
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploiting single-electron transfer in Lewis pairs for catalytic bond-forming reactions†
Yoshitaka
Aramaki
 a,
Naoki
Imaizumi
a,
Mao
Hotta
a,
Jun
Kumagai
b and
Takashi
Ooi
*ac
a,
Naoki
Imaizumi
a,
Mao
Hotta
a,
Jun
Kumagai
b and
Takashi
Ooi
*ac
aInstitute of Transformative Bio-Molecules (WPI-ITbM), Department of Molecular and Macromolecular Chemistry, Graduate School of Engineering, Nagoya University, Nagoya 464-8601, Japan. E-mail: tooi@chembio.nagoya-u.ac.jp
bInstitute of Materials and Systems for Sustainability, Nagoya University, Nagoya 464-8601, Japan
cCREST, Japan Science and Technology Agency (JST), Nagoya University, Nagoya 464-8601, Japan
First published on 20th March 2020
Abstract
A single-electron transfer (SET) between tris(pentafluorophenyl)borane (B(C6F5)3) and N,N-dialkylanilines is reported, which is operative via the formation of an electron donor–acceptor (EDA) complex involving π-orbital interactions as a key intermediate under dark conditions or visible-light irradiation depending on the structure of the aniline derivatives. This inherent SET in the Lewis pairs initiates the generation of the corresponding α-aminoalkyl radicals and their additions to electron-deficient olefins, revealing the ability of B(C6F5)3 to act as an effective one-electron redox catalyst.
Introduction
Since Gilbert N. Lewis formulated the two-electron process between an electron–pair acceptor and donor, termed the Lewis acid and base, respectively in 1923,1 the concept of Lewis pairs has been regarded as one of the most fundamental principles in chemical science. Primarily, the chemistry of Lewis pairs has been understood and developed within a direct two-electron transfer manifold to form a dative-bonded adduct (Lewis acid–base adduct or Lewis adduct). The formation of a coordination bond leads to the activation of both the Lewis acid and base, which has been exploited in various fields of chemistry, especially in synthetic chemistry, exemplified by Lewis acid catalysis involving electrophilic activation of carbonyl compounds for selective bond-forming reactions.2 Meanwhile, indirect two-electron transfer between Lewis pairs is operative in the arena of frustrated Lewis pairs (FLPs).3 In this process, Lewis acids and bases can not form a conventional Lewis adduct due to steric congestion, and thus, the resultant encounter complex acquires the capability of activating small molecules, such as dihydrogen, in a cooperative manner. On the other hand, single-electron transfer (SET) from a Lewis base to a Lewis acid to generate, in principle, a pair of a Lewis base-derived radical cation and a Lewis acid-derived radical anion has been invoked since the 1960s,4 and recent seminal studies have uncovered that SET is a viable mechanism for the reactions of frustrated and conventional Lewis pairs.5 However, the operation of this SET mechanism is limited to specific Lewis pairs, and despite its significant potential as a general means for the generation of radical–ion pairs as a reactive species, its utility in organic synthesis and catalysis remains elusive.6 This is probably due to an insufficient understanding of the possible intermediate and/or transition states of the SET process, particularly in FLPs, while the resulting radical ions have been directly observed and characterized by taking advantage of their stability owing to the steric and electronic nature pertinent to slowing back-electron transfer (BET).Under these circumstances, we paid our attention to the underlying similarity between FLPs and electron donor–acceptor (EDA) complexes (or charge-transfer (CT) complexes) as precursors of radical–ion pairs,7 considering that not only π-acceptors and donors but also σ-acceptors such as Br2,8 I2,9 NO+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 and hypervalent iodine compounds,11 and σ-donors such as cyclic alkylamines11c,12 serve as partners for EDA complexes. Upon complexation, an electron donor and an acceptor are weakly associated without the formation of a coordination bond, within an appropriate distance to realize orbital interactions for undergoing an internal SET, which could be regarded as a form of an encounter complex proposed in FLP chemistry.13 We envisaged that this interpretation of the mode of molecular association in the encounter complex could provide a clue for the understanding and generalization of the SET in Lewis pairs, which would be beneficial for its broad exploitation, specifically in the development of one-electron-mediated catalysis relevant to organic synthesis. Herein, we demonstrate that an SET between a common Lewis acid, tris(pentafluorophenyl)borane (B(C6F5)3), and simple N,N-dialkylanilines operates through the formation of an EDA complex as a key intermediate under dark conditions or visible-light irradiation depending on the structure of the aniline derivative. This inherent SET initiates the generation of the corresponding α-aminoalkyl radical and its addition to electron-deficient olefins, thereby revealing the ability of B(C6F5)3 to act as an effective one-electron redox catalyst.7b,14
10 and hypervalent iodine compounds,11 and σ-donors such as cyclic alkylamines11c,12 serve as partners for EDA complexes. Upon complexation, an electron donor and an acceptor are weakly associated without the formation of a coordination bond, within an appropriate distance to realize orbital interactions for undergoing an internal SET, which could be regarded as a form of an encounter complex proposed in FLP chemistry.13 We envisaged that this interpretation of the mode of molecular association in the encounter complex could provide a clue for the understanding and generalization of the SET in Lewis pairs, which would be beneficial for its broad exploitation, specifically in the development of one-electron-mediated catalysis relevant to organic synthesis. Herein, we demonstrate that an SET between a common Lewis acid, tris(pentafluorophenyl)borane (B(C6F5)3), and simple N,N-dialkylanilines operates through the formation of an EDA complex as a key intermediate under dark conditions or visible-light irradiation depending on the structure of the aniline derivative. This inherent SET initiates the generation of the corresponding α-aminoalkyl radical and its addition to electron-deficient olefins, thereby revealing the ability of B(C6F5)3 to act as an effective one-electron redox catalyst.7b,14
Result and discussion
At the outset of our study, we selected the commonly used B(C6F5)3 as the Lewis acid because of its ability to oxidize organic molecules5b–e and N-trimethylsilylmethylaniline derivative 1a as the Lewis base, considering its low oxidation potential as well as the susceptibility of the corresponding radical cation to irreversibly release a trimethylsilyl cation (TMS+) to generate an α-aminomethyl radical.15 Upon mixing equimolar amounts of 1a and freshly sublimed B(C6F5)3 in CH2Cl2 at room temperature, the solution colour immediately changed from colourless to blue green, and the UV-vis absorption spectrum exhibited a local absorption maximum at 648 nm with a broad shoulder (Fig. 1b). This spectrum was in good agreement with that of a mixture of 1a and AgBArf (BArf = B(3,5-(CF3)2C6H3)4), suggesting that one-electron oxidation of 1a by B(C6F5)3 had occurred. Electron spin resonance (ESR) spectroscopy experiments allowed the unambiguous assignment of the radical species as 1a˙+ by comparison with the simulated spectrum of one 14N atom, three 1H atoms in the methyl group, two 1H atoms in methylene, two 1H atoms at the ortho-position, two 1H atoms at the meta-position, and one 29Si atom with a g factor of 2.0033 > ge (Fig. 1c; see also Table S1 in the ESI† for details of the assignment). The generation of another possible radical, the neutral α-aminomethyl radical A, via the release of TMS+ from 1a˙+ was limited to an undetectable extent, judging from the comparison with the simulated spectrum of A, in which its spin density was localized on a methylene carbon atom (Fig. S6†). The stability of 1a˙+ likely stemmed from the hyper-conjugation effect of the silicon–carbon bond, contributing to the stabilization of the radical cation centre.15c,q,16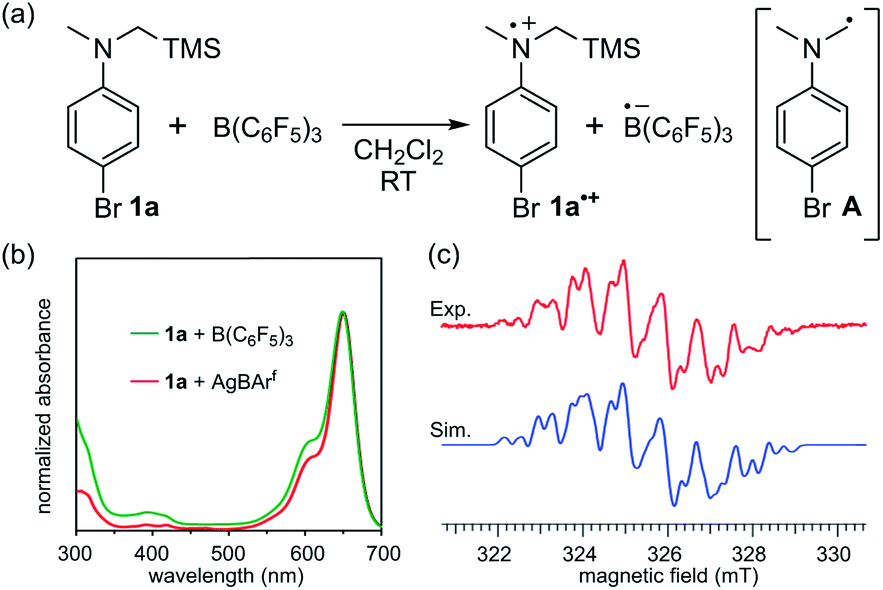 | ||
| Fig. 1 (a) SET from 1a to B(C6F5)3. (b) UV-vis absorption spectra of the mixture of B(C6F5)3 and 1a (green), and AgBArf and 1a (red). (c) Experimentally obtained ESR spectrum of the mixture in CH2Cl2 (red) and a simulated spectrum of 1a˙+ (blue). | ||
On the basis of the initial observations, we next employed para-bromo-N,N-dimethylaniline (2) as a more common, readily available Lewis base.17 In this case, an equimolar mixture of B(C6F5)3 and 2 in CH2Cl2 gave a colourless solution, the ESR analysis of which confirmed that no signal was detected. Intriguingly, however, the solution rapidly turned bright blue green upon irradiation with a 405 nm LED light source. The UV-vis absorption spectrum exhibited a characteristic absorption maximum at 613 nm with a shoulder (Fig. 2b, green), similar to that observed in the spectrum of 1a˙+ (Fig. 1b). This peak was in very good agreement with that of 2˙+ generated separately by the one-electron oxidation of 2 with AgSbF6 (Fig. 2b, red). ESR measurements under irradiation provided a well-resolved spectrum of 2˙+.18 The eight-fold-integrated spectrum after 1 h of irradiation (Fig. 2c, red) could be assigned to 2˙+, as it was in good agreement with the simulated spectrum (Fig. 2c, blue) of one 14N atom, six 1H atoms in two methyl groups, two 1H atoms at the ortho-position, and two 1H atoms at the meta-position with a g factor of 2.0029 > ge (Table S2†). The saturation of signal intensity after 18 min of irradiation (Fig. 2d) implied a reversible equilibrium for the generation of the radical–ion pair, as illustrated in Fig. 2a. While a signal corresponding to the radical anion B(C6F5)3˙− was not observed,5b–e the rapid attenuation of the signal of 2˙+ upon interruption of irradiation suggested the intervention of a BET process from the pairing radical anion, considering that the ESR signal of [2˙]+[SbF6]− in CH2Cl2 showed no decay during this time interval (Fig. S7b and c†). In addition, we confirmed that the signal reappeared immediately upon resuming light irradiation (Fig. 2d).
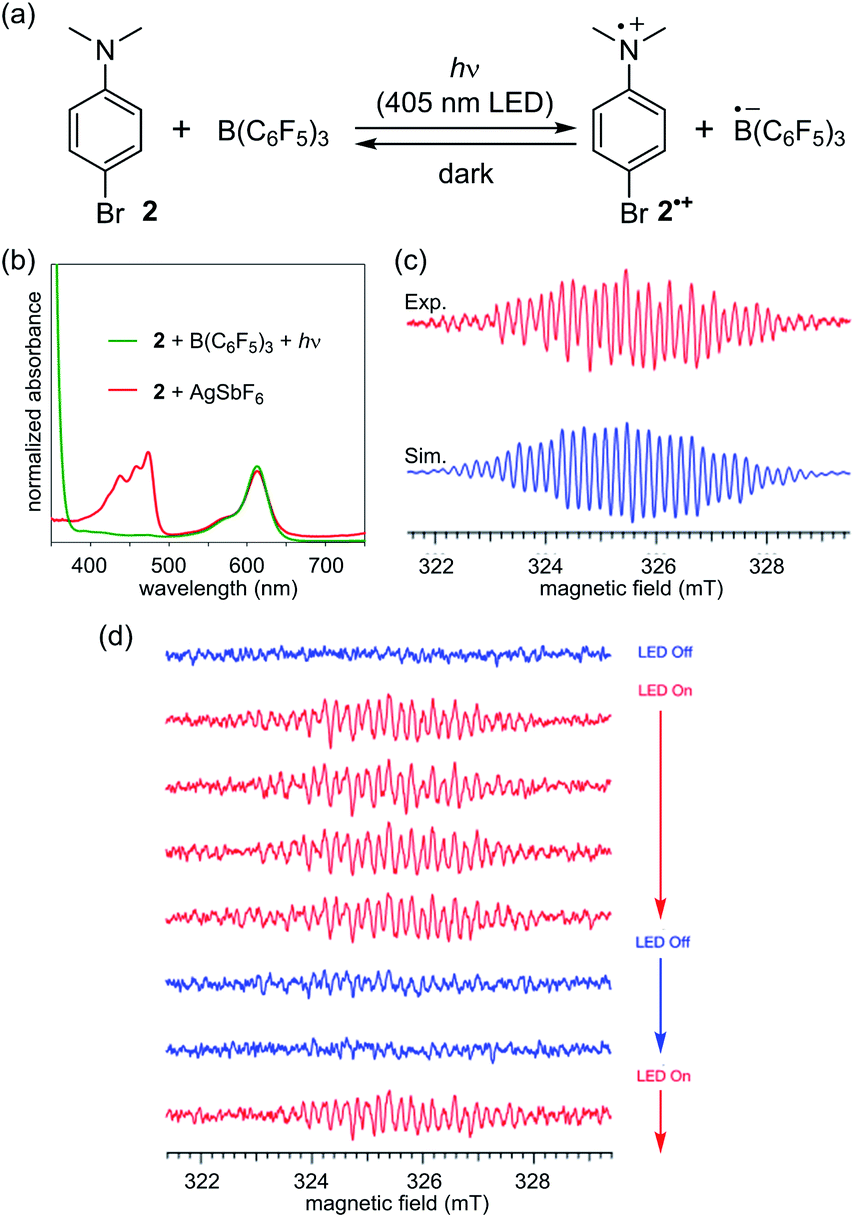 | ||
| Fig. 2 (a) Photoinduced reversible SET between B(C6F5)3 and 2. (b) UV-vis spectra of a 1:1 mixture of B(C6F5)3 and 2 after LED light irradiation (green) and AgSbF6 and 2 (red). (c) ESR spectrum after 1 h of irradiation with a LED light (red) and a simulated spectrum (blue). (d) LED on/off ESR monitoring experiment of B(C6F5)3 and 2 in CH2Cl2. The interval of each measurement is 9 min. | ||
The outcome of these investigations lead to two important considerations: (1) the origin of the difference in reactivity between 1a and 2 and (2) the role of the 405 nm light irradiation in the SET to generate a radical–ion pair [2˙]+[B(C6F5)3˙]−. The higher reactivity of 1a can be primarily accounted for by its lower oxidation potential compared to that of 2 (1a: 0.23 V, 2: 0.50 V vs. Fc/Fc+, Fig. S8†), as expected, which originates from the σ-donating effect of the C–Si bond to raise the HOMO level.16a,c In addition, the difference in the relative BET rates would be critical. We reasoned that radical cation 1a˙+ is stabilized by the β-effect of the silyl group, rendering the BET from the paired B(C6F5)3˙− slower than that in [2˙]+[B(C6F5)3˙]−. Owing to the higher energy barrier for SET and the faster BET, external energy (photoirradiation) is essential for 2 to undergo one-electron oxidation by B(C6F5)3 to generate 2˙+ in a detectable concentration. This understanding was supported by DFT calculations, which indicated that the difference in the Gibbs free energy between 2 and 2˙+ was 4.2 kcal mol−1 higher than that between 1a and 1a˙+ (see the ESI† for details of the calculation).
Notwithstanding, no absorption band was detected at approximately 405 nm in the respective absorption spectra of B(C6F5)3 and 2 (Fig. S2†), indicating that direct excitation of B(C6F5)3 and 2 is not feasible with 405 nm light. It is important to note, however, that a mixture of B(C6F5)3 and 2 exhibited very weak absorption above 405 nm (Fig. 3a), which suggested a constitutive intermolecular association between B(C6F5)3 and 2. Fortunately, an orange crystal suitable for X-ray crystallography was obtained from a pentane solution of the mixture cooled to −35 °C in an argon-purged glovebox. Single-crystal X-ray diffraction analysis revealed the three-dimensional structure of a 1:1 co-crystal (Fig. 3b), where B(C6F5)3 and 2 were alternately aligned along the a-axis with face-to-face packing between a C6F5 moiety of B(C6F5)3 and an aromatic ring of 2 (Fig. 3c). In this association, B(C6F5)3 and 2 were frustrated with the boron centre and the dimethylamino moiety, being oriented opposite to each other, and no Lewis adduct was formed. The average distance between the plane of 2 and the six carbon atoms of the C6F5 ring that constructs the columnar structure was 3.38 Å, which is close to that of the inner-sphere EDA complex (rDA ≈ 3.1 ± 0.2 Å).19 Upon further examining the conformation of B(C6F5)3, the C6F5 ring involved in the columnar structure was closer to coplanar with the sp2 hybridized boron centre (dihedral angles of C15–B1–C9–C14 and C21–B1–C9–C10 were −17.7(6)° and −15.0(6)°, respectively) compared to the other two C6F5 rings to effectively achieve the overlap of frontier orbitals with 2 (Fig. 3d, vide infra for further discussion). Furthermore, bond alternation was observed in the aniline component of 2. The N1–C3 bond length was 1.366(6) Å, and the C4–C5 and C7–C8 bond lengths were 1.372(6) and 1.368(6) Å, respectively, which were closer to a carbon–carbon double bond length than to the carbon–carbon bond length of benzene (Table S4†). These trends were similar to those reported for the co-crystal of N,N-dimethylaniline and electron-deficient hexafluorobenzene,20 indicating the presence of charge-transfer interactions. To corroborate the charge-transfer characteristics, TD-DFT calculations were conducted for the structure of a single unit of the intermolecular complex in the co-crystal (anti-complex). The lowest transition energy of this complex was calculated to be 2.74 eV, corresponding to an absorption at 455 nm, which is consistent with the observation that the crystal was orange in colour and the broad absorption band when present in solution (Fig. 3a). This excitation was assigned to the electronic transition from the HOMO of 2 to the LUMO of B(C6F5)3 (Fig. 3d), and the LUMO, which comprises a π* orbital of C6F5 and a p* orbital of boron because of the conformational coplanarity, is effectively overlapped with the HOMO of 2. This attribute appeared to be independent of the geometry of the complex, as further TD-DFT calculations for the complex with the opposite orientation of 2 (syn-complex), where the dimethylamino moiety was located close to the boron centre, indicated analogous absorption (448 nm, f = 0.02) and charge-transfer characteristics (Fig. S9†). These results suggest that the SET proceeds through the formation of an EDA complex that becomes excited upon 405 nm light irradiation, and that the possibility of a mechanism involving the homolytic cleavage of the B–N coordination bond by photoirradiation is unlikely. This is in accordance with the fact that B–N coordination bond formation between B(C6F5)3 and 2 (Lewis adduct) was not detected by 1H and 11B NMR spectroscopy, even at −90 °C (Fig. S17 and S18†), and that no absorption at 405 nm was derived from the TD-DFT calculations for the Lewis adduct (Fig. S13†). These analyses clarified the role of Lewis acid B(C6F5)3 as a π-acceptor and that of Lewis base 2 as a π-donor in this system for photoinduced SET.
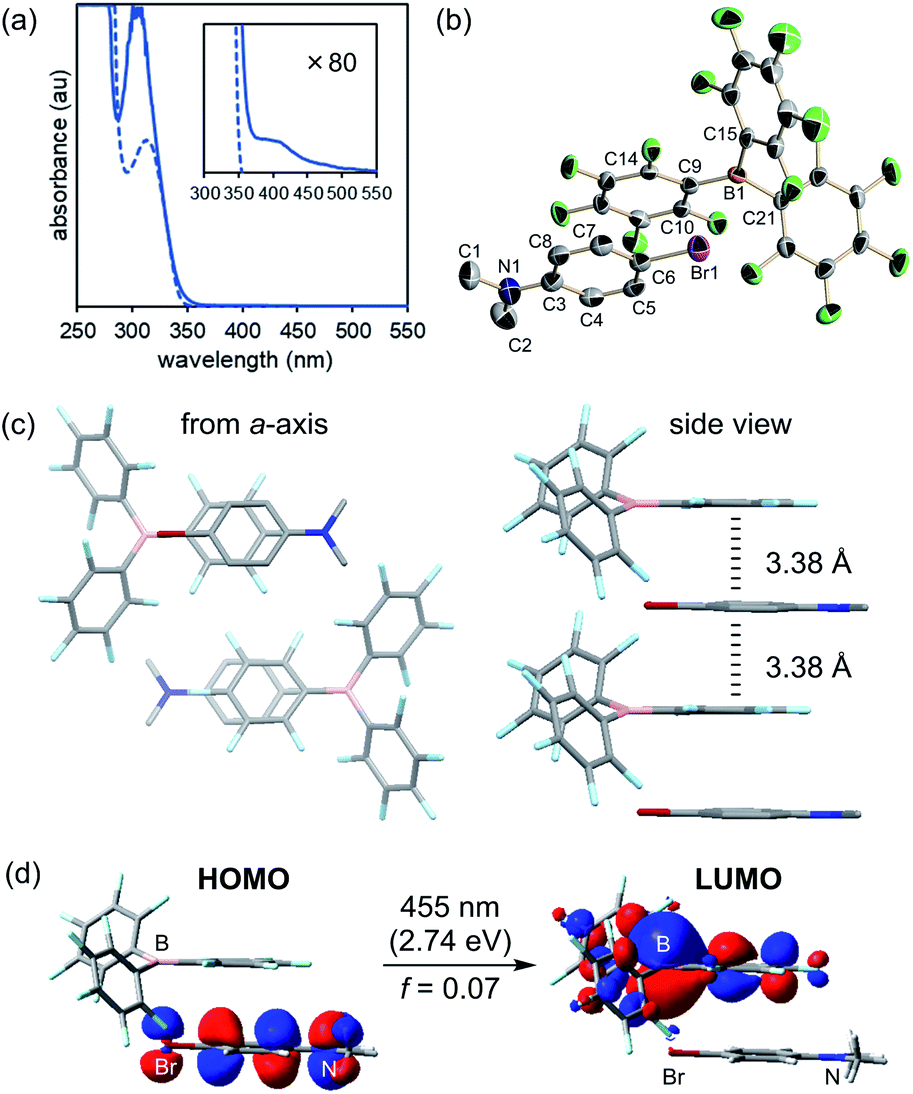 | ||
| Fig. 3 (a) UV-vis spectra of a 1:1 mixture of B(C6F5)3 and 2 (solid line), and 2 (dashed line) for 1.0 × 10−3 M with magnified spectra (inner square). (b) X-ray structure of a co-crystal of B(C6F5)3 and 2. (c) Packing structure of the co-crystal of B(C6F5)3 and 2. (d) TD-DFT calculated minimum excitation of the single complex in crystal structure (CAM-B3LYP/6-311+G(d,p)). | ||
Based on these fundamental findings, we envisioned that this unique SET process could potentially be utilized as an elementary step for effecting synthetically relevant transformations. Considering that the radical cations 1a˙+ and 2˙+ are a precursor of the nucleophilic α-aminomethyl radical,15,17 we inferred that they could be trapped by electron-deficient olefins to forge a carbon–carbon bond, providing a basis for further investigation. Thus, an excess amount of methyl vinyl ketone (3a) was added as a radical acceptor, initially, to a mixture of 1a and a catalytic quantity of B(C6F5)3 (10 mol%) in CH2Cl2. The expected bond formation indeed occurred and the corresponding radical addition product 4a was obtained in 31% yield after a standard acidic work-up and purification (Table 1, entry 1). While the use of Et2O as a solvent led to a slight improvement in chemical yield (entry 2), the efficiency was much affected by the difference in the oxidation potential of 1, as the reaction of 1b under similar conditions afforded the product 4b in 66% yield (1a: 0.23 V, 1b: 0.10 V vs. Fc/Fc+, Fig. S8†) (entry 3). However, the reactivity was still insufficient and thus, we monitored the reaction in THF-d8 by 1H NMR spectroscopy to detect possible intermediates.21 Contrary to our assumption,15e a 4b-derived TMS enol ether was not detected over the course of the reaction, and 4b was consistently observed before the acidic work-up (Fig. S15†). This profile suggested that a reaction step involving an NMR innocent species was turn-over limiting, and it could be the desilylation from 1˙+ that was a major paramagnetic species in the ESR analysis (Fig. 1c). Moreover, after the addition of the resulting α-aminomethyl radical to 3a, the transient α-carbonyl radical would undergo one-electron reduction by B(C6F5)3˙− to form an enolate ion that is protonated in situ by a trace amount of H2O or 3a. These considerations and the previous report on the effect of protic solvents for accelerating the desilylation from α-silyl amine radical cations15a–c prompted us to add MeOH primarily as a TMS trapping reagent and also as a proton source (Et2O/MeOH = 10/1), which resulted in a dramatic increase in reactivity to afford 4b in 92% yield even with reduced amounts of 3a (entry 4). In parallel, the reaction was performed in Et2O/MeOD (10/1), giving rise to 4b-d1 in 83% yield with 74% incorporation of deuterium at the internal α-position of the keto carbonyl group (Scheme 1), and no H–D exchange of isolated 4b was observed in the presence of B(C6F5)3 and MeOD in Et2O (Scheme S1†). These results strongly support the intermediacy of the enolate ion and its predominant protonation by MeOH.
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | Solvent | X | Time (h) | Yieldb (%) |
| a All reactions were performed in test tubes with septum caps and wrapped with aluminum foil in order to exclude the effect of room light irradiation with 0.1 mmol of 1 and 3a in a solvent (1 mL) in the presence of 10 mol% of B(C6F5)3 under an Ar atmosphere at room temperature. b Isolated yield. | |||||
| 1 | Br (1a) | CH2Cl2 | 10 | 38 | 31 (4a) |
| 2 | Br (1a) | Et2O | 10 | 38 | 37 (4a) |
| 3 | Me (1b) | Et2O | 10 | 38 | 66 (4b) |
| 4 | Me (1b) | Et2O/MeOH (10/1) | 3 | 16 | 92 (4b) |

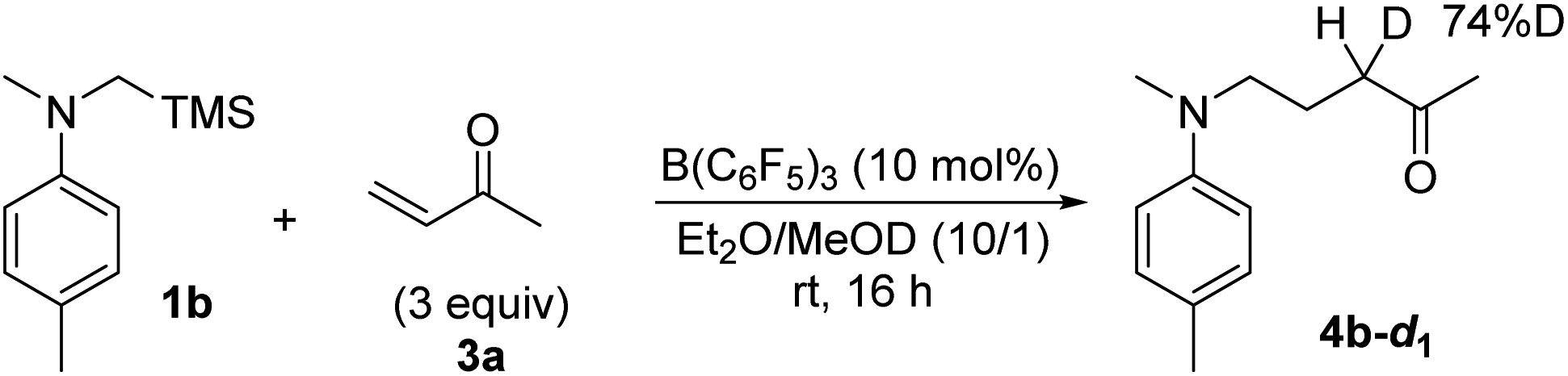 | ||
| Scheme 1 Deuterium incorporation experiment. | ||
We then moved on to an examination of the reaction between 2 and 3. In this case, treatment of a mixture of 2 and 3a with B(C6F5)3 (10 mol%) in 1,2-dichloroethane (DCE) at room temperature for 36 h showed no product formation. However, the reaction irradiated with a 405 nm LED under otherwise identical conditions gave 4a in 31% yield (Scheme 2). These observations suggested the operation of one-electron redox catalysis of B(C6F5)3 under photoirradiation.
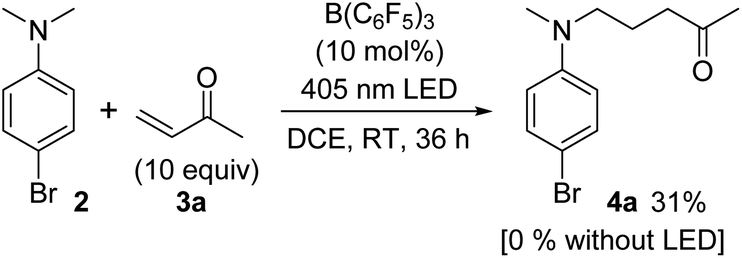 | ||
| Scheme 2 Trapping experiment of α-aminomethyl radical generated from 2 with 3a. | ||
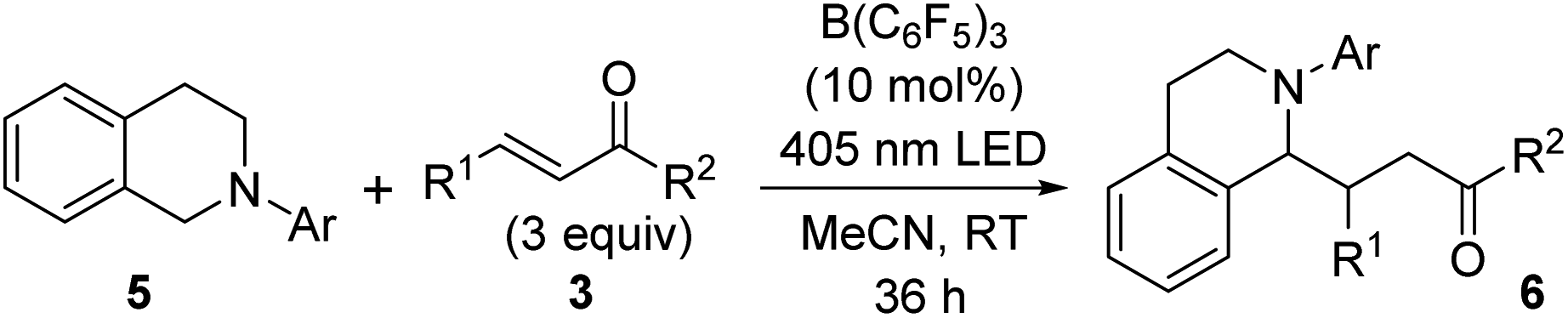
As a more suitable reaction platform for verifying this notion, we selected the coupling of N-aryltetrahydroisoquinolines 5 with α,β-unsaturated ketones, which is known to be promoted by a common photoredox catalyst.22 An initial attempt was made by irradiating a solution of N-phenyltetrahydroisoquinoline (5a) and 3a (3.0 equiv.) in DCE under the influence of B(C6F5)3 (10 mol%). This resulted in the formation of the α-coupling product 6a in 31% yield (Table S5†), and switching the solvent to acetonitrile (MeCN) delivered an improvement in the chemical yield (70%) (Table 2, entry 1). It should be noted that bond formation did not occur without light irradiation23 (entry 2), and only a trace amount of 6a was obtained in the absence of the catalyst (entry 3). In addition, the use of BPh3 as a catalyst significantly ruined the reactivity profile (entry 4), and BF3·OEt2 was ineffective (entry 5), indicating that the electron-deficient C6F5 groups are crucial for exerting sufficient catalytic activity. This information bears relevance when accounting for the electronic effect of the aryl group attached to the nitrogen atom of 5 on reaction efficiency. When para-methoxyphenyl-substituted 5b was employed as a donor component, the coupling product 6b was isolated in a higher yield (90%) (entry 6), whereas the introduction of a para-bromophenyl substituent (5c) led to a slight decrease in reactivity (entry 7). The steric demand of the aromatic appendage was also critical, as no evidence of product formation was detected with 5d bearing an ortho-tolyl group on the nitrogen (entry 8). These results support the fact that the facile formation of the EDA complex between the N-aryl moiety of 5 and B(C6F5)3 would be essential for the present catalysis. In fact, the formation of EDA complexes with 5a and 5b, but not with 5d, was suggested by UV-vis absorption spectroscopy, and the TD-DFT calculation for the complex of 5a with B(C6F5)3 also supported the charge-transfer characteristics (Fig. S4 and S11,† respectively). With respect to radical acceptors, not only simple vinyl ketones but also other enones, such as phenyl 1-propenyl ketone (3c), were tolerated (entries 9 and 10). On the other hand, less reactive acceptors, such as methyl acrylate and styrene derivatives, were not amenable to this catalytic system. Although we recognize that it is difficult to completely rule out the involvement of a radical-chain process,22b,24,26,27 the overall nature of this catalysis reflects the oxidation ability of B(C6F5)3 and a redox-neutral catalytic cycle can be operative through the transient generation of radical–ion pairs.
|
|
||||
|---|---|---|---|---|
| Entry | Ar | R1 | R2 | Yieldb (%) (dr) |
| a Unless otherwise noted, the reactions were performed with 0.1 mmol of 5 and 0.3 mmol of 3 in MeCN (1 mL) in the presence of 10 mol% of B(C6F5)3 at room temperature under 405 nm LED irradiation under an Ar atmosphere. b Isolated yield. c No LED irradiation. d Without B(C6F5)3. e With BPh3 instead of B(C6F5)3 as a catalyst. f With BF3·OEt2 instead of B(C6F5)3 as a catalyst. g Determined by 1H NMR analysis. | ||||
| 1 | Ph (5a) | H | Me (3a) | 70 (6a) |
| 2c | Ph (5a) | H | Me (3a) | 0 (6a) |
| 3d | Ph (5a) | H | Me (3a) | <5 (6a) |
| 4e | Ph (5a) | H | Me (3a) | 9 (6a) |
| 5f | Ph (5a) | H | Me (3a) | 0 (6a) |
| 6 | p-MeOC6H4 (5b) | H | Me (3a) | 90 (6b) |
| 7 | p-BrC6H4 (5c) | H | Me (3a) | 61 (6c) |
| 8 | o-MeC6H4 (5d) | H | Me (3a) | 0 (6d) |
| 9 | Ph (5a) | H | Et (3b) | 70 (6e) |
| 10 | p-MeOC6H4 (5b) | Me | Ph (3c) | 76 (1.1:1)g (6f) |
Conclusions
We have demonstrated the operation of an SET in Lewis pairs between B(C6F5)3 and simple N,N-dialkylanilines under dark or photoirradiation conditions depending on the structure of the aniline derivatives, which was verified by UV-vis and ESR spectroscopic analyses. The key intermediate of this unique SET process was revealed to be an EDA complex involving π-orbital interactions by using absorption spectra, X-ray crystallographic analysis, and DFT calculations. Furthermore, we have shown that these fundamental findings can be exploited for the development of the redox catalysis of B(C6F5)3 for synthetically relevant carbon–carbon bond formation. We anticipate that this study opens a door to a new avenue toward the understanding and exploitation of the reactivity and selectivity of radical–ion pairs generated from Lewis pairs in organic synthesis and catalysis within a single-electron transfer manifold.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is funded by the CREST-JST (JPMJCR13L2: 13418441) and Grants of JSPS for Scientific Research (KAKENHI, 19H00894 and 19K15538). We are grateful to Prof. D. Yokogawa (the University of Tokyo) for his support for computational studies.Notes and references
- G. N. Lewis, Valence and the Structure of Atoms and Molecules, The Chemical Catalogue Company, New York, 1923 Search PubMed.
- H. Yamamoto, Lewis Acids in Organic Synthesis, Wiley-VCH, Weinheim, 2000 Search PubMed.
- (a) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS PubMed; (b) D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 10018–10032 CrossRef CAS PubMed; (c) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed; (d) A. R. Jupp and D. W. Stephan, Trends Chem., 2019, 1, 35–48 CrossRef.
- (a) W. F. Forbes, P. D. Sullivan and H. M. Wang, J. Am. Chem. Soc., 1967, 89, 2705–2711 CrossRef; (b) H. van Willigen, J. Am. Chem. Soc., 1967, 89, 2229–2230 CrossRef CAS; (c) F. A. Bell, A. Ledwith and D. C. Sherrington, J. Chem. Soc. C, 1969, 2719–2720 RSC; (d) W. Schmidt and E. Steckhan, Chem. Ber., 1980, 113, 577–585 CrossRef CAS; (e) S. Dapperheld, E. Steckhan, K.-H. G. Brinkhaus and T. Esch, Chem. Ber., 1991, 124, 2557–2567 CrossRef CAS; (f) C. J. Harlan, T. Hascall, E. Fujita and J. R. Norton, J. Am. Chem. Soc., 1999, 121, 7274–7275 CrossRef CAS.
- (a) G. Ménard, J. A. Hatnean, H. J. Cowley, A. J. Lough, J. M. Rawson and D. W. Stephan, J. Am. Chem. Soc., 2013, 135, 6446–6449 CrossRef PubMed; (b) X. Zheng, X. Wang, Y. Qiu, Y. Li, C. Zhou, Y. Sui, Y. Li, J. Ma and X. Wang, J. Am. Chem. Soc., 2013, 135, 14912–14915 CrossRef CAS PubMed; (c) L. Liu, L. L. Cao, Y. Shao, G. Ménard and D. W. Stephan, Chem, 2017, 3, 259–267 CrossRef CAS; (d) Z. Dong, H. H. Cramer, M. Schmidtmann, L. A. Paul, I. Siewert and T. Müller, J. Am. Chem. Soc., 2018, 140, 15419–15424 CrossRef CAS PubMed; (e) L. L. Liu, L. L. Cao, D. Zhu, J. Zhou and D. W. Stephan, Chem. Commun., 2018, 54, 7431–7434 RSC; (f) A. Merk, H. Großekappenberg, M. Schmidtmann, M.-P. Luecke, C. Lorent, M. Driess, M. Oestreich, H. F. T. Klare and T. Müller, Angew. Chem., Int. Ed., 2018, 57, 15267–15271 CrossRef CAS PubMed; (g) Y. Kim, L. L. Liu and D. W. Stephan, Chem.–Eur. J., 2019, 25, 7110–7113 CrossRef CAS PubMed; (h) L. L. Liu and D. W. Stephan, Chem. Soc. Rev., 2019, 48, 3454–3463 RSC.
- During the preparation of this manuscript, an elegant work on the application of SET in PMes3 and B(C6F5)3 to C–C bond-forming reaction with a stoichiometric amount of the Lewis pair was reported. Y. Soltani, A. Dasgupta, T. A. Gazis, D. M. C. Ould, E. Richards, B. Slater, K. Stefkova, V. Y. Vladimirov, L. C. Wilkins, D. Willcox and R. L. Melen, Cell Reports Physical Science, 2020, 1, 100016 CrossRef.
- (a) R. S. Mulliken, J. Am. Chem. Soc., 1952, 74, 811–824 CrossRef CAS; (b) C. G. S. Lima, T. d. M. Lima, M. Duarte, I. D. Jurberg and M. W. Paixão, ACS Catal., 2016, 6, 1389–1407 CrossRef CAS.
- A. V. Vasilyev, S. V. Lindeman and J. K. Kochi, New J. Chem., 2002, 26, 582–592 RSC.
- U. M. Rabie, J. Mol. Struct., 2013, 1034, 393–403 CrossRef CAS.
- (a) E. K. Kim and J. K. Kochi, J. Am. Chem. Soc., 1991, 113, 4962–4974 CrossRef CAS; (b) S. V. Rosokha and J. K. Kochi, J. Am. Chem. Soc., 2001, 123, 8985–8999 CrossRef CAS PubMed.
- (a) T. Dohi, M. Ito, N. Yamaoka, K. Morimoto, H. Fujioka and Y. Kita, Angew. Chem., Int. Ed., 2010, 49, 3334–3337 CrossRef CAS PubMed; (b) N. Yamaoka, K. Sumida, I. Itani, H. Kubo, Y. Ohnishi, S. Sekiguchi, T. Dohi and Y. Kita, Chem.–Eur. J., 2013, 19, 15004–15011 CrossRef CAS PubMed; (c) H. Jiang, Y. He, Y. Cheng and S. Yu, Org. Lett., 2017, 19, 1240–1243 CrossRef CAS PubMed; (d) H.-Y. Tu, S. Zhu, F.-L. Qing and L. Chu, Chem. Commun., 2018, 54, 12710–12713 RSC.
- (a) S. C. Blackstock, J. P. Lorand and J. K. Kochi, J. Org. Chem., 1987, 52, 1451–1460 CrossRef CAS; (b) Y. Cheng, X. Yuan, J. Ma and S. Yu, Chem.–Eur. J., 2015, 21, 8355–8359 CrossRef CAS PubMed.
- (a) T. A. Rokob, A. Hamza, A. Stirling, T. Soós and I. Pápai, Angew. Chem., Int. Ed., 2008, 47, 2435–2438 CrossRef CAS PubMed; (b) T. A. Rokob, A. Hamza, A. Stirling and I. Pápai, J. Am. Chem. Soc., 2009, 131, 2029–2036 CrossRef CAS PubMed; (c) S. Grimme, H. Kruse, L. Goerigk and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 1402–1405 CrossRef CAS PubMed; (d) M. Pu and T. Privalov, J. Chem. Phys., 2013, 138, 154305 CrossRef PubMed; (e) L. Rocchigiani, G. Ciancaleoni, C. Zuccaccia and A. Macchioni, J. Am. Chem. Soc., 2014, 136, 112–115 CrossRef CAS PubMed.
- For seminal contributions to the exploitation of EDA-complex formation in controlling catalytic bond formations, see: (a) E. Arceo, I. D. Jurberg, A. Álvarez-Fernández and P. Melchiorre, Nat. Chem., 2013, 5, 750–756 CrossRef CAS PubMed; (b) A. Bahamonde and P. Melchiorre, J. Am. Chem. Soc., 2016, 138, 8019–8030 CrossRef CAS PubMed; (c) Z.-Y. Cao, T. Ghosh and P. Melchiorre, Nat. Commun., 2018, 9, 3274 CrossRef PubMed . For recent examples of catalysis via EDA-complex formation, see: ; (d) R. P. Shirk and S. S. V. Ramasastry, Org. Lett., 2017, 19, 5482–5485 CrossRef PubMed; (e) I. Bosque and T. Bach, ACS Catal., 2019, 9, 9103–9109 CrossRef CAS.
- (a) U. C. Yoon, J. U. Kim, E. Hasegawa and P. S. Mariano, J. Am. Chem. Soc., 1987, 109, 4421–4423 CrossRef CAS; (b) E. Hasegawa, W. Xu, P. S. Mariano, U. C. Yoon and J. U. Kim, J. Am. Chem. Soc., 1988, 110, 8099–8111 CrossRef CAS; (c) U. C. Yoon and P. S. Mariano, Acc. Chem. Res., 1992, 25, 233–240 CrossRef CAS; (d) D. W. Cho, U. C. Yoon and P. S. Mariano, Acc. Chem. Res., 2011, 44, 204–215 CrossRef CAS PubMed; (e) Y. Miyake, Y. Ashida, K. Nakajima and Y. Nishibayashi, Chem. Commun., 2012, 48, 6966–6968 RSC; (f) Y. Miyake, Y. Ashida, K. Nakajima and Y. Nishibayashi, Chem.–Eur. J., 2014, 20, 6120–6125 CrossRef CAS PubMed; (g) K. Nakajima, M. Kitagawa, Y. Ashida, Y. Miyake and Y. Nishibayashi, Chem. Commun., 2014, 50, 8900–8903 RSC; (h) D. Lenhart and T. Bach, Beilstein J. Org. Chem., 2014, 10, 890–896 CrossRef PubMed; (i) L. Ruiz Espelt, I. S. McPherson, E. M. Wiensch and T. P. Yoon, J. Am. Chem. Soc., 2015, 137, 2452–2455 CrossRef CAS PubMed; (j) K. Nakajima, Y. Ashida, S. Nojima and Y. Nishibayashi, Chem. Lett., 2015, 44, 545–547 CrossRef CAS; (k) C. Wang, Y. Zheng, H. Huo, P. Röse, L. Zhang, K. Harms, G. Hilt and E. Meggers, Chem.–Eur. J., 2015, 21, 7355–7359 CrossRef CAS PubMed; (l) D. Lenhart, A. Bauer, A. Pöthig and T. Bach, Chem.–Eur. J., 2016, 22, 6519–6523 CrossRef CAS PubMed; (m) S.-Y. Hsieh and J. W. Bode, Org. Lett., 2016, 18, 2098–2101 CrossRef CAS PubMed; (n) T. Kizu, D. Uraguchi and T. Ooi, J. Org. Chem., 2016, 81, 6953–6958 CrossRef CAS PubMed; (o) W. Ding, L.-Q. Lu, J. Liu, D. Liu, H.-T. Song and W.-J. Xiao, J. Org. Chem., 2016, 81, 7237–7243 CrossRef CAS PubMed; (p) Y. Zhao, J.-R. Chen and W.-J. Xiao, Org. Lett., 2016, 18, 6304–6307 CrossRef CAS PubMed; (q) C. Remeur, C. B. Kelly, N. R. Patel and G. A. Molander, ACS Catal., 2017, 7, 6065–6069 CrossRef CAS PubMed; (r) X. Shen, Y. Li, Z. Wen, S. Cao, X. Hou and L. Gong, Chem. Sci., 2018, 9, 4562–4568 RSC; (s) Y. Cai, Y. Tang, L. Fan, Q. Lefebvre, H. Hou and M. Rueping, ACS Catal., 2018, 8, 9471–9476 CrossRef CAS; (t) A. Casado-Sánchez, P. Domingo-Legarda, S. Cabrera and J. Alemán, Chem. Commun., 2019, 55, 11303–11306 RSC; (u) M. Grübel, C. Jandl and T. Bach, Synlett, 2019, 30, 1825–1829 CrossRef.
- (a) B. E. Cooper and W. J. Owen, J. Organomet. Chem., 1971, 29, 33–40 CrossRef CAS; (b) H. Bock and W. Kaim, Acc. Chem. Res., 1982, 15, 9–17 CrossRef CAS; (c) J. Yoshida, T. Maekawa, T. Murata, S. Matsunaga and S. Isoe, J. Am. Chem. Soc., 1990, 112, 1962–1970 CrossRef CAS.
- (a) A. McNally, C. K. Prier and D. W. C. MacMillan, Science, 2011, 334, 1114–1117 CrossRef CAS PubMed; (b) Y. Miyake, K. Nakajima and Y. Nishibayashi, J. Am. Chem. Soc., 2012, 134, 3338–3341 CrossRef CAS PubMed; (c) H. Zhou, P. Lu, X. Gu and P. Li, Org. Lett., 2013, 15, 5646–5649 CrossRef CAS PubMed; (d) C. Zhang, C. Liu, Y. Shao, X. Bao and X. Wan, Chem.–Eur. J., 2013, 19, 17917–17925 CrossRef CAS PubMed; (e) C. K. Prier and D. W. C. MacMillan, Chem. Sci., 2014, 5, 4173–4178 RSC; (f) A. Noble and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 11602–11605 CrossRef CAS PubMed; (g) X. Dai, D. Cheng, B. Guan, W. Mao, X. Xu and X. Li, J. Org. Chem., 2014, 79, 7212–7219 CrossRef CAS PubMed; (h) X. Dai, R. Mao, B. Guan, X. Xu and X. Li, RSC Adv., 2015, 5, 55290–55294 RSC; (i) D. Uraguchi, N. Kinoshita, T. Kizu and T. Ooi, J. Am. Chem. Soc., 2015, 137, 13768–13771 CrossRef CAS PubMed; (j) K. Nakajima, Y. Miyake and Y. Nishibayashi, Acc. Chem. Res., 2016, 49, 1946–1956 CrossRef CAS PubMed; (k) H. B. Hepburn and P. Melchiorre, Chem. Commun., 2016, 52, 3520–3523 RSC; (l) J. J. Murphy, D. Bastida, S. Paria, M. Fagnoni and P. Melchiorre, Nature, 2016, 532, 218 CrossRef CAS PubMed; (m) C. Wang, J. Qin, X. Shen, R. Riedel, K. Harms and E. Meggers, Angew. Chem., Int. Ed., 2016, 55, 685–688 CrossRef CAS PubMed; (n) E. Fava, A. Millet, M. Nakajima, S. Loescher and M. Rueping, Angew. Chem., Int. Ed., 2016, 55, 6776–6779 CrossRef CAS PubMed; (o) L. Li, T. Xiao, H. Chen and L. Zhou, Chem.–Eur. J., 2017, 23, 2249–2254 CrossRef CAS PubMed; (p) C.-W. Hsu and H. Sundén, Org. Lett., 2018, 20, 2051–2054 CrossRef CAS PubMed.
- The generation of paramagnetic species in the solution of B(C6F5)3 and N,N-dimethylaniline was described in W. E. Piers, Adv. Organomet. Chem., 2005, 52, 1–76 CAS ; however, the precise analysis of the paramagnetic species has not been achieved.
- S. V. Rosokha and J. K. Kochi, Acc. Chem. Res., 2008, 41, 641–653 CrossRef CAS PubMed.
- T. Dahl, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1985, 41, 931–933 CrossRef.
- Compound 4b was obtained in 57% yield with 10 equivalents of 3a when THF was used as solvent.
- (a) P. Kohls, D. Jadhav, G. Pandey and O. Reiser, Org. Lett., 2012, 14, 672–675 CrossRef CAS PubMed; (b) L. Ruiz Espelt, E. M. Wiensch and T. P. Yoon, J. Org. Chem., 2013, 78, 4107–4114 CrossRef CAS PubMed; (c) J. Xuan, T.-T. Zeng, Z.-J. Feng, Q.-H. Deng, J.-R. Chen, L.-Q. Lu, W.-J. Xiao and H. Alper, Angew. Chem., Int. Ed., 2015, 54, 1625–1628 CrossRef CAS PubMed; (d) S.-X. Lin, G.-J. Sun and Q. Kang, Chem. Commun., 2017, 53, 7665–7668 RSC; (e) J. Zheng and B. Breit, Angew. Chem., Int. Ed., 2019, 58, 3392–3397 CrossRef CAS PubMed.
- Performing the reaction with the light irradiation at fixed intervals under otherwise identical conditions revealed that the bond formation proceeded only when irradiated (Fig. S14†).
- We tried to determine the quantum yield of the reaction reported in entry 1, Table 2 by the standard ferrioxalate actinometry according to the literature procedure.25 However, the reaction did not proceed within the range of the photon flux density that could be determined by the method, which might suggest a low quantum yield value..
- (a) C. G. Hatchard and C. A. Parker, Proc. R. Soc. London, Ser. A, 1956, 235, 518–536 CrossRef CAS; (b) M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426–5434 RSC; (c) C. B. Tripathi, T. Ohtani, M. T. Corbett and T. Ooi, Chem. Sci., 2017, 8, 5622–5627 RSC.
- (a) R. Mosca, M. Fagnoni, M. Mella and A. Albini, Tetrahedron, 2001, 57, 10319–10328 CrossRef CAS; (b) N. Hoffmann, S. Bertrand, S. Marinković and J. Pesch, Pure Appl. Chem., 2006, 78, 2227–2246 CAS.
- We conducted crossover experiments with 1b and excess amount of 2 or 5a using 3a as an acceptor and B(C6F5)3 as a catalyst (10 mol%) under dark conditions (see Scheme S2† for details). In both cases, the formation of crossover product 4a or 6a was not detected, and 4b was obtained as a sole product. These results suggest that the intervention of the radical-chain process is marginal.
Footnote |
| † Electronic supplementary information (ESI) available: UV-vis absorption, ESR, electrochemical measurement, X-ray crystallography, computational studies, experimental procedures for catalytic reactions and characterization for all relevant compounds. CCDC 1986458. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc01159b |
| This journal is © The Royal Society of Chemistry 2020 |