
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A step-economic and one-pot access to chiral Cα-tetrasubstituted α-amino acid derivatives via a bicyclic imidazole-catalyzed direct enantioselective C-acylation†
Mo
Wang
ab,
Muxing
Zhou
b,
Lu
Zhang
b,
Zhenfeng
Zhang
 *b and
Wanbin
Zhang
*ab
*b and
Wanbin
Zhang
*ab
aShanghai Key Laboratory for Molecular Engineering of Chiral Drugs, School of Chemistry and Chemical Engineering, Frontiers Science Center for Transformative Molecules, Shanghai Jiao Tong University, Shanghai 200240, China. E-mail: wanbin@sjtu.edu.cn; zhenfeng@sjtu.edu.cn
bSchool of Pharmacy, Shanghai Jiao Tong University, Shanghai 200240, China
First published on 24th April 2020
Abstract
Cα-Tetrasubstituted α-amino acids are ubiquitous and unique structural units in bioactive natural products and pharmaceutical compounds. The asymmetric synthesis of these molecules has attracted a lot of attention, but a more efficient method is still greatly desired. Here we describe the first sequential four-step acylation reaction for the efficient synthesis of chiral Cα-tetrasubstituted α-amino acid derivatives from simple N-acylated amino acids via an auto-tandem catalysis using a single nucleophilic catalyst. The synthetic efficiency is improved via a direct enantioselective C-acylation; the methodology affords the corresponding Cα-tetrasubstituted α-amino acid derivatives with excellent enantioselectivities (up to 99% ee). This step-economic, one-pot, and auto-tandem strategy provides facile access to important chiral building blocks, such as peptides, serines, and oxazolines, which are often used in medicinal and synthetic chemistry.
Introduction
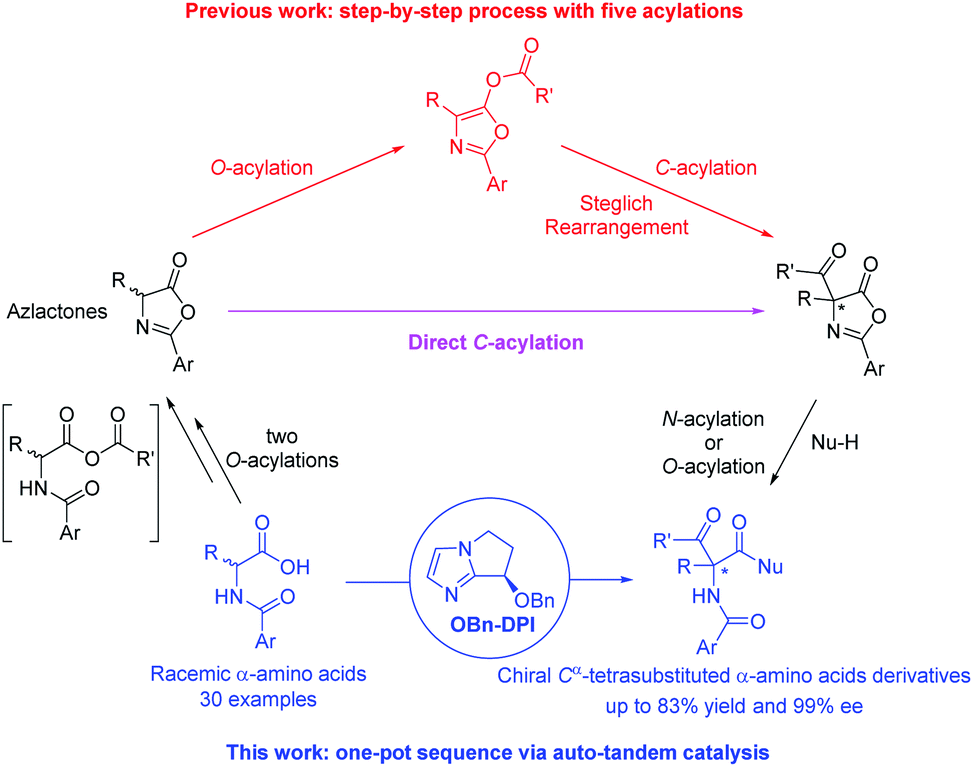
Cα-Tetrasubstituted α-amino acids are ubiquitous and unique structural units in many natural products and synthetic compounds, many of which exhibit significant biological activities and physiological effects (Fig. 1).1 Several feasible methodologies have been developed for the synthesis of Cα-tetrasubstituted α-amino acids, ranging from the use of chiral auxiliaries to the presently reported catalytic models.2 These include: (1) the enantioselective addition of alkyl, aryl, or even acyl precursors (Strecker reaction followed by hydrolysis) to ketimines,3 and; (2) the asymmetric α-alkylation, arylation, or acylation of α-substituted amino acid derivatives.4,5 However, due to the challenges imposed by Cα-tetrasubstituted α-amino acids, efficient methods are still lacking, especially for the synthesis of α-acyl-substituted Cα-tetrasubstituted α-amino acid derivatives.5 The most commonly used approach relies on the O-acylation of azlactones followed by a Lewis base-catalyzed asymmetric O- to C-acyl transfer (well-known as Steglich rearrangement, red arrows in Scheme 1). After the initial work developed by Fu et al.,5a several improvements on the asymmetric Steglich rearrangement have been reported by many other research groups.5b–k Our group has also developed an efficient bicyclic imidazole organocatalyst for this reaction, which can be easily synthesized from imidazole in only three steps.5f However, the efficiency of this transformation is hindered by the prefabrication of unstable azlactones and relatively inert O-acylated azlactones. Such inefficiencies are present in many other transformations which lead to poor practical applicability.5b,6 | ||
| Fig. 1 Examples of Cα-tetrasubstituted α-amino acid derivatives with biological activities and physiological effects. | ||
 | ||
| Scheme 1 Our new approach to enantiopure Cα-tetrasubstituted α-amino acid derivatives by a four-step acylation sequence using auto-tandem catalysis. | ||
In order to improve the synthetic efficiency, minimizing the number of steps of the synthetic sequence is important. This strategy, termed step-economy, has frequently been employed in advanced total syntheses by utilizing new reactions.7 Herein, we would like to put forward a more general approach in which a two-step rearrangement process (AB + C → AB–C → C–AB) is replaced by a direct reaction (AB + C → C–AB) by altering chemoselectivity. As the first example, we recently developed a bicyclic imidazole-catalyzed direct enantioselective C-acylation of benzofuran-2(3H)-ones and 2-oxindoles.8 For the most part, the starting material is directly converted to the C-acylated product but not via the relatively inert O-acylated intermediate. As a result, the efficiency for the synthesis of the desired products is dramatically improved compared to the corresponding Black rearrangement.9 Inspired by this, and in continuation of our interest in the construction of Cα-tetrasubstituted α-amino acids,10 we propose a direct enantioselective C-acylation of azlactones with the purpose of improving the synthetic efficiency for the preparation of 4-carboxyazlactones (pink arrow in Scheme 1).11 To further improve the synthetic efficiency, simplifying the operations via a sequential process in one pot is an applicable method. This strategy, termed pot-economy, has been widely used for the construction of multiple bonds in one pot.12 Generally, different catalysts are used for different reaction steps (tandem catalysis). Obviously, carrying out all the reaction steps with a single catalyst would be more beneficial (auto-tandem catalysis).13 This strategy, termed catalyst-economy, has often been employed in two-step sequences but seldom in multi-step sequences.13c During the initial research concerning the direct enantioselective C-acylation of azlactones, we found that the starting azlactones could be prepared via intramolecular O-acylation of in situ generated anhydrides (shown in the square brackets in Scheme 1); the produced 4-carboxyazlactones could be readily derivatized by another acylation to produce various Cα-tetrasubstituted α-amino acid derivatives. Therefore, by combining the aforementioned step-, pot-, and catalyst-economy strategies, we aimed to design a four-step acylation sequence for the efficient synthesis of chiral Cα-tetrasubstituted α-amino acid derivatives and biologically active dipeptides from simple N-acylated amino acids. The reaction would proceed via auto-tandem catalysis using a single bicyclic imidazole nucleophilic catalyst (blue part in Scheme 1). To the best of our knowledge, this type of sequential reaction that achieves excellent enantioselectivity has not been reported previously.
Results and discussion
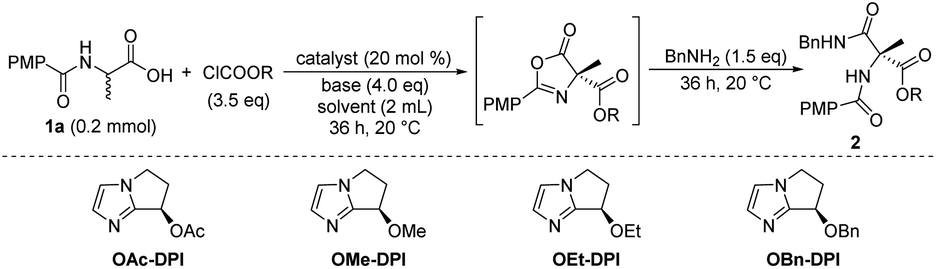
We initially focused on screening the reaction conditions for the first three steps in the sequence in one pot. (4-Methoxybenzoyl)alanine 1a was selected as the starting material and benzylamine was used as the nucleophilic reagent to derivatize the produced 4-carboxyazlactone shown in the square brackets in Table 1. Over the past decade, we have developed a series of bicyclic imidazole nucleophilic catalysts which have been successfully applied in a number of different reactions by our group5f,8,9,14 and other groups.15 We envisaged that the bicyclic imidazole would be able to catalyze all the steps in the acylation sequence. Several bicyclic imidazole catalysts were screened for the sequential acylation of 1a with benzyl chloroformate as the acylating agent and di(isopropyl)ethylamine (DIPEA) as a base to afford product 2a (Table 1, entries 1–4). Catalysts bearing an alkoxy group gave better ee than those bearing an acyloxy group, and catalyst OBn-DPI was found to give the best result. Replacing the acylating agent, benzyl chloroformate with allyl chloroformate or phenyl chloroformate, afforded the desired products in lower yield and ee (entries 5 and 6, respectively). Use of ethyl chloroformate only afforded a trace amount of the product (entry 7). The base triethylamine (TEA) was also applied in this reaction but no product was observed (entry 8). The effect of solvents was studied and toluene provided the product with the highest ee and satisfactory conversion (entries 4, 9–14). After preliminary screening, the catalyst OBn-DPI, acylating agent benzyl chloroformate, base DIPEA and solvent toluene were chosen as the optimal conditions for further study, affording the desired product in 78% yield and with 91% ee (entry 4).|
|
|||||
|---|---|---|---|---|---|
| Entrya | Catalyst | Acylating reagent | Solvent | Yieldb (%) | eec (%) |
| a Conditions: 1a (0.1 M), ClCOOR (3.5 eq.), catalyst (20 mol%), DIPEA (4.0 eq.), solvent (2 mL), 20 °C, 36 h, unless otherwise noted. b Yields were calculated from 1H NMR spectra. c The ee values were calculated from HPLC spectra. d TEA was used instead of DIPEA and only azlactone without COOBn substituent was obtained together with some NEt2COOBn. | |||||
| 1 | OAc-DPI | ClCOOBn | Toluene | 77 | 84 |
| 2 | OMe-DPI | ClCOOBn | Toluene | 62 | 87 |
| 3 | OEt-DPI | ClCOOBn | Toluene | 65 | 88 |
| 4 | OBn-DPI | ClCOOBn | Toluene | 78 | 91 |
| 5 | OBn-DPI | ClCOOallyl | Toluene | 71 | 91 |
| 6 | OBn-DPI | ClCOOPh | Toluene | 81 | 60 |
| 7 | OBn-DPI | ClCOOEt | Toluene | Trace | — |
| 8d | OBn-DPI | ClCOOBn | Toluene | — | — |
| 9 | OBn-DPI | ClCOOBn | THF | 80 | 90 |
| 10 | OBn-DPI | ClCOOBn | Dioxane | 77 | 90 |
| 11 | OBn-DPI | ClCOOBn | Et2O | 30 | 91 |
| 12 | OBn-DPI | ClCOOBn | MTBE | 66 | 91 |
| 13 | OBn-DPI | ClCOOBn | DCM | 45 | 88 |
| 14 | OBn-DPI | ClCOOBn | t-AA | 76 | 71 |
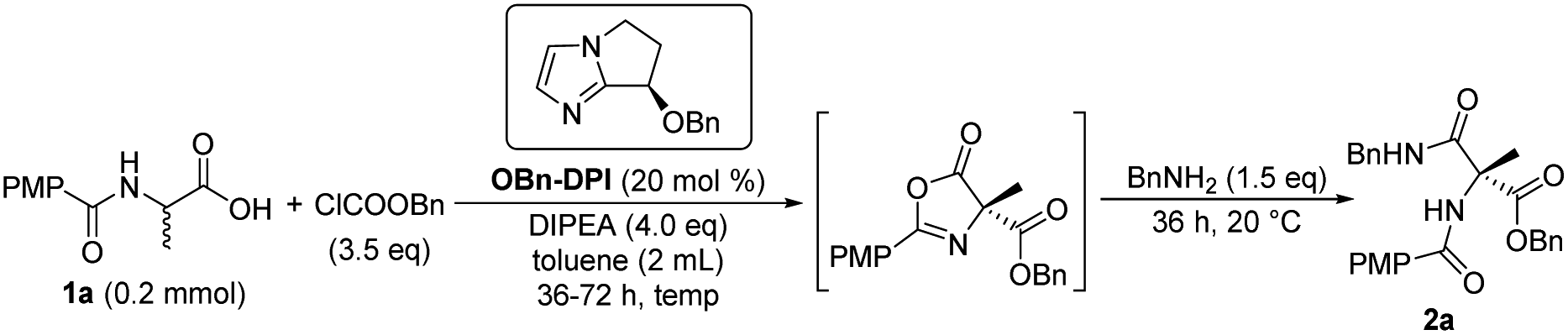
Next, the effect of reaction temperature was researched (Table 2). At a lower reaction temperature of 0 °C for 36 h, product 2a was obtained with same yield and better ee compared to that of 20 °C (entries 1 and 2). When the reaction temperature was reduced to −20 °C for 36 h, the yield of 2a was reduced to 68% while the enantioselectivity increased to 95% (entry 3). Extending the reaction time to 48 hours gave the product in an increased yield (76%, entry 4). So the reaction was conducted for a longer time (72 h) to give better yield when reaction temperature was further decreased (entries 5–7). Finally, −55 °C was found to be the optimized temperature with 78% yield and 99% ee (entry 6). Then the equivalents of ClCOOBn were investigated and it was found that 3.0 equivalents of ClCOOBn afforded the best results (entries 6, 8 and 9). Therefore, the sequential reaction of substrate 1a with 3.0 equivalents of benzyl chloroformate, 4.0 equivalents of DIPEA and 20 mol% OBn-DPI at −55 °C over 72 h was optimal for both reactivity and enantioselectivity, leading to the product 2a in 78% yield and 99% ee (entry 8).
|
|
||||
|---|---|---|---|---|
| Entrya | Temp. (°C) | Time (h) | Yieldb (%) | eec (%) |
| a Conditions: 1a (0.1 M), ClCOOBn (3.5 eq.), OBn-DPI (20 mol%), DIPEA (4.0 eq.), toluene (2 mL), unless otherwise noted. b Yields were calculated from 1H NMR spectra. c The ee values were calculated from HPLC spectra. d ClCOOBn (3.0 eq.). e ClCOOBn (2.5 eq.). | ||||
| 1 | 20 | 36 | 78 | 91 |
| 2 | 0 | 36 | 78 | 93 |
| 3 | −20 | 36 | 68 | 95 |
| 4 | −20 | 48 | 76 | 95 |
| 5 | −50 | 72 | 78 | 98 |
| 6 | −55 | 72 | 78 | 99 |
| 7 | −60 | 72 | 77 | 98 |
| 8d | −55 | 72 | 78 | 99 |
| 9e | −55 | 72 | 75 | 99 |
Having established the optimal reaction conditions, we investigated the nucleophile in the last step of the sequential process, employing (4-methoxybenzoyl)alanine 1a as the initial substrate (Scheme 2). Firstly a number of different alkyl amines and a phenyl amine were tested as nucleophiles (2a–d). Amines with less steric hindrance showed higher yields (2a, bvs.2c, d). Cholamine, bearing both amino and hydroxyl groups, was also used as the nucleophile. Due to the wide difference in the activity of the alcohols and amines for this reaction, only the product 2e was obtained, via attack of the amino group, with good yield and excellent enantioselectivity (98% ee). Secondly, excess methanol was employed as a nucleophile in this reaction to give the corresponding product 2f with similar results. In addition, a variety of amino acid esters gave their corresponding enantiomerically pure dipeptides, which will be of particular use in the fields of biology and medicine (2g–r). When using an enantiomerically pure amino acid ester which contained a chiral center, the corresponding dipeptide products bearing two stereocenters were obtained with high dr (>99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 2l–r).
:1, 2l–r).
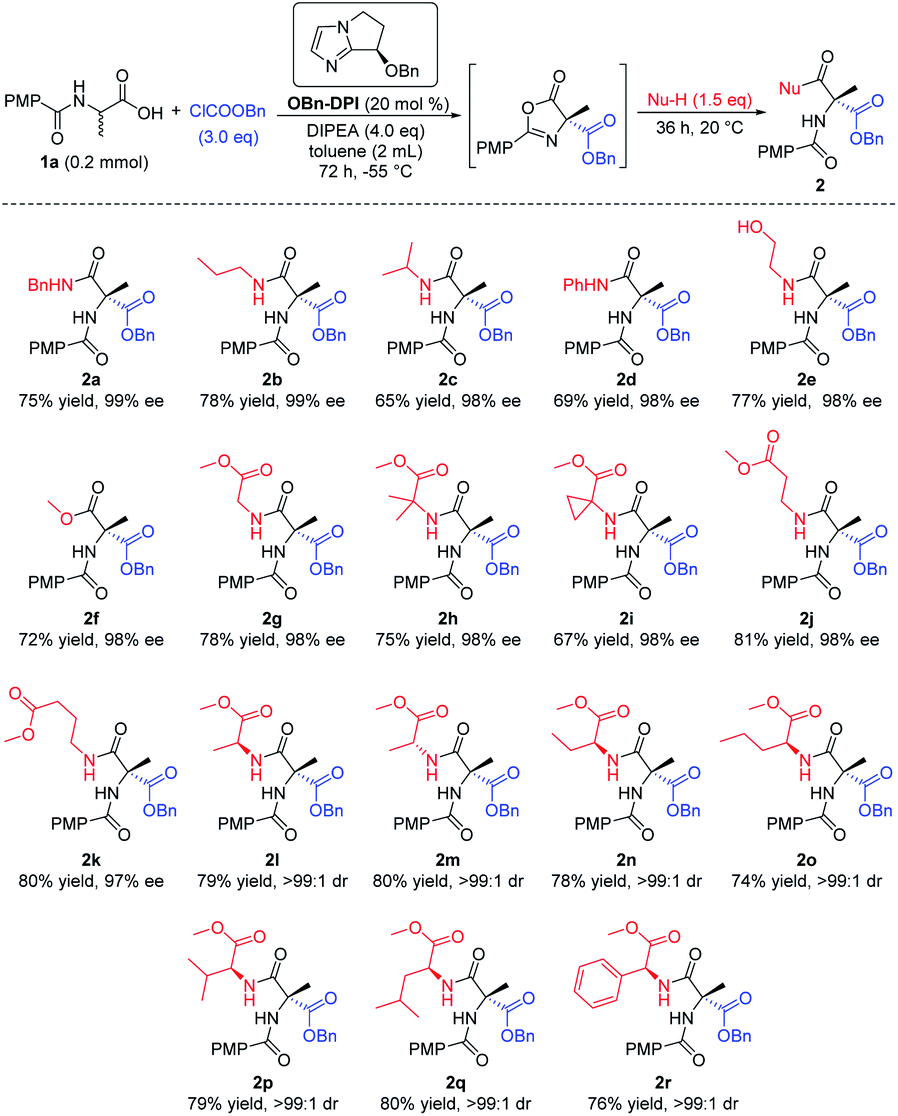 | ||
| Scheme 2 Expanding the nucleophiles. All the yields are isolated yields. 1H NMR spectroscopy of the crude reaction products was used to assess diastereoisomeric ratios (dr). | ||
Various N-protected α-amino acid substrates 1 were tested in the sequential process using methyl 3-aminopropanoate as the nucleophile (Scheme 3). Firstly, N-substituted amino acid substrates bearing a methoxy group at the para-, meta-, and ortho-positions of the phenyl ring were tested in the domino reaction (2j, 2s and 2t). All these products were obtained in good yields and with excellent enantioselectivities. The effect of different substituents at the para-position of the phenyl ring was studied (2u–x). Substrates bearing electron-donating groups such as Me or tBu, and electron-withdrawing groups such as F or Cl, all afforded the corresponding products in good yields and with excellent enantioselectivities. The presence of an electron-withdrawing group enhances the acidity of the substrate and its corresponding intermediates in the two O-acylation steps and the C-acylation step (see proposed mechanism below), thus reducing the reaction time of these substrates to 8 h, much shorter than the reaction time for substrates bearing electron-donating groups (72 h). A substrate without a substituent group on the phenyl ring also gave the corresponding product 2y in 80% yield and with 98% ee within 10 h. We then employed simple N-benzoyl amino acids to study the influence of the R group on the stereocenter (2y–ae). The substrate bearing a phenyl substituent gave the corresponding product 2ae with only 57% ee, and the substrate bearing an isopropyl group only gave the O-acylated byproduct. All other tested substrates gave the corresponding products in good yields and with excellent enantioselectivities. It's worth mentioning that products 2ab and 2ac were both obtained with 99% ee.
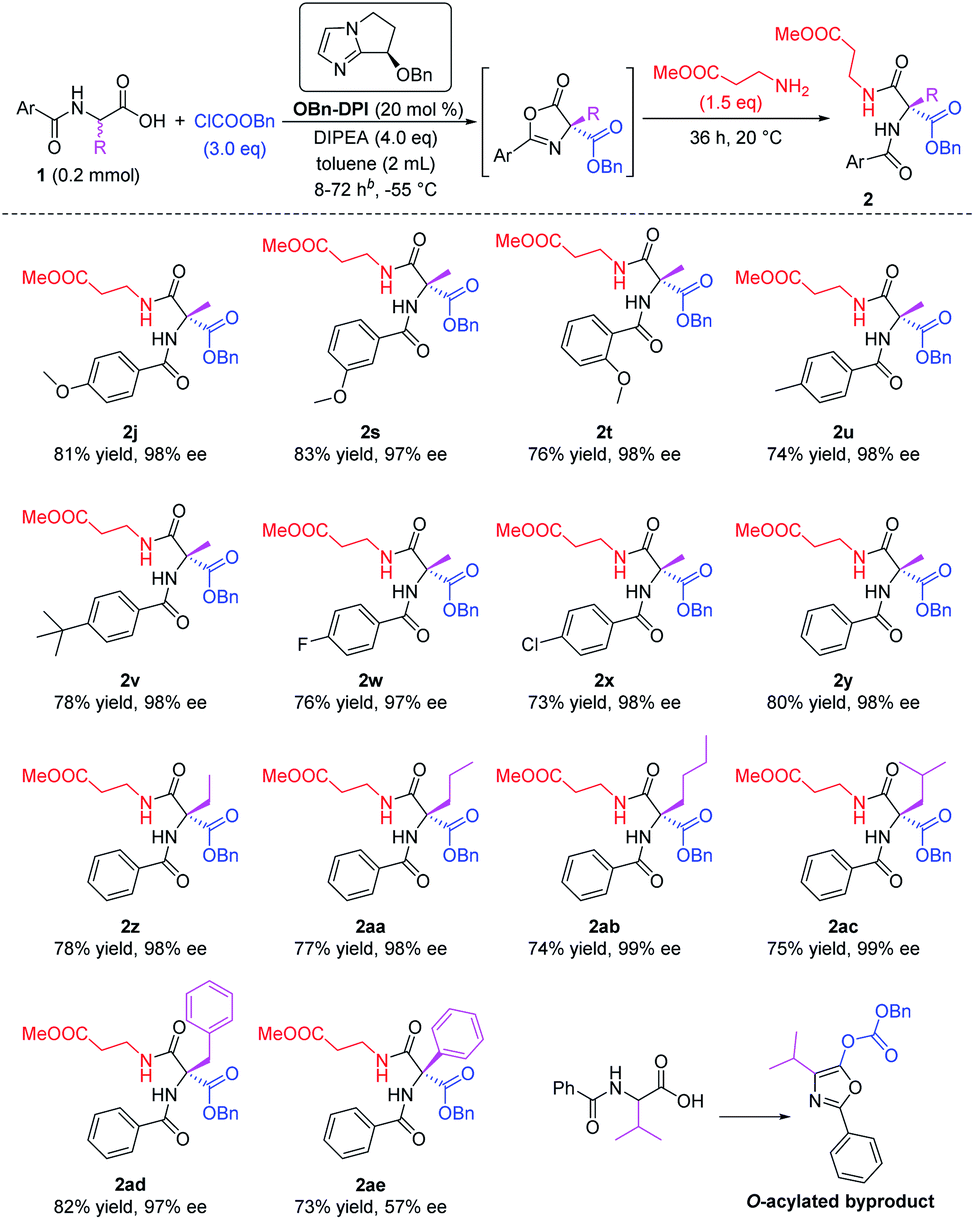 | ||
| Scheme 3 Expanding the racemic amino acid substrates. All the yields are isolated yields. Reaction time for 2s–v is 72 h, reaction time for 2w–x is 8 h, reaction time for 2y–ad is 10 h. | ||
Having explored the substrate scope of both nucleophiles and amino acids, we focused on elucidating the effect of the catalyst OBn-DPI in each step of the acylation sequence (Scheme 4). Firstly, benzoylalanine 1h was reacted with 1.0 equivalent of benzyl chloroformate in the presence or absence of catalyst OBn-DPI to test the effect of the catalyst in the first two O-acylation steps (step I and step II). In the presence of the catalyst, 1h afforded the cyclized product 3 in 63% yield at −55 °C in 10 min; however in the absence of catalyst, 1h only gave the desired product 3 in 35% yield. We next subjected 3 to reaction with benzyl chloroformate in the presence or absence of catalyst OBn-DPI to test effect of the catalyst in the third C-acylation step (step III). In the presence of the catalyst, 3 afforded C-acylated product 4 in 93% yield in 10 h, whereas in the absence of catalyst, 3 was unable to afford C-acylated product 4 giving O-acylated compound 5 in 26% yield. Compound 5 was then employed as a reactant at −55 °C in the presence of the catalyst OBn-DPI; no rearrangement product 4 was observed after 24 h. These experimental results strongly support that 3 undergoes a direct C-acylation to afford 4. Finally, 4 was reacted with an amino acid ester in order to test the effect of the catalyst in the last step (step IV). In the presence of the catalyst, 4 afforded final product 2y in 85% yield at 20 °C in 18 h, whereas 2y was only obtained in 46% yield in the absence of catalyst.
 | ||
| Scheme 4 Experimental studies on the effect of catalyst OBn-DPI and direct C-acylation. Yields were calculated from 1H NMR spectra. | ||
Based on these results, a mechanism for the four-step acylation sequence has been proposed (Scheme 5). First, the catalyst OBn-DPI and benzyl chloroformate generate active species A. In the presence of DIPEA, N-substituted amino acid substrates 1, such as 1h, attack A to generate the intermediate mixed anhydride B. Reaction of B and OBn-DPI gives intermediate C which generates azlactone 3via an intramolecular cyclization. In step III, azlactone 3 attacks the active species A to form C-acylated azlactone 4via an enantioselective direct C-acylation. This chiral compound 4 and catalyst OBn-DPI form active intermediate D, which is attacked by the amino acid ester to further generate valuable dipeptide 2yvia N-acylation. Experimental studies suggest that OBn-DPI acts as a nucleophilic catalyst in all the steps of the acylation sequence with compound 4 being formed from direct C-acylation.
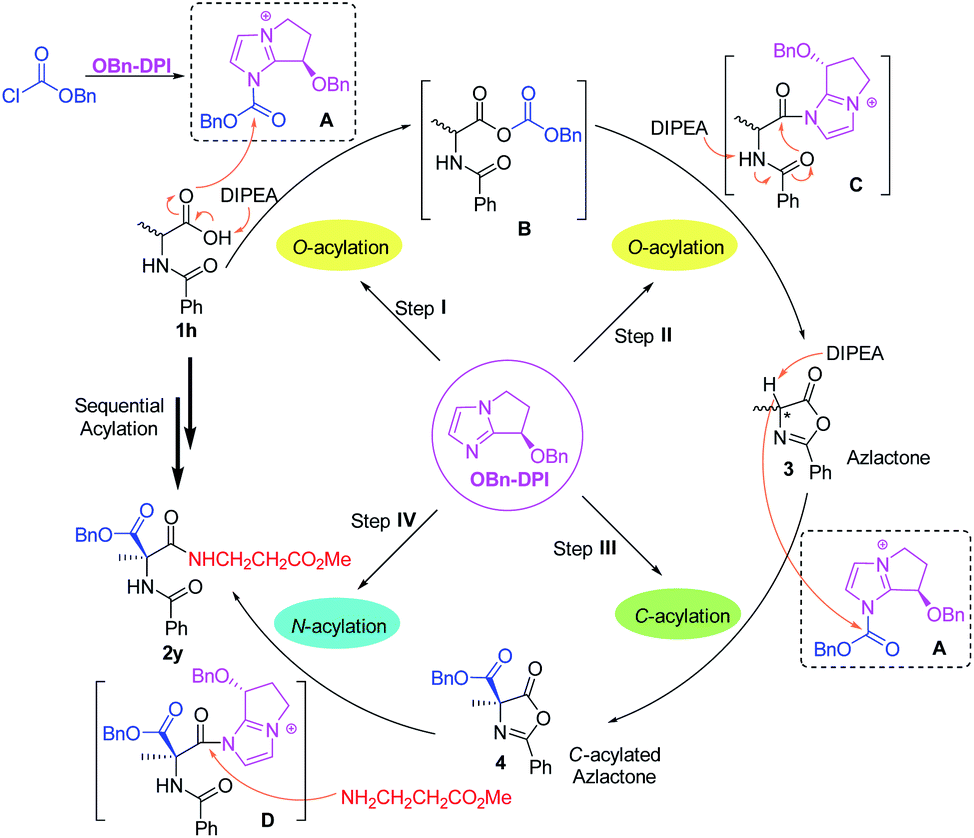 | ||
| Scheme 5 Proposed mechanism for the four-step acylation sequence via auto-tandem catalysis (the anion is omitted in A, B, C, and D for clarity). | ||
Small peptides play a significant role in many biological, pharmaceutical, and environmental applications.16 Therefore the stereoselective synthesis of such biomolecules is of great importance. Here, we describe the concise synthesis of small optically active peptides from the sequentially acylated product 2n (Scheme 6). Firstly, Cα-tetrasubstituted α-amino acid 6 could be obtained from hydrogenation of 2n under 3 atm hydrogen pressure with Pd(OH)2/C in quantitative yield. Then, coupling of 6 with several different amino acids or dipeptides afforded a variety of valuable small chiral peptides in good yields (7a–g). In particular, coupling of 6 with enantiomerically pure amino acids or dipeptides which contained a chiral center afforded products bearing three stereocenters with high dr (7b–d, 7f–g).
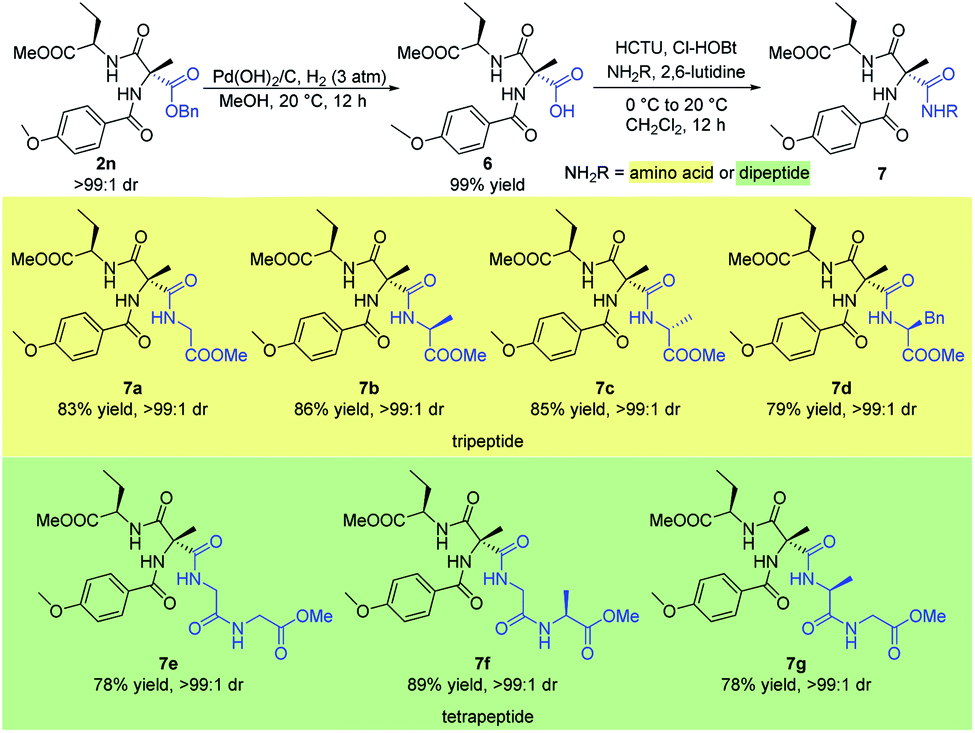 | ||
| Scheme 6 Synthesis of small peptide. All the yields are isolated yields. 1H NMR spectroscopy of the crude reaction products was used to assess diastereoisomeric ratios (dr). | ||
Furthermore, the utility of this acylation sequence was demonstrated via the enantiodivergent synthesis of α-methyl serine from 2f by manipulating the different reactivities of the two ester groups (Scheme 7). Hydrogenation of the benzyl ester group of 2f with Pd(OH)2/C in MeOH at 20 °C for 12 h provided 8 in quantitative yield. Reduction of 8 with LiBH4 in THF at ambient temperature for 12 h afforded the (R)-isomer of N-protected α-methyl serine, (R)-10, in 83% yield. Hydrolysis of the methyl ester group of 2f with KOH in THF/H2O at 20 °C provided 9 in 78% yield, which was reduced with LiBH4 to give the (S)-isomer of N-protected α-methyl serine, (S)-10, in 85% yield.
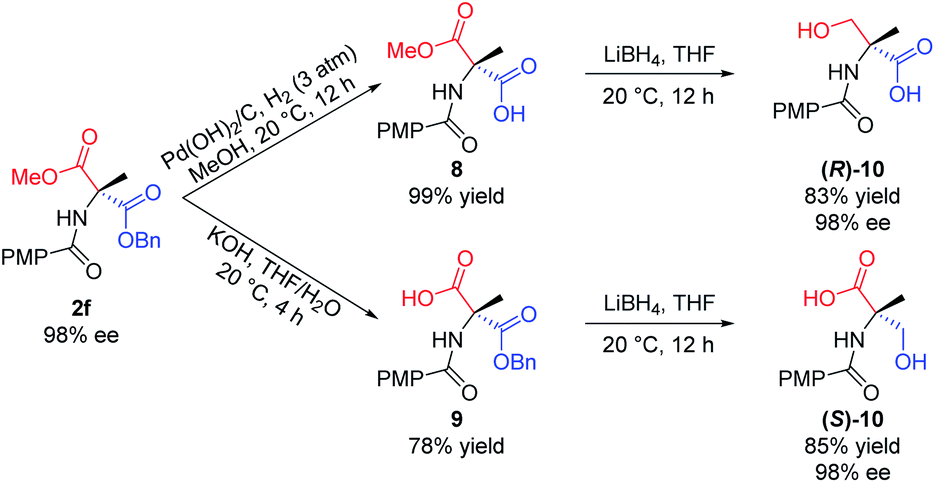 | ||
| Scheme 7 Enantiodivergent synthesis of α-methyl serine. All the yields are isolated yields. | ||
In addition, we realized the synthesis of 4,4′-disubstituted oxazoline from 2a, as shown in Scheme 8. Reduction of 2a with LiBH4 in THF provided the corresponding alcohol 11. Mesylation of 11 with methanesulfonyl chloride in DCM at 0 °C in the presence of triethylamine, followed by intramolecular cyclization at 20 °C in one pot, finally afforded oxazoline 12 in 94% yield (74% overall from 2a). This method allows for the practical synthesis of chiral oxazolines, which can be used as chiral ligands for asymmetric catalysis.17
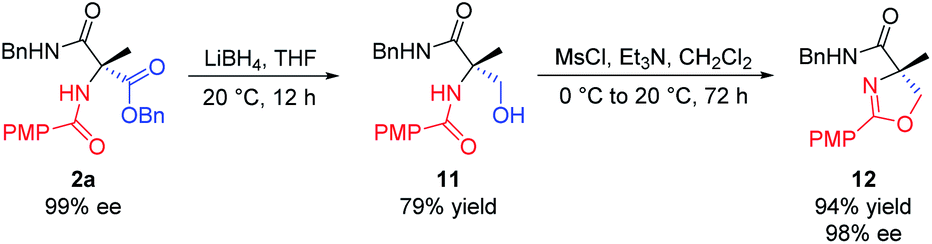 | ||
| Scheme 8 Synthesis of 4,4′-disubstituted oxazoline. All the yields are isolated yields. | ||
Experimental
Under a N2 atmosphere, the substrate 1h (0.2 mmol, 44.7 mg), the catalyst OBn-DPI (0.04 mmol, 8.6 mg) and DIPEA (0.8 mmol, 132.2 μL) were dissolved in anhydrous toluene (2 mL) and cooled to −55 °C in a dry two-necked flask. ClCOOallyl (0.6 mmol, 84.5 μL) was then added and the vial was sealed with a septum. The reaction mixture was stirred at −55 °C for 10 h and then the temperature was gradually raised to 20 °C. Methyl 3-aminopropionate hydrochloride (0.3 mmol, 41.9 mg) and DIPEA (0.3 mmol, 49.6 μL) were dissolved in anhydrous toluene (1 mL) in another dry flask and stirred for 10 min. Then the mixture was transferred into the former reaction flask and the reaction mixture was stirred at 20 °C for 36 h. The reaction mixture was quenched with 0.2 M HCl (5 mL) and extracted with DCM (5 mL × 3). The combined organic phases were dried over Na2SO4. After filtration, the residue was purified by column chromatography (petroleum ether/ethyl acetate) to give the corresponding product 2y. The ee value was determined by chiral HPLC analysis after purification by column chromatography (petroleum ether/ethyl acetate).Conclusions
In summary, the first four-step sequential acylation reaction for the concise asymmetric synthesis of Cα-tetrasubstituted α-amino acid derivatives via auto-tandem catalysis has been successfully developed. This step-economic, one-pot, and auto-tandem strategy is promoted by a direct enantioselective C-acylation. Through four acylations catalyzed by a single chiral bicyclic imidazole, the simple N-acylated amino acids could be smoothly converted to the corresponding Cα-tetrasubstituted α-amino acid derivatives with excellent enantioselectivities (up to 99% ee). Significantly, the obtained products, particularly dipeptides, are potentially biologically active compounds. These products can be further transformed to other biomolecules and important chiral building blocks such as small peptides, α-substituted serines and 4,4′-disubstituted oxazolines.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This work was partially supported by National Natural Science Foundation of China (No. 21572131, 21620102003 and 91856106) and Shanghai Municipal Education Commission (No. 201701070002E00030). We thank the Instrumental Analysis Center of Shanghai Jiao Tong University.Notes and references
- (a) H. Bräuner-Osborne, J. Egebjerg, E. Ø. Nielsen, U. Madsen and P. Krogsgaard-Larsen, J. Med. Chem., 2000, 43, 2609–2645 CrossRef PubMed; (b) V. R. Macherla, S. S. Mitchell, R. R. Manam, K. A. Reed, T.-H. Chao, B. Nicholson, G. Deyanat-Yazdi, B. Mai, P. R. Jensen, W. F. Fenical, S. T. C. Neuteboom, K. S. Lam, M. A. Palladino and B. C. M. Potts, J. Med. Chem., 2005, 48, 3684–3687 CrossRef CAS PubMed; (c) J.-F. Liu, Z.-Y. Jiang, R.-R. Wang, Y.-T. Zheng, J.-J. Chen, X.-M. Zhang and Y.-B. Ma, Org. Lett., 2007, 9, 4127–4129 CrossRef CAS PubMed; (d) C. D. Hupp and J. J. Tepe, Org. Lett., 2008, 10, 3737–3739 CrossRef CAS PubMed; (e) À. Pericas, A. Shafir and A. Vallribera, Org. Lett., 2013, 15, 1448–1451 CrossRef PubMed.
- For selected reviews, see: (a) S. H. Kang, S. Y. Kang, H.-S. Lee and A. J. Buglass, Chem. Rev., 2005, 105, 4537–4558 CrossRef CAS PubMed; (b) K. Bera and I. N. N. Namboothiri, Asian J. Org. Chem., 2014, 3, 1234–1260 CrossRef CAS; (c) A. E. Metz and M. C. Kozlowski, J. Org. Chem., 2015, 80, 1–7 CrossRef CAS PubMed; (d) H. Jiang, Y. Jin and J. Lin, Mini-Rev. Org. Chem., 2017, 14, 434–447 CAS.
- (a) Y.-L. Liu, T.-D. Shi, F. Zhou, X.-L. Zhao, X. Wang and J. Zhou, Org. Lett., 2011, 13, 3826–3829 CrossRef CAS PubMed; (b) D. Zhang, J. Zhou, F. Xia, Z. Kang and W. Hu, Nat. Commun., 2015, 6, 5801 CrossRef CAS PubMed; (c) T. Takeda, A. Kondoh and M. Terada, Angew. Chem., Int. Ed., 2016, 55, 4734–4737 CrossRef CAS PubMed; (d) H.-Y. Wang, C.-W. Zheng, Z. Chai, J.-X. Zhang and G. Zhao, Nat. Commun., 2016, 7, 12720 CrossRef PubMed; (e) R.-R. Liu, J.-P. Hu, J.-J. Hong, C.-J. Lu, J.-R. Gao and Y.-X. Jia, Chem. Sci., 2017, 8, 2811–2815 RSC; (f) M. Quan, L. Tang, J. Shen, G. Yang and W. Zhang, Chem. Commun., 2017, 53, 609–612 RSC; (g) Z. Ling, S. Singh, F. Xie, L. Wu and W. Zhang, Chem. Commun., 2017, 53, 5364–5367 RSC.
- Selected examples for asymmetric α-alkylation and arylation of α-substituted amino acid derivatives, see: (a) B. M. Trost and X. Ariza, Angew. Chem., Int. Ed., 1997, 36, 2635–2637 CrossRef CAS; (b) S. Cabrera, E. Reyes, J. Alemán, A. Milelli, S. Kobbelgaard and K. A. Jørgensen, J. Am. Chem. Soc., 2008, 130, 12031–12037 CrossRef CAS PubMed; (c) D. Uraguchi, Y. Ueki and T. Ooi, J. Am. Chem. Soc., 2008, 130, 14088–14089 CrossRef CAS PubMed; (d) M. Terada, H. Tanaka and K. Sorimachi, J. Am. Chem. Soc., 2009, 131, 3430–3431 CrossRef CAS PubMed; (e) S. Dong, X. Liu, X. Chen, F. Mei, Y. Zhang, B. Gao, L. Lin and X. Feng, J. Am. Chem. Soc., 2010, 132, 10650–10651 CrossRef CAS PubMed; (f) B. M. Trost and L. C. Czabaniuk, J. Am. Chem. Soc., 2012, 134, 5778–5781 CrossRef CAS PubMed; (g) W.-Q. Zhang, L.-F. Cheng, J. Yu and L.-Z. Gong, Angew. Chem., Int. Ed., 2012, 51, 4085–4088 CrossRef CAS PubMed; (h) X. Wei, D. Liu, Q. An and W. Zhang, Org. Lett., 2015, 17, 5768–5771 CrossRef CAS PubMed; (i) T. Wang, Z. Yu, D. L. Hoon, C. Y. Phee, Y. Lan and Y. Lu, J. Am. Chem. Soc., 2016, 138, 265–271 CrossRef CAS PubMed; (j) D. Uraguchi, K. Yoshioka and T. Ooi, Nat. Commun., 2017, 8, 14793 CrossRef; (k) D. Leonard, J. W. Ward and J. Clayden, Nature, 2018, 562, 105–109 CrossRef CAS; (l) J. Kikuchi and M. Terada, Angew. Chem., Int. Ed., 2019, 58, 8458–8462 CrossRef CAS PubMed.
- Selected examples for asymmetric α-acylation of α-substituted amino acid derivatives, see: (a) J. C. Ruble and G. C. Fu, J. Am. Chem. Soc., 1998, 120, 11532–11533 CrossRef CAS; (b) S. A. Shaw, P. Aleman and E. Vedejs, J. Am. Chem. Soc., 2003, 125, 13368–13369 CrossRef CAS PubMed; (c) S. A. Shaw, P. Aleman, J. Christy, J. W. Kampf, P. Va and E. Vedejs, J. Am. Chem. Soc., 2006, 128, 925–934 CrossRef CAS PubMed; (d) C. Joannesse, C. P. Johnston, C. Concellón, C. Simal, D. Philp and A. D. Smith, Angew. Chem., Int. Ed., 2009, 48, 8914–8918 CrossRef CAS PubMed; (e) D. Uraguchi, K. Koshimoto, S. Miyake and T. Ooi, Angew. Chem., Int. Ed., 2010, 49, 5567–5569 CrossRef PubMed; (f) Z. Zhang, F. Xie, J. Jia and W. Zhang, J. Am. Chem. Soc., 2010, 132, 15939–15941 CrossRef CAS PubMed; (g) C. K. De, N. Mittal and D. Seidel, J. Am. Chem. Soc., 2011, 133, 16802–16805 CrossRef CAS PubMed; (h) C.-T. Chen, C.-C. Tsai, P.-K. Tsou, G.-T. Huang and C.-H. Yu, Chem. Sci., 2017, 8, 524–529 RSC; (i) T. Cruchter, M. G. Medvedev, X. Shen, T. Mietke, K. Harms, M. Marsch and E. Meggers, ACS Catal., 2017, 7, 5151–5162 CrossRef CAS; (j) T. Yamamoto, R. Murakami and M. Suginome, J. Am. Chem. Soc., 2017, 139, 2557–2560 CrossRef CAS PubMed; (k) M.-S. Xie, Y.-F. Zhang, M. Shan, X.-X. Wu, G.-R. Qu and H.-M. Guo, Angew. Chem., Int. Ed., 2019, 58, 2839–2843 CrossRef CAS PubMed.
- I. D. Hills and G. C. Fu, Angew. Chem., Int. Ed., 2003, 42, 3921–3924 CrossRef CAS PubMed.
- (a) P. A. Wender, V. A. Verma, T. J. Paxton and T. H. Pillow, Acc. Chem. Res., 2008, 41, 40–49 CrossRef CAS PubMed; (b) T. Newhouse, P. S. Baran and R. W. Hoffmann, Chem. Soc. Rev., 2009, 38, 3010–3021 RSC; (c) J. Rittle, M. J. Field, M. T. Green and F. A. Tezcan, Nat. Chem., 2019, 11, 434–441 CrossRef CAS PubMed.
- M. Wang, X. Zhang, Z. Ling, Z. Zhang and W. Zhang, Chem. Commun., 2017, 53, 1381–1384 RSC.
- M. Wang, Z. Zhang, S. Liu, F. Xie and W. Zhang, Chem. Commun., 2014, 50, 1227–1230 RSC.
- (a) Q. Shao, L. Wu, J. Chen, I. D. Gridnev, G. Yang, F. Xie and W. Zhang, Adv. Synth. Catal., 2018, 360, 4625–4633 CrossRef CAS; (b) P. Liu, X. Huo, B. Li, R. He, J. Zhang, T. Wang, F. Xie and W. Zhang, Org. Lett., 2018, 20, 6564–6568 CrossRef CAS PubMed; (c) X. Huo, R. He, J. Fu, J. Zhang, G. Yang and W. Zhang, J. Am. Chem. Soc., 2017, 139, 9819–9822 CrossRef CAS PubMed; (d) X. Huo, J. Zhang, J. Fu, R. He and W. Zhang, J. Am. Chem. Soc., 2018, 140, 2080–2084 CrossRef CAS PubMed.
- An NHC-catalyzed conversion from N-acylated amino acids to C-carboxyazlactones via O- to C-carboxyl transfer was reported with less than 15% ee: (a) C. D. Campbell, N. Duguet, K. A. Gallagher, J. E. Thomson, A. G. Lindsay, A. C. O'Donoghue and A. D. Smith, Chem. Commun., 2008, 3528–3530 RSC; (b) C. D. Campbell, C. J. Collett, J. E. Thomson, A. M. Z. Slawin and A. D. Smith, Org. Biomol. Chem., 2011, 9, 4205–4218 RSC.
- For selected examples, see: (a) B.-C. Hong, A. Raja and V. M. Sheth, Synthesis, 2015, 47, 3257–3285 CrossRef CAS; (b) Y. Hayashi, Chem. Sci., 2016, 7, 866–880 RSC; (c) T. Kurose, C. Tsukano and Y. Takemoto, Org. Lett., 2017, 19, 4762–4765 CrossRef CAS PubMed.
- For selected reviews, see: (a) D. E. Fogg and E. N. dos Santos, Coord. Chem. Rev., 2004, 248, 2365–2379 CrossRef CAS; (b) J.-C. Wasilke, S. J. Obrey, R. T. Baker and G. C. Bazan, Chem. Rev., 2005, 105, 1001–1020 CrossRef CAS PubMed; (c) C. Grondal, M. Jeanty and D. Enders, Nat. Chem., 2010, 2, 167–178 CrossRef CAS PubMed; (d) B.-L. Lu, L. Dai and M. Shi, Chem. Soc. Rev., 2012, 41, 3318–3339 RSC; (e) X. Zeng, Chem. Rev., 2013, 113, 6864–6900 CrossRef CAS PubMed. For selected papers, see: (f) D. Enders, M. R. M. Hüttl, C. Grondal and G. Raabe, Nature, 2006, 441, 861–863 CrossRef CAS PubMed; (g) N. Shindoh, Y. Takemoto and K. Takasu, Chem. - Eur. J., 2009, 15, 12168–12179 CrossRef CAS PubMed; (h) L. Li and S. B. Herzon, Nat. Chem., 2013, 6, 22–27 CrossRef PubMed.
- Representative papers published by our group: (a) S. Liu, Z. Zhang, F. Xie, N. A. Butt, L. Sun and W. Zhang, Tetrahedron: Asymmetry, 2012, 23, 329–332 CrossRef CAS; (b) Z. Zhang, M. Wang, F. Xie, H. Sun and W. Zhang, Adv. Synth. Catal., 2014, 356, 3164–3170 CrossRef CAS; (c) L. Zhang, M. Wang, M. Zhou, Z. Zhang, M. Muraoka and W. Zhang, Asian J. Org. Chem., 2019, 8, 1024–1028 CrossRef; (d) M. Zhou, E. He, L. Zhang, J. Chen, Z. Zhang, Y. Liu and W. Zhang, Org. Chem. Front., 2019, 6, 3969–3972 RSC.
- Representative papers published by other groups: (a) D. A. DiRocco, Y. Ji, E. C. Sherer, A. Klapars, M. Reibarkh, J. Dropinski, R. Mathew, P. Maligres, A. M. Hyde, J. Limanto, A. Brunskill, R. T. Ruck, L.-C. Campeau and I. W. Davies, Science, 2017, 356, 426–430 CrossRef CAS PubMed; (b) D. A. Glazier, J. M. Schroeder, S. A. Blaszczyk and W. Tang, Adv. Synth. Catal., 2019, 361, 3729–3732 CrossRef CAS.
- For selected reviews, see: (a) S. A. Sieber and M. A. Marahiel, Chem. Rev., 2005, 105, 715–738 CrossRef CAS PubMed; (b) I. W. Hamley, Chem. Rev., 2017, 117, 14015–14041 CrossRef CAS PubMed. Selected papers: (c) K. Haslinger, M. Peschke, C. Brieke, E. Maximowitsch and M. J. Cryle, Nature, 2015, 521, 105–109 CrossRef CAS PubMed; (d) X. Zhang, G. Lu, M. Sun, M. Mahankali, Y. Ma, M. Zhang, W. Hua, Y. Hu, Q. Wang, J. Chen, G. He, X. Qi, W. Shen, P. Liu and G. Chen, Nat. Chem., 2018, 10, 540–548 CrossRef CAS PubMed; (e) C. P. Ting, M. A. Funk, S. L. Halaby, Z. Zhang, T. Gonen and W. A. van der Donk, Science, 2019, 365, 280–284 CrossRef CAS PubMed.
- For selected reviews, see: (a) G. C. Hargaden and P. J. Guiry, Chem. Rev., 2009, 109, 2505–2550 CrossRef CAS PubMed; (b) G. Yang and W. Zhang, Chem. Soc. Rev., 2018, 47, 1783–1810 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc00808g |
| This journal is © The Royal Society of Chemistry 2020 |