
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInhibiting cancer metabolism by aromatic carbohydrate amphiphiles that act as antagonists of the glucose transporter GLUT1†
Alexandra
Brito
 abcd,
Patrícia M. R.
Pereira
c,
Diana
Soares da Costa
ab,
Rui L.
Reis
abe,
Rein V.
Ulijn
dfg,
Jason S.
Lewis
chijk,
Ricardo A.
Pires
*abe and
Iva
Pashkuleva
*ab
abcd,
Patrícia M. R.
Pereira
c,
Diana
Soares da Costa
ab,
Rui L.
Reis
abe,
Rein V.
Ulijn
dfg,
Jason S.
Lewis
chijk,
Ricardo A.
Pires
*abe and
Iva
Pashkuleva
*ab
a3B's Research Group, I3Bs – Research Institute on Biomaterials, Biodegradables and Biomimetics, University of Minho, Headquarters of the European Institute of Excellence on Tissue Engineering and Regenerative Medicine, AvePark, Parque de Ciência e Tecnologia, Zona Industrial da Gandra, 4805-017 Barco, Guimarães, Portugal. E-mail: rpires@i3bs.uminho.pt; pashkuleva@i3bs.uminho.pt
bICVS/3Bs – PT Government Associate Laboratory, Braga/Guimarães, Portugal
cDepartment of Radiology, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA
dAdvanced Science Research Center (ASRC) at the Graduate Center, City University of New York (CUNY), 85 St Nicholas Terrace, New York, New York 10031, USA
eThe Discoveries Centre for Regenerative and Precision Medicine Headquarters at University of Minho, Avepark, 4805-017 Barco, Guimarães, Portugal
fDepartment of Chemistry, Hunter College, City University of New York, 695 Park Avenue, New York 10065, USA
gPhD Programs in Biochemistry and Chemistry, The Graduate Center of the City University of New York, New York 10016, USA
hDepartment of Radiology, Weill Cornell Medical College, New York, NY 10065, USA
iMolecular Pharmacology Program, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA
jDepartment of Pharmacology, Weill Cornell Medical College, New York, NY 10065, USA
kRadiochemistry and Molecular Imaging Probes Core, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA
First published on 20th March 2020
Abstract
We report on aromatic N-glucosides that inhibit selectively the cancer metabolism via two coexistent mechanisms: by initial deprivation of the glucose uptake through competitive binding in the glucose binding pocket of GLUT1 and by formation of a sequestering nanoscale supramolecular network at the cell surface through localized (biocatalytic) self-assembly. We demonstrate that the expression of the cancer associated GLUT1 and alkaline phosphatase are crucial for the effectiveness of this combined approach: cancer cells that overexpress both proteins are prompter to cell death when compared to GLUT1 overexpressing cells. Overall, we showcase that the synergism between physical and biochemical deprivation of cancer metabolism is a powerful approach for development of effective anticancer therapies.
Introduction
Cancer is a major health problem for modern society, causing 9.6 million deaths worldwide in 2018 and the 5 year prevalence was estimated to be 43.8 million.1 These statistics show that the conventional therapies are not sufficiently effective. There are two main reasons that limit the efficacy of existing treatments - the great complexity and consequent limited understanding of cancer cells, and their resistance to the currently used therapeutics.2–4 Common strategies for targeting the inhibition of specific molecular pathways often have only temporary success followed by a tumor relapse.4 The use of a combined approach with several drugs can give rise to higher efficiency and avoidance of chemoresistance but so far, the results did not achieve the required efficacy.2 Herein, we propose an alternative approach to cancer management that uses a single, multifunctional but yet specific molecule that targets a common cancer hallmark – their accelerated metabolism associated with a higher demand for nutrients and energy – via two synergistic pathways, namely by formation of a nanoscale supramolecular network around cancer cells and by the blockage of glucose transportation (Fig. 1).4,5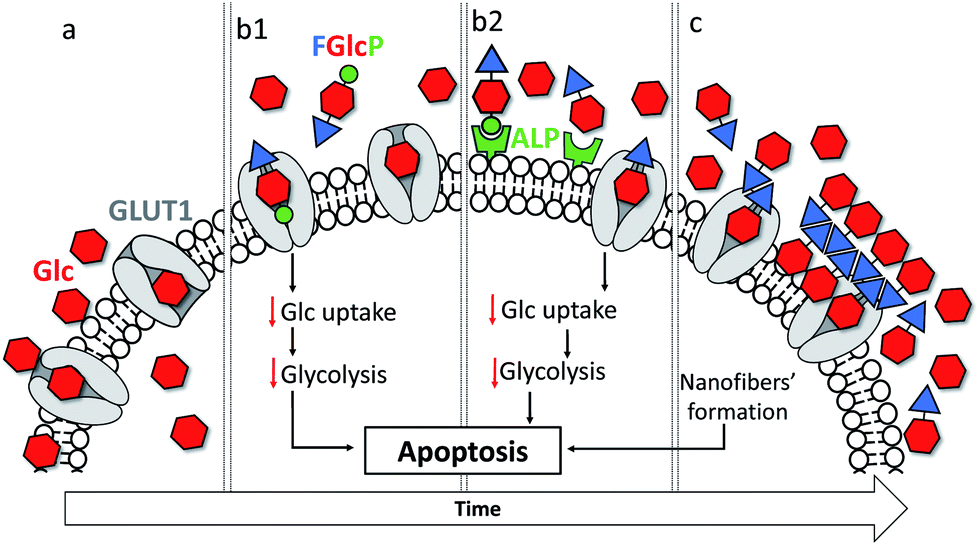 | ||
| Fig. 1 Schematic presentation of the mechanism of action of N-fluorenylmethyloxycarbonyl-glucosamine-6-phosphate (FGlcP): (a) the increased glucose (Glc) metabolism is targeted via concomitant (b) blocking of the overexpressed glucose transporters (GLUT1) with (b1) FGlcP or/and (b2) its dephosphorylated analogue generated in situ and (c) formation of nanonet by self-assembly of this analogue. | ||
The altered cancer metabolism has inspired the development of various therapies.6–11 Among them, biocatalytic self-assembly (BSA) has emerged as a biochemical approach that makes use of pathologically overexpressed enzymes (typically MMPs or phosphatases) to transform specifically designed precursors into self-assembling molecules and to trigger their assembly in situ, i.e. in close proximity of cancer cells.2,6,12–20 Several studies suggested a mechanism that involves an initial internalization of the precursor by the cells, followed by intracellular enzymatic transformation and assembly that leads to cellular death.6,18,21 Most of the described precursors are based on short self-assembling peptides that are functionalized with enzymatic sensitive moieties.11,12,17,19–21 We recently demonstrated the use of a glucose derivative, N-fluorenylmethyloxycarbonyl-glucosamine-6-phosphate (FGlcP), as an alternative to peptide-based systems in phosphatase-triggered BSA cancer management.7 For this system, we did not observe any intracellular assembly but formation of a nanoscale supramolecular network around osteosarcoma cells that overexpress membrane-bound alkaline phosphatase (ALP). The system proved to be effective in killing osteosarcoma cells. The amphiphile structure is based on glucose (Glc), which led us to hypothesize that FGlcP might act not only as a BSA precursor but also as an antagonist of the Glc transport, inhibiting aerobic glycolysis. Herein we investigate this hypothesis and demonstrate that a single compound, FGlcP, acts as an efficient cancer antimetabolite by concomitant blocking the glucose transporter 1 (GLUT1) via specific interactions with them and formation of a nanonet serving not only as a physical barrier between the cancer cells and their environment, but also as a reservoir of GLUT inhibitor (Fig. 1).
Results
Molecular design and synthesis of aromatic N-glucosides
We designed and synthesized three derivatives (Fig. 2) of Glc (1a) aiming to establish reliable controls and to investigate the influence of different functional components (aromatic, glucose, charged groups) in the interactions with GLUT1. FGlcP (1b) is a BSA precursor that is transformed into the self-assembling molecule N-fluorenylmethyloxycarbonyl-glucosamine (FGlc, 1c) upon phosphatase action (Fig. 1(b2)).7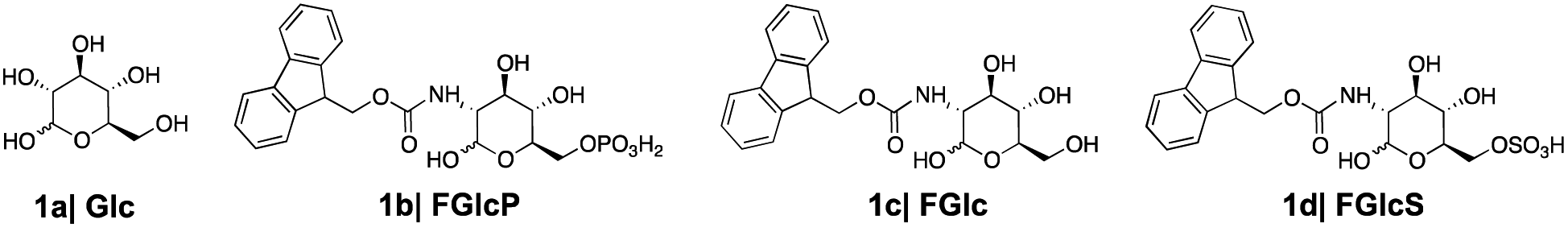 | ||
| Fig. 2 Glucose (1a) and aromatic N-glucosides (1b–d) synthesized and used in this study. | ||
FGlc does not contain a charged group and thus, it has reduced solubility in aqueous media. Previously, we have shown that 1c can self-assemble into nanofibers above a critical concentration that can be achieved upon heating or enzymatic action.7,22,23N-fluorenylmethyloxycarbonyl glucosamine-6-sulphate (FGlcS, 1d) is an analogue of FGlcP, in which the phosphate group is replaced by a negatively charged sulphate. From a mechanistic point of view, the main difference between 1b and 1d is that the sulfate derivative is not susceptible to phosphatase transformation and the subsequent self-assembly (Fig. S4†), i.e. we hypothesize that it will act solely as GLUT1 antagonist. All compounds were synthesized using a previously established procedure.7,22,23
In silico analysis shows that 1a–d bind to GLUT1
Overexpression of GLUT1 has been associated with different tumor types.9,10,24 We evaluated the differential interactions of the compounds 1a–d with GLUT1 by a computational molecular-docking process using the web-based SwissDock program (Tables 1 and S1†).25,26 The parameters were set for blind docking, i.e. we did not define a priori any specific region for binding. Docking studies with Glc (1a) were also carried out using these parameters to confirm the fidelity of the generated outputs by comparison with published data.8,27,28 The results demonstrated spontaneous complexation between GLUT1 and all compounds 1a–d: Gibbs free energy for all studied systems was negative (ΔG < 0) but the absolute values are larger for the amphiphiles 1b–d when compared to 1a (Table 1), indicating a higher affinity of these derivatives to the transporter. Van der Waals interactions (ΔGvdw) have the greatest contribution to this spontaneous process (InterFull values are the same as ΔGvdw) especially in the case of aromatic N-glucosides (1avs.1b–d) suggesting involvement of additional π–π and CH–π interactions due to the introduced Fmoc functionality. Modification with polar groups reduces additionally the ΔGvdw (1cvs.1b and 1d) but also the solvation energy (ΔGligsolvpol) evidencing formation of H-bonding between the GLUT1 and phosphate (1b) or sulfate (1d) groups. Of note, all compounds 1a–d bind to the same pocket via interactions with amino acids that are crucial for the Glc complexation with GLUT1 (Fig. 3 and Table S1†).8,27,28 Altogether the in silico data indicated that the studied compounds can deprive the Glc transport via competitive binding to GLUT1 and their antagonist activity follows the order: 1b > 1d > 1c.| Comp | ΔG (kcal mol−1) | ΔGvdw (kcal mol−1) | ΔGligsolvnonpol (kcal mol−1) | ΔGligsolvpol (kcal mol−1) |
|---|---|---|---|---|
| a ΔG: Gibbs energy; ΔGvdw: energy of van der Waals interactions; ΔGligsolvnonpol: solvation energy due to non-polar interactions; ΔGligsolvpol: solvation energy due to polar interactions. | ||||
| 1a | −6.77 | −36.32 | 6.48 | −13.50 |
| 1b | −10.09 | −68.89 | 10.36 | −24.31 |
| 1c | −8.33 | −44.94 | 9.39 | −16.74 |
| 1d | −9.04 | −55.70 | 10.57 | −23.14 |
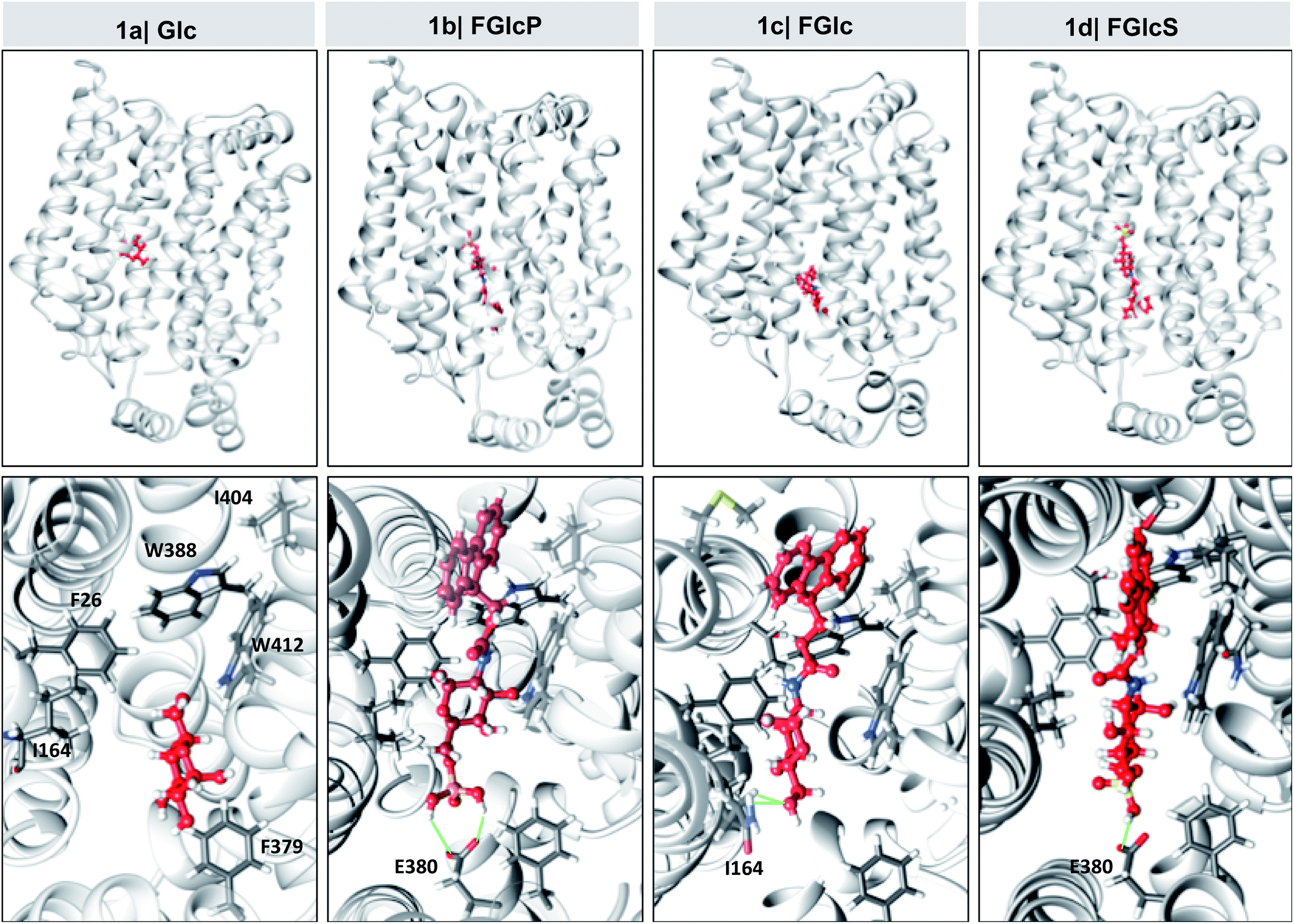 | ||
| Fig. 3 In silico models for the interactions between the synthesized aromatic N-glucosides (1b–d, red) and GLUT1 (grey). The upper row shows top score clusters (with the lowest FullFitness) of the full-size complex where the extracellular segment of GLUT1 is oriented upwards and the intracellular one downwards. The bottom row represents the magnification of the glucose-binding pocket in GLUT1. Hydrogen bonds are denotated with green lines. | ||
Selection of cancer cell lines
To test the effect of the aromatic carbohydrate amphiphiles we selected an osteosarcoma cell line (SaOs2) and a mammary gland/breast cancer cell line (MDA-MB-468) based on an initial search in the Cancer Cell Line Encyclopedia (CCLE) for cell lineages with high expression of solute carrier family 2 member 1 gene (SLC2A1) encoding GLUT1.29,30 We confirmed the expression of GLUT1 in SaOs2 and MDA-MB-468 cancer cells at transcriptional and protein levels (Fig. S5†).PCR analysis demonstrated that the selected cell lines express the SLC2A1 gene (Fig. S5a†). Immunostaining confirmed the expression of GLUT1 for both cell lines (Fig. S5b†) but flow cytometry analysis demonstrated 2-fold larger population of cells expressing GLUT1 in SaOs2 compared with MDA-MB-468 (Fig. S5c†). In addition, SaOs2 overexpress membrane-bound alkaline phosphatase that can trigger BSA on their surface while MDA-MB-468 present 3-fold less expression of this enzyme (Fig. S5d†).7 These differences are important for the experimental design as they might enable us to distinguish the two possible mechanisms by which the amphiphiles can affect the metabolism of the cancer cells – BSA and inhibition of GLUT1 as well as any cooperative effect between these mechanisms.
Aromatic N-glucosides decrease Glc uptake by tumor cells
We used 2-(N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino)-2-deoxyglucose (NBDG) for visualization and quantification of the cells' glucose uptake in the presence and absence of compounds 1b–d.31,32 Initially, the rate of NBDG uptake was studied for different incubation periods and concentrations using previously established protocols.31,33 A maximal fluorescence (indicating maximal NBDG uptake) was observed when cells were cultured in medium supplemented with 0.02 mM NBDG for 30 min, which were the conditions used below.We designed a competitive assay in which the selected cell lines were incubated with NBDG in the presence of one of the compounds 1b–d, i.e. conditions at which NBDG will compete with the aromatic N-glucosides for GLUT1. We observed that significantly less NBDG was taken up by both of the tested cell lines (Fig. 4) suggesting that 1b–d act as GLUT1 antagonists, in agreement with the in silico models. To demonstrate whether this approach is applicable in clinically relevant scenario, we also performed molecular imaging with the clinically established fluorodeoxyglucose (18F-FDG) positron emission tomography (PET). Our results showed that PET is suitable for monitoring the effect of the aromatic N-glucosides on the Glc distribution in vivo and we observed deprivation of the NBDG uptake at the tumor site upon supplementation with 1b (Fig. S6†).
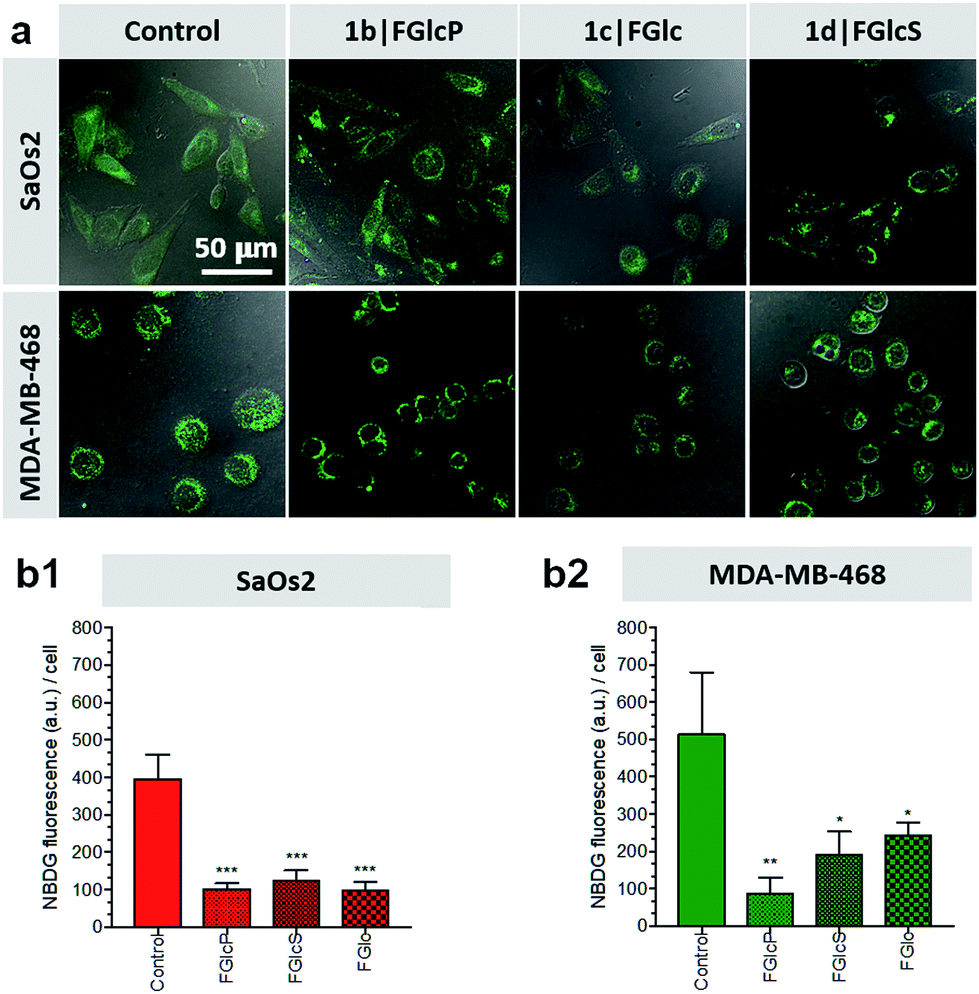 | ||
| Fig. 4 Aromatic N-glucosides 1b–d block glucose uptake by tumor cells: (a) representative fluorescent confocal scanning microscopy images showing uptake of 2-(N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino)-2-deoxyglucose (NBDG, green) by the SaOs2 and MDA-MB-468 cancer cells in the presence of aromatic N-glucosides 1b–d: cells were incubated with 0.02 mM of NBDG in the presence of 0.5 mM of the aromatic N-glucoside and visualized after 30 min; (b) NBDG uptake calculated from the fluorescence images; statistics were calculated using the t-test *: p < 0.05; **: p < 0.01; ***p < 0.001. | ||
The effect of the aromatic N-glucosides is mediated by GLUT1
GLUT1 expression was quantified by western blot analysis (Fig. 5a). We performed cell surface biotinylation to distinguish GLUT1 expressed at the cell surface and responsible for the Glc uptake from the total GLUT1 expression. The results demonstrated that cancer cells' incubation with aromatic N-glucosides significantly reduces the transporter expression on the cell surface but does not affect the total GLUT1 expression (Fig. 5a). These data agree with the formation of a strong complex between the aromatic N-glucosides and GLUT1 with consecutive blockage of the transporter (Fig. 5d). Further confirmation of this mechanism of action was derived by SLC2A1 knockdown. We used a pool of three target-specific nucleotides 19–25 (nt) small-interfering RNA (siRNA). A maximal SLC2A1 suppression (>50%) was observed 48 h after post-transfection for the SaOs2 cells (Fig. 5b) and 24 h for the MDA-MB-468 (Fig. S7†). The transfected cells displayed a markedly decreased sensitivity to the compounds 1b–d confirming that their activity is mediated by GLUT1 (Fig. 5c).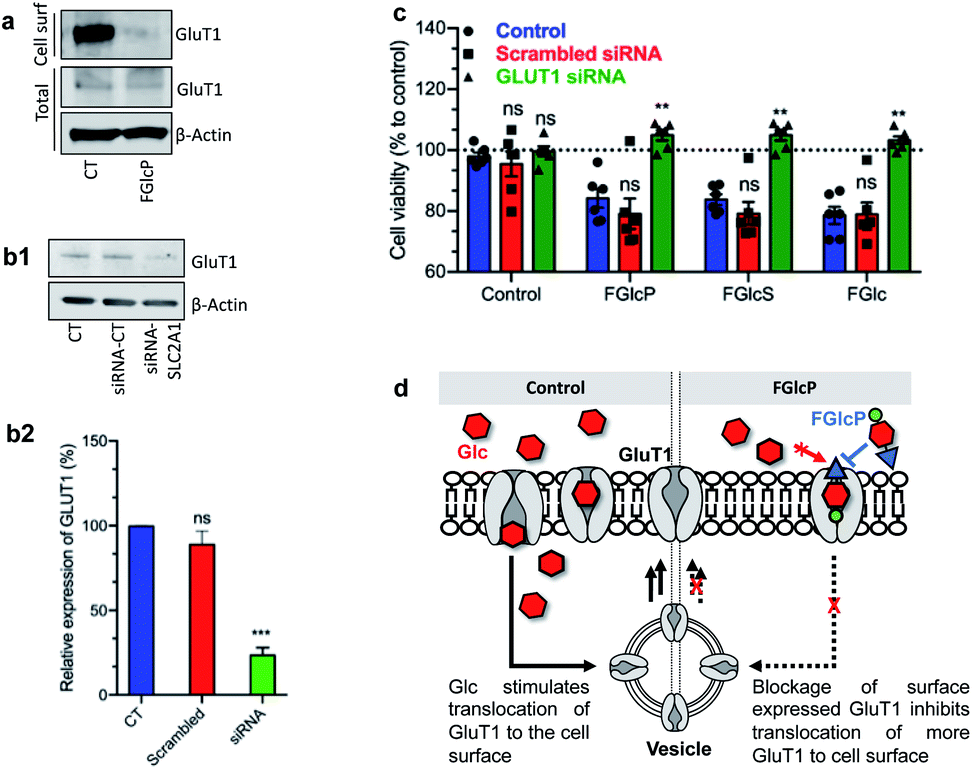 | ||
| Fig. 5 Influence of GLUT1 expression on the effect of aromatic N-glucosides on SaOs2 viability. (a) Representative western blot analysis showing changes in GLUT1 expressed on the cell surface (extracts obtained by cell-surface biotinylation) and the total GLUT1 in SaOs2 cells without (CT) and with supplementation of 1b. (b1) Representative western blot analysis and (b2) the respective densitometric analysis of GLUT1 expression by SaOs2 cells without transfection (CT) and after transfection with scrambled siRNA (siRNA-CT) or specific GLUT1 siRNA (siRNA-SLC2A1); (c) cell viability of the transfected SaOs2 cells after incubation with 0.5 mM aromatic N-glucosides 1b–d for 1 h (quantification was performed after 24 h). (d) Schematic presentation of the GLUT1 transduction role. ns, non-significant; **p < 0.05; ***p < 0.001. Data for MDA-MB-468 are provided in Fig. S3.† | ||
Concomitant saturation binding experiments using NBDG at different concentrations without and in the presence of the respective aromatic N-glucosides were performed to determine the dissociation constant (KdNBDG) and the number of the binding sites (Bmax) for each cell line (Table 2 and Fig. S8†).
| Cells | Comp | B max (GLUT1/cell × 108) | K d NBDG (μM) |
|---|---|---|---|
| SaOs2 | NBDG | 1.70 | 0.99 |
| 1b | 0.75 | 0.14 | |
| 1c | 0.58 | 0.32 | |
| 1d | 0.76 | 0.38 | |
| MDA-MB-468 | NBDG | 3.80 | 0.28 |
| 1b | 2.42 | 0.19 | |
| 1c | 2.73 | 0.16 | |
| 1d | 2.80 | 0.11 |
The results supported the proposed mechanism (Fig. 5d) and showed that supplementation with 1b–d decreased the expression of GLUT1 on the cell surface (Bmax, Table 2) and the NBDG binding by GLUT1 (KdNBDG, Table 2). Of note, the used method does not allow comparison of the obtained KdNBDG (defined as a concentration at which NBDG binds to half of the available GLUT1) for SaOs2 and MDA-MB-468 cells because of the different Bmax values but the effect of 1b–d on each cell line can be compared (similar Bmax).34 For SaOs2 cells, we obtained the smallest KdNBDG in the presence of 1b which means that this N-glucosamine blocks more efficiently the GLUT1 on the cell surface and less NBDG is needed to saturate the available GLUT1. In the case of MDA-MB-468 cells, the smallest KdNBDG was obtained for the 1d.
N-glucosides-mediated GLUT1 inhibition leads to cell death via an apoptotic mechanism
We observed that cancer cells incubated with the compounds 1b–d have lower metabolic activity and cell proliferation, leading to an overall decrease in cell survival over time (Fig. 6a, S9 and S10†). This effect was not observed for prechondrocyte ATDC5 cells, which remained viable and proliferated in the presence of 1b and 1d (Fig. S11†).Confirmation that this response is due to the GLUT1 inhibition and consecutive deprivation of the Glc uptake was obtained by quantification of the lactate production in the studied cells. Lactate is the last metabolite of glycolysis in cancer cells and indeed, its production was significantly inhibited (>60%) by treatment with 1b (Fig. 6b). We also investigated the type of cell death induced by this treatment. We studied the effect of 1b–d in a presence of an apoptosis (Z-Fa-FMK) or a necroptosis (Nec-1) inhibitor.35,36 We observed that Z-Fa-FMK rescues the SaOs2 and MDA-MB-468 cells treated by 1b–d (Fig. 6c) while the addition of Nec-1 has no effect on cell survival (Fig. S12†). These results demonstrate that the aromatic N-glucosides activate apoptotic pathways in the cells overexpressing GLUT1. This result was also confirmed by the expression of the apoptosis-associated caspases 8 and 9.37,38 Caspases are initially produced as inactive monomeric procaspases that require dimerization and often cleavage for activation.38 We found that the apoptosis pathways are activated by cleavage of procaspase 8 (53 kDa) to shorter motifs (43 kDa, 18 kDa) and procaspase 9 (47 kDa) to oligomers of 25 kDa (Fig. S13†).
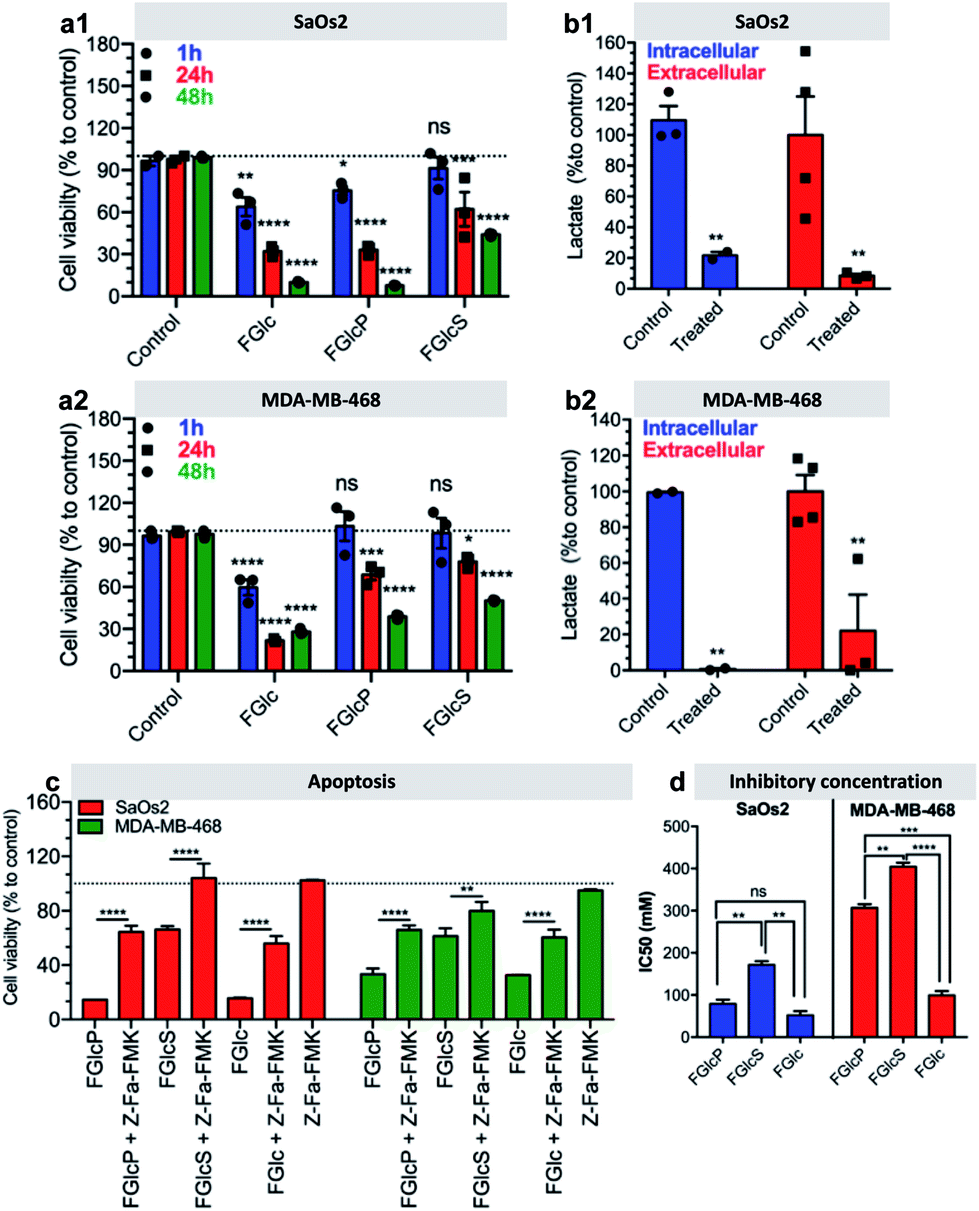 | ||
| Fig. 6 Aromatic N-glucosides deprive glycolysis and induce cell death. (a) Viability of (a1) SaOs2 and (a2) MDA-MB-468 cells after different incubation times with 0.5 mM 1b–d; (b) intra- and extracellular lactate production by (b1) SaOs2 and (b2) MDA-MB-468 cells prior and after treatment with 1b; (c) effect of the apoptosis inhibitor Z-Fa-FMK on cell viability (24 h) in the presence of the compounds 1b–d (0.5 mM); (d) inhibitory concentrations of the studied N-glucosides determined for SaOs2 and MDA-MB-468 (24 h). All results are normalized to the control – cell culture without aromatic N-glucosides. ns statistically non-significant difference; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. | ||
Discussion
Cancer cells have a high energy demand that is associated with an increased glucose uptake (Warburg effect) and overexpression of glucose transporter proteins (GLUTs). The elevated consumption of Glc assures an immediate production of adenosine triphosphate, but also increases the availability of biosynthetic intermediates that support the highly proliferative nature of cancer cells.39–41 As a result, cancer cells are very sensitive to Glc deprivation, which can prevent proliferation and induce cell death.41 Glc is delivered inside cancer cells via the GLUTs, among which GLUT1 is upregulated in most of the solid tumours.9,10,41 GLUTs are therefore suitable targets for the treatment of different types of cancers. Herein, we designed and synthetized several aromatic N-glucosides 1b–d and investigated their potential to deprive the glycolysis in cancer cells. The molecular design of 1b–d combines several functional elements: the aromatic Fmoc-functionality, which provides amphiphilic character and possibility for self-assembly via π–π interactions and the Glc portion that can participate in H-bonding and CH–π interactions and attribute GLUT antagonist characteristics are common features for all studied compounds. FGlc (1c) that contains only these two functional moieties has poor solubility in aqueous media (dimethyl sulfoxide at a concentration below 0.005% was used as a co-solvent). Thus, negatively charged polar group were incorporated within the structure of 1c generating 1b and 1d aiming to improve their water solubility. The polar phosphate group of 1b imparts also enzyme sensitivity: FGlcP (1b) is a BSA precursor that generates the self-assembling FGlc (1c) upon ALP action (Fig. 1b) causing cell death by formation of a pericellular supramolecular nanonet (Fig. 1c and S4†).7,17In silico studies confirmed that all aromatic N-glucosides 1b–d bind to the Glc pocket. The significant decrease in ΔGvdw for these compounds when compared to Glc (1a) indicate that the aromatic portion (Fmoc) can interact via CH–π and π–π interactions with the aromatic rings of e.g. Trp388 and Phe26 in the glucose-binding pocket causing conformational changes that can disturb the GLUT1 function (the inward opening, Fig. S14†).27,28 On the other hand, the lower ΔG of the compounds 1b–d indicates that even if the inward opening is not compromised, they might not be released because of the strong bonds formed with the GLUT1.
The effect of the aromatic N-glucosides on Glc transport was studied in vitro using two human cancer cell lines. Because SaOs2 cells express on their surface both ALP and GLUT1 proteins, it is challenging to distinguish the effect of BSA, triggered by ALP versus the GLUT1 antagonist effect of 1b–c. We and others previously demonstrated that BSA is a time and concentration (both enzyme and precursor) dependent process - short incubation times and low concentrations result in formation of very sparse and thus likely permeable supramolecular nanoscale network (Fig. S4†).7,42 Thus, we have selected one additional human cancer cell line, MDA-MB-468 that expresses 3-fold less ALP on their surface (Fig. S5†). We have also shortened the experimental time to 30 min and lowered the concentration of the aromatic N-glucosides to 0.5 mM (versus 7 h and 1 mM when the significant effect of BSA on cell viability was observed7) aiming to avoid BSA of 1b and to determine the GLUT1 antagonist effect of the aromatic N-glucosides. As in a previous study, we have used ATDC5 prechondrocytes for comparative purposes, i.e. to demonstrate the specificity of the aromatic N-glucosides towards cancer cells (Fig. S11†).7 At the studied conditions (30 min, 0.5 mM concentration of N-glucosides), we observed very similar response by the two cancer cell lines: the compounds 1b–d inhibit Glc transport in SaOs2 and MDA-MB-469 (Fig. 4), while ATDC5 were not affected (Fig. S11†). This response was directly related with the GLUT1 expression: viability of SaOs2 and MDA-MB-468 with depleted GLUT1 was not affected by the aromatic N-glucosides similarly to ATDC5 that do not express GLUT1 on the surface. It is expected that the reduction of the Glc uptake will result in the translocation of more GLUT1 to the cell membrane.41 Surprisingly, we observed exactly the opposite effect (Fig. 4a, Bmax in Table 2). The decrease of GLUT1 translocation to the membrane is indicative about the mechanism of action: GLUT1 proteins bind the aromatic N-glucosides but do not release them intracellularly, i.e. it is in a permanent ON mode and thus does not mediate correctly the need of Glc (Fig. 4 and S14B†). As a result, cells do not respond to the Glc deprivation because they cannot sense it via GLUT1. Another indicative result about the mechanism of action is the obtained KdNBDG (Table 2) and IC50 values (Fig. 6d and S9†). KdNBDG were determined 30 min after supplementation of 1b–d and showed that in the case of MDA-MB-468 the sulphated 1d is the most efficient GLUT1 antagonist. Different result was obtained for SaOs2: lowest KdNBDG were obtained for 1b that act simultaneously as a GLUT1 antagonist and as a substrate for biocatalytic self-assembly locally, i.e. at the cell surface where ALP is located, thus creating a physical barrier. 1c can self-assemble too but it forms bulk (throughout the solution) and not local (on the cell surface) nanonet and thus, longer time (or higher concentration) is needed to achieve the same blocking efficacy.
Indeed, the IC50 values determined after 24 h of cells exposition to 1b–d, were lowest for the self-assembling FGlc (1c) either for SaOs2 or for MDA-MB-468 cells. A similarly low value was obtained for 1b but only for SaOs2 cells confirming the above-mentioned mechanism, i.e. that the precursor 1b is already transformed by the ALP of SaOs2 into 1c and a nanonet is generated around SaOs2 cells.7 Because MDA-MB-468 cells have lower expression of ALP this transformation does not occur and the IC50 value of 1b is 4-fold higher for these cells. 1d that is solely involved in the GLUT1 blocking because does not self-assemble into nanofibers (Fig. S4†)22 and is not susceptible to BSA presented the highest IC50 values for both cell lines. These results demonstrate that the formation of a nanonet around the cells not only sequesters them, but also add the inhibitory effect of the aromatic N-glucosides most probably by maintaining these compounds at high concentration in the pericellular space, i.e. close to GLUT1. In such scenario, the self-assembling 1c has the greatest effect but its application can be hampered by the poor solubility in aqueous media. The precursor 1b is an excellent alternative for tumors that overexpress phosphatases on their surface. Besides having the same activity as 1c, it uses two molecular targets (ALP and GLUT1), thus enhancing its selectivity.
Conclusions
Our study demonstrates that the aromatic N-glucosides 1b and 1d have a potent anticancer activity due to their ability to inhibit Glc transportation and GLUT1 expression via two synergistic physical and biochemical mechanisms: blockage of GLUT1 (1b and 1d) and/or formation of a supramolecular net around the cells (1b).Ethical statement
The animal experiments in this study were performed in strict accordance with the guidelines for the care and use of laboratory animals and approved by the Institutional Animal Care and Use Committee of Memorial Sloan Kettering Cancer Center (NY, USA).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the EU's H2020 program (Forecast 668983; THE DISCOVERIES CTR 739572) and the Portuguese FCT (BD/113794/2015; BPD/85790/2012; PTDC/NAN-MAT/28468/2017 CANCER_CAGE) for the financial support. The authors gratefully acknowledge members of the MSKCC Small Animal Imaging Core Facility, the Radiochemistry and Molecular Imaging Probe Core (both supported by NIH P30 CA008748) as well as NIH R35 CA232130 (JSL). PMRP acknowledges the Tow Foundation Postdoctoral Fellowship from the MSK Center for Molecular Imaging and Nanotechnology. AB is grateful to the Portuguese League Against Cancer for her Fellowship. RVU acknowledges support from the US Army Research 719 Office (proposal number 69180-CH). We thank Luca Gasperini for his help in the quantification of NBDG fluorescence images using the Cell Profiler software and Andreia Carvalho for providing comments and suggestions that were helpful in acquiring data for this manuscript.Notes and references
- WHO (WHO), Latest global cancer data: Cancer burden rises to 18.1 million new cases and 9.6 million cancer deaths in 2018, International Agency for research in cancer, 2018 Search PubMed.
- J. Zhou and B. Xu, Bioconjugate Chem., 2015, 26, 987–999 CrossRef CAS PubMed.
- R. A. Pires, Y. M. Abul-Haija, R. L. Reis, R. V. Ulijn and I. Pashkuleva, Hydrogels: Design, Synthesis and Application in Drug Delivery and Regenerative Medicine, 2018, p. 170 Search PubMed.
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef CAS PubMed.
- R. A. Cairns, I. S. Harris and T. W. Mak, Nat. Rev. Cancer, 2011, 11, 85–95 CrossRef CAS PubMed.
- Z. M. Yang, K. M. Xu, Z. F. Guo, Z. H. Guo and B. Xu, Adv. Mater., 2007, 19, 3152–3156 CrossRef CAS.
- R. A. Pires, Y. M. Abul-Haija, D. S. Costa, R. Novoa-Carballal, R. L. Reis, R. V. Ulijn and I. Pashkuleva, J. Am. Chem. Soc., 2015, 137, 576–579 CrossRef CAS PubMed.
- K. Kapoor, J. S. Finer-Moore, B. P. Pedersen, L. Caboni, A. Waight, R. C. Hillig, P. Bringmann, I. Heisler, T. Muller, H. Siebeneicher and R. M. Stroud, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 4711–4716 CrossRef CAS PubMed.
- D. A. Chan, P. D. Sutphin, P. Nguyen, S. Turcotte, E. W. Lai, A. Banh, G. E. Reynolds, J. T. Chi, J. Wu, D. E. Solow-Cordero, M. Bonnet, J. U. Flanagan, D. M. Bouley, E. E. Graves, W. A. Denny, M. P. Hay and A. J. Giaccia, Sci. Transl. Med., 2011, 3, 94ra70 CAS.
- C. Granchi, S. Fortunato and F. Minutolo, Medchemcomm, 2016, 7, 1716–1729 RSC.
- J. Zhou, X. W. Du, N. Yamagata and B. Xu, J. Am. Chem. Soc., 2016, 138, 3813–3823 CrossRef CAS PubMed.
- Z. Q. Q. Feng, H. M. Wang, X. Y. Chen and B. Xu, J. Am. Chem. Soc., 2017, 139, 15377–15384 CrossRef CAS PubMed.
- Q. Yao, Z. Huang, D. Liu, J. Chen and Y. Gao, Adv. Mater., 2018, 31, 1804814 CrossRef PubMed.
- D. Kalafatovic, M. Nobis, N. Javid, P. W. J. M. Frederix, K. I. Anderson, B. R. Saunders and R. V. Ulijn, Biomater. Sci., 2015, 3, 246–249 RSC.
- D. Kalafatovic, M. Nobis, J. Y. Son, K. I. Anderson and R. V. Ulijn, Biomaterials, 2016, 98, 192–202 CrossRef CAS PubMed.
- J. Son, D. Kalafatovic, M. Kumar, B. Yoo, M. A. Cornejo, M. Contel and R. V. Ulijn, ACS Nano, 2019, 13, 1555–1562 CrossRef CAS PubMed.
- Y. Kuang, J. F. Shi, J. Li, D. Yuan, K. A. Alberti, Q. B. Xu and B. Xu, Angew. Chem., Int. Ed., 2014, 53, 8104–8107 CrossRef CAS PubMed.
- H. B. Shi, R. T. K. Kwok, J. Z. Liu, B. G. Xing, B. Z. Tang and B. Liu, J. Am. Chem. Soc., 2012, 134, 17972–17981 CrossRef CAS PubMed.
- C. F. Anderson and H. G. Cui, Ind. Eng. Chem. Res., 2017, 56, 5761–5777 CrossRef CAS PubMed.
- A. G. Cheetham, Y. A. Lin, R. Lin and H. G. Cui, Acta Pharmacol. Sin., 2017, 38, 874–884 CrossRef CAS PubMed.
- Y. Kuang and B. Xu, Angew. Chem., Int. Ed., 2013, 52, 6944–6948 CrossRef CAS PubMed.
- A. Brito, Y. M. Abul-Haija, D. S. da Costa, R. Novoa-Carballal, R. L. Reis, R. V. Ulijn, R. A. Pires and I. Pashkuleva, Chem. Sci., 2019, 10, 2385–2390 RSC.
- L. S. Birchall, S. Roy, V. Jayawarna, M. Hughes, E. Irvine, G. T. Okorogheye, N. Saudi, E. De Santis, T. Tuttle, A. A. Edwards and R. V. Ulijn, Chem. Sci., 2011, 2, 1349–1355 RSC.
- S. Rastogi, S. Banerjee, S. Chellappan and G. R. Simon, Cancer Lett., 2007, 257, 244–251 CrossRef CAS PubMed.
- A. Grosdidier, V. Zoete and O. Michielin, Proteins: Struct., Funct., Bioinf., 2007, 67, 1010–1025 CrossRef CAS PubMed.
- A. Grosdidier, V. Zoete and O. Michielin, J. Comput. Chem., 2011, 32, 2149–2159 CrossRef CAS PubMed.
- D. Deng, P. C. Sun, C. Y. Yan, M. Ke, X. Jiang, L. Xiong, W. L. Ren, K. Hirata, M. Yamamoto, S. L. Fan and N. Yan, Nature, 2015, 526, 391–396 CrossRef CAS PubMed.
- D. Deng, C. Xu, P. C. Sun, J. P. Wu, C. Y. Yan, M. X. Hu and N. Yan, Nature, 2014, 510, 121–125 CrossRef CAS PubMed.
- J. Barretina, G. Caponigro, N. Stransky, K. Venkatesan, A. A. Margolin, S. Kim, C. J. Wilson, J. Lehar, G. V. Kryukov, D. Sonkin, A. Reddy, M. W. Liu, L. Murray, M. F. Berger, J. E. Monahan, P. Morais, J. Meltzer, A. Korejwa, J. Jane-Valbuena, F. A. Mapa, J. Thibault, E. Bric-Furlong, P. Raman, A. Shipway, I. H. Engels, J. Cheng, G. Y. K. Yu, J. J. Yu, P. Aspesi, M. de Silva, K. Jagtap, M. D. Jones, L. Wang, C. Hatton, E. Palescandolo, S. Gupta, S. Mahan, C. Sougnez, R. C. Onofrio, T. Liefeld, L. MacConaill, W. Winckler, M. Reich, N. X. Li, J. P. Mesirov, S. B. Gabriel, G. Getz, K. Ardlie, V. Chan, V. E. Myer, B. L. Weber, J. Porter, M. Warmuth, P. Finan, J. L. Harris, M. Meyerson, T. R. Golub, M. P. Morrissey, W. R. Sellers, R. Schlegel and L. A. Garraway, Nature, 2012, 483, 603–607 CrossRef CAS PubMed.
- N. Stransky, M. Ghandi, G. V. Kryukov, L. A. Garraway, J. Lehar, M. Liu, D. Sonkin, A. Kauffmann, K. Venkatesan, E. J. Edelman, M. Riester, J. Barretina, G. Caponigro, R. Schlegel, W. R. Sellers, F. Stegmeier, M. Morrissey, A. Amzallag, I. Pruteanu-Malinici, D. A. Haber, S. Ramaswamy, C. H. Benes, M. P. Menden, F. Iorio, M. R. Stratton, U. McDermott, M. J. Garnett, J. Saez-Rodriguez and C. Drug Sensitivity, L. Cancer Cell, I. Broad, R. Novartis Inst Biomed, S. Genomics Drug, H. Massachusetts Gen, L. European Mol Biol, I. European Bioinformatics and I. Wellcome Trust Sanger, Nature, 2015, 528, 84–87 CrossRef CAS PubMed.
- K. Yamada, M. Nakata, N. Horimoto, M. Saito, H. Matsuoka and N. Inagaki, J. Biol. Chem., 2000, 275, 22278–22283 CrossRef CAS PubMed.
- C. H. Zou, Y. J. Wang and Z. F. Shen, J. Biochem. Biophys. Methods, 2005, 64, 207–215 CrossRef CAS PubMed.
- K. Yamada, M. Saito, H. Matsuoka and N. Inagaki, Nat. Protoc., 2007, 2, 753–762 CrossRef CAS PubMed.
- P. Hein, M. C. Michel, K. Leineweber, T. Wieland, N. Wettschureck and S. Offermanns, in Practical Methods in Cardiovascular Research, ed. S. Dhein, F. Wilhelm Mohr and M. Delmar, Springer, Berlin, Heidelberg, 2005, ch. 6, pp. 723–783, DOI:10.1007/b137833.
- F. J. Lopez-Hernandez, M. A. Ortiz, Y. Bayon and F. J. Piedrafita, Mol. Cancer Ther., 2003, 2, 255–263 CAS.
- N. Takahashi, L. Duprez, S. Grootjans, A. Cauwels, W. Nerinckx, J. B. DuHadaway, V. Goossens, R. Roelandt, F. Van Hauwermeiren, C. Libert, W. Declercq, N. Callewaert, G. C. Prendergast, A. Degterev, J. Yuan and P. Vandenabeele, Cell Death Dis., 2012, 3, 891–899 Search PubMed.
- Y. Q. Wu, D. Zhao, J. N. Zhuang, F. Q. Zhang and C. Xu, PLoS One, 2016, 11, e0168268 CrossRef PubMed.
- F. Pirnia, E. Schneider, D. C. Betticher and M. M. Borner, Cell Death Differ., 2002, 9, 905–914 CrossRef CAS PubMed.
- R. A. Gatenby and R. J. Gillies, Nat. Rev. Cancer, 2004, 4, 891–899 CrossRef CAS PubMed.
- P. P. Hsu and D. M. Sabatini, Cell, 2008, 134, 703–707 CrossRef CAS PubMed.
- M. L. Macheda, S. Rogers and J. D. Best, J. Cell. Physiol., 2005, 202, 654–662 CrossRef CAS PubMed.
- J. Zhou, X. W. Du and B. Xu, Angew. Chem., Int. Ed., 2016, 55, 5770–5775 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc00954g |
| This journal is © The Royal Society of Chemistry 2020 |