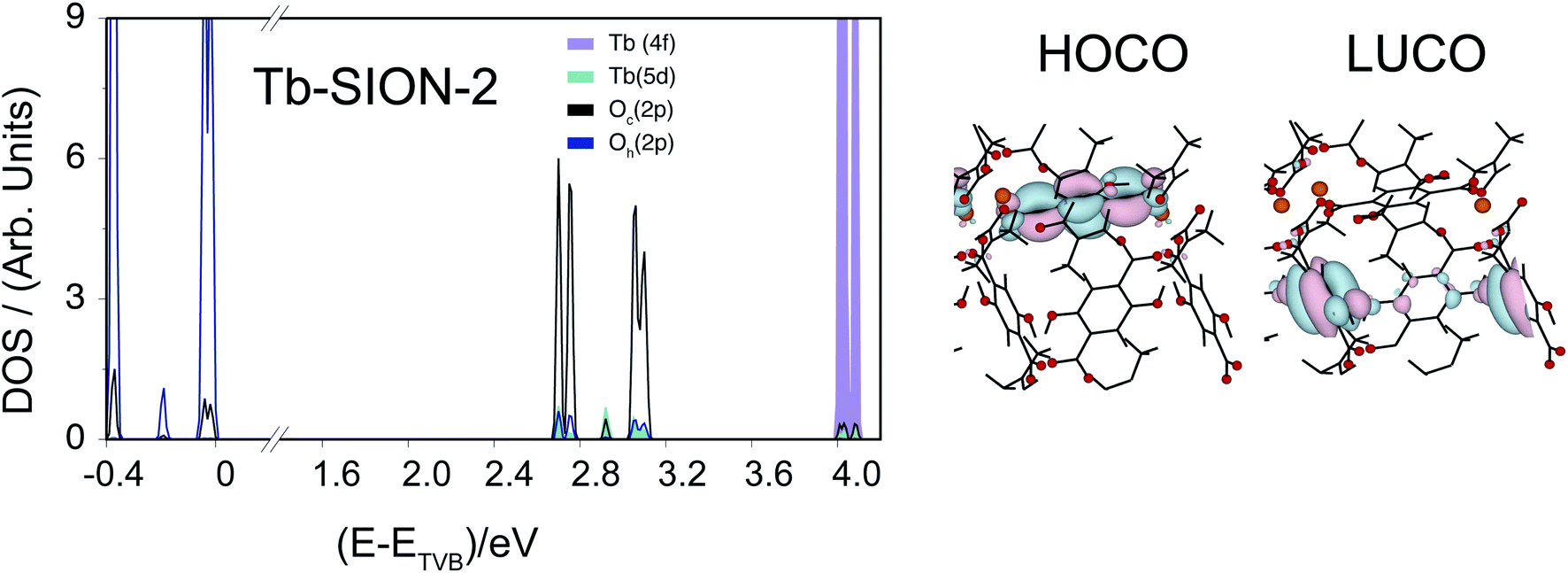 | ||
| Fig. 4 Projected density of states (pDOS) and crystalline orbitals of Tb-SION-2. Here, the HOCO and LUCO of Tb-SION-2, which have respectively π and π* character, are centered on different DHBDC ligands, lying on crystal planes perpendicular to each other. Color scheme for the orbital plots: red, O; orange, Tb; black, C. | ||
The electronic properties of the Ln-SION-1 family are remarkably different. In Fig. 5 we present the band structure and HOCO/LUCO orbitals for Ce-, Tb-, and Lu-SION-1 (with the rest of the Ln-SION-1 series illustrated in ESI† Section 8). Ce-SION-1 is a typical example of the early Ln, while Lu-SION-1 is a representative case of late lanthanides, with Tb-SION-1 half way through the Ln-SION-1 series. The calculated electronic structure of Ce-SION-1 showed that the HOCOs correspond to the ligand π orbitals, while the LUCOs have a predominantly Ln (4f) character. This can also be inferred from the corresponding projected DOS in Fig. 5. Since the Ln (4f) orbitals are more compact, they can not overlap in space with the ligand (π*) orbitals. In contrast, the Ln (5d) orbitals are more diffused, which allows for some spatial overlapping with the π* orbitals. The mixing between the π* and 5d orbitals in the conduction band of Ce-SION-1 gives rise to a new π → π*/5d transition, which appears in the visible part of the spectrum as a shallow shoulder at ∼500 nm in the absorption spectrum.
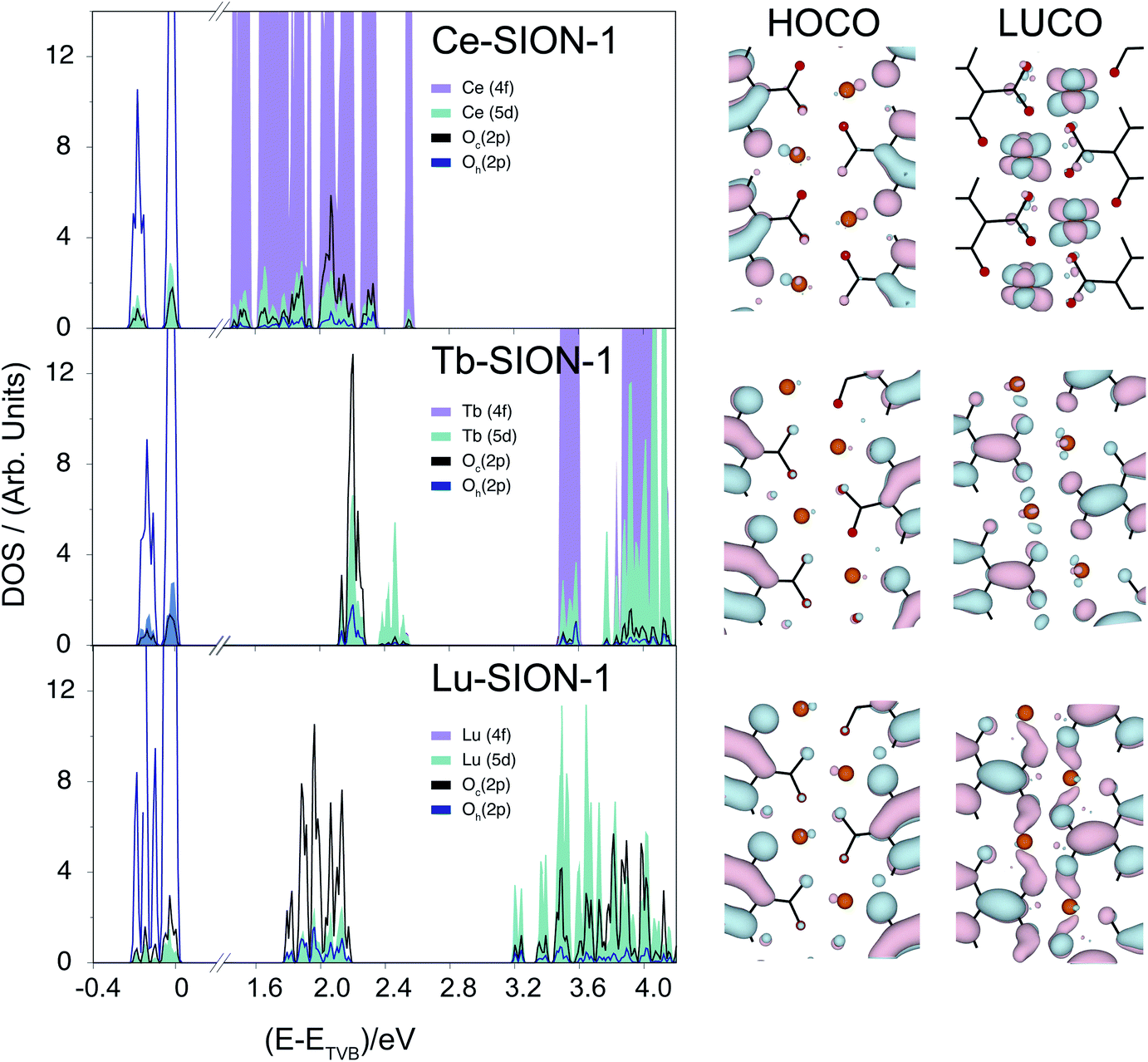 | ||
| Fig. 5 Projected density of states (pDOS) and crystalline orbitals of Ce-SION-1, Tb-SION-1, and Lu-SION-1. The LUCOs show a mixed Ln(5d)/DOBDC(π*) character that increases while moving along the lanthanide series (Ce → Lu). Color scheme for the orbital plots: red, O; orange, Ln; black, C. | ||
When moving through the Ln(III) ion series from Ce to Lu, the Ln ionic radii decreases due to the filling and weaker shielding of the inner 4f orbitals. This phenomenon, commonly known as the lanthanide contraction, reduces the bond length between the Ln(III) and the oxido O atom of DOBDC ligand and, as a consequence, increases the spatial overlap between the (π*) and 5d orbitals within the 1-dimensional chains. The increase of mixed character in the LUCO orbitals of Ln-SION-1 structure from Tb to Lu is shown in Fig. 5. This explains the observed increase of intensity of the visible absorption band in Tb-SION-1 with respect to Ce-SION-1, and more dramatically in Lu-SION-1 (Fig. 2). For Dy-SION-1 the Ln–O distances are optimal for an ideal overlap of 5d and π* orbitals, and once this distance is optimal, no increase in absorption intensity for the late lanthanides is observed.
Detailed inspection of the optical spectra showed that in all cases the absorption starts at a comparable wavelength (ca. 575 nm), suggesting an optical gap of ca. 2.1–2.2 eV for the entire Ln-SION-1 series. This is also what our DFT calculations showed when considering the lowest π → π*/5d transition (see ESI† Section 9). Note that the optical gap does not correspond to the HOCO/LUCO energy difference, but to the energy difference between the π and π*/5d band peaks. Altogether, our results nicely illustrate that a combined orbital and band structure analysis is required to study the hybrid organic–inorganic character of MOFs.
To assess the photoconductive properties of these materials, current–voltage (I–V) measurements were performed for Ce-SION-1 and Yb-SION-1 under inert and dark conditions (ESI† Section 10). The observed non-linear I–V curves are typical of semiconducting materials, supporting our predictions on photoconductivity. However, the I–V curves cannot distinguish the two materials in terms of magnitude and more evidence is required to characterize their dielectric behaviour.
Further evidence of electron mobility could be provided by photocatalysis experiments. We therefore tested whether the material could photocatalytically produce hydrogen, with an appropriate co-catalyst and sacrificial reagent in a water solution (see ESI† Section 11). Consistent with a semiconductor like behavior, Yb-SION-1 was shown to generate detectable amounts of hydrogen upon visible light irradiation, in contrast to Ce-SION-1, in which no hydrogen was detected. The absence of hydrogen production in Ce-SION-1 could be originated in the inefficient electron mobility and migration to the crystal surface. In contrast Yb-SION-1 has the highest dispersion band in the series, associated with a high electron mobility required to produce H2. Despite not likely being to lead to commercially viable process, these experimental results do confirm our theoretical predictions.
Further improvement of the photocatalytic performance would involve a better understanding of the individual photo activated processes (e.g., the characterisation of the dynamic of the excited states), which include the lifetime and mobility of the charge-carriers and their transfer to the co-catalyst.
Conclusion
In this work, we presented a combined experimental and computational study of the optical and electronic properties of two new classes of lanthanides MOFs, Ln-SION-1 and Ln-SION-2, each having the same linker and Ln but differing in their crystal geometry. The intriguing aspects of the two classes of MOFs is that one class, Ln-SION-2, has the traditional behavior of Ln-materials in which the optical absorption properties are dominated by the light-active linkers. The behavior of Ln-SION-1 is completely different; in this material the linker and the lanthanide do interact through efficient orbital mixing between the π and π*/5d orbitals, which allowed us to tune the optical absorption properties by selecting different lanthanides.In addition, this set of experimental data allowed us to systematically compare the band structure predictions of these materials. Given that there is little experimental data on these type systems, this comparison allowed us to demonstrate that current state-of-the-art DFT calculations can make reliable predictions of the optical absorption properties of these materials. From a computational point of view we showed that one needs to combine band-structure calculations with crystal orbital analysis to obtain a complete picture of the electronic properties of these fascinating materials.
That we could confirm experimentally that the late lanthanides are weakly conductive is on the one hand an encouraging result. On the other hand the fact that we could not make a quantitative prediction on the conductivity and that we need experiments to asses this, already indicates where we need to improve our theoretical understanding.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
KCS thanks Swiss National Science Foundation (SNF) for funding under the Ambizione Energy Grant n.PZENP2_166888 and National Center of Competence in Research (NCCR), Materials' Revolution: Computational Design and Discovery of Novel Materials (MARVEL), of the Swiss National Science Foundation (SNSF) for funding – DD4.5. CPI is grateful to the EU for a Marie Curie Fellowship (705861 – ASPAir, H2020-MSCA-IF-2015). GC is supported by SNF under the Grant n.IZKSZ2_162130 and Project Funding (200021_172759). The authors thank Dr T. N. Nguyen and Dr B. Vlaisavljevic for useful discussions. This work was supported by the Swiss National Supercomputing Centre (CSCS) under project no. s611.Notes and references
- O. K. Farha, I. Eryazici, N. C. Jeong, B. G. Hauser, C. E. Wilmer, A. A. Sarjeant, R. Q. Snurr, S. T. Nguyen, A. O. Yazaydin and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 15016–15021 CrossRef CAS PubMed.
- P. Li, N. A. Vermeulen, C. D. Malliakas, D. A. Gomez-Gualdron, A. J. Howarth, B. L. Mehdi, A. Dohnalkova, N. D. Browning, M. O'Keeffe and O. K. Farha, Science, 2017, 356, 624–627 CrossRef CAS PubMed.
- J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed.
- J. A. Mason, M. Veenstra and J. R. Long, Chem. Sci., 2014, 5, 32–51 RSC.
- M. P. Suh, H. J. Park, T. K. Prasad and D.-W. Lim, Chem. Rev., 2012, 112, 782–835 CrossRef CAS PubMed.
- M. Ranocchiari and J. A. van Bokhoven, Phys. Chem. Chem. Phys., 2011, 13, 6388–6396 RSC.
- Y. B. Huang, J. Liang, X. S. Wang and R. Cao, Chem. Soc. Rev., 2017, 46, 126–157 RSC.
- A. H. Chughtai, N. Ahmad, H. A. Younus, A. Laypkov and F. Verpoort, Chem. Soc. Rev., 2015, 44, 6804–6849 RSC.
- L. L. Wu, J. F. Zhao, H. Wang and J. Y. Wang, CrystEngComm, 2016, 18, 4268–4271 RSC.
- J. J. Gassensmith, J. Y. Kim, J. M. Holcroft, O. K. Farha, J. F. Stoddart, J. T. Hupp and N. C. Jeong, J. Am. Chem. Soc., 2014, 136, 8277–8282 CrossRef CAS PubMed.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed.
- Z. C. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC.
- S. E. Miller, M. H. Teplensky, P. Z. Moghadam and D. Fairen-Jimenez, Interface Focus, 2016, 6, 20160027 CrossRef PubMed.
- L. R. Mingabudinova, V. V. Vinogradov, V. A. Milichko, E. Hey-Hawkins and A. V. Vinogradov, Chem. Soc. Rev., 2016, 45, 5408–5431 RSC.
- Y. J. Cui, H. Xu, Y. F. Yue, Z. Y. Guo, J. C. Yu, Z. X. Chen, J. K. Gao, Y. Yang, G. D. Qian and B. L. Chen, J. Am. Chem. Soc., 2012, 134, 3979–3982 CrossRef CAS PubMed.
- W. Zhang and R. G. Xiong, Chem. Rev., 2012, 112, 1163–1195 CrossRef CAS PubMed.
- M. Kurmoo, Chem. Soc. Rev., 2009, 38, 1353–1379 RSC.
- D. Tiana, C. H. Hendon and A. Walsh, Chem. Commun., 2014, 50, 13990–13993 RSC.
- C. H. Hendon, D. Tiana and A. Walsh, Phys. Chem. Chem. Phys., 2012, 14, 13120–13132 RSC.
- D. Tiana, C. H. Hendon, A. Walsh and T. P. Vaid, Phys. Chem. Chem. Phys., 2014, 16, 14463–14472 RSC.
- L. Sun, M. G. Campbell and M. Dinca, Angew. Chem., Int. Ed., 2016, 55, 3566–3579 CrossRef CAS PubMed.
- P. Ramaswamy, N. E. Wong and G. K. H. Shimizu, Chem. Soc. Rev., 2014, 43, 5913–5932 RSC.
- S. Horike, D. Umeyama and S. Kitagawa, Acc. Chem. Res., 2013, 46, 2376–2384 CrossRef CAS PubMed.
- A. Morozan and F. Jaouen, Energy Environ. Sci., 2012, 5, 9269–9290 RSC.
- S. I. Weissman, J. Chem. Phys., 1942, 10, 214–217 CrossRef CAS.
- W. J. Evans, Polyhedron, 1987, 6, 803–835 CrossRef CAS.
- J.-C. G. Bunzli and S. V. Eliseeva, Lanthanide Luminescence: Photophysical, Analytical and Biological Aspects, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, pp. 1–45 Search PubMed.
- J. C. G. Bunzli, Chem. Rev., 2010, 110, 2729–2755 CrossRef PubMed.
- J. Feng and H. J. Zhang, Chem. Soc. Rev., 2013, 42, 387–410 RSC.
- X. Qin, X. W. Liu, W. Huang, M. Bettinelli and X. G. Liu, Chem. Rev., 2017, 117, 4488–4527 CrossRef CAS PubMed.
- A. Vogler and H. Kunkely, Inorg. Chim. Acta, 2006, 359, 4130–4138 CrossRef CAS.
- S. L. Anderson, A. Gladysiak, P. G. Boyd, C. P. Ireland, P. Mieville, D. Tiana, B. Vlaisavljevich, P. Schouwink, W. van Beek, K. J. Gagnon, B. Smit and K. C. Stylianou, CrystEngComm, 2017, 19, 3407–3413 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystal structures for Tb-SION-1 and Tb-SION-2, identifiers 1503702 and 1503701 respectively. See DOI: 10.1039/d0sc00740d |
| ‡ These authors contributed equally to this work. |
| § Current address: Department of Chemistry, Oregon State University, Corvallis, OR 97331, USA. |
| This journal is © The Royal Society of Chemistry 2020 |