
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of poly(disulfide)s with narrow molecular weight distributions via lactone ring-opening polymerization†
Sungwhan
Kim
a,
Kamila I.
Wittek
b and
Yan
Lee
 *a
*a
aDepartment of Chemistry, College of Natural Sciences, Seoul National University, Seoul 08826, Korea. E-mail: gacn@snu.ac.kr
bDepartment of Chemistry, Johannes Gutenberg-University Mainz, Duesbergweg 10-14, 55128 Mainz, Germany
First published on 16th April 2020
Abstract
We report the first example of controlled polymerization of poly(disulfide)s with narrow molecular weight distributions. 1,4,5-oxadithiepan-2-one (OTP), a disulfide-containing 7-membered ring lactone, was polymerized by using the diphenylphosphate (DPP) catalyzed lactone ring-opening polymerization method. The polymerization proceeded in a living manner, and the resulting polymers displayed very narrow polydispersity index (PDI) values below 1.1 and excellent backbone degradability responding to reducing conditions and UV irradiation.
Introduction
The disulfide bond is often regarded as a dynamic covalent bond with conditional reactivity or meta-stability.1 It can sufficiently maintain molecular integrity, being a reasonably strong bond with typical bond dissociation energies of 60 kcal mol−1. On the other hand, it can be easily cleaved and exchanged with other bonds in the presence of physical (i.e. light,2 heat,3 mechanical force,4etc.) or chemical (i.e. radicals,5 nucleophiles,6etc.) stimuli overcoming the dissociation energy barrier. In particular, the reversibility of oxidative coupling and reductive degradation between a disulfide and two thiols is a key factor in the structural integrity and controlled reactivity of proteins.7,8The intriguing properties of disulfides have been popularly studied for application in the fields of polymer and materials science. Disulfide acts as a cross-linker between polymeric chains to improve the stiffness of polymeric networks, as shown in vulcanized rubber9 or disulfide-based hydrogels.10 Various pharmaceuticals can be conjugated to polymers or proteins via disulfide bonds for drug delivery.11,12 When appropriate signals are applied to the disulfide-based materials in a spatiotemporal manner, the disulfide bonds degrade either to disintegrate the polymer network structure or to release the active pharmaceuticals from the delivery carrier. UV-responsive self-healing gels,13 nanogels,14 and antibody–drug conjugates12 responding to high glutathione concentrations around cancerous tissues or in the cytosol are all on the frontier of materials and pharmaceutical sciences.
Notwithstanding the recent rapid progress in polymer science, the synthesis of fine polymers containing disulfide backbones is very limited, probably due to the vulnerability of disulfide bonds under most polymerization conditions. Oxidative coupling of disulfhydryl monomers,15 condensation polymerization of disulfide-pre-containing monomers16 and copolymerization of dihalide-containing monomers with sodium disulfide17 are representative strategies for the synthesis of poly(disulfide)s. However, the aforementioned step-growth polymerization methods inevitably lead to polymers only with a broad distribution of molecular weights (polydispersity index (PDI) > 2). Chain-growth polymerization has been attempted to obtain poly(disulfide)s with more narrow molecular weight distributions (MWDs), but only with limited improvement. For instance, ring-opening polymerization of strained disulfide monomers via thiol–disulfide exchange often produced mixtures of linear and cyclic polymers and even catenanes with broad MWDs.18–22 The formation of these complex product mixtures may be attributed to the limited selectivity of the thiol–disulfide exchange between disulfides in the monomer and the polymer backbone. Various reaction conditions were examined to overcome this problem and obtain linear polymers as a major product, but undesired cyclic oligomers were still present with PDI > 1.4.22 Ring-opening metathesis polymerization (ROMP), which is orthogonal with the thiol–disulfide exchange reaction, was also applied for the synthesis of poly(disulfide)s. However, undesirable interactions between the ruthenium catalyst and disulfides caused not only a large PDI > 1.5 but also a failure in homopolymerization of the disulfide monomer.23 From the previous results, we inferred that it is critical to choose polymerization conditions where unwanted thiol–disulfide exchange and catalyst–disulfide interactions are inhibited for obtaining fine poly(disulfide)s with narrow MWDs.
Results and discussion
In this research work, we selected lactone polymerization for the synthesis of poly(disulfide)s since numerous catalysts for the controlled polymerization of poly(lactone)s have already been established.24,25 As for the monomer, we designed 1,4,5-oxadithiepan-2-one (OTP), an analogue of ε-caprolactone (εCL), where two of the five methylene groups are replaced with a disulfide. We reasoned that the 7-membered ring structure may provide sufficient ring strain for polymerization (Scheme 1). OTP was prepared from the catalytic intramolecular oxidation26 of an α,ω-disulfhydryl ester formed from 2-mercaptoethanol and 2-thioglycolic acid (see the ESI†). We have screened various catalysts ranging from bases to Lewis and Brønsted acids for the OTP polymerization, but the disulfide bonds were vulnerable under most of the reaction conditions, as the catalysts induced undesirable nucleophilic attack on disulfides by enhancing the electrophilicity of disulfides or nucleophilicity of nucleophiles. Among them, diphenylphosphate (DPP), a Brønsted acid with a pKa of 3.88 in DMSO,27 showed remarkable tolerance to disulfides but strong catalytic activity for the polymerization of OTP. Therefore, we used DPP as the catalyst in all the polymerization reactions below.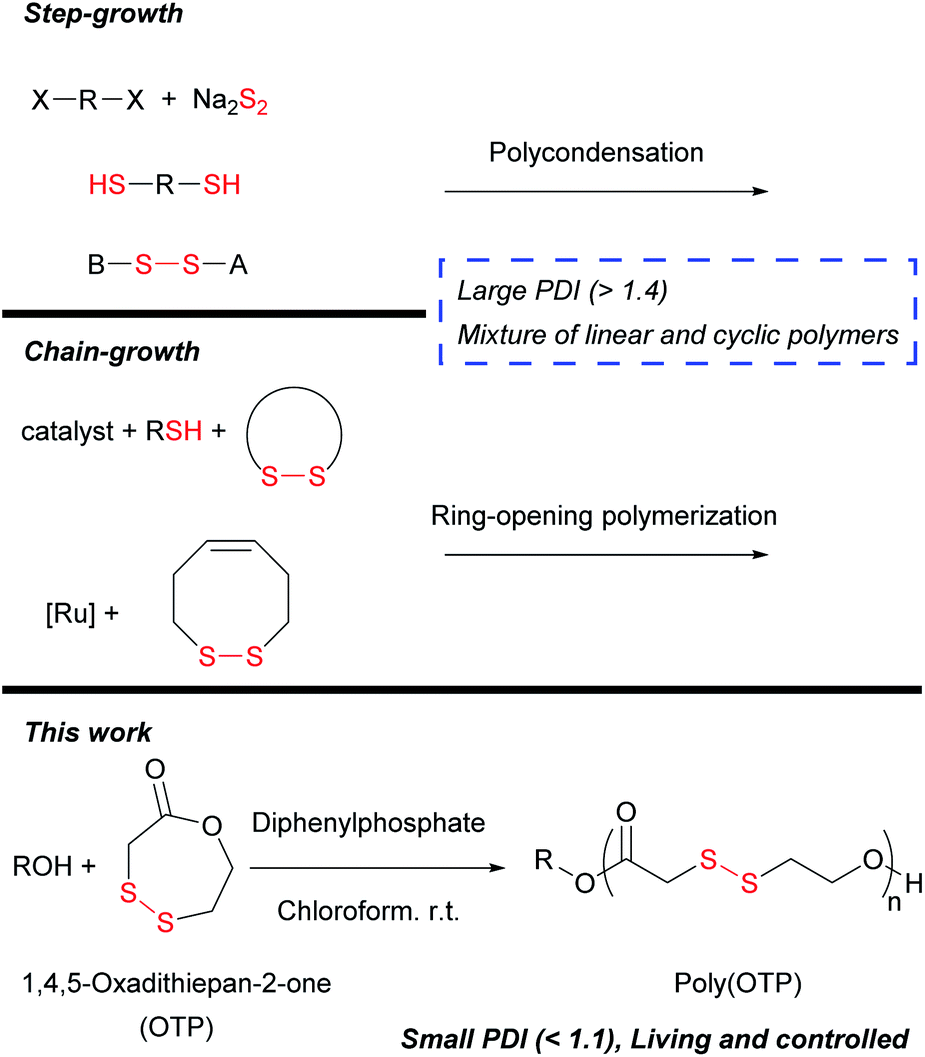 | ||
| Scheme 1 Polymerization of 1,4,5-oxadithiepan-2-one (OTP). | ||
The polymerization of OTP was carried out with catalytic DPP and benzyl alcohol (BnOH) as the initiator (I) at various [OTP]/[I] ratios (from [OTP]/[I] = 20 to 180). The general procedure for the polymerization is described in detail in the ESI.† We chose chloroform as the reaction solvent due to the solubility issue of the OTP polymer with a high sulfur content of approximately 43% (w/w). The MWDs were examined by size exclusion chromatography (SEC) (Table 1 and S1, ESI†). Surprisingly, the polymers (P1–P5) were found to have very narrow MWDs (PDI < 1.1) throughout the wide range of the monomer (M) to initiator ratio. The PDI values of poly(OTP)s are even smaller than those of poly(εCL) or poly(δ-valerolactone) in previous reports where the same catalyst was utilized.25Mn values from the SEC analysis were in reasonable agreement with the target Mn. Additionally, we could confirm the preservation of a well-oriented head-to-tail backbone without disulfide exchange and fidelity of the terminal benzyl group in poly(OTP)s by MALDI-MS and 1H NMR study (Fig. S7 and S13–S17, ESI†).
| Entry | Initiator | Target [M]/[I] | Time | Conversion (%)a | M n,target. (kDa) | M n,SEC (kDa) | PDI |
|---|---|---|---|---|---|---|---|
| a Determined by 1H NMR spectra. b Determined by size exclusion chromatography (SEC), with chloroform as the eluent, calibrated with polystyrene (PS) standards. c Determined by SEC, with tetrahydrofuran as the eluent, calibrated with PS standards. | |||||||
| P1 | BnOH | 20 | 18 | >99 | 3.11 | 3.08b | 1.07b |
| P2 | BnOH | 40 | 24 | >99 | 6.12 | 5.71b | 1.05b |
| P3 | BnOH | 80 | 48 | >99 | 12.1 | 11.2b | 1.05b |
| P4 | BnOH | 120 | 72 | >99 | 18.1 | 17.0b | 1.04b |
| P5 | BnOH | 180 | 96 | 99 | 21.7 | 22.6b | 1.03b |
| P6 | Propargyl alcohol | 50 | 32 | >99 | 7.57 | 7.82b | 1.05b |
| P7 | 2-Propanol | 50 | 32 | >99 | 7.57 | 8.79b | 1.05b |
| P8 | mPEO-OH (1 kDa) | 50 | 32 | >99 | 8.51 | 9.22c | 1.05c |
| P9 | mPEO-OH (2 kDa) | 80 | 48 | >99 | 14.0 | 13.1c | 1.04c |
To widen the applicability of our method, we investigated various alcohols for initiation. Propargyl alcohol also produced poly(OTP) (P6) with desired molecular weights and a narrow PDI (Fig. S2, ESI†), which gives access to future end group functionalization via the alkyne–azide click reaction.28 Next, when we utilized 2-propanol as the initiator, we could still obtain a narrow MWD (P7, PDI = 1.05) (Fig. S3, ESI†) although secondary alcohols are generally recognized to be unsuitable for rapid initiation.29 We assumed that the initiation rate of OTP, even by secondary alcohols, is significantly faster than the propagation rate, thus leading to the low PDI. Furthermore, ω-methoxy-capped polyethylene oxide (mPEO) was also used as a macroinitiator for the synthesis of a block copolymer. The polymerization resulted in mPEO-b-poly(OTP) with narrow MWDs (P8 and P9) (Fig. S4 and S5, ESI†). These well-defined amphiphilic block copolymers with narrow MWDs would form controllable polymeric nanostructures with disulfide-based responsiveness for future applications.
We then examined whether OTP is polymerized in a living/controlled manner. The polymerization kinetics of poly(OTP) were measured by 1H NMR and SEC (Fig. 1). We could observe a linear relationship between Mnversus the conversion ratio and a decrease in the PDI (Mw/Mn) as the reaction progressed. Also, by plotting −ln([M]/[M]0) versus reaction time, we could obtain a strict linear relationship, which suggests that the polymerization rate is proportional to the OTP concentration in the first order.
 | ||
| Fig. 1 Controlled ring-opening polymerization of OTP. (a) Mn and PDI of poly(OTP) plotted against conversion. [OTP]/[BnOH] = 40, Mn and PDI were determined by SEC (chloroform as the eluent, PS standards). The conversion was determined by 1H NMR. (b) −ln([M]/[M]0) plotted against reaction time. [M]/[M]0 was determined by 1H NMR. (c) SEC chromatograms of poly(OTP)s changing over reaction time. | ||
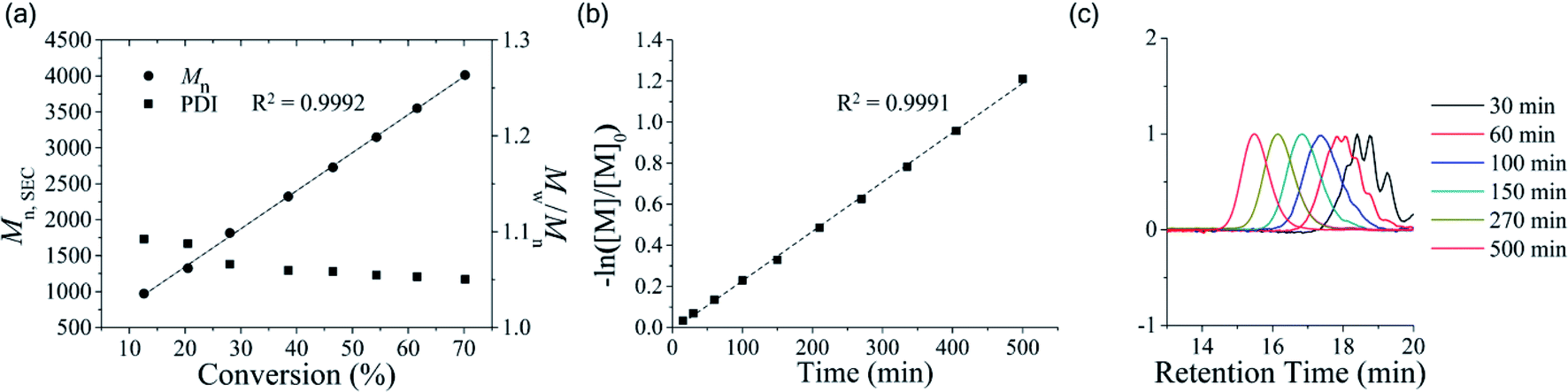
We also performed one-pot post-polymerization with additional OTP feed to prove the livingness of the polymeric end group. First, we conducted polymerization at a [M]/[I] ratio of 25 for 24 h, where the full conversion of the monomer was confirmed by 1H NMR. The solution was further stirred without quenching for an additional 24 h. There was still no sign of backbiting or peak broadening in the SEC chromatogram even after 24 h from monomer depletion (Fig. 2a, black curve). Then, we added the second monomer solution feed ([M]/[I] = 25) to the unquenched polymer solution and stirred the mixture for an additional 48 h. We confirmed the second full consumption of the monomer by 1H NMR. The SEC chromatogram clearly showed the peak shift with an almost 2-fold increase of Mn from 3.63 kDa to 6.56 kDa, as well as the maintenance of the low PDI < 1.05 (Fig. 2a, red curve), which implies that the chain end of poly(OTP) is still “living”.
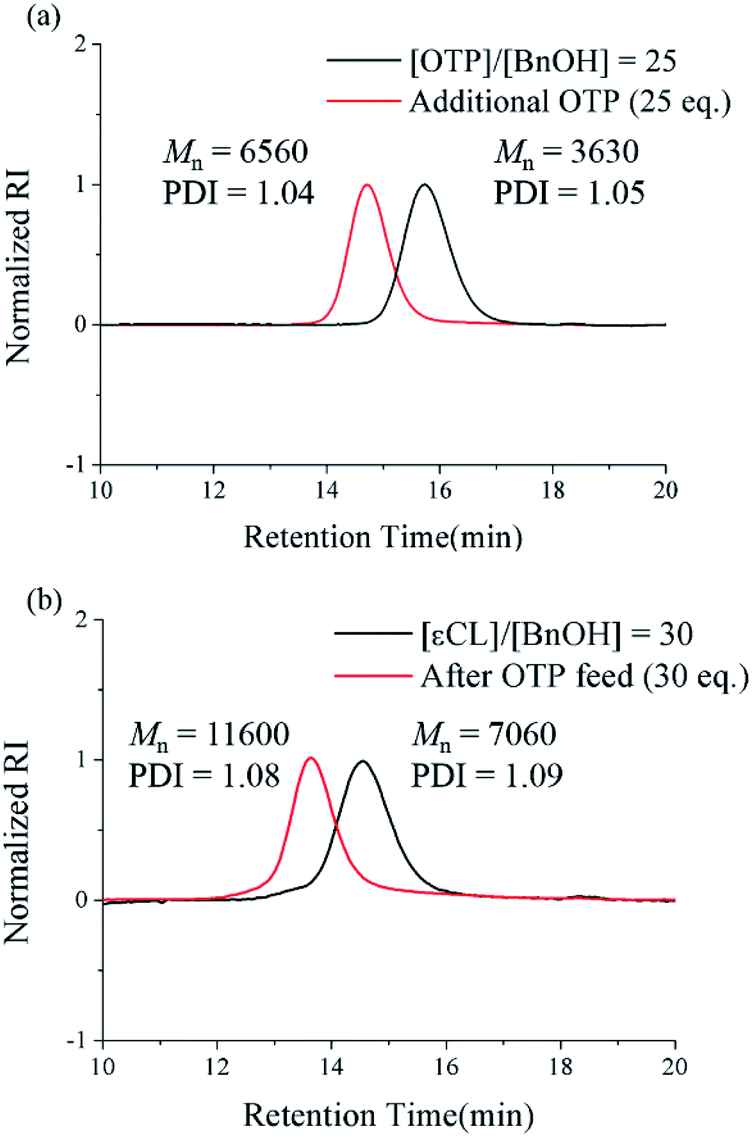 | ||
| Fig. 2 Successive one-pot post-polymerization of OTP. (a) The SEC chromatograms of poly(OTP)s prepared by one-pot post-homopolymerization. The first feed ([OTP]/[BnOH] = 25) and the second feed ([OTP]/[BnOH] = 25). (b) The SEC chromatograms of poly(εCL) and poly(εCL)-b-poly(OTP) prepared by one-pot post-copolymerization. The first feed ([εCL]/[BnOH] = 30) and the second feed ([OTP]/[BnOH] = 30). The black and red curves indicate the molecular weight distributions of polymers from the polymerization with only the first feed and with both the first and the second feeds, respectively. | ||
The results above encouraged us to attempt a successive ring-opening polymerization of εCL and OTP in a one-pot manner for the preparation of poly(εCL)-b-poly(OTP). After the first polymerization of the poly(εCL) block at a [εCL]/[I] ratio of 30 for 24 h, where the full conversion of εCL was confirmed by 1H NMR, the second OTP monomer was injected rapidly at an [OTP]/[I] ratio of 30. After the second polymerization proceeded for 48 h, we could observe that the SEC peak had shifted from the initial peak of poly(εCL) and that Mn showed an increase of 4.5 kDa (Fig. 2b). The PDI value was also maintained below 1.09 during the successive one-pot polymerization. In addition, the peak corresponding to the methylene proton at the chain end (–CH2OH) of poly(εCL) at 3.65 ppm (ref. 25) shifted to 4.15 ppm corresponding to those next to ester (–CH2OOC–) in the 1H NMR spectrum after the polymerization (see the ESI†), implying the successful one-pot post-polymerization with OTP from poly(εCL).
The disulfide backbone of poly(OTP) is expected to have degradability responding to various chemical and physical stimuli. As a representative chemical stimulus to trigger the disulfide metathesis in poly(OTP), we chose D,L-dithiothreitol (DTT), a reagent well known to induce thiol–disulfide exchange reactions by forming a stable intramolecular cyclic disulfide while reducing other disulfide substrates.30P3 (Mn = 11.2 kDa) was dissolved in chloroform and DTT was added to the solution at a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 relative to the disulfide content of P3. After incubation at ambient temperature for 24 h, the backbone of poly(OTP) was almost completely degraded to oligomers, as shown in the SEC chromatogram in Fig. 3a. Poly(OTP) showed the characteristic degradability of disulfide polymers responding to the reducing conditions. Then, we also examined the backbone degradation of poly(OTP) by UV irradiation, which is known to facilitate disulfide metathesis by generating thiyl radicals via the homolytic cleavage of disulfide.2 After irradiation with UV light (λmax = 357 nm, 5 W cm−2), the initial peak of P2 (Mn = 5.71 kDa) in the SEC chromatogram gradually decreased in intensity with an increase in retention time, while small peaks appeared with increasing intensity in the oligomer region (Fig. 3b). The results clearly exhibit the UV-responsive degradability of the poly(OTP) backbone. In addition, the disulfide bonds in the poly(OTP) backbone are quite stable even at 100 °C, where the broadening of the MWD was observed probably due to transesterification, but start to show exchange reactions to produce a mixture of head-to-head, head-to-tail and tail-to-tail disulfide bonds with larger broadening of the MWD at 120 °C (Fig. S11 and S12, ESI†).
:1 relative to the disulfide content of P3. After incubation at ambient temperature for 24 h, the backbone of poly(OTP) was almost completely degraded to oligomers, as shown in the SEC chromatogram in Fig. 3a. Poly(OTP) showed the characteristic degradability of disulfide polymers responding to the reducing conditions. Then, we also examined the backbone degradation of poly(OTP) by UV irradiation, which is known to facilitate disulfide metathesis by generating thiyl radicals via the homolytic cleavage of disulfide.2 After irradiation with UV light (λmax = 357 nm, 5 W cm−2), the initial peak of P2 (Mn = 5.71 kDa) in the SEC chromatogram gradually decreased in intensity with an increase in retention time, while small peaks appeared with increasing intensity in the oligomer region (Fig. 3b). The results clearly exhibit the UV-responsive degradability of the poly(OTP) backbone. In addition, the disulfide bonds in the poly(OTP) backbone are quite stable even at 100 °C, where the broadening of the MWD was observed probably due to transesterification, but start to show exchange reactions to produce a mixture of head-to-head, head-to-tail and tail-to-tail disulfide bonds with larger broadening of the MWD at 120 °C (Fig. S11 and S12, ESI†).
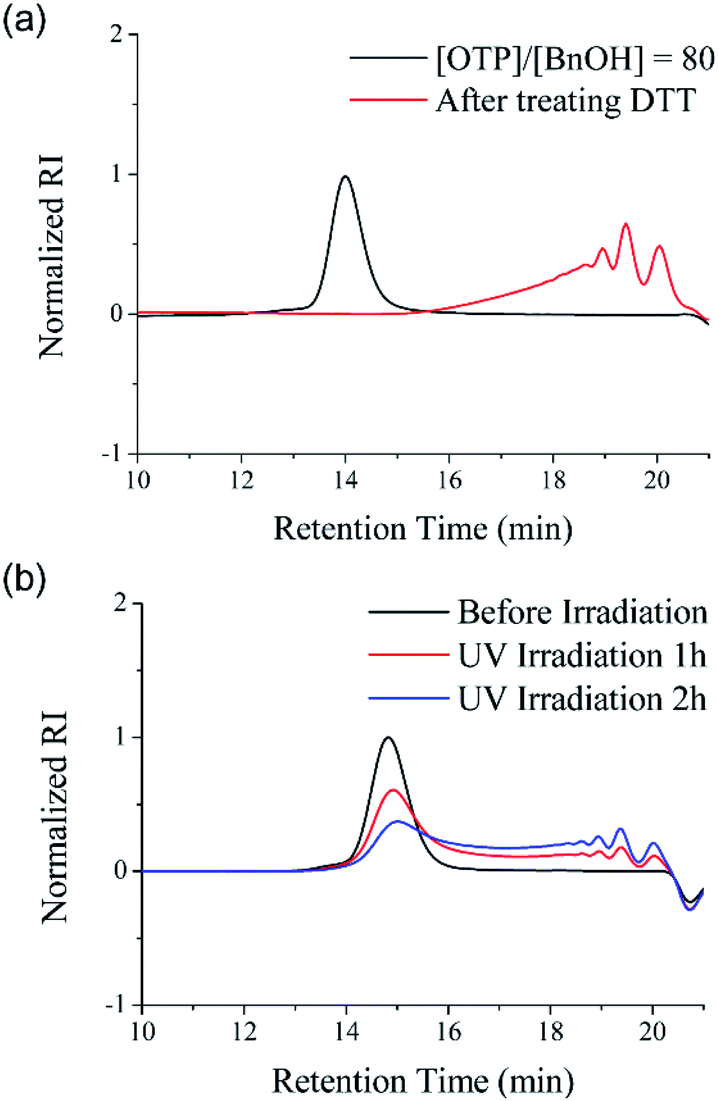 | ||
| Fig. 3 Stimuli-responsive degradability of poly(OTP)s. (a) The change of the SEC chromatogram of poly(OTP) (P3) before and after treatment with DTT (1 eq. of the disulfide bonds in P3). (b) The change of the SEC chromatogram of poly(OTP) (P2) before and after UV irradiation (5 W cm−2). The black curves indicate the molecular weight distributions of poly(OTP)s before stimuli-triggering. | ||
Conclusions
In summary, we have demonstrated the first example of controlled living polymerization of disulfide-backbone polymers with narrow MWDs (PDI < 1.1). We could successfully synthesize poly(OTP)s from various alcohol initiators and their block copolymers with PEO and poly(εCL) through the combination of a newly designed 7-membered disulfide-containing lactone, OTP, and a lactone-activating catalyst unreactive to disulfides. The poly(OTP)s showed characteristic degradability of disulfide-bearing compounds under exposure to thiols and UV irradiation. Thanks to the simple introduction of a poly(disulfide) backbone with a controlled length, the OTP polymerization can be a very useful tool for the development of smart materials with stimuli-responsive reversible degradability, especially in the biomedical fields. More importantly, we believe that poly(OTP)s can be applicable to supramolecular chemistry for formation of delicate nanostructures requiring narrow MWDs of the components, where the application of poly(disulfide)s with attractive stimuli-responsiveness has been difficult due to broad MWDs until now.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a National Research Foundation of Korea Grant funded by the Korean Government (NRF-2015R1C1A1A01052185 and NRF-2017M3A9E9072938).Notes and references
- S. P. Black, J. K. M. Sanders and A. R. Stefankiewicz, Chem. Soc. Rev., 2014, 43, 1861 RSC.
- (a) J. Cao and D.-C. Chen, Phys. Chem. Chem. Phys., 2019, 21, 4176 RSC; (b) F. Klepel and B. J. Ravoo, Org. Biomol. Chem., 2017, 15, 3840 RSC.
- U. Lafont, H. van Zeijl and S. van der Zwaag, ACS Appl. Mater. Interfaces, 2012, 4, 6280 CrossRef CAS PubMed.
- T. J. Kucharski, Z. Huang, Q.-Z. Yang, Y. Tian, N. C. Rubin, C. D. Concepcion and R. Boulatov, Angew. Chem., Int. Ed., 2009, 48, 7040 CrossRef CAS PubMed.
- (a) V. Favaudon, H. Tourbez, C. Houée-Levin and J.-M. Lhoste, Biochemistry, 1990, 29, 10978 CrossRef CAS PubMed; (b) C. H. Sohn, J. Gao, D. A. Thomas, T.-Y. Kim, W. A. Goddard III and J. L. Beauchamp, Chem. Sci., 2015, 6, 4550 RSC.
- (a) R. Caraballo, M. Rahm, P. Vongvilai, T. Brinck and O. Ramström, Chem. Commun., 2008, 6603 RSC; (b) D. L. Rabenstein and K. H. Weaver, J. Org. Chem., 1996, 61, 7391 CrossRef CAS PubMed.
- M. V. Trivedi, J. S. Laurence and T. J. Siahaan, Curr. Protein Pept. Sci., 2009, 10, 614 CrossRef CAS PubMed.
- K. S. Siddiqui, A. Poljak, M. Guilhaus, G. Feller, S. D'Amico, C. Gerday and R. Cavicchioli, J. Bacteriol., 2005, 187, 6206 CrossRef CAS PubMed.
- M. Akiba and A. S. Hashim, Prog. Polym., Sci., 1997, 22, 475 CrossRef CAS.
- (a) X. Z. Shu, Y. Liu, Y. Luo, M. C. Roberts and G. D. Prestwich, Biomacromolecules, 2002, 3, 1304 CrossRef CAS PubMed; (b) H. Yu, Y. Wang, H. Yang, K. Peng and X. Zhang, J. Mater. Chem. B, 2017, 5, 4121 RSC.
- (a) S. Lv, Z. Tang, D. Zhang, W. Song, M. Li, J. Lin, H. Liu and X. Chen, J. Controlled Release, 2014, 194, 220 CrossRef CAS PubMed; (b) W. Chen, L. Ali Shah, M. Siddiq, J. Hu and D. Yang, RSC Adv., 2015, 5, 7559 RSC.
- J. D. Sadowsky, T. H. Pillow, J. Chen, F. Fan, C. He, Y. Wang, G. Yan, H. Yao, Z. Xu, S. Martin, D. Zhang, P. Chu, J. dela Cruz-Chuh, A. O'Donohue, G. Li, G. Del Rosario, J. He, L. Liu, C. Ng, D. Su, G. D. Lewis Phillips, K. R. Kozak, S.-F. Yu, K. Xu, D. Leipold and J. Wai, Bioconjugate Chem., 2017, 28, 2086 CrossRef CAS PubMed.
- B. D. Fairbanks, S. P. Singh, C. N. Bowman and K. S. Anseth, Macromolecules, 2011, 44, 2444 CrossRef CAS PubMed.
- S. A. Elkassih, P. Kos, H. Xiong and D. J. Siegwart, Biomater. Sci., 2019, 7, 607 RSC.
- (a) E. Q. Rosenthal, J. E. Puskas and C. Wesdemiotis, Biomacromolecules, 2012, 13, 154 CrossRef CAS PubMed; (b) N. V. Tsarevsky and K. Matyjaszewski, Macromolecules, 2002, 35, 9009 CrossRef CAS.
- M. Ou, R. Xu, S. H. Kim, D. A. Bull and S. W. Kim, Biomaterials, 2009, 30, 5804 CrossRef CAS PubMed.
- G. R. Pettit and G. R. Petit III., J. Chem. Educ., 1978, 55, 472 CrossRef CAS.
- X. Zhang and R. M. Waymouth, J. Am. Chem. Soc., 2017, 139, 3822 CrossRef CAS PubMed.
- E.-K. Bang, G. Gasparini, G. Molinard, A. Roux, N. Sakai and S. Matile, J. Am. Chem. Soc., 2013, 135, 2088 CrossRef CAS PubMed.
- H. Ishida, A. Kisanuki and K. Endo, Polym. J., 2009, 41, 110 CrossRef CAS.
- K. Endo, T. Shiroi, N. Murata, G. Kojima and T. Yamanaka, Macromolecules, 2004, 37, 3143 CrossRef CAS.
- Y. Liu, Y. Jia, Q. Wu and J. S. Moore, J. Am. Chem. Soc., 2019, 141, 17075 CrossRef CAS PubMed.
- C.-C. Chang and T. Emrick, Macromolecules, 2014, 47, 1344 CrossRef CAS.
- For various examples of living polymerization of lactones, see: (a) N. Nomura, A. Taira, T. Tomioka and M. Okada, Macromolecules, 2000, 33, 1497 CrossRef CAS; (b) R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2006, 128, 4556 CrossRef CAS PubMed; (c) Y. Shibasaki, H. Sanada, M. Yokoi, F. Sanda and T. Endo, Macromolecules, 2000, 33, 4316 CrossRef CAS; (d) S. Slomkowski, Macromol. Symp., 1996, 103, 213 CAS; (e) S. Gazeau-Bureau, D. Delcroix, B. Martin-Vaca, D. Boourissou, C. Navarro and S. Magnet, Macromolecules, 2008, 41, 3782 CrossRef CAS.
- (a) K. Makiguchi, T. Satoh and T. Kakuchi, Macromolecules, 2011, 44, 1999 CrossRef CAS; (b) D. Delcroix, A. Couffin, N. Susperregui, C. Navarro, L. Maron, B. Martin-Vaca and D. Bourissou, Polym. Chem., 2011, 2, 2249 RSC.
- M. Kirihara, Y. Asai, S. Ogawa, T. Noguchi, A. Hatano and Y. Hirai, Synthesis, 2007, 21, 3286 CrossRef.
- P. Christ, A. G. Lindsay, S. S. Vormittag, J.-M. Neudörfl, A. Berkessel and A. C. O'Donoghue, Chem.- Eur. J., 2011, 17, 8524 CrossRef CAS PubMed.
- M. Degirmenci, P. Atuk Besli and N. Genli, J. Polym. Res., 2014, 21, 540 CrossRef.
- J. Zhao, D. Pahovnik, Y. Gnanou and N. Hadjichristidis, Macromolecules, 2014, 47, 3814 CrossRef CAS.
- W. W. Cleland, Biochemistry, 1964, 3, 480 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc00834f |
| This journal is © The Royal Society of Chemistry 2020 |