
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
General and selective synthesis of primary amines using Ni-based homogeneous catalysts†
Kathiravan
Murugesan
 ,
Zhihong
Wei
,
Vishwas G.
Chandrashekhar
,
Haijun
Jiao
*,
Matthias
Beller
* and
Rajenahally V.
Jagadeesh
*
,
Zhihong
Wei
,
Vishwas G.
Chandrashekhar
,
Haijun
Jiao
*,
Matthias
Beller
* and
Rajenahally V.
Jagadeesh
*
Leibniz-Institut für Katalyse e. V., Albert Einstein-Str. 29a, 18059 Rostock, Germany. E-mail: haijun.jiao@catalysis.de; matthias.beller@catalysis.de; jagadeesh.rajenahally@catalysis.de
First published on 25th March 2020
Abstract
The development of base metal catalysts for industrially relevant amination and hydrogenation reactions by applying abundant and atom economical reagents continues to be important for the cost-effective and sustainable synthesis of amines which represent highly essential chemicals. In particular, the synthesis of primary amines is of central importance because these compounds serve as key precursors and central intermediates to produce value-added fine and bulk chemicals as well as pharmaceuticals, agrochemicals and materials. Here we report a Ni-triphos complex as the first Ni-based homogeneous catalyst for both reductive amination of carbonyl compounds with ammonia and hydrogenation of nitroarenes to prepare all kinds of primary amines. Remarkably, this Ni-complex enabled the synthesis of functionalized and structurally diverse benzylic, heterocyclic and aliphatic linear and branched primary amines as well as aromatic primary amines starting from inexpensive and easily accessible carbonyl compounds (aldehydes and ketones) and nitroarenes using ammonia and molecular hydrogen. This Ni-catalyzed reductive amination methodology has been applied for the amination of more complex pharmaceuticals and steroid derivatives. Detailed DFT computations have been performed for the Ni-triphos based reductive amination reaction, and they revealed that the overall reaction has an inner-sphere mechanism with H2 metathesis as the rate-determining step.
Introduction
Catalytic reductive aminations and hydrogenations constitute essential processes widely applied in research laboratories and industries for the synthesis of fine and bulk chemicals as well as molecules used in life sciences.1 In particular, the selective and efficient synthesis of amines by applying these processes starting from inexpensive and easily available starting materials, and green and abundant reagents using non-noble metal-based catalysts continues to be an important goal of chemical research. In general, amines are highly essential fine and bulk chemicals used in chemistry, medicine, biology and materials.2 Noteworthily, amine functionalities constitute integral parts of a large number of life science molecules and play significant roles in their activities.2 As an example, more than 75% of 200 top selling drugs of the year 2018 contain amine/nitrogen moieties.2e Among different kinds of amines, primary amines are highly valued because these compounds serve as key precursor and central intermediates for the synthesis of advanced chemicals, pharmaceuticals, agrochemicals and materials. The reductive amination of carbonyl compounds with ammonia3 and the hydrogenation of nitroarenes4 are found to be more expedient processes to synthesize benzylic, aliphatic and aromatic primary amines.3,4 Notably for the advancement of more sustainable and cost-effective synthesis of this class of amines, the development of base metal catalysts is highly desired and continues to attract scientific interest. Catalytic reductive aminations, especially for the synthesis of primary amines are challenging processes and are often non-selective and suffer from side reactions such as over alkylation and reduction to the corresponding alcohols.3 Hence in order to perform these reactions in an efficient and highly selective manner the development of suitable catalysts is of central importance. Regarding potential catalysts for reductive amination to prepare primary amines from carbonyl compounds using ammonia and molecular hydrogen, mainly heterogeneous catalysts based on Rh,3k,s Ru,3k–m,q,r Ni3d,h,n and Co3a are applied. Compared to that of heterogeneous materials, the design of homogeneous catalysts for this reaction is highly challenging due to the inactivation of metal complexes in the presence of ammonia by forming Werner-amine type complexes and common problems such as formation of secondary and/or tertiary amines and over hydrogenation of carbonyl compounds to corresponding alcohols associated with reductive aminations.3 Nevertheless, in recent years, a few Rh,3i Ru,3c,f,g,j Ir3p and Co3e-based complexes have been established to catalyze the synthesis of primary amines from carbonyl compounds and ammonia. Despite these achievements, still the development of base metal homogeneous catalysts for the synthesis of benzylic and aliphatic primary amines is highly desired and attracts scientific interest.The catalytic hydrogenation of nitroarenes represents an indispensable and widely applied process for the synthesis of aromatic amines (anilines).1c,d,4 In general this reaction mainly relies on heterogeneous catalysts. Unfortunately, homogeneous catalysts for the hydrogenation of nitro compounds are scarcely explored and using them remains a challenge.4c–f Hence the development of suitable homogeneous catalysts, especially based on non-noble metals, for the synthesis of functionalized anilines continues to be important is also of significant interest. Here, we report a Ni-based complex for both reductive amination of carbonyl compounds with ammonia and hydrogenation of nitroarenes for the general synthesis of all kinds of primary amines.
Ni-based complexes are well known to catalyze a number of synthetic processes5–11 including hydrogenations,6 CH-activations,7 coupling reactions8 and amination of alcohols,8,9 as well as polymerizations10 and photo-redox reactions.11 The key to success for this Ni-catalysis in organic synthesis is the use of a broad variety of complexes based on specific ligands.5–11 Although Ni-based homogeneous catalysts are well recognized for a variety of reactions, they are still underdeveloped for reductive aminations as well as for the hydrogenation of nitro compounds.5–11 To the best of our knowledge until now there has been no homogeneous Ni-based catalyst known for both of these reactions to synthesize primary amines. Certainly, there is potential interest in the development of homogeneous Ni-based reductive amination and hydrogenation catalysts for the general and selective synthesis of primary amines. In addition to synthetic applications, it is also important to know the mode of action of Ni-complexes, formation of catalytically active species, and reactivity and selectivity towards reductive amination as well as their compatibility with ammonia. In this respect, here we report a Ni-linear triphos (bis(diphenylphosphinoethyl)phenylphosphine) complex as the first homogeneous Ni-based catalyst for both reductive amination and hydrogenation of nitroarenes. Remarkably, this Ni-triphos complex enabled the synthesis of functionalized and structurally diverse benzylic, heterocyclic, and aliphatic linear and branched primary amines as well as aromatic primary amines starting from inexpensive and easily accessible carbonyl compounds (aldehydes and ketones) and nitroarenes using ammonia and molecular hydrogen. In addition, we also performed DFT studies to know the active catalytic species and mode of reactivity as well as to propose the plausible Ni-based reductive amination mechanism.
Results and discussion
Design of the Ni-catalyst for the reductive amination reaction
In recent years triphos-based non-noble metal complexes have emerged as promising catalysts for hydrogenation6a,12a–c,12f–k and amination reactions.3e,12d,e Due to the strong coordination nature to the central metal atom, triphos-based complexes are suitable to activate hydrogen and ammonia. Inspired by previous amination3e,12d,e and hydrogenation 6a,12a–c,12f–k processes, we started to design tridentate-Ni complexes for the synthesis of different kinds of primary amines by reductive amination of carbonyl compounds and hydrogenation of nitroarenes.First we tested the combination of Ni(BF4)2·6H2O and linear triphos (L1; bis(diphenylphosphinoethyl) phenylphosphine) as the in situ catalyst for the reductive amination of veratraldehyde 1 (3,4-dimethoxy benzaldehyde) to veratrylamine 2 (3,4-dimethoxy benzylamine) in the presence of ammonia and molecular hydrogen as a benchmark reaction (Table 1). Gratifyingly, this in situ complex exhibited high activity and selectivity for the reductive amination of veratraldehyde 1 and obtained 96% of the desired product, veratrylamine 2 (Table 1, entry 1). In order to know the reactivity of other tridentate ligands, we tested different PNP, POP and NNN ligands (Table 1, entries 2 to 7). All these tested tridentate ligands (L2–L7) showed poor activity with a conversion of up to 25% and in none of these cases the formation of primary amine 1 was observed (Table 1, entries 2 to 7). Notably, the Ni-triphos system was well tolerated with ammonia and due to strong coordination, and this PPP-ligand was found to be appropriate to avoid the formation of a Werner-type ammonia complex. Applying this active system (Ni(BF4)2–L1), next we performed the optimization of the benchmark reaction by evaluating the effect of hydrogen pressure, reaction temperature and catalyst loading (Table 1; entries 8–10). These results revealed that in order to achieve the best yield of 2, a 40 bar hydrogen pressure, 5 bar ammonia pressure, 100 °C reaction temperature and 4 mol% catalyst (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Ni(BF4)2·6H2O–L1) are required. Further to know the effect of solvents, different polar (MeOH, EtOH, t-BuOH, t-amyl alcohol, TFE) and non-polar solvents (toluene, THF) were tested (Table S1†). Among these solvents, trifluroethanol (TFE) was found to be the best solvent. However, in other tested solvents, we did not observe the formation of the desired product, primary amine 2.
:1 Ni(BF4)2·6H2O–L1) are required. Further to know the effect of solvents, different polar (MeOH, EtOH, t-BuOH, t-amyl alcohol, TFE) and non-polar solvents (toluene, THF) were tested (Table S1†). Among these solvents, trifluroethanol (TFE) was found to be the best solvent. However, in other tested solvents, we did not observe the formation of the desired product, primary amine 2.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Ligand | Conv. (%) | Yield (%) | |||
| 2 | 3 | 4 | 5 | |||
| a Reaction conditions: 0.5 mmol veratraldehyde, 4 mol% Ni(BF4)2·6H2O, 4 mol% ligand, 5 bar NH3, 40 bar H2 2 mL trifluoroethanol (TFE), 100 °C, 15 h, and GC yields using n-hexadecane as the standard. b Same as ‘a’ with 20 bar H2. c Same as ‘a’ at 80 °C. d Same as ‘a’ with 3 mol% catalyst. | ||||||
| 1a | L1 | >99 | 96 | — | 3 | — |
| 2a | L2 | 15 | — | — | 13 | — |
| 3a | L3 | 23 | — | — | 21 | — |
| 4a | L4 | 20 | — | — | 18 | — |
| 5a | L5 | 12 | — | — | 10 | — |
| 6a | L6 | 5 | — | — | 3 | — |
| 7a | L7 | 5 | — | — | 3 | — |
| 8b | L1 | >99 | 92 | — | 6 | — |
| 9c | L1 | >99 | 91 | — | 7 | — |
| 10d | L1 | >99 | 93 | — | 5 | — |
| 11a | — | 10 | — | — | 8 | — |

|
||||||
After having obtained the best results with the in situ generated, Ni(BF4)2·6H2O–L1 system, we were interested in preparing a molecularly defined complex and testing its activity in the model reaction. Unfortunately, we could not isolate Ni(BF4)2–L1 complex due its stability problem. However, the NiCl2–L1 complex was prepared, isolated and tested in the benchmark reaction (see the ESI†)6a. Noteworthily, this defined complex also exhibited similar activity to the in situ Ni(BF4)2·6H2O–L1 complex and obtained 97% of veratrylamine 2. In addition, the in situ NiCl2–L1 complex also showed good activity and obtained 90% of the desired primary amine 2. We attempted to isolate a nickel hydride complex; however, we could not due to its highly unstable nature. In order to know the nature of the reaction, catalytic poisoning tests were performed with Hg and PPh3 (Table S3†). Addition of either Hg or PPh3 to the reaction under standard conditions did not affect either the activity or selectivity of the active Ni-complex. These results showed that the reaction proceeds via homogeneous catalysis.
DFT computational study
Parallel to our experimental studies we carried out detailed density functional theory computations on the reductive amination reaction mechanisms. Since both in situ Ni(BF4)2·6H2O-triphos (L1) and the well-defined NiCl2–L1 complexes showed the same activity and selectivity, we used this well-defined complex as the pre-catalyst, which can be converted with NH3 to the active catalyst bearing the Ni–H bond [L1NiH]+(I). In our computations we used the real-size catalyst as well as phenylmethanimine (Ph–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH) for benzaldehyde as the substrate for the reaction. To evaluate the effect of the van der Waals dispersion correction (GD3BJ) and solvation (SMD) of 2,2,2-trifluoroethanol (TFE) we tested different combinations and several functionals. Based on these testing results, we discussed our results including dispersion and solvation (B3PW91–GD3BJ–SMD). The computational details for the models and methods are listed in the ESI(S7).†
NH) for benzaldehyde as the substrate for the reaction. To evaluate the effect of the van der Waals dispersion correction (GD3BJ) and solvation (SMD) of 2,2,2-trifluoroethanol (TFE) we tested different combinations and several functionals. Based on these testing results, we discussed our results including dispersion and solvation (B3PW91–GD3BJ–SMD). The computational details for the models and methods are listed in the ESI(S7).†
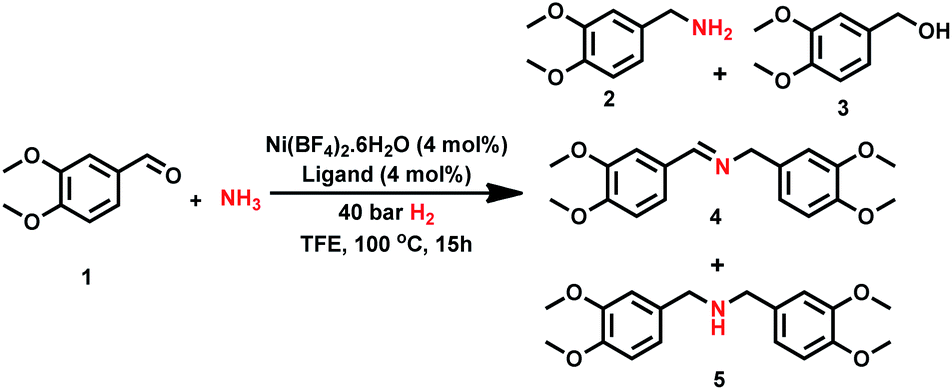
Since complex I can have fac (facial) and mer (meridional) conformations, we first computed their relative energy (Fig. 1). It is found that the I-mer is more stable than I-fac by 4.6 kcal mol; and I-mer should be the dominant isomer under equilibrium (>99.9%). Since complex I has 16 valence electrons, we tested the stability of the triplet state, and the singlet state is more stable than the corresponding triplet state by 51.8 and 50.2 kcal mol−1 for the fac- and mer-coordination, respectively. Under the coordination of imines, however, the complex (M1-fac-syn) from I-fac is more stable than that (M1-mer) from I-mer by 3.2 kcal mol−1 (>99%), indicating that M1-fac-syn should be the major component in the reaction medium and this indicates the stability change upon substrate coordination. Indeed, imine coordination to I-mer is slightly endergonic by 0.2 kcal mol−1, while imine coordination to I-fac is exergonic by 7.6 kcal mol−1. It is also noted that M1-fac-syn is more stable than M1-fac-anti by 2.4 kcal mol−1.
 | ||
| Fig. 1 Relative energies (kcal mol−1) of the Ni-catalyst and Ni-complexes. | ||
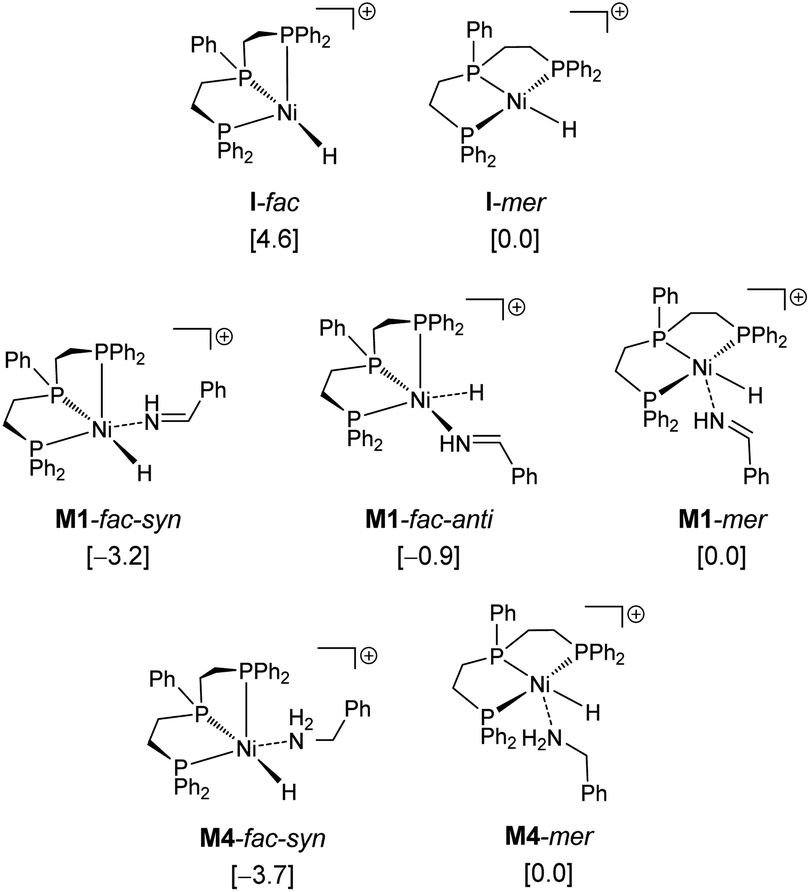
Therefore, we used M1-fac-syn for our computations. It is also noted that only the potential energy surface of the fac-coordination has been obtained; and it is not possible to have the potential energy surface for the mer-coordination. For the mer-coordination, only the complexes of imine and amine coordination have been found and they have higher energy and are less stable than the fac-coordination. The computed potential free energy surface is shown in Fig. 2.
 | ||
| Fig. 2 Potential free energy surface (kcal mol−1) with the reference of I-mer, Ph–CHNH and H2. | ||
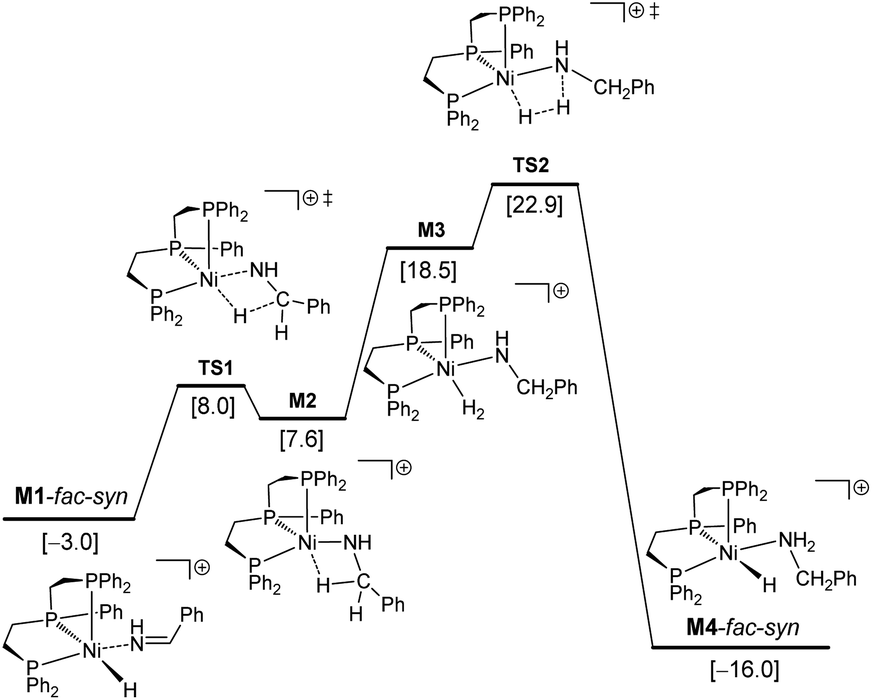
In M1-fac-syn, imine coordination is the nitrogen lone pair instead of the expected CN double bond. Starting from M1-fac-syn, the first step is the transfer of the hydride to the carbon center of the CN double bond via the transition state (TS1) due to the different electronegativities of C and N atoms. This hydride transfer results in the formation of an amido complex with C–H agostic interaction with the Ni center, M2. This hydride transfer needs a free energy barrier of 11.0 kcal mol−1 and is endergonic by 10.6 kcal mol−1. The next step is the breaking of the C–H agostic interaction via H2 coordination with the formation of intermediate M3, and this step is endergonic by 10.9 kcal mol−1. In M3, molecular H2 coordination has been found instead of oxidative addition. Having formed M3 with molecular H2 coordination, the next step is metathesis (hydrogenolysis) instead of reductive elimination and the formed amine (Ph–CH2–NH2) still coordinates to the Ni center, M4. This step needs a free energy barrier of 4.4 kcal mol−1 and is strongly exergonic by 34.5 kcal mol−1.
On the basis of I-mer and Ph–CHNH, the apparent free energy barrier is 22.9 kcal mol−1, and this barrier is in reasonable agreement with an applied high reaction temperature of 100–120 °C and a long reaction time of 24 hours. In addition, the endergonic molecular H2 coordination and the high apparent barrier reasonably explain the need for a high H2 pressure of 40–50 bar. It is noted that the computed apparent barrier is 41.6 kcal mol−1 in the gas phase, and 41.9 kcal mol−1 under the consideration of the solvation effect. In the case of van der Waals dispersion correction, the apparent barrier becomes 19.2 in the gas phase. In the case of solvation and dispersion corrections, the apparent barrier is 22.9 kcal mol−1. This demonstrates the effect of dispersion correction. In addition, we tested other functional methods including solvation and dispersion corrections and found that the apparent barrier is 28.1, 21.1, 25.4 and 27.8 kcal mol−1 for B3LYP, BP86, M06L and MN15, respectively.
Having studied the catalytically active species by DFT calculations, we proposed the plausible mechanism for the Ni-triphos catalyzed reductive amination of carbonyl compounds with ammonia and molecular hydrogen (Scheme 1). After the generation of the active catalyst with the Ni–H functionality (I), the mechanism has three main steps, (i) substrate coordination (II) followed by the Ni–H selective insertion into the CN double bond (TS1) and the formation of agostically interacting intermediate (III); and (ii) H2 coordination (IV) followed by H2 metathesis (TS2) and the formation of amine coordinated intermediate (V). The final step is the release of the amine and regeneration of the active catalyst. Overall, the reaction has an inner-sphere mechanism and the rate-determining step is H2 metathesis, and the apparent barrier is about 23 kcal mol−1. All this reasonably explains the need for a high H2 pressure of 40–50 bar and the long reaction time (Table 1).
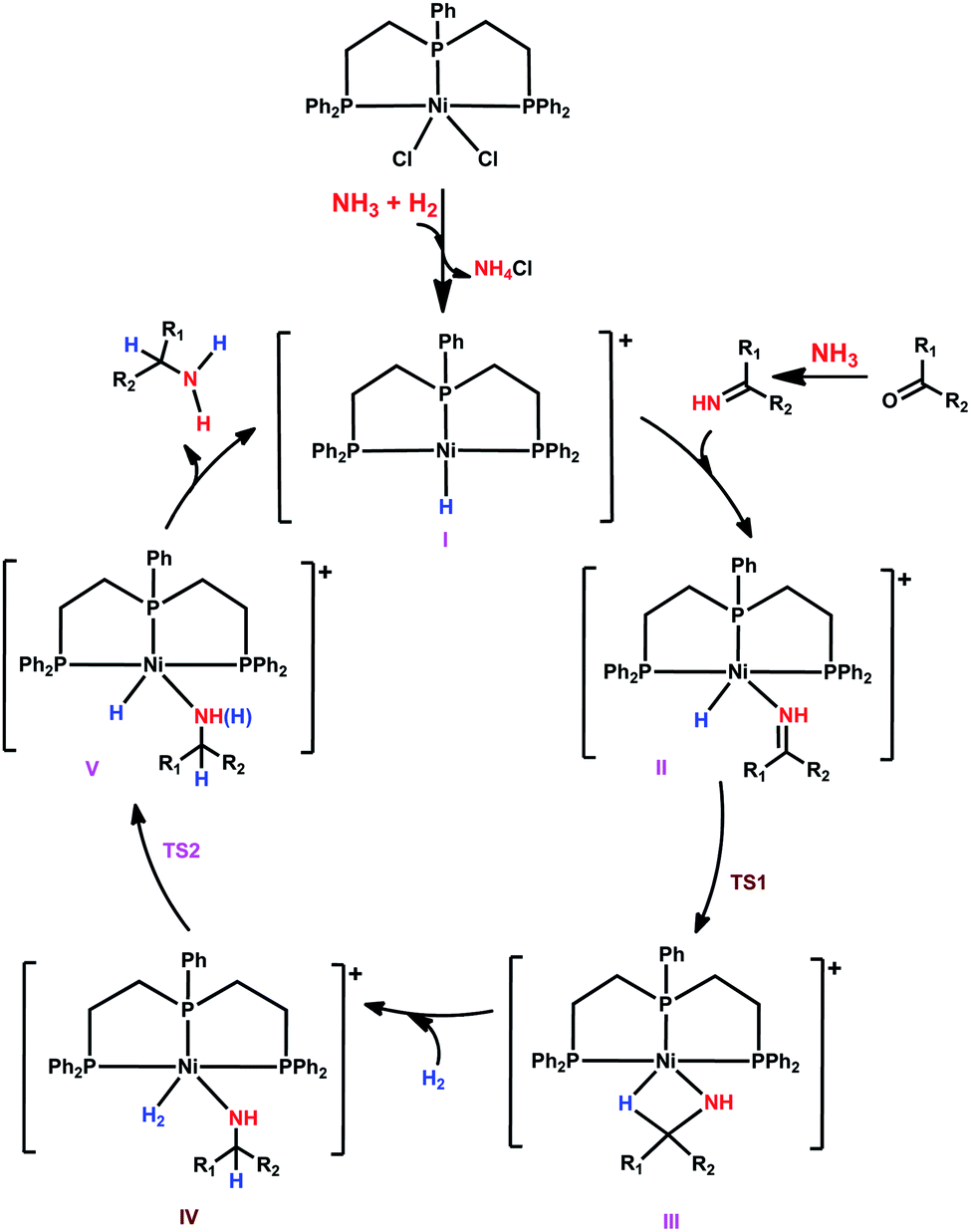 | ||
| Scheme 1 Plausible reaction mechanism for the NiCl2-triphos catalyzed reductive amination for the synthesis of primary amines. | ||
Synthesis of benzylic and aliphatic linear primary amines
After having investigated the nickel-triphos (L1) as the most active catalyst system, we explored its (in situ system) general applicability for the preparation of various primary amines starting from carbonyl compounds. As shown in Schemes 2 and 3 this Ni-triphos catalyst allowed for the amination of both aldehydes and ketones with ammonia in the presence of molecular hydrogen and obtained structurally diverse and functionalized linear and branched primary amines. Simple aldehydes and substrate bearing electron -donating and -withdrawing groups including halide substituted ones were reacted smoothly and the corresponding primary amines were obtained in good to excellent yields (Scheme 2, products 6–11). For any given amination/hydrogenation catalyst, achieving a high degree of chemoselectivity is challenging and important in organic synthesis and drug discovery.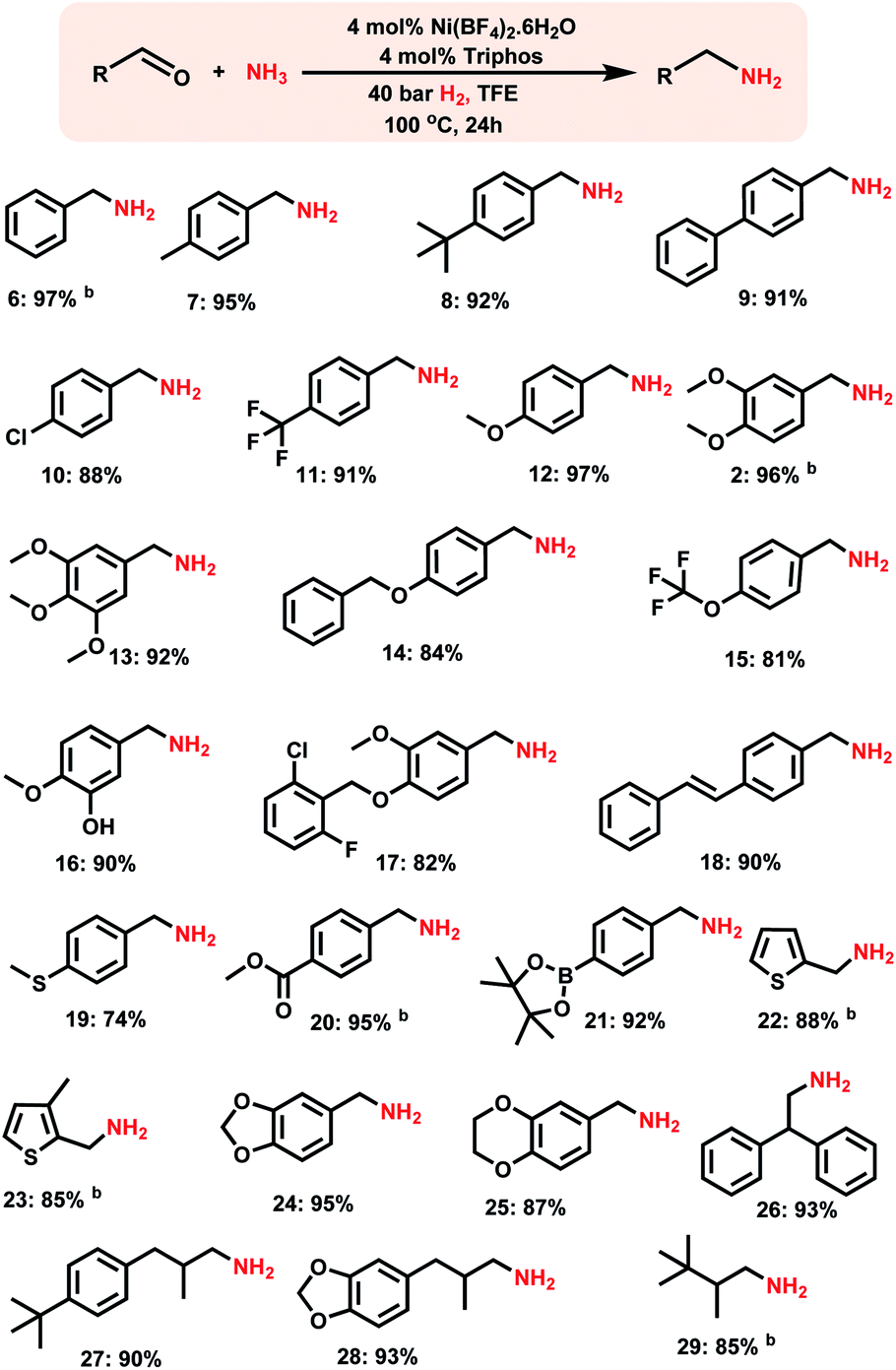 | ||
| Scheme 2 Synthesis of linear primary amines using the Ni-triphos complex.a aReaction conditions: 0.5 mmol aldehyde, 4 mol% Ni(BF4)2·6H2O, 4 mol% triphos (L1), 5–7 bar NH3, 40 bar H2, 2 mL degassed trifluoroethanol (TFE), 100 °C, 24 h, and isolated yields. bGC yields using n-hexadecane as the standard. Isolated as free amines and converted to hydrochloride salts. The corresponding hydrochloride salts were subjected to NMR analysis. | ||
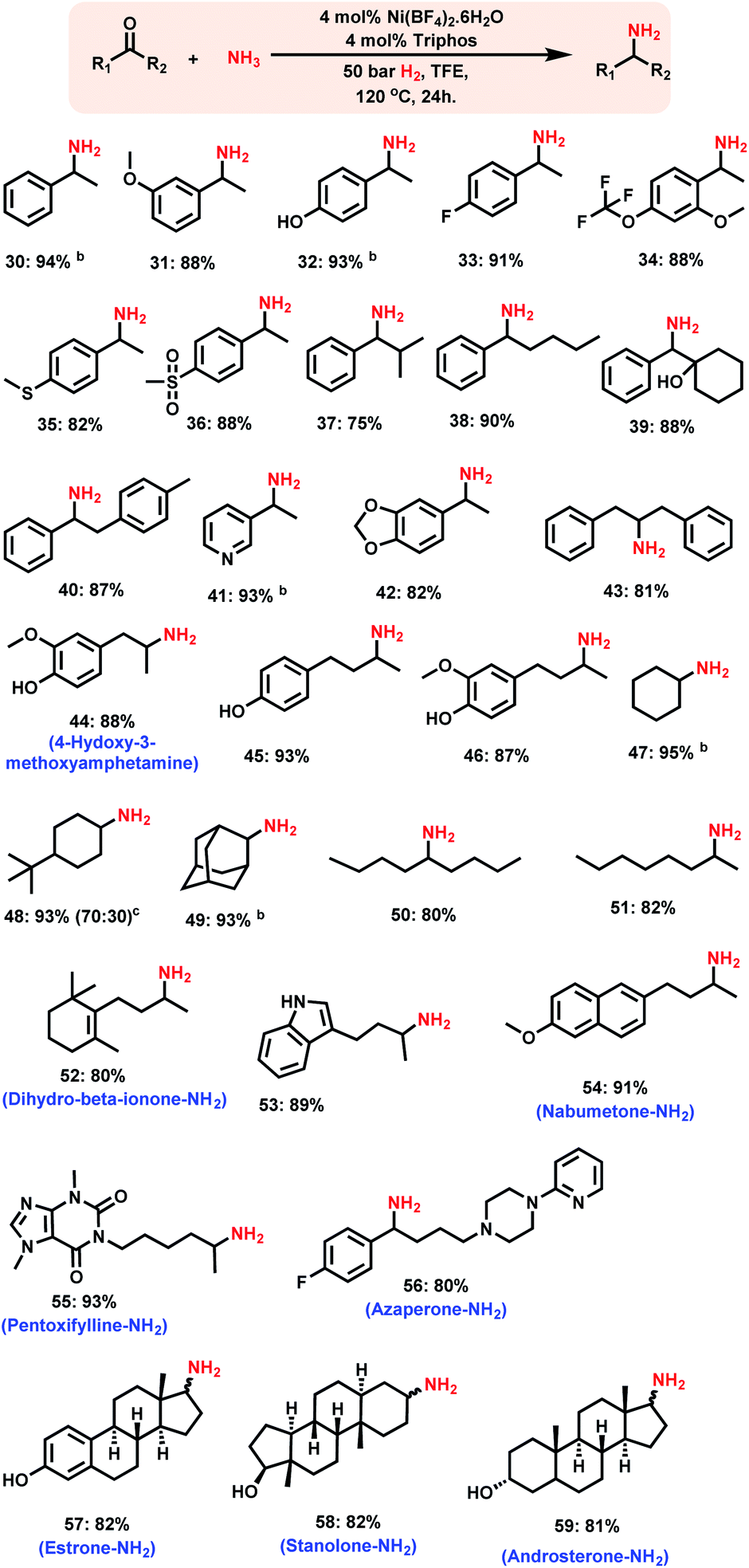 | ||
| Scheme 3 Nickel-triphos catalysed synthesis of branched primary amines.a aReaction conditions: 0.5 mmol ketone, 4 mol% Ni(BF4)2·6H2O, 4 mol% triphos (L1), 5–7 bar NH3, 50 bar H2, 2 mL degassed trifluoroethanol (TFE), 120 °C, 24 h, and isolated yields. bGC yields using n-hexadecane as the standard. Isolated as free amines and converted to hydrochloride salts. The corresponding hydrochloride salts were subjected to NMR analysis. | ||
To demonstrate this aspect, reductive amination of various functionalized aldehydes was performed. Interestingly, the functionalized aldehydes containing ether, phenolic, C–C double bond, ester, boronic acid ester and thioether groups were all highly selectively aminated and the corresponding linear primary amines were obtained in up to 96% yield, without the reduction of other functional groups (Scheme 2, products 12–21). In addition, heterocyclic primary amines were prepared in up to 95% yield (Scheme 2, products 22–25 and 28). Primary amines of 3,4-methylenedioxy and benzo-1,4-dioxane, which represent versatile motifs in many drugs and natural products, were prepared in up to 96% yield (Scheme 2, products 24 to 25 and 28). Further, aliphatic aldehydes, which are difficult to react were also aminated and the corresponding primary amines were obtained in good to excellent yields (Scheme 2; products 26–29).
Synthesis of branched primary amines from ketones
Compared to that of linear primary amines, the synthesis of branched primary amines starting from ketones is more challenging, because the reduction of corresponding imines from ketones is more difficult than that of the imines of corresponding aldehydes.Remarkably, the Ni-triphos complex is highly active and selective for the reductive amination of ketones with ammonia and hydrogen. As a result, all kinds of ketones were efficiently aminated to produce corresponding branched primary amines in high yields (Scheme 3). In addition to the simple and functionalized ketones, the ones bearing easily coordinating groups such as –NH, and –OH phenolic groups as well as pyridines to metals were also smoothly reacted with ammonia and gave corresponding primary amines in up to 94% yield (Scheme 3, products 41, 44–46, 53 and 56). In addition, the synthesis of various aliphatic branched primary amines, which are difficult to prepare,3i,n was performed with different ketones using this Ni-triphos system (Scheme 3, products 44–51). A more valuable application of this Ni-based protocol has been demonstrated by performing the amination of structurally complex life science molecules and steroid derivatives (Scheme 3). Gratifyingly, by applying the Ni-triphos catalyst the –NH2 moiety has been introduced in nabumetone, pentoxifylline, azaperone, estrone, androsterone and stanolone (Scheme 3, products 54–60).
Synthesis of aromatic primary amines by Ni-catalyzed hydrogenation of nitroarenes
The design of homogeneous catalysts for the hydrogenation of nitroarenes to anilines continues to be challenging.4c–f Here we explored the applicability of Ni-triphos as a homogeneous catalyst for the hydrogenation of nitroarenes. Advantageously this in situ generated Ni–L1 complex also exhibited excellent activity for the hydrogenation of nitroarenes (Scheme 4).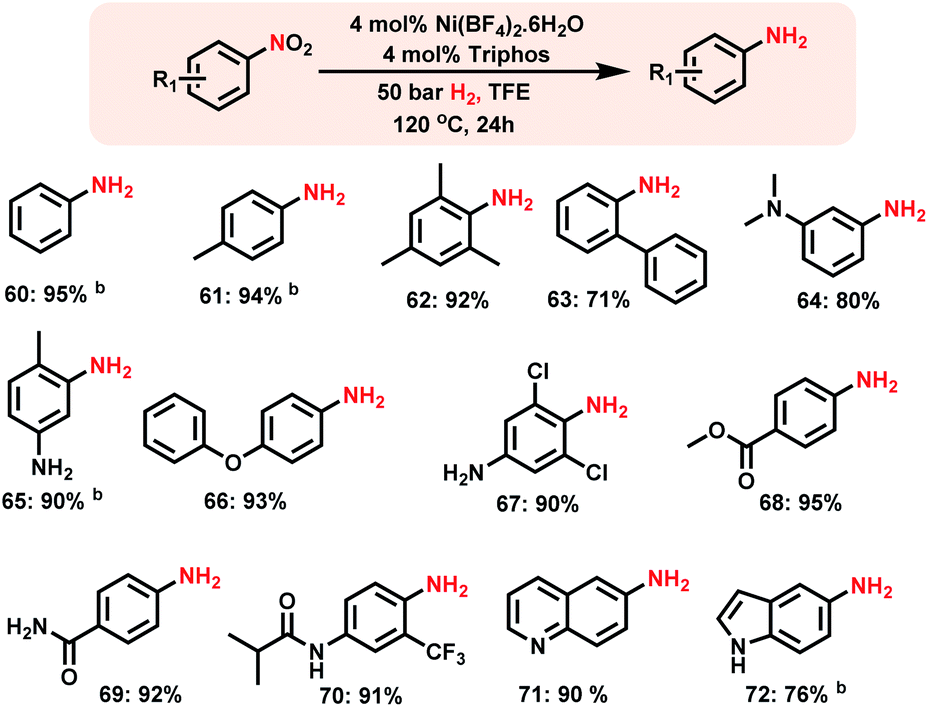 | ||
| Scheme 4 Homogeneous Ni-catalyzed synthesis of aromatic primary amines.a aReaction conditions: 0.5 mmol substrate, 4 mol% Ni(BF4)2·6H2O, 4 mol% triphos (L1), 50 bar H2, 2 mL degassed trifluoroethanol (TFE), 120 °C, 24 h, and bGC yields using n-hexadecane as the standard. | ||
Nitrobenzenes containing both electron-donating and -withdrawing groups were selectively hydrogenated to obtain corresponding anilines in up to 95% yield (Scheme 4, products 62 to 72). Functionalized nitroarenes containing esters, amides and ethers as well as halide groups were hydrogenated to anilines by tolerating these functional groups without being reduced (Scheme 4, products 66–70).
Conclusions
In conclusion, for the first time we introduced a homogeneous Ni-based catalyst for both reductive amination of carbonyl compounds and hydrogenation of nitroarenes to prepare all kinds of primary amines. The key to success for this synthesis is the use of a linear triphos-ligated Ni-complex. By applying this Ni-based homogeneous catalyst, starting from inexpensive and easily available carbonyl compounds and nitroarenes using abundant and atom economical reagents such as ammonia and molecular hydrogen, commercially and industrially important as well as pharmaceutically relevant aromatic, heterocyclic, and aliphatic primary amines were synthesized in good to excellent yields. DFT computations revealed that the overall reaction has an inner-sphere mechanism with H2 metathesis as the rate-determining step and this reasonably explains the need for the high H2 pressure and long reaction time on the basis of the computed apparent barriers.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We gratefully acknowledge the European Research Council, (EU project 670986-NoNaCat), the State of Mecklenburg-Vorpommern and the Leibniz Foundation (ZW, Leibniz Competition, SAW-2016-LIKAT-1). We thank the analytical staff of the Leibniz-Institute for Catalysis, Rostock for their excellent service.Notes and references
- (a) S. Gomez, J. A. Peters and T. Maschmeyer, Adv. Synth. Catal., 2002, 344, 1037–1057 CrossRef CAS; (b) A. Heshmatollah, Y. Hossein and S. Fatemeh, Curr. Org. Chem., 2015, 19, 1021–1049 CrossRef; (c) M. Orlandi, D. Brenna, R. Harms, S. Jost and M. Benaglia, Org. Process Res. Dev., 2018, 22, 430–445 CrossRef CAS; (d) D. Formenti, F. Ferretti, F. K. Scharnagl and M. Beller, Chem. Rev., 2019, 119, 2611–2680 CrossRef CAS PubMed; (e) F. Pohlki and S. Doye, Chem. Soc. Rev., 2003, 32, 104–114 RSC; (f) D. B. Bagal and B. M. Bhanage, Adv. Synth. Catal., 2015, 357, 883–900 CrossRef CAS; (g) D. B. Bagal and B. M. Bhanage, Recent Advances in Transition Metal-Catalyzed Hydrogenation of Nitriles, Adv. Synth. Catal., 2015, 357, 883–900 CrossRef CAS; (h) J. G. de Vries and C. J. Elsevier, in Handbook of Homogeneous Hydrogenation, Wiley-VCH, NewYork, 2007 Search PubMed; (i) S. Nishimura, Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis, Wiley, New York, 2001 Search PubMed; (j) F. Shi and X. Cui, in Catalytic Amination for N-Alkyl Amine Synthesis, Academic Press, 2018 Search PubMed; (k) Reductive amination review, https://erowid.org/archive/rhodium/chemistry/reductive.amination.html, 2004 Search PubMed; (l) T. Yan, B. L. Feringa and K. Barta, Nat. Commun., 2014, 5, 5602 CrossRef CAS PubMed; (m) T. Yan, B. L. Feringa and K. Barta, Sci. Adv., 2017, 3, eaao6494 CrossRef PubMed; (n) T. Yasukawa, R. Masuda and S. Kobayashi, Nat. Catal., 2019, 2, 1088–1092 CrossRef CAS; (o) C. Gunanathan and D. Milstein, Angew. Chem. Int. Ed., 2008, 47, 8661–8664 CrossRef CAS PubMed.
- (a) S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS PubMed; (b) V. Froidevaux, C. Negrell, S. Caillol, J.-P. Pascault and B. Boutevin, Chem. Rev., 2016, 116, 14181–14224 CrossRef CAS PubMed; (c) S. A. Lawrence, Amines: Synthesis, Properties and Applications, Cambridge University Press, Cambridge, UK, 2004 Search PubMed; (d) A. Ricci, Amino Group Chemistry: From Synthesis to the Life Sciences, Wiley VCH, Weinheim, 2008 Search PubMed; (e) https://njardarson.lab.arizona.edu/sites/njardarson.lab.arizona.edu/files/2018Top200PharmaceuticalRetailSalesPosterLowResFinalV2.pdf .
- (a) R. V. Jagadeesh, K. Murugesan, A. S. Alshammari, H. Neumann, M.-M. Pohl, J. Radnik and M. Beller, Science, 2017, 358, 326–332 CrossRef CAS PubMed; (b) K. Murugesan, T. Senthamarai, M. Sohail, A. S. Alshammari, M.-M. Pohl, M. Beller and R. V. Jagadeesh, Chem. Sci., 2018, 9, 8553–8560 RSC; (c) T. Senthamarai, K. Murugesan, J. Schneidewind, N. V. Kalevaru, W. Baumann, H. Neumann, P. C. J. Kamer, M. Beller and R. V. Jagadeesh, Nat. Commun., 2018, 9, 4123 CrossRef PubMed; (d) K. Murugesan, M. Beller and R. V. Jagadeesh, Angew. Chem., Int. Ed., 2019, 58, 5064–5068 CrossRef CAS PubMed; (e) K. Murugesan, Z. Wei, V. G. Chandrashekhar, H. Neumann, A. Spannenberg, H. Jiao, M. Beller and R. V. Jagadeesh, Nat. Commun., 2019, 10, 5443 CrossRef PubMed; (f) J. Gallardo-Donaire, M. Ernst, O. Trapp and T. Schaub, Adv. Synth. Catal., 2016, 358, 358–363 CrossRef CAS; (g) J. Gallardo-Donaire, M. Hermsen, J. Wysocki, M. Ernst, F. Rominger, O. Trapp, A. S. K. Hashmi, A. Schäfer, P. Comba and T. Schaub, J. Am. Chem. Soc., 2018, 140, 355–361 CrossRef CAS PubMed; (h) G. Hahn, P. Kunnas, N. de Jonge and R. Kempe, Nat. Catal., 2018, 2, 71–77 CrossRef; (i) T. Gross, A. M. Seayad, M. Ahmad and M. Beller, Org. Lett., 2002, 4, 2055–2058 CrossRef CAS PubMed; (j) X. Tan, S. Gao, W. Zeng, S. Xin, Q. Yin and X. Zhang, J. Am. Chem. Soc., 2018, 140, 2024–2027 CrossRef CAS PubMed; (k) M. Chatterjee, T. Ishizaka and H. Kawanami, Green Chem., 2016, 18, 487–496 RSC; (l) B. Dong, X. Guo, B. Zhang, X. Chen, J. Guan, Y. Qi, S. Han and X. Mu, Catalysts, 2015, 5, 2258–2270 CrossRef CAS; (m) S. Nishimura, K. Mizuhori and K. Ebitani, Res. Chem. Intermed., 2016, 42, 19–30 CrossRef CAS; (n) J. Krupka, L. Dluhoš and L. Mrózek, Chem. Eng. Technol., 2017, 40, 870–877 CrossRef CAS; (o) Z. Wang, Mignonac reaction, in Comprehensive organic name reactions and reagents, John Wiley & Sons, New Jersey, 2010 CrossRef; (p) T. Riermeier, K.-J. Haack, U. Dingerdissen, A. Boerner, A. V. Tararov and R. Kadyrov, Method for producing amines by homogeneously catalyzed reductive amination of carbonyl compounds, Weniger, US, 2005, vol. 6, pp. 884–887B1 Search PubMed; (q) T. Komanoya, T. Kinemura, Y. Kita, K. Kamata and M. Hara, J. Am. Chem. Soc., 2017, 139, 11493–11499 CrossRef CAS PubMed; (r) G. Liang, A. Wang, L. Li, G. Xu, N. Yan and T. Zhang, Angew. Chem., Int. Ed., 2017, 56, 3050–3054 CrossRef CAS PubMed; (s) J. Bódis, L. Lefferts, T. E. Müller, R. Pestman and J. A. Lercher, Catal. Lett., 2005, 104, 23–28 CrossRef.
- (a) T. Schwob and R. Kempe, Angew. Chem., Int. Ed., 2016, 55, 15175–15179 CrossRef CAS PubMed; (b) R. V. Jagadeesh, A.-E. Surkus, H. Junge, M.-M. Pohl, J. Radnik, J. Rabeah, H. Huan, V. Schünemann, A. Brückner and M. Beller, Science, 2013, 342, 1073–1076 CrossRef CAS PubMed; (c) J. F. Knifton, J. Org. Chem., 1976, 41, 1200–1206 CrossRef CAS; (d) R. M. Deshpande, A. N. Mahajan, M. M. Diwakar, P. S. Ozarde and R. V. Chaudhari, J. Org. Chem., 2004, 69, 4835–4838 CrossRef CAS PubMed; (e) G. Wienhöfer, M. Baseda-Krüger, C. Ziebart, F. A. Westerhaus, W. Baumann, R. Jackstell, K. Junge and M. Beller, Chem. Commun., 2013, 49, 9089–9091 RSC; (f) Z.-J. Yao, J.-W. Zhu, N. Lin, X.-C. Qiao and W. Deng, Dalton Trans., 2019, 48, 7158–7166 RSC; (g) G. Hahn, J.-K. Ewert, C. Denner, D. Tilgner and R. Kempe, ChemCatChem, 2016, 8, 2461–2465 CrossRef CAS; (h) P. Ryabchuk, G. Agostini, M.-M. Pohl, H. Lund, A. Agapova, H. Junge, K. Junge and M. Beller, Sci. Adv., 2018, 4, eaat0761 CrossRef PubMed; (i) Y. Hu, Y. Yu, X. Zhao, H. Yang, B. Feng, H. Li, Y. Qiao, L. Hua, Z. Pan and Z. Hou, Sci. China Chem., 2010, 53, 1541–1548 CrossRef CAS; (j) S. Pisiewicz, D. Formenti, A.-E. Surkus, M.-M. Pohl, J. Radnik, K. Junge, C. Topf, S. Bach-mann, M. Scalone and M. Beller, ChemCatChem, 2016, 8, 129–134 CrossRef CAS; (k) F. Yang, M. Wang, W. Liu, B. Yang, Y. Wang, J. Luo, Y. Tang, L. Hou, Y. Li, Z. i. Li, B. Zhang, W. Yang and Y. Li, Green Chem., 2019, 21, 704–711 RSC; (l) N. Mahata, A. F. Cunha, J. J. M. Órfão and J. L. Figueiredo, Appl. Catal. Gen., 2018, 351, 204–209 CrossRef; (m) W. She, T. Qi, M. Cui, P. Yan, S. W. Ng, W. Li and G. Li, ACS Appl. Mater. Interfaces, 2018, 10, 14698–14707 CrossRef CAS PubMed.
- (a) S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef CAS PubMed; (b) J. A. Garduno, A. Arevalo and J. J. Garcia, Dalton Trans., 2015, 44, 13419–13438 RSC.
- (a) M. Beller, K. Murugesan, C. B. Bheeter, P. R. Linnebank, A. Spannenberg, J. N. H. Reek, R. V. Jagadeesh and M. Beller, ChemSusChem, 2019, 12, 3363–3369 CrossRef; (b) N. G. Léonard and P. J. Chirik, ACS Catal., 2018, 8, 342–348 CrossRef; (c) T. J. Mooibroek, E. C. M. Wenker, W. Smit, I. Mutikainen, M. Lutz and E. Bouwman, Inorg. Chem., 2013, 52, 8190–8201 CrossRef CAS; (d) K. V. Vasudevan, B. L. Scott and S. K. Hanson, Eur. J. Inorg. Chem., 2012, 2012, 4898–4906 CrossRef CAS; (e) I. M. Angulo, A. M. Kluwer and E. Bouwman, Chem. Commun., 1998, 2689–2690 RSC; (f) S. Chakraborty, P. Bhattacharya, H. Dai and H. Guan, Acc. Chem. Res., 2015, 48, 1995–2003 CrossRef CAS PubMed.
- (a) S. A. Johnson, Dalton Trans., 2015, 44, 10905–10913 RSC; (b) Y. Nakao, Shokubai, 2008, 50, 705–709 CAS; (c) A. C. Williams, in Nickel-catalyzed C–H activation, CRC Press, 2015, pp. 113–144 Search PubMed; (d) F. Juliá-Hernández, T. Moragas, J. Cornella and R. Martin, Nature, 2017, 545, 84 CrossRef PubMed; (e) N. A. Harry, S. Saranya, S. M. Ujwaldev and G. Anilkumar, Catal. Sci. Technol., 2019, 9, 1726–1743 RSCV. G. Landge, C. H. Shewale, G. Jaiswal, M. K. Sahoo, S. P. Midya and E. Balaraman, Catal. Sci. Technol., 2016, 6, 1946–1951 RSC.
- (a) M. Henrion, V. Ritleng and M. J. Chetcuti, ACS Catal., 2015, 5, 1283–1302 CrossRef CAS; (b) R. Shi, Z. Zhang and X. Hu, Acc. Chem. Res., 2019, 52, 1471–1483 CrossRef CAS PubMed; (c) C. M. Lavoie and M. Stradiotto, ACS Catal., 2018, 8, 7228–7250 CrossRef CAS; (d) L. Guo and M. Rueping, Acc. Chem. Res., 2018, 51, 1185–1195 CrossRef CAS PubMed; (e) M. O. Konev and E. R. Jarvo, Angew. Chem., Int. Ed., 2016, 55, 11340–11342 CrossRef CAS PubMed; (f) C. Liu, S. Tang, D. Liu, J. Yuan, L. Zheng, L. Meng and A. Lei, Angew. Chem., Int. Ed., 2012, 51, 3638–3641 CrossRef CAS PubMed.
- (a) J. B. Sweeney, A. K. Ball, P. A. Lawrence, M. C. Sinclair and L. J. Smith, Angew. Chem., Int. Ed., 2018, 57, 10202–10206 CrossRef CAS PubMed; (b) M. Vellakkaran, K. Singh and D. Banerjee, ACS Catal., 2017, 7, 8152–8158 CrossRef CAS.
- (a) F. AlObaidi, Z. Ye and S. Zhu, Polymer, 2004, 45, 6823–6829 CrossRef CAS; (b) R. P. Quirk and W. Yu, High Perform. Polym., 2005, 17, 349–360 CrossRef CAS; (c) D. G. Yakhvarov, K. R. Basvani, M. K. Kindermann, A. B. Dobrynin, I. A. Litvinov, O. G. Sinyashin, P. G. Jones and J. Heinicke, Eur. J. Inorg. Chem., 2009, 1234–1242 CrossRef CAS.
- (a) J. A. Milligan, J. P. Phelan, S. O. Badir and G. A. Molander, Angew. Chem., Int. Ed., 2019, 58, 6152–6163 CrossRef CAS PubMed; (b) J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS; (c) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed.
- (a) Z. Liu, Z. Yang, P. Wang, X. Yu, Y. Wu, H. Wang and Z. Liu, ACS Sustainable Chem. Eng., 2019, 7, 18236–18241 CrossRef CAS; (b) L. Iffland, A. Khedkar, A. Petuker, M. Lieb, F. Wittkamp, M. van Gastel, M. Roemelt and U.-P. Apfel, Organometallics, 2019, 38, 289–299 CrossRef CAS; (c) F. Ferretti, F. K. Scharnagl, A. Dall'Anese, R. Jackstell, S. Dastgir and M. Beller, Catal. Sci. Technol., 2019, 9, 3548–3553 RSC; (d) B. Emayavaramban, P. Chakraborty and B. Sundararaju, ChemSusChem, 2019, 12, 3089–3093 CrossRef CAS PubMed; (e) W. Liu, B. Sahoo, A. Spannenberg, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2018, 57, 11673–11677 CrossRef CAS PubMed; (f) A. Cavaille, B. Joyeux, N. Saffon-Merceron, N. Nebra, M. Fustier-Boutignon and N. Mezailles, Chem. Commun., 2018, 54, 11953–11956 RSC; (g) J. Schneidewind, R. Adam, W. Baumann, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2017, 56, 1890–1893 CrossRef CAS PubMed; (h) J. R. Cabrero-Antonino, R. Adam, V. Papa, M. Holsten, K. Junge and M. Beller, Chem. Sci., 2017, 8, 5536–5546 RSC; (i) T. M. Buscagan, P. H. Oyala and J. C. Peters, Angew. Chem., Int. Ed., 2017, 56, 6921–6926 CrossRef CAS PubMed; (j) T. J. Korstanje, J. Ivar van der Vlugt, C. J. Elsevier and B. de Bruin, Science, 2015, 350, 298–302 CrossRef CAS PubMed; (k) F. K. Scharnagl, M. F. Hertrich, G. Neitzel, R. Jackstell and M. Beller, Adv. Synth. Catal., 2019, 361, 374–379 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc01084g |
| This journal is © The Royal Society of Chemistry 2020 |