
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExpedient synthesis of conjugated triynes via alkyne metathesis†
Idriss
Curbet
,
Sophie
Colombel-Rouen
 ,
Romane
Manguin
,
Anthony
Clermont
,
Alexandre
Quelhas
,
Daniel S.
Müller
,
Thierry
Roisnel
,
Olivier
Baslé‡
,
Yann
Trolez
* and
Marc
Mauduit
*
,
Romane
Manguin
,
Anthony
Clermont
,
Alexandre
Quelhas
,
Daniel S.
Müller
,
Thierry
Roisnel
,
Olivier
Baslé‡
,
Yann
Trolez
* and
Marc
Mauduit
*
Univ Rennes, Ecole Nationale Supérieure de Chimie de Rennes, CNRS, ISCR – UMR 6226, F-35000 Rennes, France. E-mail: marc.mauduit@ensc-rennes.fr; yann.trolez@ensc-rennes.fr
First published on 27th April 2020
Abstract
The first synthesis of conjugated triynes by molybdenum-catalysed alkyne metathesis is reported. Strategic to the success of this approach is the utilization of sterically-hindered diynes that allowed for the site-selective alkyne metathesis to produce the desired conjugated triyne products. The steric hindrance of the alkyne moiety was found to be crucial in preventing the formation of diyne byproducts. This novel synthetic strategy was amenable to self- and cross-metathesis providing straightforward access to the corresponding symmetrical and dissymmetrical triynes with high selectivity.
Introduction
Polyynes are an important class of natural products which display a broad array of biological activities such as antibacterial, antimicrobial, antitumor and anticancer properties.1 Conjugated diynes and triynes (e.g. ivorenolide B 1 (ref. 2) and ichthyothereol 2 (ref. 3)) represent the major part of this class of compounds that can be isolated from plants, fungi or bacteria (Fig. 1a). While the diyne core is easily accessible,4 the triyne moiety appears more difficult to access and represents a synthetic challenge.5 Several methods have been developed to access symmetrical and unsymmetrical triynes.6 For instance, Tykwinski and co-workers recently reported an elegant synthetic strategy involving a Pd-catalysed cross-coupling between 3 and various terminal alkynes that lead to the desired triynes 4 (Fig. 1b).7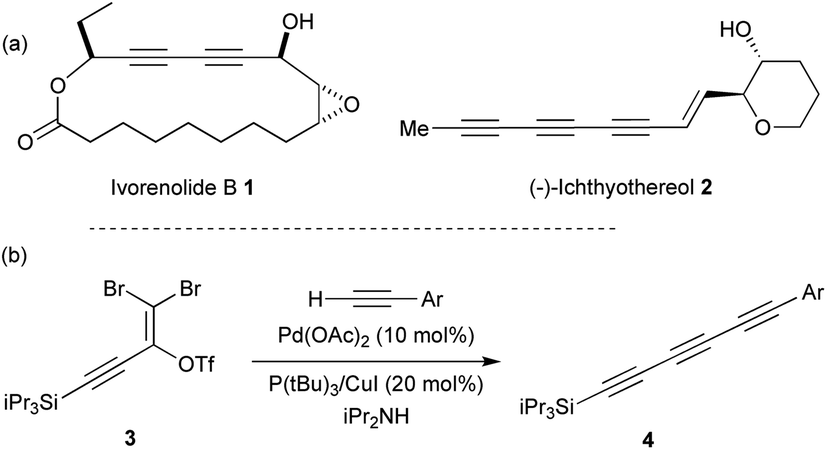 | ||
| Fig. 1 (a) Examples of natural products containing diynes and triynes. (b) Synthesis of triynes as reported by Tykwinski and co-workers. | ||
Despite the mentioned advances in this field, the development of versatile and efficient methodologies is still necessary. Several well-defined W- and Mo-based catalysts (e.g.Cat-1 and Cat-2, Fig. 2) can promote metathesis reactions of alkynes, allowing for the formation of new carbon–carbon triple bonds.8 Logically, extension of this methodology to triyne substrates would represent a promising and straightforward way to synthesize the highly desirable 1,3,5-hexatriyne core.
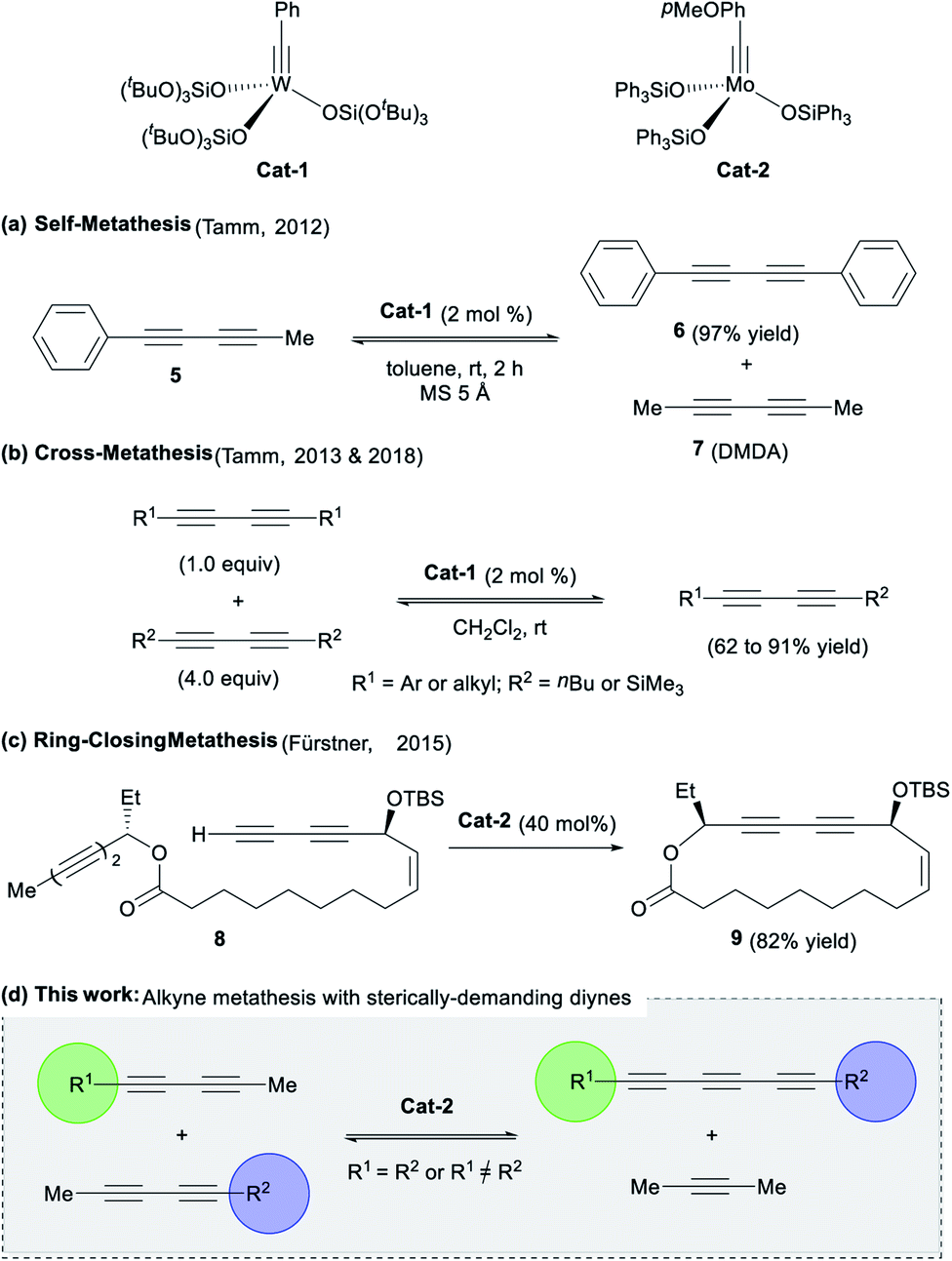 | ||
| Fig. 2 (a–c) Previous works on diynes metathesis catalysed by Mo- or W-catalysts and (d) the proposed concept for the access of conjugated triynes (This work). | ||
This strategy was recently attempted by Tamm and co-workers using a tungsten-alkyne complex Cat-1, which unexpectedly led to the exclusive formation of symmetrical diynes (e.g.5) and related dimethyldiacetylene 7 (Fig. 2a). The anticipated conjugated triynes could not be isolated.9 In 2013, the authors extended the methodology to diyne cross-metathesis (DYCM) (Fig. 2b)10,11 resulting in unsymmetrical diynes products along with traces of the triyne products upon prolonged reaction time. In line with these pioneering works, Fürstner and coworkers also observed a similar reactivity in the catalytic ring-closing metathesis (RCM) of a bis-diyne 8 with molybdenum-alkyne complex Cat-2 that produced the corresponding cyclized diyne 9 (Fig. 2c), a key intermediate in the total synthesis of ivorenolide B 1 (Fig. 1).12 In order to enforce the appropriate C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C triple bond to react, it was hypothesized that the introduction of bulky groups would influence the regioselectivity13 of diyne metathesis reactions favouring the triyne products over the diyne products as previously reported (Fig. 2d).9–12 We would like now to report on a methodology that allows for the unprecedented selective formation of symmetrical and dissymmetrical conjugated triynes from alkyne metathesis.
C triple bond to react, it was hypothesized that the introduction of bulky groups would influence the regioselectivity13 of diyne metathesis reactions favouring the triyne products over the diyne products as previously reported (Fig. 2d).9–12 We would like now to report on a methodology that allows for the unprecedented selective formation of symmetrical and dissymmetrical conjugated triynes from alkyne metathesis.
Results and discussion
Investigations began with the synthesis of diynes 13a–e bearing various substituents following a robust methodology from the literature (Scheme 1).14 Starting from terminal alkynes 10, the Pd-catalysed Sonogashira coupling with (E)-1,2-dichloroethylene 11 resulted in the corresponding chloro-enynes 12. The latter was then reacted with n-BuLi and iodomethane to produce the expected diynes 13a–e in moderate to good overall yields (Scheme 1).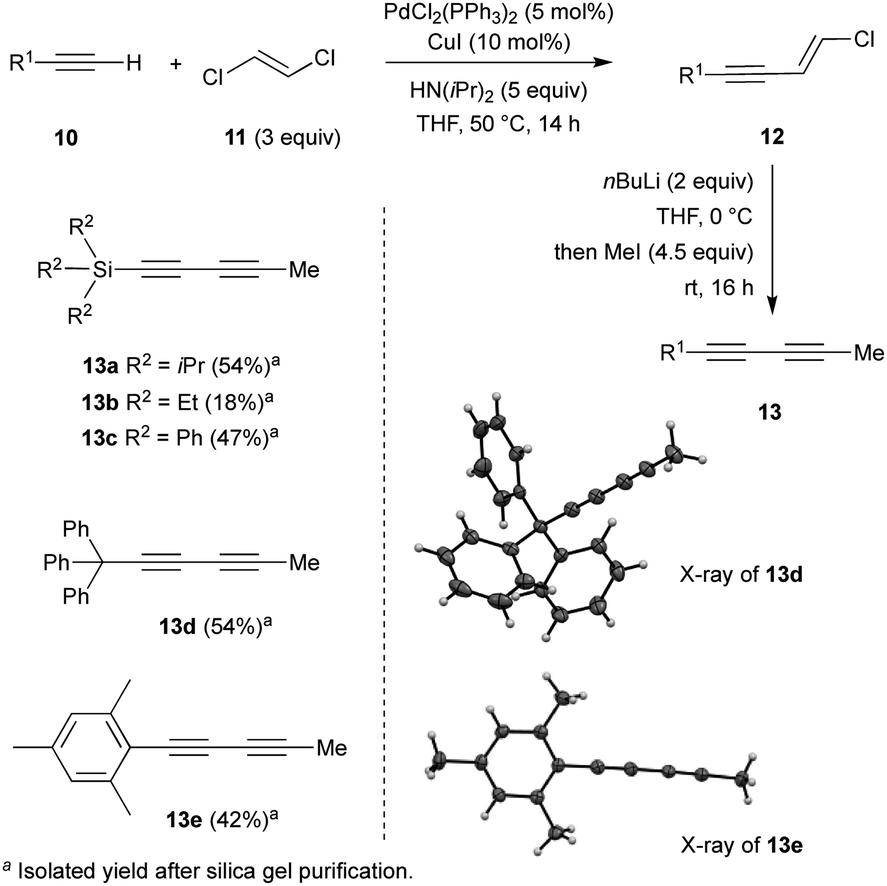 | ||
| Scheme 1 Synthetic access of sterically-demanding dienes 13a–e. | ||
Additionally, structures of diynes 13d and 13e were confirmed by single crystal X-ray diffraction studies (Scheme 1). The reactivity and behaviour of these more or less sterically-demanding diynes were then assessed in alkyne self- and cross-metathesis. First, we investigated the self-metathesis of silylated diynes 13a–c,f (Table 1). Triisopropylsilylpentadiyne 13a was exposed to alkyne metathesis catalyst Cat-2 (3 mol%) at 40 °C over three hours. While a 50% conversion was observed, only traces of desired triyne 14a (1%) were detected by GC analysis (entry 1). Inspired by the work of Fürstner and co-workers,12c the addition of 4 and 5 Å molecular sieves (MS) in the reaction media to trap 2-butyne could not only increase the conversion up to 96% but also allow for the formation of the desired symmetrical (TIPS)2triyne 14a in excellent 86% GC yield (Table 1, entry 2). Nevertheless, symmetrical (TIPS)2diyne 15a was also obtained in a respective triyne/diyne ratio of 86![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :14 (see ESI,† for details). Interestingly, at lower catalyst loading (2 and 1 mol%, entries 3 and 5) as well as at ambient temperature (entry 4), Cat-2 remained quite productive reaching respectively 80, 59 and 74% GC yields.
:14 (see ESI,† for details). Interestingly, at lower catalyst loading (2 and 1 mol%, entries 3 and 5) as well as at ambient temperature (entry 4), Cat-2 remained quite productive reaching respectively 80, 59 and 74% GC yields.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Diynes | Cat-2 (mol%) | Conv.b (%) | Ratio 14:15c |
Yield 14b | Yield 15b |
| a Reaction conditions: diyne (0.04 mmol), catalyst (3 mol%), MS 4 Å/5 Å (40 mg), toluene (0.35 mL), 40 °C, 3 h, under Ar. b Determined by GC-analysis with acetophenone as internal standard. c Determined by GC-analysis. d Performed without MS 4 Å/5 Å. e Performed at 20 °C. f Performed at 0.55 mmol-scale. g Isolated yield after silica gel chromatography. h Performed at 20 °C over 1 h. i Performed at 0.26 mmol-scale. j Based on the recovered starting material. k Determined by quantitative 13C NMR spectroscopy. l Estimated yield from an isolated mixture of 14c + 15c. | ||||||
| 1d | 13a | 3 | 50 | Nd | 1 | — |
| 2 | 13a | 3 | 96 | 86:14 |
86 | — |
| 3 | 13a | 2 | 91 | 90:10 |
80 | — |
| 4e | 13a | 2 | 89 | 90:10 |
74 | — |
| 5 | 13a | 1 | 75 | 95:5 |
59 | — |
| 6f | 13a | 3 | 96 | 81:19 |
63g | — |
| 7h | 13f | 3 | 91 | 0:100 |
— | 48 |
| 8 | 13b | 3 | >98 | 2:98 |
— | 56 |
| 9i | 13c | 3 | 95j | 40:60k |
12kl | 19kl |
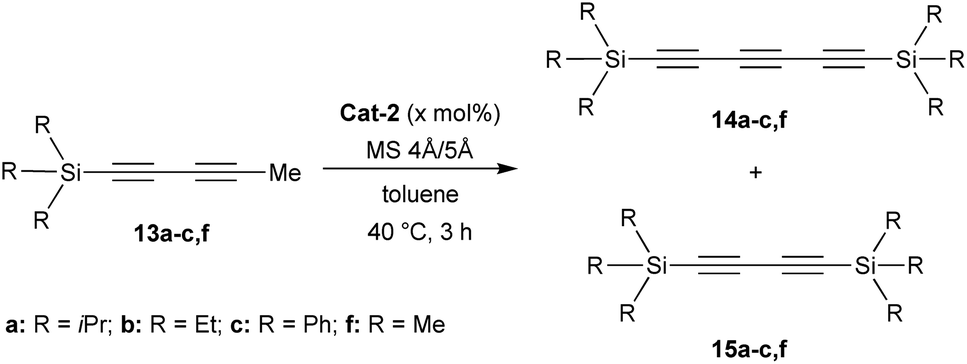
More importantly, the selectivity increased up to 95:5 with the lowest catalyst loading, although the GC yield dropped to 59% for triyne 14a (entry 5). By increasing the scale of the reaction, it was possible to isolate 14a in good 63% isolated yield after silica gel purification (entry 6). After demonstrating that the presence of a bulky triisopropylsilyl (TIPS) group allowed for the formation of the triyne as the major metathesis product, it was investigated whether less sterically-hindered Si-substituent such as trimethylsilyl (TMS), triethylsilyl (TES) or triphenylsilyl (TPS) groups would give preferentially rise to the symmetrical diyne or the triyne product. Hence, TMS-diyne 13f was synthesized according to a protocol from the literature and exposed to the aforementioned conditions (3 mol%, 40 °C, 3 h).15 Mo-based complex Cat-2 appeared quite productive, although symmetrical (TMS)2diyne 15f was exclusively formed (ratio 14f:15f = 0:100) in 48% GC yield (Table 1, entry 7), which is consistent with previous observations from the Tamm group (Scheme 1b).11 A similar behaviour was also observed with TES-diyne 13b (ratio 14b:15b = 2:98) that led to symmetrical (TES)2diyne 15b in a moderate 56% GC yield (entry 8). Regarding diyne 13c featuring a bulkier Ph3Si group, the expected symmetrical (TPS)2triyne 14c was formed more significantly but (TPS)2diyne 15c remained predominant (ratio 14c:15c = 40:60, entry 9).16 Moreover, X-ray diffraction analysis could be done allowing to confirm the solid-state structure of desired triyne 14c that exhibited usual geometrical features for this kind of compounds6b (Fig. 3).
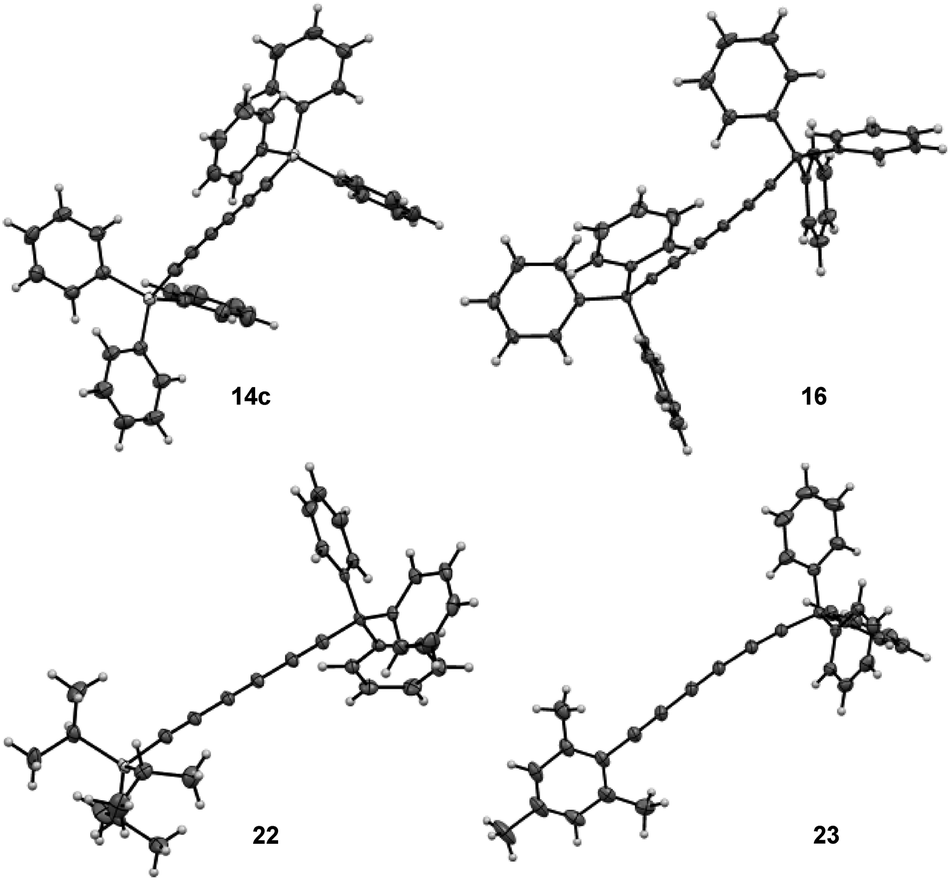 | ||
| Fig. 3 Solid-state structures of symmetrical triynes 14c and 16 and dissymmetrical triynes 22 and 23 from single crystal X-ray diffraction. | ||
Encouraged by the promising good result reached with TIPS substituent, we decided to investigate other substituents such as triphenylmethyl (trityl) or mesityl (Mes) groups (Scheme 2). To our delight, as seen with the TIPS-substrate 13a, Cat-2 demonstrated efficient catalytic activity in the self-metathesis of the trityl-substituted diyne 13d resulting in the desired symmetrical triyne product 16 in 54% isolated yield (Scheme 2a).
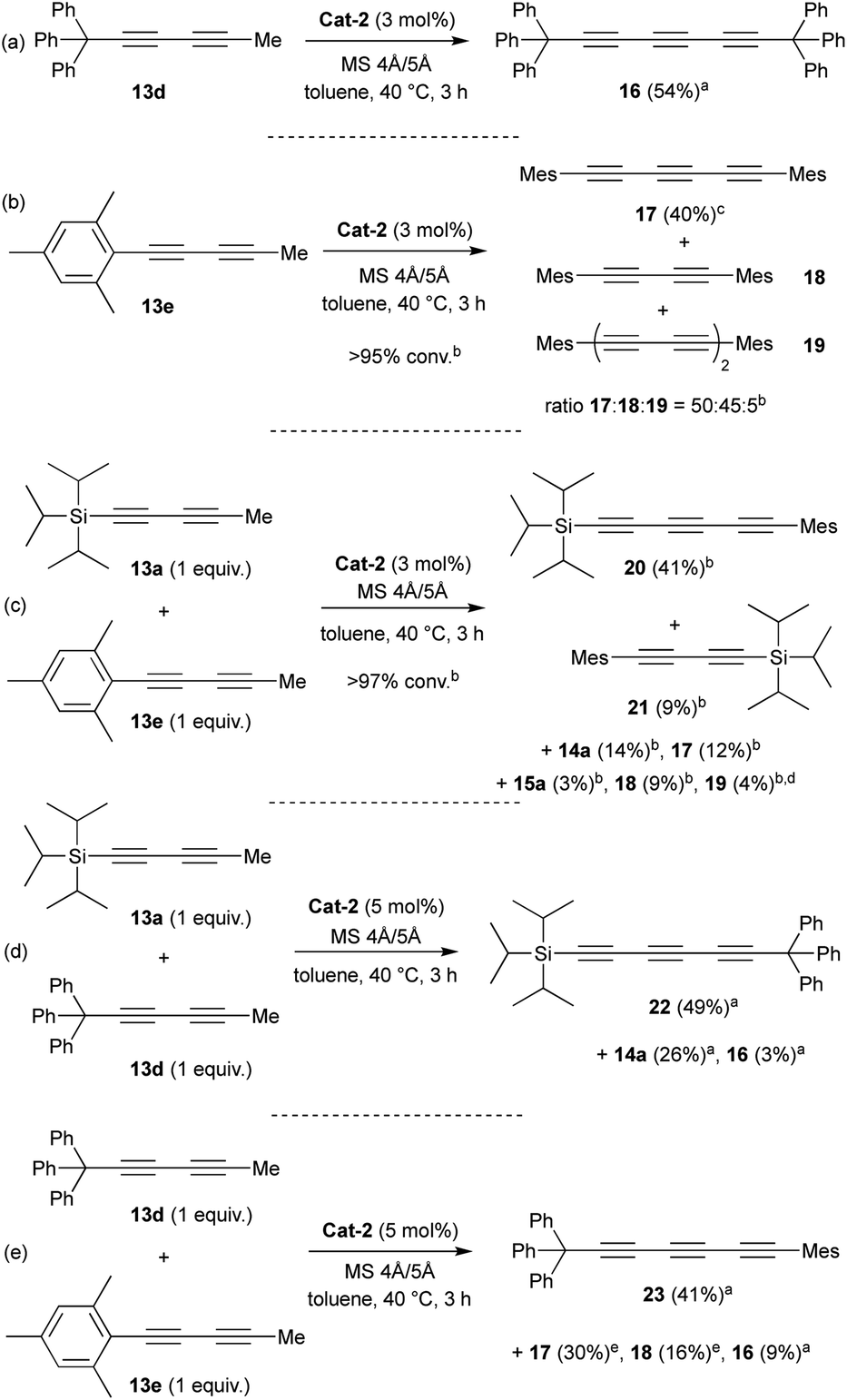 | ||
| Scheme 2 Scope of self- and cross-metathesis catalysed by Mo-complex Cat-2. aIsolated yield after silica gel chromatography. bDetermined by GC-MS analysis. cYield determined by GC-MS analysis with n-dodecane as the internal standard. d(TIPS)2tetrayne and TIPS/Mes-tetrayne were also detected (1–3%). eYield from an inseparable isolated mixture of 17 + 18 (ratio = 55/45). | ||
Furthermore, X-ray diffraction analysis unambiguously confirmed the structure of trityl-substituted triyne 16 (Fig. 3). Additionally, the stability of trityl-triyne 16 toward the Mo-benzylidyne catalyst was evaluated, as the competitive conversion of triyne to higher polyynes was suspected. Triyne 16 was thus exposed to Mo-Cat 2 (3 mol%) at 40 °C over 3 hours. Interestingly, 96% of the starting material were recovered. It is worth to notice that a similar behaviour was also observed with a mixture of triyne/diyne 14a/15a (81/19 ratio) without any alteration of the triyne/diyne ratio (see ESI for details†). Those additional experiments highlighted the remarkable stability of the triynes into the reactive media. Mo-benzylidyne catalyst was also efficient toward mesityl-substituted diyne 13e affording the expected symmetrical triyne 17 in moderate 40% GC-MS yield (Scheme 2b). However, GC-MS analysis evidenced a significant formation of symmetrical diyne 18 but also some traces of tetrayne 19 (ratio 17:18:19 = 50:45:5).17 The steric hindrance of the mesityl group appeared thus less suitable to promote the selective formation of the triyne metathesis product. The more challenging cross-metathesis reaction was then studied. First, by reacting TIPS- and Mes-diynes 13a and 13e, the formation of the highly desirable dissymmetrical triyne 20 as the major product was observed, reaching 41% GC yield (Scheme 2c). Unsurprisingly, some amounts of symmetrical triynes 14a and 17 were also observed (14 and 12%, resp.), as well as the dissymmetrical diyne 21 (9%) and its symmetrical congeners 15 and 18 (3 and 9%, resp.). Of note, few traces (1–4%) of symmetrical/dissymmetrical tetraynes were also detected (see ESI for details†). Secondly, with trityl-diyne 13d as a partner, TIPS-diyne 13a led to the expected dissymmetrical triyne 22 in 49% isolated yield while Mes-diyne 13e produced the corresponding dissymmetrical triyne 23 in 41% isolated yield (Scheme 2d and e). Again, some amounts of symmetrical triynes/diynes were also formed. Furthermore, solid-state structures of these challenging dissymmetrical triynes 22 and 23 were also confirmed by X-ray diffraction analysis (Fig. 3).
In order to show the synthetic potential of this methodology to furnish valuable triyne building-blocks, the post-transformation of dissymmetrical triyne 22 was then explored. As depicted in Scheme 3, the TIPS fragment was successfully removed using silver fluorine followed by direct iodination of the resulting deprotected alkyne in the presence of N-iodosuccinimide (NIS). The expected 7-iodohepta-2,4,6-triyne-1-trityl 24 was isolated in good 69% isolated yield. Considering the aforementioned experimental results, a plausible reaction pathway for the self-metathesis of sterically-hindered diynes is suggested in Scheme 4. Depending on the steric hindrance brought by the RL-substituent of the diyne, and according to the established mechanism of the alkyne metathesis,9–11,18 a catalytic cycle could be proposed. If RL is large enough, Mo-based complex Cat-2 reacts preferably with the less hindered δ,γ-CC triple bond to form the metallacyclobutadiene MCBD-α leading to propagating species PSγ. The latter is then engaged through the catalytic cycle A by reacting again towards the less hindered CC triple bond to produce the valuable symmetrical (γ,γ)-triyne P1. In opposition, if PSγ reacts with the more hindered internal α,β-alkyne, a catalytic cycle B already described by Tamm and co-workers is involved and explains the formation of the undesired diyne product P3.11
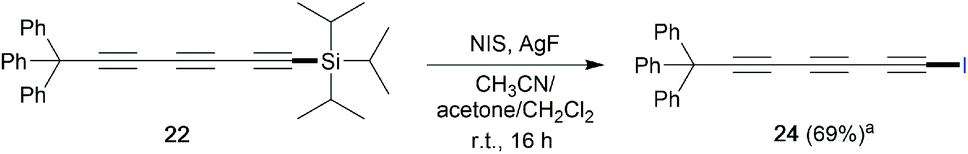 | ||
| Scheme 3 Post-functionalization of dissymmetrical triyne 22 (a isolated yield after silica gel purification). | ||
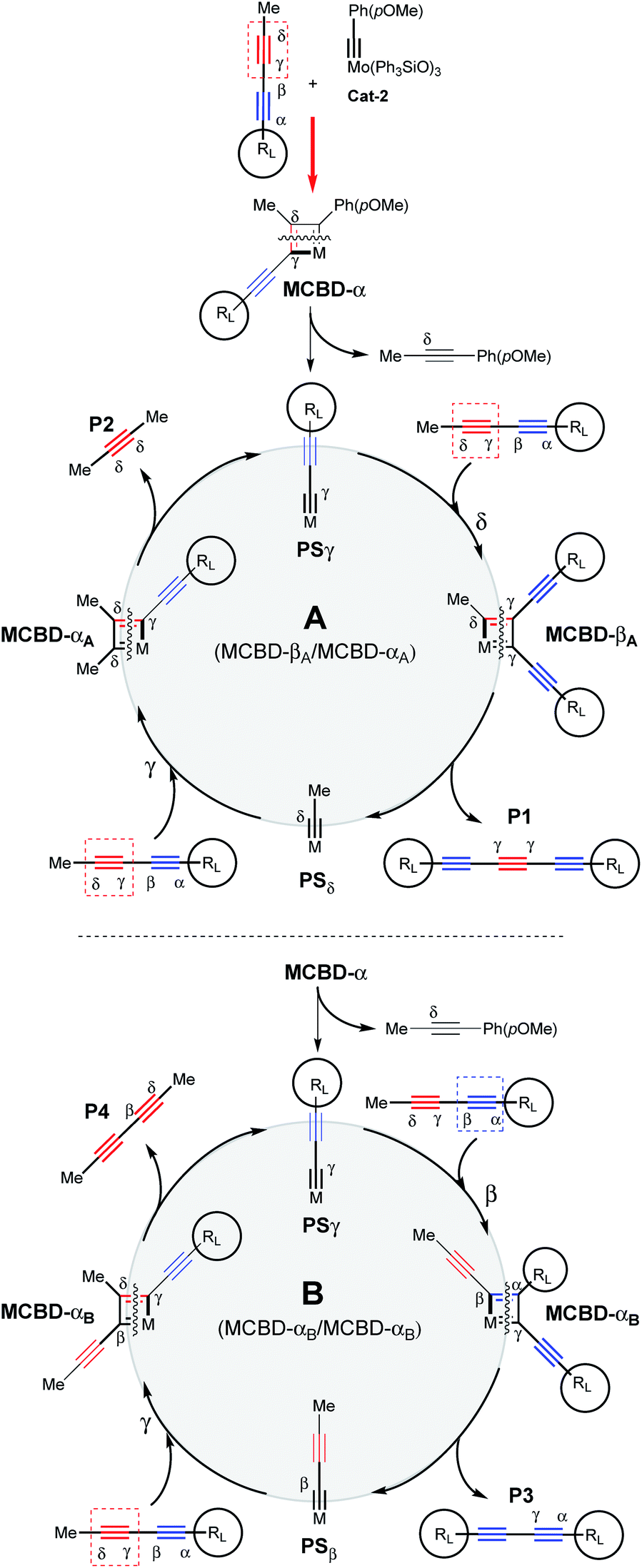 | ||
| Scheme 4 Proposed mechanism for the self-metathesis of sterically-hindered diynes catalysed by Mo-complex Cat-2 leading to conjugated triynes (cycle A) or diynes (cycle B). Reversibility is expected for each step. Only productive metathesis steps are represented. | ||
Conclusions
In conclusion, it was demonstrated herein that the utilization of sterically-hindered diynes can modify the selectivity of alkyne metathesis to favour the formation of the desired triynes. Therefore, for the first time, the synthesis of symmetrical and dissymmetrical conjugated triynes by self- and cross-metathesis was successfully achieved. By involving an efficient molybdenum benzylidyne complex,19 remarkable triyne:diyne ratios were reached (up to >95:5) affording expected triynes in moderate to good yields (up to 86%). These pioneer results pave the way to further developments with the quest for more efficient alkyne-metathesis complexes demonstrating higher selectivity and productivity.20
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the CNRS, the Ecole Nationale Supérieure de Chimie de Rennes (grant to RM and AC) and the Ministère de l’Enseignement Supérieur, de la Recherche et de l’Innovation (grant to RM and AQ). This work was also supported by the region Bretagne (SAD 2016 No. 9639 – RZSELECT; grant to DSM) and the FASO (grant to IC and DSM). Prof. A. Fürstner is acknowledged for the generous gift of molybdenum benzylidyne complex. We are grateful to Elsa Caytan and the PRISM core facility (Biogenouest©, UMS, Biosit, Université de Rennes 1) for quantitative 13C NMR experiences.Notes and references
- For a recent review, see: A. L. K. Shi Shun and R. R. Tykwinski, Angew. Chem., Int. Ed., 2006, 45, 1034 CrossRef PubMed.
- Y. Wang, Q.-F. Liu, J.-J. Xue, Y. Zhou, H.-C. Yu, S.-P. Yang, B. Zhang, J.-P. Zuo, Y. Li and J.-M. Yue, Org. Lett., 2014, 16, 2062 CrossRef CAS PubMed.
- S. C. Cascon, W. B. Mors, B. M. Tursch, R. T. Aplin and L. J. Durham, J. Am. Chem. Soc., 1965, 87, 5237 CrossRef CAS PubMed.
- For a recent review, see: W. Shi and A. Lei, Tetrahedron Lett., 2014, 55, 2763 CrossRef CAS.
- For a recent review, see: M. Jevric and M. B. Nielsen, Asian J. Org. Chem., 2015, 4, 286 CrossRef CAS.
- (a) E. Jahnke and R. R. Tykwinski, Chem. Commun., 2010, 46, 3235 RSC; (b) W. A. Chalifoux and R. R. Tykwinski, Nat. Chem., 2010, 2, 967 CrossRef CAS PubMed; (c) E. Métay, Q. Hu and E. Negishi, Org. Lett., 2006, 8, 5773 CrossRef PubMed; (d) R. Decicco, A. Black, L. Li and N. S. Goroff, Eur. J. Org. Chem., 2012, 4699 CrossRef CAS.
- K. Azyat, E. Jahnke, T. Rankin and R. R. Tykwinski, Chem. Commun., 2009, 433 RSC.
- For recent reviews, see: (a) A. Fürstner, Angew. Chem., Int. Ed., 2013, 52, 2794 CrossRef PubMed; (b) H. Ehrhorn and M. Tamm, Chem.–Eur. J., 2019, 25, 3190 CAS; (c) A. Fürstner, Handbook of Metathesis, ed. R. H. Grubbs, A. G. Wnezel, D. J. O'Leary and E. Khosravi, 2nd edn, Wiley-VCH, Weinheim, Germany, 2015, ch. 6 Search PubMed.
- S. Lysenko, J. Volbeda, P. G. Jones and M. Tamm, Angew. Chem., Int. Ed., 2012, 51, 6757 CrossRef CAS PubMed.
- S. T. Li, T. Schnabel, S. Lysenko, K. Brandhorst and M. Tamm, Chem. Commun., 2013, 49, 7189 RSC.
- T. M. Schnabel, D. Melcher, K. Brandhorst, D. Bockfeld and M. Tamm, Chem.–Eur. J., 2018, 24, 9022 CrossRef CAS PubMed.
- (a) F. Ungeheuer and A. Fürstner, Chem.–Eur. J., 2015, 21, 11387 CrossRef CAS PubMed; (b) S. Schaubach, K. Gebauer, F. Ungeheuer, L. Hoffmeister, M. K. Ilg, C. Wirtz and A. Fürstner, Chem.–Eur. J., 2016, 22, 8494 CrossRef CAS PubMed For pioneer development of Mo-alkylidyne Cat-2, see: (c) J. Heppekausen, R. Stade, R. Goddard and A. Fürstner, J. Am. Chem. Soc., 2010, 132, 11045 CrossRef CAS PubMed; (d) J. Heppekausen, R. Stade, A. Kondoh, G. Seidel, R. Goddard and A. Fürstner, Chem.–Eur. J., 2012, 18, 10281 CrossRef CAS PubMed.
- We previously reported that sterically-hindered alkynes could promote high regioselectivity in asymmetric conjugate addition of enynones, see: M. Tissot, D. Poggiali, H. Hénon, D. Müller, L. Guénée, M. Mauduit and A. Alexakis, Chem.–Eur. J., 2012, 18, 8731 CrossRef CAS PubMed.
- N. Kerisit, R. Ligny, E. S. Gauthier, J.-P. Guegan, L. Toupet, J.-C. Guillemin and Y. Trolez, Helv. Chim. Acta, 2019, 102, e1800232 CrossRef.
- V. Fiandanese, D. Bottalico, G. Marchese and A. Punzi, Tetrahedron, 2006, 62, 5126 CrossRef CAS.
- Due to their higher molecular weight, all metathesis reactions with 13d were monitored by quantitative 13C-NMR spectroscopy.
- Tetrayne 19 resulted from the self-metathesis of triyne 17.
- T. J. Katz and J. McGinnis, J. Am. Chem. Soc., 1975, 97, 1592 CrossRef CAS.
- (a) W. Zhang, S. Kraft and J. S. Moore, J. Am. Chem. Soc., 2004, 126, 329 CrossRef CAS PubMed; (b) D. E. Gross and J. S. Moore, Macromolecules, 2011, 44, 3685 CrossRef CAS; (c) P. Persich, J. Llaveria, R. Lhermet, T. de Haro, R. Stade, A. Kondoh and A. Fürstner, Chem.–Eur. J., 2013, 19, 13047 CrossRef CAS PubMed.
- (a) M. Koy, I. Elser, J. Meisner, W. Frey, K. Wurst, J. Kästner and M. R. Buchmeiser, Chem.–Eur. J., 2017, 23, 15484 CrossRef CAS PubMed; (b) J. Hillenbrand, M. Leutzsch and A. Fürstner, Angew. Chem., Int. Ed., 2019, 58, 15690 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR spectra. CCDC 1908076, 1908077, 1966970, 1908075, 1915266 and 1920652. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc01124j |
| ‡ Present address: LCC-CNRS, Université de Toulouse, CNRS, UPR, Toulouse, France. |
| This journal is © The Royal Society of Chemistry 2020 |