
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct synthesis of amides from nonactivated carboxylic acids using urea as nitrogen source and Mg(NO3)2 or imidazole as catalysts†‡
A. Rosie
Chhatwal§
a,
Helen V.
Lomax§
b,
A. John
Blacker
 *c,
Jonathan M. J.
Williams
a and
Patricia
Marcé
*a
*c,
Jonathan M. J.
Williams
a and
Patricia
Marcé
*a
aDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: pmv21@bath.ac.uk
bCentre for Sustainable Chemical Technologies, University of Bath, Claverton Down, Bath, BA2 7AY, UK
cInstitute of Process Research & Development, School of Chemistry, University of Leeds, Woodhouse Lane, Leeds LS2 9JT, UK
First published on 19th May 2020
Abstract
A new method for the direct synthesis of primary and secondary amides from carboxylic acids is described using Mg(NO3)2·6H2O or imidazole as a low-cost and readily available catalyst, and urea as a stable, and easy to manipulate nitrogen source. This methodology is particularly useful for the direct synthesis of primary and methyl amides avoiding the use of ammonia and methylamine gas which can be tedious to manipulate. Furthermore, the transformation does not require the employment of coupling or activating agents which are commonly required.
Introduction
The importance of the amide functional group emerges from their presence in many crucial compounds such as proteins, fabrics, fertilizers, insecticides, plastics, drugs, and in a vast number of synthetic structures. For this reason, it is very relevant to develop new methods for the efficient synthesis of amides.Traditional methods require the transformation of the acid into the corresponding acid chloride, to use the Schotten–Baumann reaction, or coupling reagents commonly used in peptide synthesis.1–4 Although these methods produce amides under mild reaction conditions and good yields, stoichiometric amounts of activating reagent are required, and an equivalent of waste is generated, making these low-atom economy processes. Besides, the removal of the corresponding by-product can be tedious increasing the cost of the transformation. New methodologies described for the synthesis of amides5,6 involve the use of catalysts, and employ starting materials such as esters,7–17 aldehydes,18–27 alcohols,28–33 nitriles,34–45 and oximes.46–56 The catalysts are mainly based on expensive metals such as rhodium, ruthenium, iridium, and palladium. Although the use of cheaper metals such as copper, iron, titanium, hafnium and zirconium have been recently reported.5,6,44,56–60 Transamination61–64 reactions to convert primary amides into more complex amides, and the acylation of amines to produce secondary amides are also important transformations reported in this field.65
The direct synthesis of secondary amides from nonactivated carboxylic acids is an important transformation that has been less exploited and studied.57,64,66 Secondary and tertiary amides can be obtained by condensation of the acid and the amine, but the competing acid–base reaction makes this coupling challenging, overcome by forcing conditions.4 Thermal amidations in the absence of a catalyst have been previously reported67–72 and are favoured by the use of apolar solvents such as toluene.71
The direct synthesis of primary amides by this methodology is more challenging due to the low nucleophilic nature of the nitrogen source, and the use of coupling reagents is often required. The use of catalysts is an attractive approach for the direct formation of primary amides. The most relevant methodologies reported in this regard involve the use of enzymes such as lipases,73–82 boric acids,83–89 Group IV metals such as zirconium, titanium,90,91 and heterogeneous catalyst as ZrOCl2·8H2O and CAN combined with microwave radiation (Scheme 1). Although, the latter methodologies have been reported to be difficult to reproduce.90
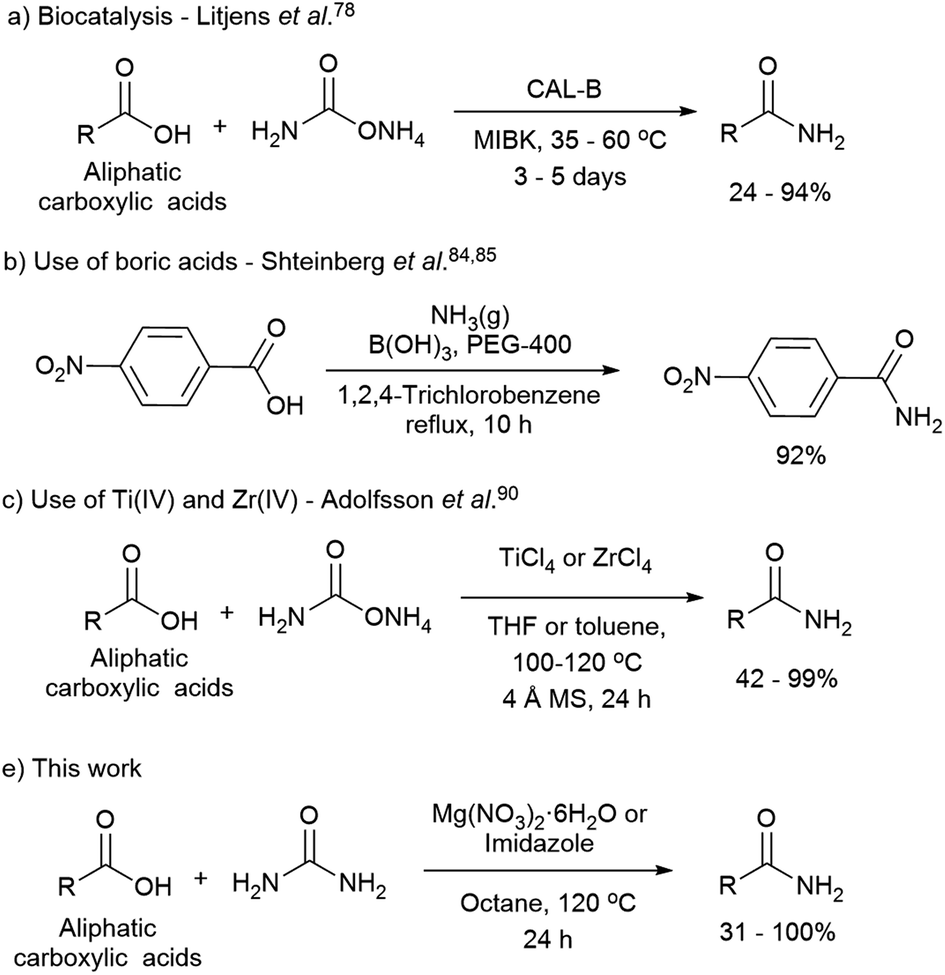 | ||
| Scheme 1 Relevant direct primary amide formation from carboxylic acids. | ||
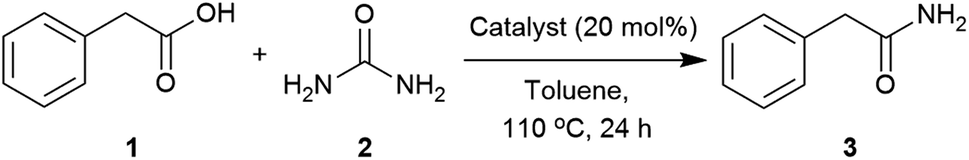
The number of catalytic protocols reported for the synthesis of primary amides is still limited.43,51,92–98 In this work, we present a new protocol for the synthesis of amides from nonactivated carboxylic acids by direct coupling using a low-cost, readily available, and easy to manipulate catalyst and nitrogen source.
Results and discussion
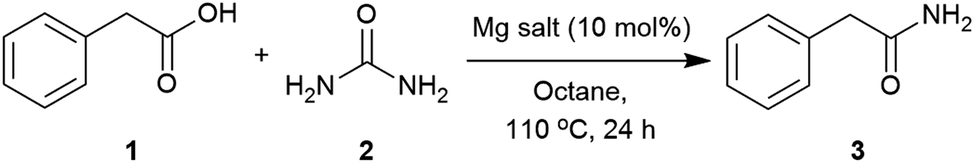
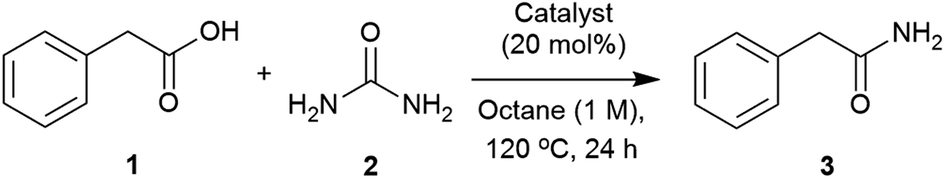
Our initial investigation identified urea as able to transform phenylacetic acid (1) into 2-phenylacetamide (3) (Table S1, see ESI‡). A catalyst screen was carried out to meet the abovementioned requirements (Table 1). Compared to the control, Group IV metals such as titanium and zirconium (Table 1, entries 2 and 3) improved the conversion,90 whilst others such as Ni(NO3)2, ZnCl2, iodide salts and protic acids showed little effect. Interestingly, Mg(OAc)2, imidazole and DMAP presented similar activities to those found with Zr(IV) and Ti(IV). Considering these results, and the low cost, availability and stability of magnesium salts and imidazole, the reaction conditions were further optimised using these two catalysts.|
|
||
|---|---|---|
| Entry | Catalyst | Conversionb (%) |
| a Reaction conditions: phenylacetic acid (1 mmol), urea (1 mmol), catalyst (20 mol%), PhMe (1 mL), 110 °C, 24 h. b Conversions were determined by analysis of the crude 1H NMR spectra. | ||
| 1 | — | 12 |
| 2 | Cp 2 ZrCl 2 | 57 |
| 3 | Ti(O i Pr) 4 | 57 |
| 4 | Ni(NO3)2·6H2O | 32 |
| 5 | ZnCl2 | 10 |
| 6 | LiBr | 17 |
| 7 | Sc(OTf)3 | 20 |
| 8 | Mg(OAc) 2 ·4H 2 O | 54 |
| 9 | AgI | 8 |
| 10 | KI | 15 |
| 11 | pTSA | 8 |
| 12 | Zn(OAc)2·2H2O | 18 |
| 13 | InCl3 | 7 |
| 14 | NaI | 11 |
| 15 | Acetic acid | 12 |
| 16 | Nitric acid | 9 |
| 17 | CaI2 | 10 |
| 18 | Imidazole | 58 |
| 19 | DMAP | 56 |
Magnesium salts as catalyst
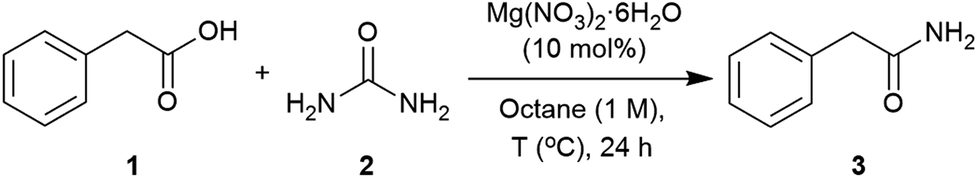
Applying the conditions used in the initial catalyst screen, the most suitable solvent was determined (Table S2, see ESI‡). Dipolar aprotic solvents, DMF and DMSO, showed low conversions,71,99 whilst polar solvents such as CPME (cyclopentylmethylether), isoamyl alcohol and butyronitrile revealed reasonable conversions. The use of non-polar solvents such as toluene and octane showed the highest conversions into the corresponding amide.100 These solvents enabled higher temperatures, and melting of the starting materials (1 = 76 °C and 2 = 133 °C) was observed to give a second liquid phase. This polar dispersed phase might dissolve the magnesium salt catalyst, and the high concentrations in the droplets are expected to facilitate the reaction.101–103 Octane was selected to screen other magnesium salts (Table 2). All those tested showed a good catalytic activity with the best conversions achieved using Mg(OAc)2·4H2O, MgCl2 and Mg(NO3)2·6H2O. When magnesium acetate was used, detailed analysis of the crude reaction revealed the formation of acetamide as a by-product.104 Mg(NO3)2·6H2O was chosen as the most appropriate catalyst for further optimisation of the reaction conditions. The use of 2 equivalents of urea at 120 °C were found to give an optimal 93% conversion to the amide (Table 3, entries 3). Three equivalents of urea had a detrimental effect on the formation of 2-phenylacetamide (Table 3, entries 3 and 4). Varying the catalyst loading and reaction concentration, the best conditions identified were using 10 mol% of Mg(NO3)2·6H2O with a 1 M concentration in octane (Tables S6 and S7, see ESI‡).|
|
||
|---|---|---|
| Entry | Mg catalyst | Conversionb (%) |
| a Reaction conditions: phenylacetic acid (1 mmol), urea (1 mmol), Mg catalyst (10 mol%), octane (1 mL), 110 °C, 24 h. b Conversions were determined by analysis of the crude 1H NMR spectra. | ||
| 1 | — | 26 |
| 2 | Mg(OAc) 2 ·4H 2 O | 68 |
| 3 | Mg turnings | 51 |
| 4 | Mg(NO 3 ) 2 ·6H 2 O | 64 |
| 5 | MgO | 54 |
| 6 | Mg(OTf)2 | 61 |
| 7 | MgCl 2 ·6H 2 O | 65 |
| 8 | MgSO4 | 50 |
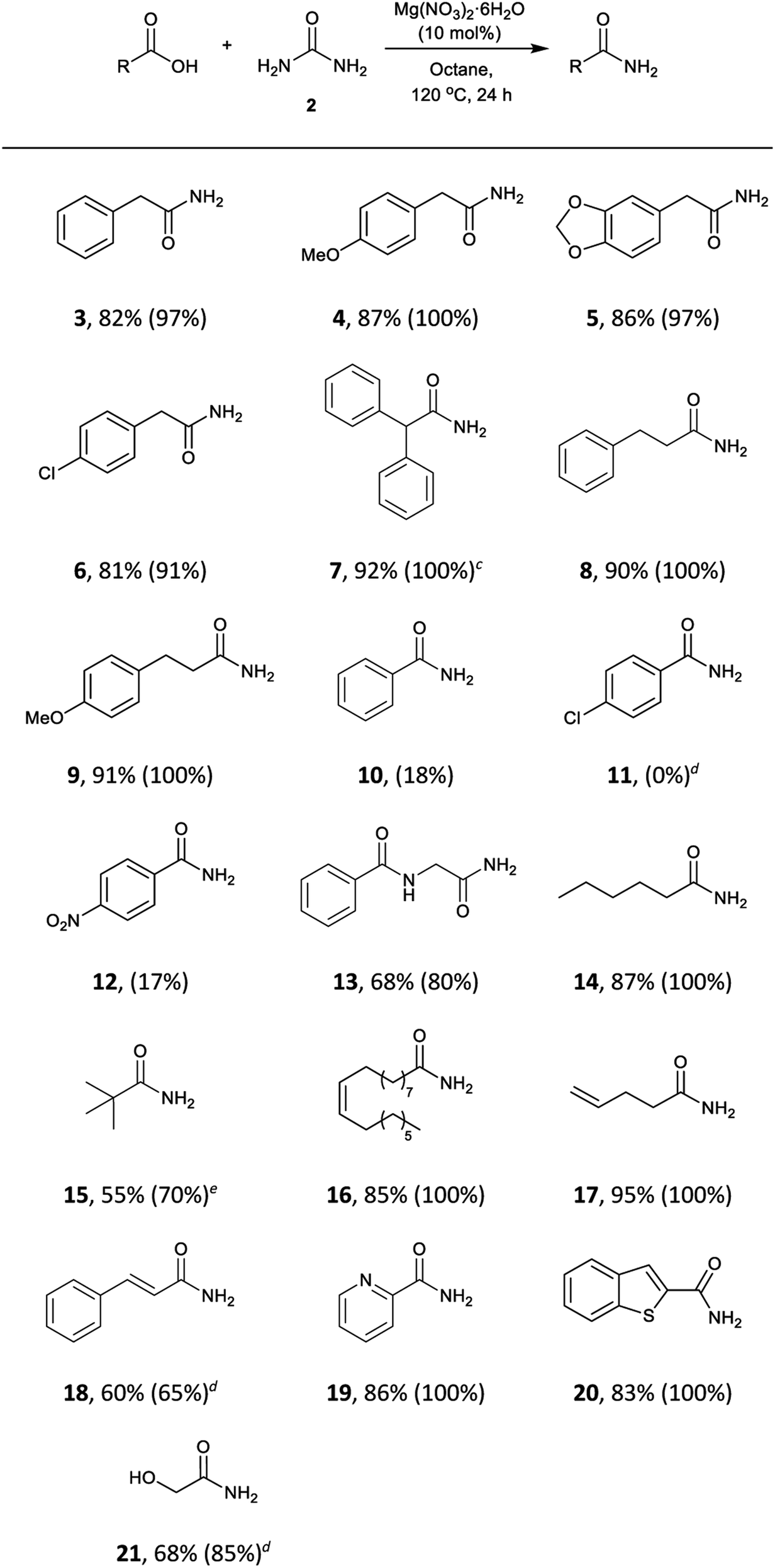
The scope of the reaction was subjected to study. This methodology turned out to be effective for a wide range of aliphatic and phenylacetic acids (Table 4). Phenylacetic acids bearing electron-withdrawing and electron-donating groups were converted into the corresponding amides (3, 4, 5, and 6) in 81–87% isolated yields. The sterically demanding substrate diphenylacetic acid was also successfully transformed into diphenylacetamide (7) in 92% yield, and aliphatic acids (8, 9, 13, 14, and 15), with internal (16) and terminal (17) double bonds were also well-tolerated. Aliphatic acids containing conjugated double bonds (18) were more challenging substrates and lower conversions were observed even at elevated temperatures. The precursor benzoic acid (10) and substituted acids 11 and 12 showed very low reactivity under the reaction conditions, perhaps due to the delocalisation of electrons and subsequent decrease of electrophilicity of the carboxyl group. Surprisingly, heterocycles such as 2-picolinic acid and benzothiophene-2-carboxylic acid gave the amides 19 and 20 in excellent yields. Furthermore, the hydroxyl group in glycolic acid was also tolerated to give 21 in 68% yield.
| a Reaction conditions: carboxylic acid (3 mmol), urea (6 mmol), Mg(NO3)2·6H2O (10 mol%), octane (3 mL), 120 °C, 24 h. b Isolated yields, conversions were determined by analysis of the crude 1H NMR spectra and are shown in parentheses. c 1 mmol scale. d 130 °C. e Reaction at 130 °C did not improve the conversion. Longer reaction times did not show a substantial increase in the conversion. |
|---|
|
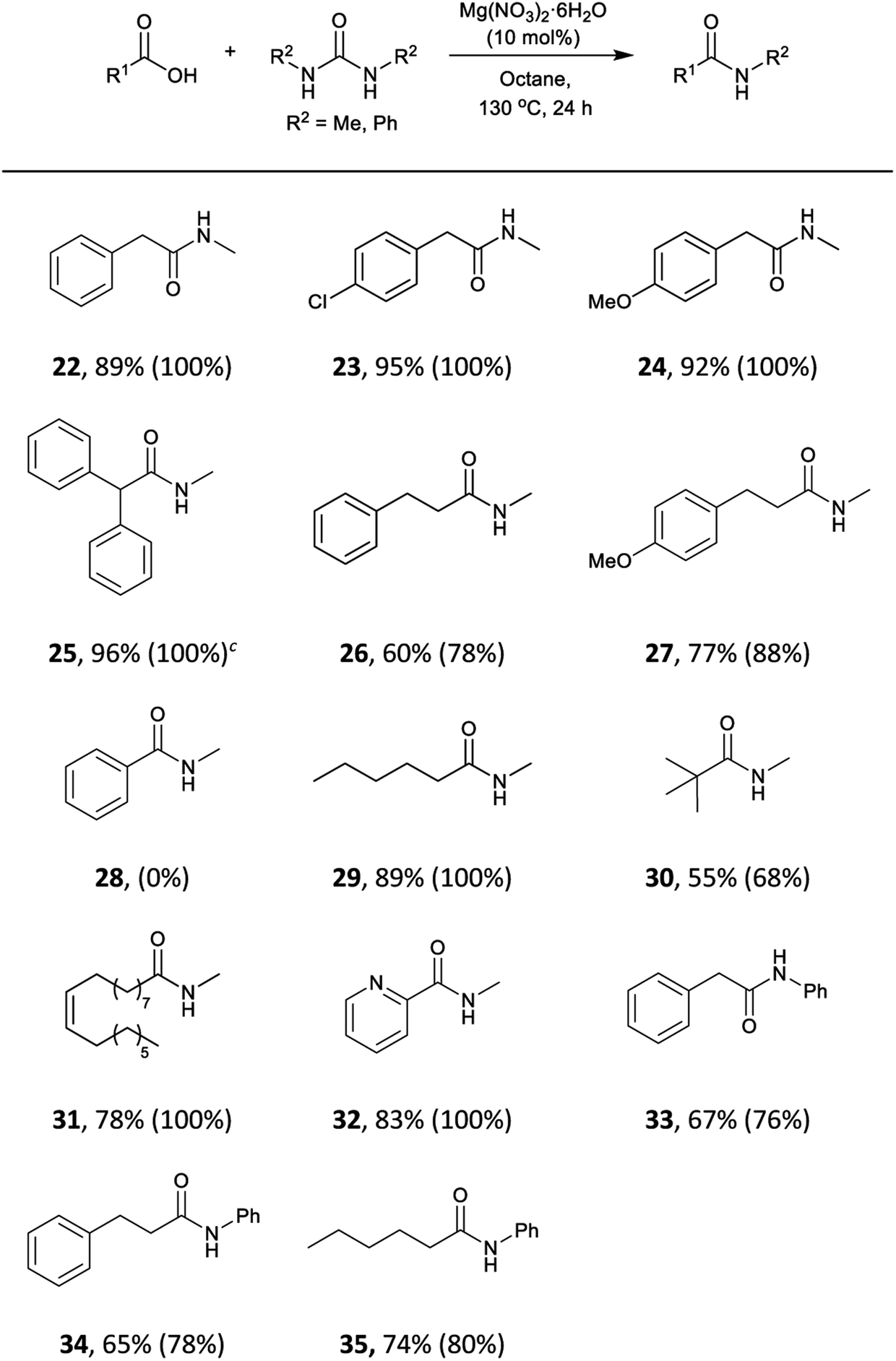
We envisaged that our methodology could be also applied to the synthesis of secondary amides. N-Methyl amides are commonly obtained by direct coupling with methylamine gas and few alternatives methods are available.105–108 The use of N,N′-dimethylurea could be particularly useful, due to its availability and simpler handling. The scope of the reaction with this and N,N′-diphenylurea was tested with aliphatic and phenylacetic acids (Table 5). A wide range of aliphatic and phenylacetic acids was converted into the corresponding amides. For instance, N-methyl amides 22, 23, 24 and 25 were isolated in 89–96% yield. The method could be extended to aliphatic acids, and amides 26, 27, 29, 30 and 31 were obtained in good yields. On the other hand, less electrophilic benzoic acid (28) was unreactive under these conditions. Three acids were also tested with N,N′-diphenylurea giving satisfactory yields of N-phenylacetamides 33, 34 and 35.
Imidazole as catalyst
The initial screen showed the potential of imidazole and DMAP as catalysts for this transformation (Table 1). So, our attention was turned into the inexpensive and readily available imidazole organocatalyst. Using the previously determined conditions, phenylacetic acid and urea were reacted to give phenylacetamide (3) in 78% conversion (Table 6, entry 4). Further improvement was achieved with 1.5 equivalents of urea, but higher loadings failed to obtain better conversions. Increasing the temperature to 120 °C led to the optimal reaction conditions (Table 6, entry 5).109,110|
|
||||
|---|---|---|---|---|
| Entry | Imidazole (equiv.) | Conversionb (%) | ||
| Urea | ||||
| 1.0 equiv. | 1.5 equiv. | 2.0 equiv. | ||
| a Reaction conditions: phenylacetic acid (1 mmol), urea and catalyst in octane (1 mL), 110 °C, 24 h. b Conversions were determined by analysis of 1H NMR spectra. c The temperature was increased to 120 °C. | ||||
| 1 | 0 | 24 | 26 | |
| 2 | 0.1 | 71 | 72 | |
| 4 | 0.2 | 78 | 86 | 84 |
| 5 | 0.3 | 85 | ||
| 6c | 0.2 | 96 | ||
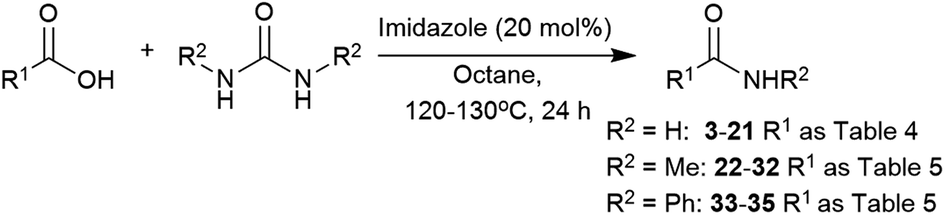
Using the optimised conditions, the substrate scope of the carboxylic acid was investigated (Table 6). Phenylacetic acid and hydrocinnamic acid proceed to their corresponding amides 3 and 8 in 91% and 97%, respectively. The presence of electron-donating and -withdrawing groups at the para position had little effect on the conversion, and amides 4, 5, 6, and 9 were obtained in high yields. Aliphatic groups were also well-tolerated and hexanamide (13) was produced in 89% yield. The presence of bulky substituents in the aromatic or aliphatic chain had a detrimental effect, and diphenylacetamide (7) was produced in 65% yield, 27% less than with Mg(NO2)2·6H2O. On the other hand, pivalamide (15) was obtained in 60% yield. Oleic acid gave 16 in 91% yield with no alteration of the double bond. Similar to the observations in Tables 4 and 5, the conjugated carboxylic acids did not perform well, with benzoic and cinnamic acids giving 10, 11, 12 and 18 in low conversions. 2-Picolinic acid and benzothiophene-2-carboxylic acid showed conversions into amides 19 and 20 of 65% and 100%, respectively, whilst glycolic acid was also converted into 21 in 61% yield, showing this catalyst also tolerates hydroxyl groups.
N,N′-Dimethylurea and N,N′-diphenylurea were also explored to synthesise secondary amides using imidazole (Table 7). In this case, 2 equivalents of urea and 130 °C were required to drive the reaction towards the formation of the amide (Table S13, see ESI‡). Both phenylacetic acid and hydrocinnamic acid gave 22 and 23 in 84% and 80% yields, respectively. In contrast, amidation of hexanoic acid into N-methylhexamide (29) gave only 70% yield, 19% less than the metal catalyst. Pivalic acid was converted into the amide 30 in only 38% yield, again indicating a steric problem. As with the other reactions, benzoic acid did not perform well in these conditions. Imidazole was an effective catalyst for making 2-picolinamide (32), obtained in 76% yield.
|
|
|||
|---|---|---|---|
| Compound R2 = Ha | Yieldc % (conv. %) | Compound R2 = Me or Phb | Yieldc % (conv. %) |
| a Reaction conditions: carboxylic acid (3 mmol), urea (4.5 mmol), imidazole (20 mol%), octane (3 mL), 120 °C, 24 h. b Reaction conditions: carboxylic acid (3 mmol), urea (6 mmol), imidazole (20 mol%), octane (3 mL), 130 °C, 24 h. c Isolated yields, conversions were determined by analysis of the crude 1H NMR spectra and are shown in parentheses. | |||
| 3 | 91 (100) | 22 | 84 (100) |
| 4 | 91 (100) | 23 | 93 (100) |
| 5 | 90 (97) | 24 | 95 (100) |
| 6 | 96 (100) | 25 | 89 (100) |
| 7 | 65 (73) | 26 | 80 (100) |
| 8 | 97 (100) | 27 | 93 (100) |
| 9 | 90 (97) | 28 | (25) |
| 10 | (30) | 29 | 70 (88) |
| 11 | (18) | 30 | 38 (50) |
| 12 | (33) | 31 | 60 (84) |
| 13 | 74 (86) | 32 | 76 (84) |
| 14 | 89 (100) | 33 | 55 (72) |
| 15 | 60 (72) | 34 | 68 (85) |
| 16 | 91 (100) | 35 | 63 (78) |
| 18 | 52 (63) | ||
| 19 | 42 (65) | ||
| 20 | 79 (100) | ||
| 21 | 61 (78) | ||
When N,N′-diphenylurea was used to synthesise N-phenylamides with imidazole catalyst, lower conversions were obtained. Phenylacetic acid, hydrocinnamic acid and hexanoic acid gave the corresponding amides 33, 34 and 35 in 55%, 68% and 63% yields respectively.
Mechanistic insights
A slow uncatalysed reaction between phenylacetic acid and urea was observed (Tables 1 and 2, entries 1), however, the addition of a suitable Lewis acid or an organocatalyst considerably improves the rates and conversions. Since the reaction mechanism was unclear, three models were proposed (Scheme 2): (1) decomposition of urea and direct amidation; (2) magnesium salt facilitates the formation of an N-acylurea intermediate which can be hydrolysed to produce the amide, and an unstable carbamic acid, followed by decarboxylation of the later. To explain the observed products, reaction with the more substituted urea nitrogen is required; (3) condensation of the carboxylic acid with imidazole to form N-acyl imidazolium, this activated amide would then react with urea to produce an N-acylurea intermediate, again, breaking down to form ammonia and carbon dioxide.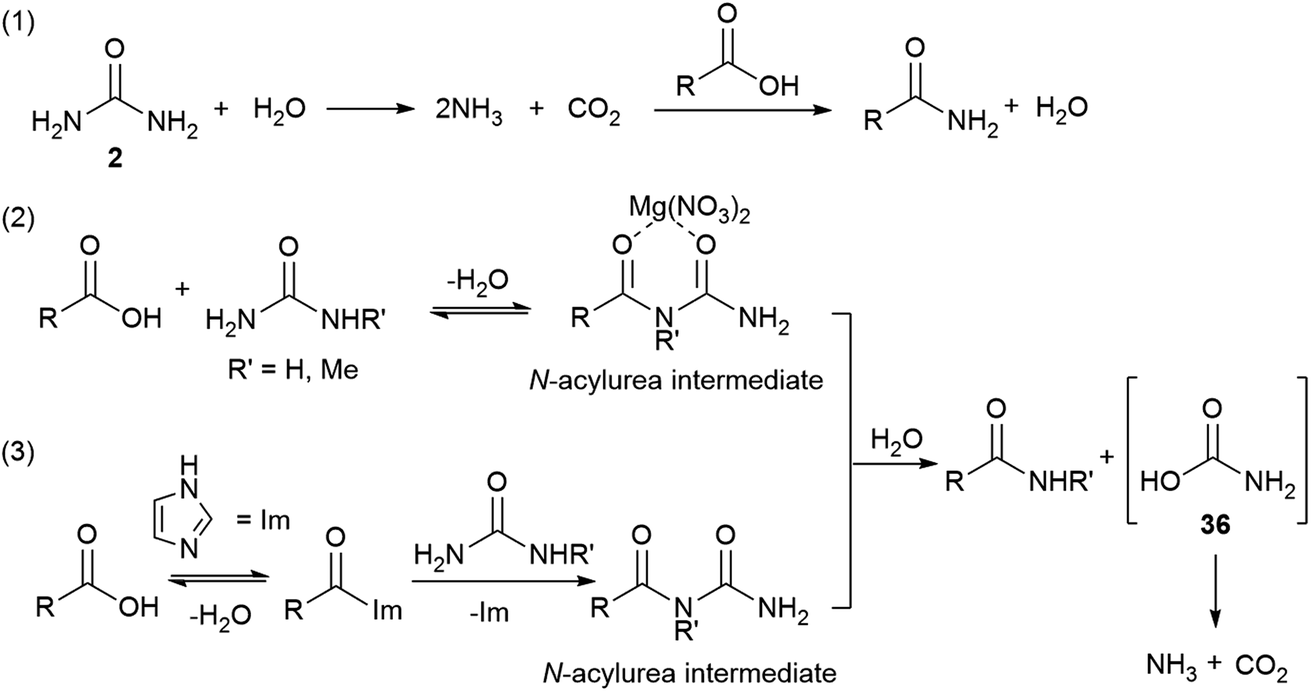 | ||
| Scheme 2 Plausible mechanisms for the formation of amides. | ||
According to mechanism (1), the decomposition of urea into ammonia and CO2 is reported to take place at temperatures above 152 °C;111 even though the temperatures in our reactions are not as high as these, the possibility of a catalysed urea degradation was investigated. Urea was treated with either imidazole or Mg(NO3)2·6H2O under the reaction conditions for 24 hours. The analysis of the reaction crudes by 1H NMR showed the presence of urea indicating no degradation. Furthermore, gravimetric analysis, before and after reaction, gave 96% urea recovery, suggesting no degradation had taken place. The reaction was repeated with N-methylurea, N,N′-dimethylurea and N,N′-diphenylurea, and in each case >92% of the starting material was recovered. To investigate this further, phenylacetic acid (1) was exposed to the reaction conditions using aniline as the nitrogen source, which might be formed during the degradation of N,N′-diphenylurea (Scheme 3, eqn (1)). Similar conversions were observed when the reaction was carried out with and without imidazole or magnesium catalyst, suggesting it is not involved in the direct coupling.112 However, when phenylacetic acid (1) was reacted with N,N′-diphenylurea, the uncatalysed reaction gave no product, but in the catalysed reactions conversions of 40–51% were observed (Scheme 3, eqn (2)); notably less than direct amidation (70–77% conversion) but expected, as anilines are well known to undergo direct amidation.72
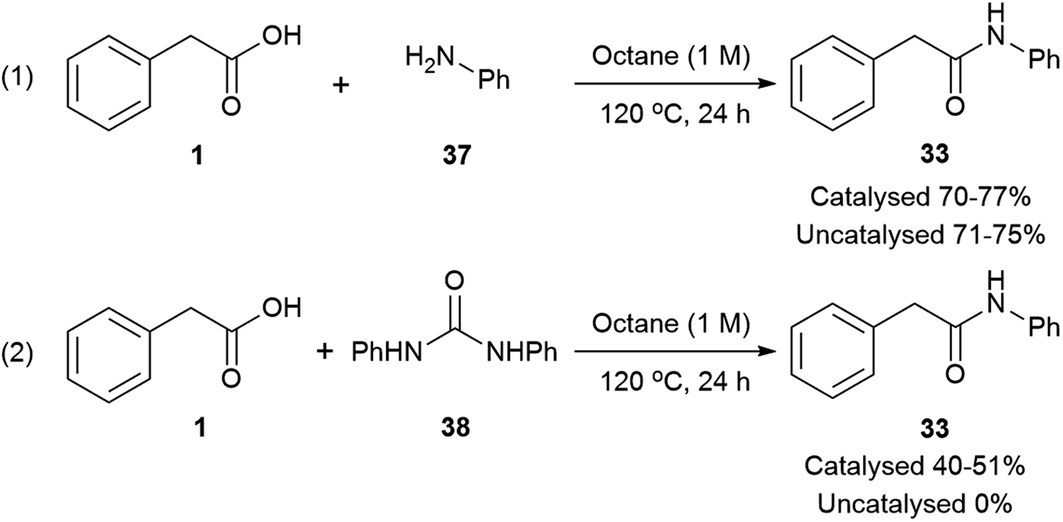 | ||
| Scheme 3 Conversions on the direct amidation of phenylacetic acid vs. amidation using N,N′-diphenylurea. | ||
A previous report describes the CO2 evolved in the acidolysis of ureas using 13C-labelled carboxylic acids derives from the urea, and support other research that invokes a carbamic acid intermediate.113,114
Mechanisms (2) and (3) were tested with unsymmetrical substituted ureas. The highly hindered N,N,N′N′-tetramethylurea did not form the tertiary amide 40 even at 130 °C, indicating that the steric bulk interferes (Scheme 4, eqn (1)). However, the reaction between phenylacetic acid and N-methylurea or N,N-dimethylurea gave the secondary amides 22 an 40 in 66% conversion with MgNO3·6H2O and 77% with imidazole, along with traces of the primary amide (Scheme 4, eqn (2) and (3), Table S14, see ESI‡). In order to check the formation of ammonia during the reaction a litmus paper test was conducted. A colour change from yellow to blue was observed confirming the formation of a basic gas. These results might indicate that unsymmetrical ureas react to give the most substituted amide, and presumably carbamic acid which decomposes liberating carbon dioxide and ammonia.113,114 Water is required for this reaction and may come from the MgNO3·6H2O, or during condensation of acid with the imidazole. In either case a thermally unstable (N-alkyl)carbamic acid is implicated,115 (Scheme 2, eqn (2) and (3)). MgNO3·6H2O might coordinate to the 1,3-dicarbonyl, activating it to urea condensation.116 With imidazole, the direct reaction with urea was discounted, however its reaction with carboxylic acids and esters, in solvent under thermal conditions is reported.117–119 Protonated N-acyl imidazoles are known to react with amines and thiols, so it is reasonable that urea, may react to form the N-acylurea intermediate.115,120
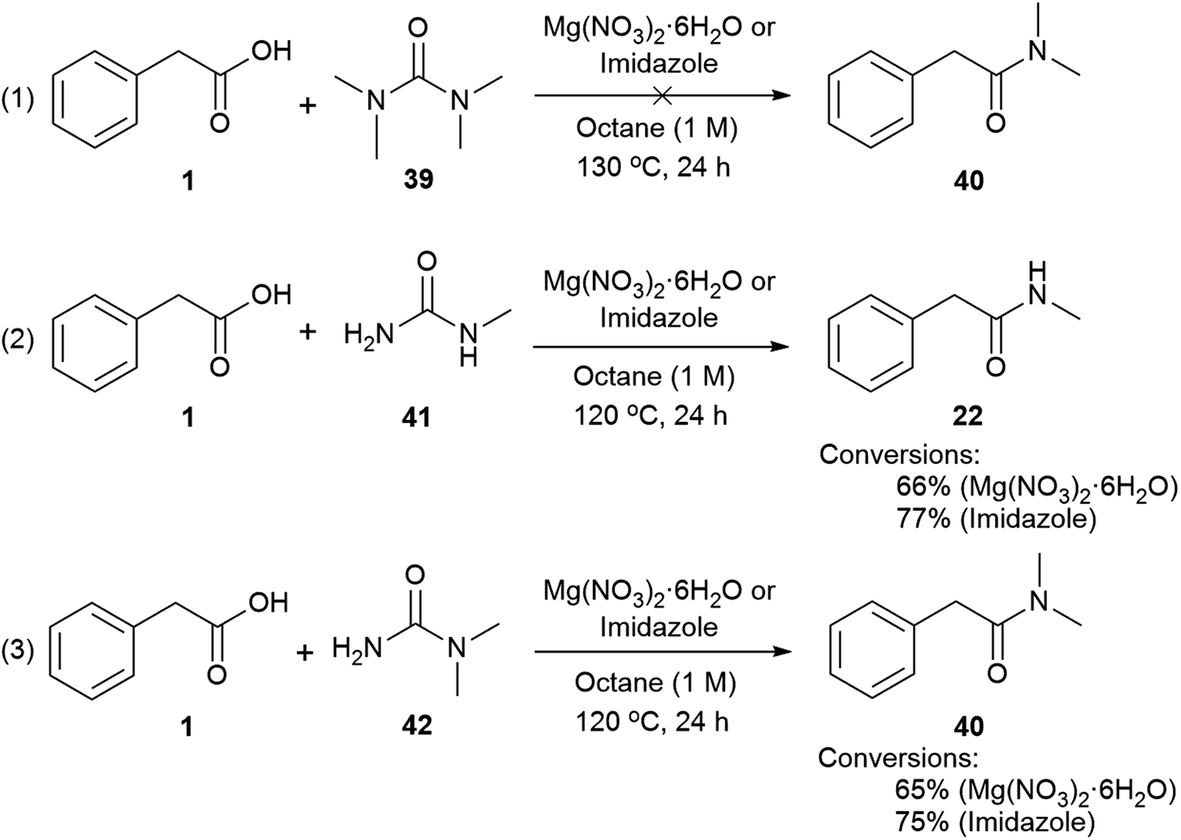 | ||
| Scheme 4 Use of tetrasubstituted and asymmetric ureas. | ||
The reaction mixture was analysed by HRMS after 6 hours and a species with m/z of 201.0636 was found, which corresponds with the sodium adduct of the N-acylurea intermediate (theoretical m/z 201.0640 [M + Na]+). To test the reactivity of the N-acylurea intermediate, N-pivaloylurea was synthesised and subjected to different reaction conditions,121 and conversion to pivalamide was analysed by 1H NMR (Table 8).
|
|
||||
|---|---|---|---|---|
| Entry | Mg(NO3)2·6H2O (10 mol%) | Imidazole (20 mol%) | Water (2 equiv.) | Conversionb (%) |
| a Reaction conditions: N-carbamoylpivalamide (1 mmol), water (2 mmol), imidazole (20 mol%) or Mg(NO3)2·6H2O (10 mol%), octane (1 mL), 120 °C, 24 h. b Conversions were determined by analysis of 1H NMR spectra. | ||||
| 1 | ✗ | ✗ | ✗ | 0 |
| 2 | ✗ | ✗ | ✓ | 6 |
| 3 | ✓ | ✗ | ✗ | 6 |
| 4 | ✗ | ✓ | ✗ | 4 |
| 5 | ✓ | ✗ | ✓ | 25 |
| 6 | ✗ | ✓ | ✓ | 14 |
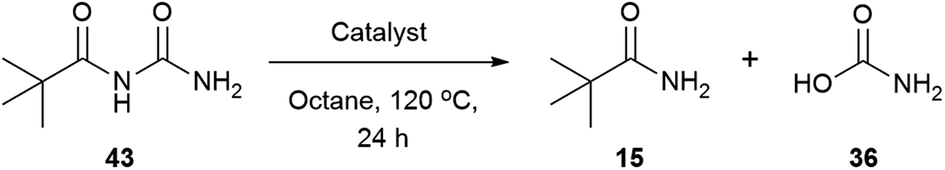
In the absence of the catalyst and water, only the starting material was recovered (Table 8, entry 1). The addition of two equivalents of water produced the pivalamide with 6% conversion (Table 8, entry 2). The presence of the catalyst did not improve the reaction outcome (Table 8, entries 3 and 4), whilst either catalyst alone failed to improve the conversions (Table 8, entries 5 and 6). However, when the catalyst and water were combined, 25% and 14% of amide were detected (Table 8, entries 5 and 6). The conversions are less than pivalic acid and urea, (55% and 60% to 15, Tables 4 and 7). To dismiss a substrate effect, N-phenylacetylurea was synthesised and subjected to the same study, and similar behaviour was observed.121,122 Since N-alkylcarbamic acids decompose readily in acidic media,114 1 equivalent of hydrocinnamic acid was added, but also gave similar low conversions. These results suggested that other physico-chemical effects may be playing a role. Solubility studies indicated that phenylacetic acid is soluble in octane at 120 °C while urea and 2-phenylacetamide (3) are not. When all the starting materials were mixed together in octane at 120 °C a biphasic system was obtained, and at the end of the reaction a white solid corresponding to 2-phenylacetamide (3) was formed. In these conditions an on-solvent system may be occurring in which the urea, carboxylic acid and catalyst are at high concentration and form a polar, hydrogen bonded structure that facilitates the reaction.101,102 Besides, the liberation of ammonia and CO2 along with the precipitation of 2-phenylacetamide (3) might be the driving force of this transformation. The lack of solubility of the N-acylurea intermediate in octane could explain the slow reactivity observed in Table 8.
To investigate further the role of imidazole in this transformation, N-1-methylimidazole and 2-methylimidazole were used as catalysts (Table 9). A significant decrease in conversion, from 86% to 37%, was observed when 2-methylimidazole was employed (Table 9, entry 4). Whereas, N-1-methylimidazole gave 66% conversion, only slightly less than imidazole (86%) (Table 9, entry 3). These results suggest that the mode of activation is through the nitrogen lone pair, as the 2-methyl group blocks this position. This is supported by the similar catalytic activity of DMAP (Table 9, entry 5), that also has a sp2 nitrogen with a lone pair of electrons. The mechanism by which the reaction proceeds is still unclear and further studies are still undergoing to understand the reaction pathway.
Conclusions
A new method for the direct synthesis of primary amides from nonactivated carboxylic acids has been described, in which wasteful activating reagents are avoided. The methodology reports the use of cheap and readily available catalysts such as Mg(NO3)2·6H2O and imidazole, with urea as an atom-efficient nitrogen source. The process has been shown to produce not only primary, but secondary and a tertiary amides from readily available ureas. The method shows a broad scope of reaction, although conjugated carboxylic acids do not perform well. The reaction mechanism has been studied, and initial results point to the involvement of an N-acylurea intermediate, although, the pathway of its decomposition to the product remains unclear. Further studies are still undergoing to propose a more plausible mechanism.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the EPSRC for funding through the UK Catalysis Hub and the Doctoral Training Centre in Sustainable Chemical Technologies for studentship (H. V. L.). We would also like to acknowledge the NMR and MS facilities provided through the Material and Chemical Characterisation (MC2) at the University of Bath.Notes and references
- A. El-Faham and F. Albericio, Chem. Rev., 2011, 111, 6557–6602 CrossRef CAS.
- S.-Y. Han and Y.-A. Kim, Tetrahedron, 2004, 60, 2447–2467 CrossRef CAS.
- E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC.
- C. A. G. N. Montalbetti and V. Falque, Tetrahedron, 2005, 61, 10827–10852 CrossRef CAS.
- C. L. Allen and J. M. J. Williams, Chem. Soc. Rev., 2011, 40, 3405–3415 RSC.
- B. N. Atkinson, A. R. Chhatwal and J. M. J. Williams, in Synthetic Methods in Drug Discovery, vol. 2, 2016, pp. 413–453 Search PubMed.
- R. Arora, S. Paul and R. Gupta, Can. J. Chem., 2005, 83, 1137–1140 CrossRef CAS.
- B. Gnanaprakasam and D. Milstein, J. Am. Chem. Soc., 2011, 133, 1682–1685 CrossRef CAS PubMed.
- Y. Ishii, M. Takeno, Y. Kawasaki, A. Muromachi, Y. Nishiyama and S. Sakaguchi, J. Org. Chem., 1996, 61, 3088–3092 CrossRef CAS PubMed.
- B. C. Ranu and P. Dutta, Synth. Commun., 2003, 33, 297–301 CrossRef CAS.
- C. Han and J. A. Porco, Org. Lett., 2007, 9, 1517–1520 CrossRef CAS PubMed.
- C. Han, J. P. Lee, E. Lobkovsky and J. A. Porco, J. Am. Chem. Soc., 2005, 127, 10039–10044 CrossRef CAS PubMed.
- N. Caldwell, C. Jamieson, I. Simpson and T. Tuttle, Org. Lett., 2013, 15, 2506–2509 CrossRef CAS PubMed.
- M. Movassaghi and M. A. Schmidt, Org. Lett., 2005, 7, 2453–2456 CrossRef CAS PubMed.
- M. K. Kiesewetter, M. D. Scholten, N. Kirn, R. L. Weber, J. L. Hedrick and R. M. Waymouth, J. Org. Chem., 2009, 74, 9490–9496 CrossRef CAS.
- C. Sabot, K. A. Kumar, S. Meunier and C. Mioskowski, Tetrahedron Lett., 2007, 48, 3863–3866 CrossRef CAS.
- E. C. de Lima, C. C. de Souza, R. de O. Soares, B. G. Vaz, M. N. Eberlin, A. G. Dias and P. R. R. Costa, J. Braz. Chem. Soc., 2011, 22, 2186–2190 CrossRef.
- J. A. Thomson and L. L. Schafer, Dalton Trans., 2012, 41, 7897 RSC.
- H. U. Vora and T. Rovis, J. Am. Chem. Soc., 2007, 129, 13796–13797 CrossRef CAS PubMed.
- Z. Liu, J. Zhang, S. Chen, E. Shi, Y. Xu and X. Wan, Angew. Chem., Int. Ed., 2012, 51, 3231–3235 CrossRef CAS PubMed.
- S. C. Ghosh, J. S. Y. Ngiam, A. M. Seayad, D. T. Tuan, C. L. L. Chai and A. Chen, J. Org. Chem., 2012, 77, 8007–8015 CrossRef CAS PubMed.
- W.-J. Yoo and C.-J. Li, J. Am. Chem. Soc., 2006, 128, 13064–13065 CrossRef CAS PubMed.
- Y. Suto, N. Yamagiwa and Y. Torisawa, Tetrahedron Lett., 2008, 49, 5732–5735 CrossRef CAS.
- O. Saidi, M. J. Bamford, A. J. Blacker, J. Lynch, S. P. Marsden, P. Plucinski, R. J. Watson and J. M. J. Williams, Tetrahedron Lett., 2010, 51, 5804–5806 CrossRef CAS.
- R. Cadoni, A. Porcheddu, G. Giacomelli and L. De Luca, Org. Lett., 2012, 14, 5014–5017 CrossRef CAS PubMed.
- A. Porcheddu and L. De Luca, Adv. Synth. Catal., 2012, 354, 2949–2953 CrossRef CAS.
- Y. Li, F. Jia and Z. Li, Chem.–Eur. J., 2013, 19, 82–86 CrossRef CAS PubMed.
- K. Shimizu, K. Ohshima and A. Satsuma, Chem.–Eur. J., 2009, 15, 9977–9980 CrossRef CAS.
- X. Bantreil, C. Fleith, J. Martinez and F. Lamaty, ChemCatChem, 2012, 4, 1922–1925 CrossRef CAS.
- X.-F. Wu, M. Sharif, A. Pews-Davtyan, P. Langer, K. Ayub and M. Beller, Eur. J. Org. Chem., 2013, 2783–2787 CrossRef CAS.
- B. Kang, Z. Fu and S. H. Hong, J. Am. Chem. Soc., 2013, 135, 11704–11707 CrossRef CAS PubMed.
- S. C. Ghosh, S. Muthaiah, Y. Zhang, X. Xu and S. H. Hong, Adv. Synth. Catal., 2009, 351, 2643–2649 CrossRef CAS.
- A. J. A. Watson, A. C. Maxwell and J. M. J. Williams, Org. Lett., 2009, 11, 2667–2670 CrossRef CAS PubMed.
- A. Guérinot, S. Reymond and J. Cossy, Eur. J. Org. Chem., 2012, 19–28 CrossRef.
- S. Davulcu, C. L. Allen, K. Milne and J. M. J. Williams, ChemCatChem, 2013, 5, 435–438 CrossRef CAS.
- C. L. Allen, A. A. Lapkin and J. M. J. Williams, Tetrahedron Lett., 2009, 50, 4262–4264 CrossRef CAS.
- S. Murahashi, T. Naota and E. Saito, J. Am. Chem. Soc., 1986, 108, 7846–7847 CrossRef CAS PubMed.
- Y.-M. Liu, L. He, M.-M. Wang, Y. Cao, H.-Y. He and K.-N. Fan, ChemSusChem, 2012, 5, 1392–1396 CrossRef CAS PubMed.
- M. Tamura, H. Wakasugi, K. Shimizu and A. Satsuma, Chem.–Eur. J., 2011, 17, 11428–11431 CrossRef CAS PubMed.
- A. Goto, K. Endo and S. Saito, Angew. Chem., Int. Ed., 2008, 47, 3607–3609 CrossRef CAS PubMed.
- T. J. Ahmed, S. M. M. Knapp and D. R. Tyler, Coord. Chem. Rev., 2011, 255, 949–974 CrossRef CAS.
- V. Y. Kukushkin and A. J. L. Pombeiro, Inorg. Chim. Acta, 2005, 358, 1–21 CrossRef CAS.
- P. Marcé, J. Lynch, A. J. Blacker and J. M. J. Williams, Chem. Commun., 2016, 52, 1436–1438 RSC.
- E. L. Downs and D. R. Tyler, Coord. Chem. Rev., 2014, 280, 28–37 CrossRef CAS.
- D. D. Sanz Sharley and J. M. J. Williams, Tetrahedron Lett., 2017, 58, 4090–4093 CrossRef CAS.
- H.-J. Pi, J.-D. Dong, N. An, W. Du and W.-P. Deng, Tetrahedron, 2009, 65, 7790–7793 CrossRef CAS.
- L. Yadav, R. Patel and V. Srivastava, Synthesis, 2010, 1771–1776 CrossRef CAS.
- C. L. Allen, R. Lawrence, L. Emmett and J. M. J. Williams, Adv. Synth. Catal., 2011, 353, 3262–3268 CrossRef CAS.
- N. A. Owston, A. J. Parker and J. M. J. Williams, Org. Lett., 2007, 9, 3599–3601 CrossRef CAS PubMed.
- C. L. Allen, S. Davulcu and J. M. J. Williams, Org. Lett., 2010, 12, 5096–5099 CrossRef CAS PubMed.
- R. R. Gowda and D. Chakraborty, Eur. J. Org. Chem., 2011, 2011, 2226–2229 CrossRef.
- N. A. Owston, A. J. Parker and J. M. J. Williams, Org. Lett., 2007, 9, 73–75 CrossRef CAS PubMed.
- H. Fujiwara, Y. Ogasawara, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2007, 46, 5202–5205 CrossRef CAS PubMed.
- S. K. Sharma, S. D. Bishopp, C. Liana Allen, R. Lawrence, M. J. Bamford, A. A. Lapkin, P. Plucinski, R. J. Watson and J. M. J. Williams, Tetrahedron Lett., 2011, 52, 4252–4255 CrossRef CAS.
- C. L. Allen, C. Burel and J. M. J. Williams, Tetrahedron Lett., 2010, 51, 2724–2726 CrossRef CAS.
- P. Crochet and V. Cadierno, Chem. Commun., 2015, 51, 2495–2505 RSC.
- H. Lundberg, F. Tinnis, N. Selander and H. Adolfsson, Chem. Soc. Rev., 2014, 43, 2714–2742 RSC.
- R. García-Álvarez, P. Crochet and V. Cadierno, Green Chem., 2013, 15, 46–66 RSC.
- V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479 CrossRef CAS PubMed.
- K. Ishihara, Tetrahedron, 2009, 65, 1085–1109 CrossRef CAS.
- S. E. Eldred, D. A. Stone, S. H. Gellman and S. S. Stahl, J. Am. Chem. Soc., 2003, 125, 3422–3423 CrossRef CAS PubMed.
- B. N. Atkinson, A. R. Chhatwal, H. V. Lomax, J. W. Walton and J. M. J. Williams, Chem. Commun., 2012, 48, 11626 RSC.
- M. Zhang, S. Imm, S. Bähn, L. Neubert, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2012, 51, 3905–3909 CrossRef CAS PubMed.
- R. M. Lanigan and T. D. Sheppard, Eur. J. Org. Chem., 2013, 33, 7453–7465 CrossRef.
- D. D. Sanz Sharley and J. M. J. Williams, Chem. Commun., 2017, 53, 2020–2023 RSC.
- H. Lundberg and H. Adolfsson, ACS Catal., 2015, 5, 3271–3277 CrossRef CAS.
- H. Lundberg, F. Tinnis and H. Adolfsson, Chem.–Eur. J., 2012, 18, 3822–3826 CrossRef CAS PubMed.
- M. Rahman, D. Kundu, A. Hajra and A. Majee, Tetrahedron Lett., 2010, 51, 2896–2899 CrossRef CAS.
- C. Grosjean, J. Parker, C. Thirsk and A. R. Wright, Org. Process Res. Dev., 2012, 16, 781–787 CrossRef CAS.
- H. Charville, D. Jackson, G. Hodges and A. Whiting, Chem. Commun., 2010, 46, 1813 RSC.
- C. L. Allen, A. R. Chhatwal and J. M. J. Williams, Chem. Commun., 2012, 48, 666–668 RSC.
- L. Gooßen, D. Ohlmann and P. Lange, Synthesis, 2009, 2009, 160–164 CrossRef.
- T. Nuijens, E. Piva, J. A. W. Kruijtzer, D. T. S. Rijkers, R. M. J. Liskamp and P. J. L. M. Quaedflieg, Tetrahedron Lett., 2012, 53, 3777–3779 CrossRef CAS.
- M. Kidwai, R. Poddar and P. Mothsra, Beilstein J. Org. Chem., 2009, 5, 1–7 Search PubMed.
- R. V. Ulijn, B. Baragaña, P. J. Halling and S. L. Flitsch, J. Am. Chem. Soc., 2002, 124, 10988–10989 CrossRef CAS PubMed.
- M. Fernández-Pérez and C. Otero, Enzyme Microb. Technol., 2001, 28, 527–536 CrossRef.
- M. A. P. J. Hacking, H. Akkus, F. van Rantwijk and R. A. Sheldon, Biotechnol. Bioeng., 2000, 68, 84–91 CrossRef CAS.
- M. J. J. Litjens, A. J. J. Straathof, J. A. Jongejan and J. J. Heijnen, Tetrahedron, 1999, 55, 12411–12418 CrossRef CAS.
- V. Čeřovský and M.-R. Kula, Angew. Chem., Int. Ed., 1998, 37, 1885–1887 CrossRef.
- T. Maugard, M. Remaud-Simeon, D. Petre and P. Monsan, Tetrahedron, 1997, 53, 5185–5194 CrossRef CAS.
- B. Tuccio, E. Ferré and L. Comeau, Tetrahedron Lett., 1991, 32, 2763–2764 CrossRef CAS.
- F. Servat, D. Montet, M. Pina, P. Galzy, A. Arnaud, H. Ledon, L. Marcou and J. Graille, J. Am. Oil Chem. Soc., 1990, 67, 646–649 CrossRef CAS.
- P. Nelson and A. Pelter, J. Chem. Soc., 1965, 5142 RSC.
- L. Y. Shteinberg, Russ. J. Org. Chem., 2003, 39, 972–974 CrossRef CAS.
- L. Y. Shteinberg, Russ. J. Appl. Chem., 2005, 78, 1715–1717 CrossRef CAS.
- T. Mohy El Dine, W. Erb, Y. Berhault, J. Rouden and J. Blanchet, J. Org. Chem., 2015, 80, 4532–4544 CrossRef CAS PubMed.
- N. Gernigon, R. M. Al-Zoubi and D. G. Hall, J. Org. Chem., 2012, 77, 8386–8400 CrossRef CAS PubMed.
- R. M. Lanigan, P. Starkov and T. D. Sheppard, J. Org. Chem., 2013, 78, 4512–4523 CrossRef CAS PubMed.
- P. Starkov and T. D. Sheppard, Org. Biomol. Chem., 2011, 9, 1320 RSC.
- F. Tinnis, H. Lundberg and H. Adolfsson, Adv. Synth. Catal., 2012, 354, 2531–2536 CrossRef CAS.
- M. Hosseini-Sarvari, E. Sodagar and M. M. Doroodmand, J. Org. Chem., 2011, 76, 2853–2859 CrossRef CAS PubMed.
- U. S. Pat., 2109941A.
- A. A. Bakibaev, V. K. Gorshkova, O. V. Arbit, V. D. Filimonov and A. S. Saratikov, Pharm. Chem. J., 1994, 28, 335–338 CrossRef.
- A. Khalafi-Nezhad, B. Mokhtari and M. N. Soltani Rad, Tetrahedron Lett., 2003, 44, 7325–7328 CrossRef CAS.
- K. Yamaguchi, H. Kobayashi, T. Oishi and N. Mizuno, Angew. Chem., Int. Ed., 2012, 51, 544–547 CrossRef CAS PubMed.
- M. Rezaei, K. Amani and K. Darvishi, Catal. Commun., 2017, 91, 38–42 CrossRef CAS.
- F.-L. Yang, X. Zhu, D.-K. Rao, X.-N. Cao, K. Li, Y. Xu, X.-Q. Hao and M.-P. Song, RSC Adv., 2016, 6, 37093–37098 RSC.
- S. Rostamnia, E. Doustkhah and B. Zeynizadeh, J. Mol. Catal. A: Chem., 2016, 416, 88–95 CrossRef CAS.
- L. Y. Shteinberg, S. A. Kondratov and S. M. Shein, Russ. J. Org. Chem., 2005, 41, 304–305 CrossRef CAS.
- Other solvent screen was performed at 80 °C but low conversions were observed.
- M. C. Etter, M. Zia-Ebrahimi, Z. Urbañczyk-Lipkowska and T. W. Panunto, J. Am. Chem. Soc., 1990, 112, 8415–8426 CrossRef CAS.
- A. O. F. Jones, C. K. Leech, G. J. McIntyre, C. C. Wilson and L. H. Thomas, CrysEngCommon, 2014, 16, 8177–8184 RSC.
- The reaction in neat was tested using Mg(NO3)2 giving similar results.
- The amide was formed the by the reaction of the acetate ligand with the urea.
- A. Khalafi-Nezhad, A. Parhami, M. N. Soltani Rad and A. Zarea, Tetrahedron Lett., 2005, 46, 6879–6882 CrossRef CAS.
- A. R. Hajipour and M. Ghasemi, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2001, 40B, 504–507 CAS.
- A. Zare, A. Hasaninejad, A. R. Moosavi-Zare, A. Parhami an. P. and A. Khalafi-Nezhad, Asian J. Chem., 2009, 21, 1090–1096 CAS.
- V. K. Das, R. R. Devi and A. J. Thakur, Appl. Catal., A, 2013, 456, 118–125 CrossRef CAS.
- A screen of other nitrogen sources was performed to ensure that urea was still the best when imidazole was employed as catalyst (Table S9, see ESI‡).
- The concentration, solvent and temperature were studied showing that the optimal conditions were using octane, 1 M at 120 °C (Tables S10–S12, see ESI‡).
- P. M. Schaber, J. Colson, S. Higgins, D. Thielen, B. Anspach and J. Brauer, Thermochim. Acta, 2004, 424, 131–142 CrossRef CAS.
- J. R. Cabrero-Antonino, R. Adam, K. Junge and M. Beller, Catal. Sci. Technol., 2016, 6, 7956–7966 RSC.
- E. Cherbuliez and F. Landolt, Helv. Chim. Acta, 1946, 29, 1438–1446 CrossRef CAS.
- H. R. V Arnstein and R. Bentley, J. Chem. Soc., 1951, 3509–3510 Search PubMed.
- W. Ried and H. Nenninger, Synthesis, 1990, 167–170 CrossRef CAS.
- T. Maki, K. Ishihara and H. Yamamoto, Synlett, 2004, 1355–1358 CAS.
- I. Aiad, D. Emam, A. El-Deeb and E. Abd-Alrahman, J. Surfactants Deterg., 2013, 16, 927–935 CrossRef CAS.
- M. Yamashita, H. Murai, A. Mittra, T. Yoshioka, I. Kawasaki, M. Gotoh, T. Higashi, R. Hatsuyama and S. Ohta, Heterocycles, 1998, 48, 2327–2337 CrossRef CAS.
- The direct coupling of imidazole with the carboxylic acid under our reaction conditions was performed showing no conversion. We believe that the presence of urea is crucial to enhance the hydrogen bonding among the reagents and promote their reactivity.
- W. P. Jencks and J. Carriuolo, J. Biol. Chem., 1958, 234, 1280–1285 Search PubMed.
- D. Kaufmann, M. Bialer, J. A. Shimshoni, M. Devor and B. Yagen, J. Med. Chem., 2009, 52, 7236–7248 CrossRef CAS PubMed.
- J. D. Taylor, G. B. Trimitsis, T. Hudlicky and J. F. Wolfe, J. Org. Chem., 1973, 38, 1236–1238 CrossRef CAS.
Footnotes |
| † The paper is dedicated to the memory of Prof. J. M. J. Williams. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures and spectroscopic characterisation of all organic compounds. See DOI: 10.1039/d0sc01317j |
| § These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |