
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLuminescent tungsten(VI) complexes as photocatalysts for light-driven C–C and C–B bond formation reactions†
Daohong
Yu
 ab,
Wai-Pong
To
b,
Glenna So Ming
Tong
b,
Liang-Liang
Wu
b,
Kaai-Tung
Chan
b,
Lili
Du
b,
David Lee
Phillips
b,
Yungen
Liu
a and
Chi-Ming
Che
*abc
ab,
Wai-Pong
To
b,
Glenna So Ming
Tong
b,
Liang-Liang
Wu
b,
Kaai-Tung
Chan
b,
Lili
Du
b,
David Lee
Phillips
b,
Yungen
Liu
a and
Chi-Ming
Che
*abc
aDepartment of Chemistry, Southern University of Science and Technology, Shenzhen, Guangdong 518055, China. E-mail: cmche@hku.hk
bState Key Laboratory of Synthetic Chemistry, HKU-CAS Joint Laboratory on New Materials, Department of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong, China
cHKU Shenzhen Institute of Research and Innovation Shenzhen, Guangdong 518055, China
First published on 25th May 2020
Abstract
The realization of photocatalysis for practical synthetic application hinges on the development of inexpensive photocatalysts which can be prepared on a large scale. Herein an air-stable, visible-light-absorbing photoluminescent tungsten(VI) complex which can be conveniently prepared at the gram-scale is described. This complex could catalyse photochemical organic transformation reactions including borylation of aryl halides, such as aryl chloride, reductive coupling of benzyl bromides for C–C bond formation, reductive coupling of phenacyl bromides, and decarboxylative coupling of redox-active esters of alkyl carboxylic acid with high product yields and broad functional group tolerance.
Introduction
Transition metal photocatalysis has become an intensively investigated area and has developed rapidly as a useful strategy for a broad range of organic transformations under mild conditions.1 Most of the photocatalysts employed in these studies are usually based on platinum group metals such as RuII and IrIII due to the efficient light-absorption properties of their complexes in the visible spectral region and accessibility to attain triplet excited states with a sufficient lifetime for bimolecular energy transfer and electron transfer reactions upon photo-excitation. Non-precious metal complexes such as those of Cu,2 Ni,3 Zr,4 Mo,5 Cr,6 Fe,7 and Ce8 have also been studied and applied in light-driven organic transformation reactions. Meanwhile, earth-abundant tungsten(VI) oxides such as WO3 and polyoxotungstate9 display rich photoluminescence, an example of which is (n-Bu4N)4[W10O32] photocatalysed C–H bond functionalization.10 Nonetheless, tungsten oxide has low solubility in common organic solvents and the decatungstate anion only absorbs in the UV spectral region up to 400 nm.11 In this regard, the design and modification of tungsten oxide coordinated with ligands are appealing. Tungsten, though having a large spin–orbit coupling constant (ζ = 2433 cm−1) for efficient intersystem crossing from the singlet to the triplet excited state, has rarely been explored in the design of photo-functional molecular materials. In the literature, several luminescent W0 isocyanides12 have been found as “proof-of-concept” strong photo-reductants.Recently, we reported air-stable luminescent W(VI) complexes and described their applications in organic light-emitting diodes as well as photo-sensitization of singlet oxygen and reduced oxygen species (O2˙−).13 Herein the broad applications of W(VI) oxo complexes in visible-light driven coupling reactions for C–C bond formation are described. Given the ease of preparation and facile tuning of the photophysical properties of the Schiff base ligand scaffold, we further modified the structure of luminescent W(VI) Schiff base complexes to increase their absorption in the visible spectral region and to modulate the excited state reduction potential by introducing an electron-donating group into the ligand.14,15 These complexes (Fig. 1, W1a–W1c) display photoluminescence with quantum yields of up to 0.13 and lifetimes of up to 83.6 μs in dichloromethane solution at room temperature. With these complexes as the photocatalyst, reactions such as borylation of aryl halides, including aryl chloride, and C–C reductive coupling of benzyl bromides were achieved in moderate to good product yields under blue LED light irradiation.
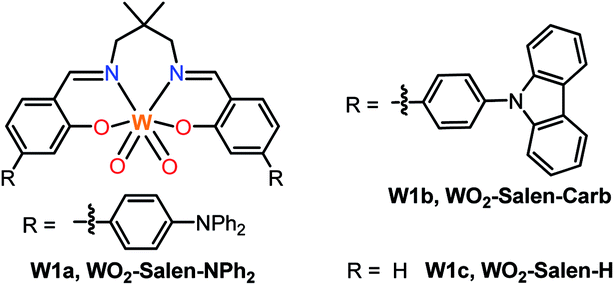 | ||
| Fig. 1 Tungsten(VI) complexes W1a–W1c studied in this work. | ||
Results and discussion
Synthesis and characterization
Schiff base ligands were synthesized by condensation reaction of the respective salicylaldehyde with 2,2-dimethylpropane-1,3-diamine. Strongly electron-donating diphenylamine (–NPh2) substituents are present on the phenoxide moiety of W1a so as to generate an intraligand charge transfer (ILCT) excited state with large molar absorptivity in the visible spectral region and with the photo-generated “hole” localized mainly on the –NPh2 groups for improving the stability of the metal complex in the course of photocatalysis. The W(VI) complexes were synthesized by refluxing a mixture of the ligand and [W(eg)3] (eg = 1,2-ethanediolato) in MeOH/CHCl3 (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and obtained as a purified product in 68–90% yields. The synthesis of W1a could be scaled up to give 1 g of the target complex in a one-pot reaction. These complexes are air- and moisture-stable yellow solids. They exhibit good solubility in CH2Cl2, THF and DMF and excellent stability in these solvents in the absence of light. The photo-stability of these W(VI) complexes in DCM and DMF was examined. Light irradiation (450 nm LED) of W1a in [D2]DCM or [D7]DMF in the absence of oxygen for 48 hours led to a slight change in the 1H NMR spectrum (Fig. S1†). However, W1b and W1c were found to decompose completely under the same conditions, suggesting that the presence of NPh2 provides a higher photo-stability to W1a.
:1) and obtained as a purified product in 68–90% yields. The synthesis of W1a could be scaled up to give 1 g of the target complex in a one-pot reaction. These complexes are air- and moisture-stable yellow solids. They exhibit good solubility in CH2Cl2, THF and DMF and excellent stability in these solvents in the absence of light. The photo-stability of these W(VI) complexes in DCM and DMF was examined. Light irradiation (450 nm LED) of W1a in [D2]DCM or [D7]DMF in the absence of oxygen for 48 hours led to a slight change in the 1H NMR spectrum (Fig. S1†). However, W1b and W1c were found to decompose completely under the same conditions, suggesting that the presence of NPh2 provides a higher photo-stability to W1a.
Spectroscopic and photophysical properties
The spectroscopic and excited state properties of these W(VI) complexes have been examined (Table 1 and S1†). In CH2Cl2, complex W1a shows two intense (3.84–4.98 × 104 M−1 cm−1), broad absorption bands with maxima at 297 and 407 nm, the latter of which ends at ∼510 nm, while W1b and W1c exhibit intense (1.82–5.71 × 104 M−1 cm−1) absorption bands at 250–320 nm and a relatively weak band (5.5–11.4 × 103 M−1 cm−1; shoulder for W1b) at 402–420 nm (Fig. 2). Complex W1b exhibits additional vibronic-structured absorption at ∼290 and 340 nm, ascribable to the absorption of the carbazole unit on the ligand. These absorption bands are assigned to the 1IL (singlet intraligand) transitions of the ligands, whereas the intense absorption band of W1a at 407 nm is proposed to have significant ILCT characteristics due to the electron-rich diphenylamine unit on the ligand. The strong absorptivity of W1a at a wavelength >400 nm is advantageous for this complex to catalyse visible light driven reactions when compared to W1b and W1c. Upon light excitation, W1a exhibits a broad, structureless emission with a peak maximum at 608 nm and with an emission quantum yield of 0.11 and lifetime of 4.6 μs. W1b and W1c exhibit photoluminescence with quantum yields of 0.13 and 0.03, respectively, and much longer emission lifetimes of 74.9 and 83.8 μs, respectively. The much higher emission quantum yields of W1a and W1b compared to W1c indicate that installing an amino substituent at the 4-position of the phenolate moiety can enhance the photoluminescence quantum yield of this class of complexes. While their emission profiles appear broad and structureless, a shoulder can be observed at ∼500 nm. By examining their emission in aerated solutions (Fig. 3), it was found that these complexes exhibit prompt fluorescence which appears as a broad band with peak maxima at 509 and 513 nm, respectively. Their fluorescence emission quantum yields are estimated to be 0.006 and 0.001, respectively. The excitation spectra obtained at these emission wavelengths are highly similar to the absorption spectra of the respective complexes, confirming that this fluorescence originates from the W(VI) complexes (Fig. S3†). The long emission lifetimes of W1b and W1c also render their phosphorescence completely quenched under aerated conditions. Together with the minor solvent effect (<7 nm) on the emission for W1b and W1c (Table S1†), the emissions of W1b and W1c originate from 3IL excited states. For W1a, a broad emission band with a peak maximum at 605 nm and an emission lifetime of 0.54 μs was observed under aerated conditions, implying that its emission was not completely quenched in the presence of dissolved oxygen (Fig. S4†). Femtosecond time-resolved emission (fs-tre) measurements (Fig. 4a) revealed that W1a shows a broad, structureless fluorescence peak with the peak maximum at ∼560 nm at the early stage (within 1 ps); this fluorescence then showed a dynamic Stokes shift to ∼620 nm in about 3 ps and decayed with a time constant of 324 ps. The steady state emission of W1a exhibits significant positive solvatochromism (λmax = 510 nm in toluene, 572 nm in THF, and 608 nm in CH2Cl2; Fig. 4b and Table S1†), and the emission quantum yield and lifetime of W1a even reach 0.28 and 22.1 μs in degassed CHCl3. The positive solvatochromism is suggestive of substantial charge-transfer characteristics in its emissive excited state and its emission in toluene originates predominantly from the 3ILCT excited state according to the DFT calculation in our previous work.15 In toluene, W1a shows a structured emission with the peak maximum at 510 nm and a shoulder at 552 nm with a quantum yield and lifetime of 0.04 and 67.6 μs, respectively, which corresponds to a radiative decay rate constant (kr) of 600 s−1. But upon changing the solvent to CHCl3 and CH2Cl2, its emission maximum red-shifts to 583 and 608 nm, and kr increases to 1.27 × 104 and 2.39 × 104 s−1, which are, respectively, 21 and 40 times larger than those in toluene (Table S1†). Such an increase in kr implies a change in the emission origin to a faster radiative decay mechanism, possibly with the involvement of TADF.15 A similar emission peak maximum observed with fs-tre (∼620 nm), which represents the S1 → S0 prompt fluorescence, and steady state emission measurement (608 nm) also support such a possibility. It is noted that although both W1a and W1b are appended with amino-substituents, W1a exhibits an emission with a larger kr of 2.39 × 104 s−1 and with the involvement of TADF while W1b displays long-lived 3IL emission. One possible reason accounting for this difference is the much higher-lying HOMO of W1a (0.31 V higher than that of W1b; Table 2) resulting in the 1/3ILCT excited states being lower-lying than the 3IL excited state. A similar finding for the difference in the luminescence properties between complexes having carbazole and diphenylamine, respectively, was observed in pincer Au(III) aryl complexes as well.16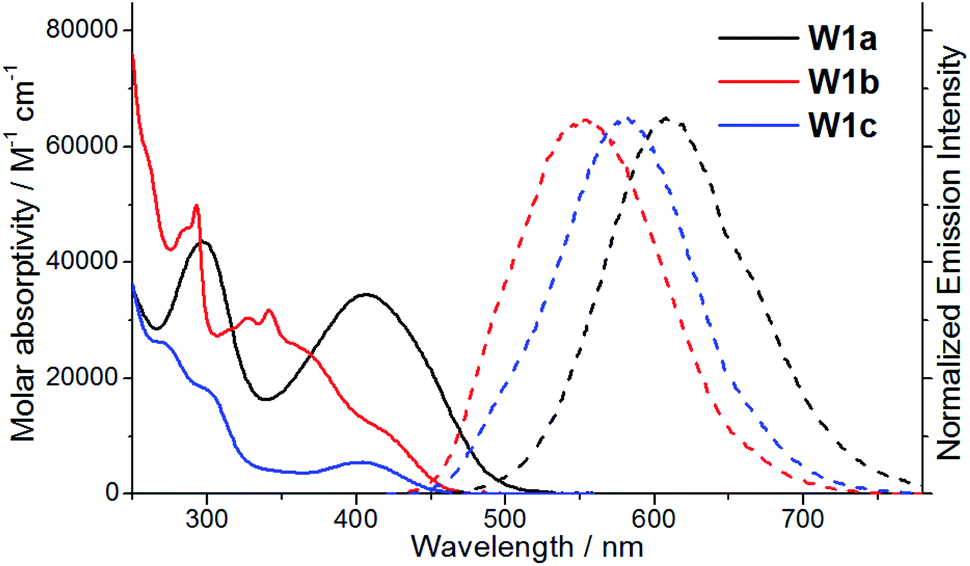 | ||
| Fig. 2 UV-vis absorption (solid line) and emission (dashed line) spectra of W1a–W1c in degassed dichloromethane solutions at room temperature. | ||
| Absorption | Emission | |
|---|---|---|
| λ abs [nm] (ε [×103 mol−1 dm3 cm−1])a | λ em [nm] (Φ; τ [μs]; kr [103 s−1]) | |
| a Data were measured in degassed CH2Cl2 at 298 K. Emission quantum yields (Φ) were estimated with [Ru(2,2′-bipyridine)3](PF6)2 in degassed CH3CN as the standard (Φ = 0.062). The radiative decay rate constant is estimated using kr = Φ/τ. b Values taken from ref. 15. | ||
| W1a | 297 (43.7), 407 (34.6) | 608 (0.11; 4.6; 23.9)b |
| W1b | 261 (57.1), 285 (45.9), 292 (49.8), 327 (30.4), 341 (31.9), 365 (sh, 25.0), 420 (sh, 10.6) | 553 (0.13; 74.9; 1.7) |
| W1c | 270 (26.2), 299 (18.2), 402 (5.5) | 582 (0.03; 83.6; 0.4)b |
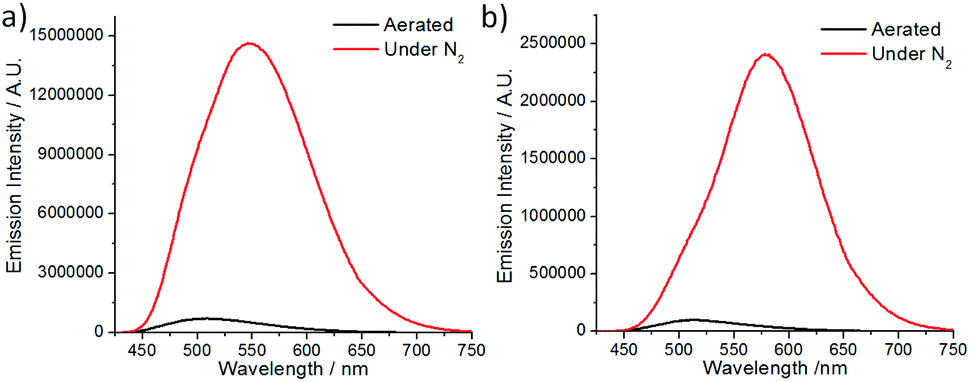 | ||
| Fig. 3 Emission spectra of (a) W1b and (b) W1c in CH2Cl2 under aerated conditions and under N2. | ||
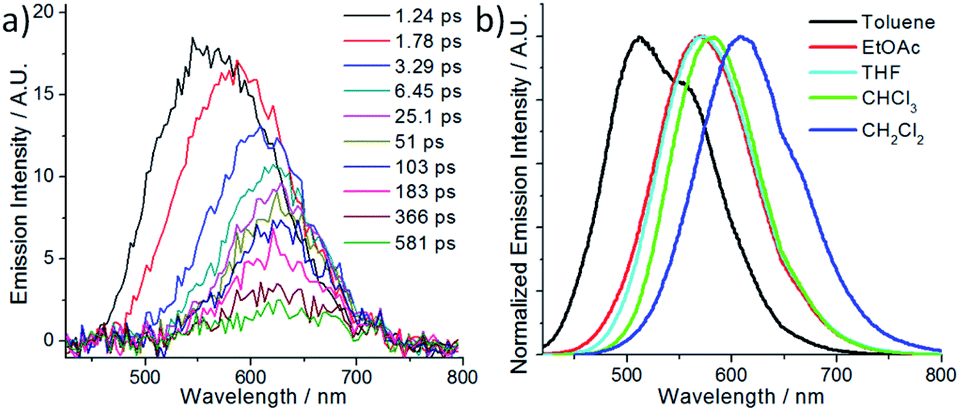 | ||
| Fig. 4 (a) Femtosecond time-resolved emission spectra of W1a in CH2Cl2 and (b) emission of W1a in different solvents (adapted with permission from ref. 15. Copyright 2019, Wiley-VCH Verlag GmbH & Co. KGaA). | ||
DFT/TDDFT calculations
In most cases, the charge transfer absorption band is usually weak due to spatial separation of the donor and acceptor orbitals which minimizes the overlap between the two wavefunctions, and hence the transition dipole moment.15 However, it is observed herein that the first absorption peak of W1a, which has been assigned as derived from the S0 → 1ILCT transition, is rather intense (ε in the order of 104 M−1 cm−1). This seems quite unusual, especially when compared with the first absorption peak of W1c, which has been assigned to the S0 → 1IL transition with ε in the order of 103 M−1 cm−1. Time-dependent density functional theory (TDDFT) calculations at their respective optimized S0 geometries have been performed to understand this phenomenon. The computed first absorption band is at 370 and 390–443 nm in toluene for complexes W1c and W1a, respectively, which is in reasonable agreement with the experimentally observed absorption band (Fig. S15† displays the simulated absorption spectra of W1a and W1c). In particular, the computed first absorption band of W1a is more intense than that of W1c, as reflected by the absorption band oscillator strengths (f = 0.078 and 0.31–0.49 for W1c and W1a, respectively), which is also in line with experimental observations. These lend support to the reliability of the computational results for rationalization of the experimental data. The energy of the oxide(O2−)-to-tungsten charge transfer (LMCT) transition of W1c and W1a was estimated using TDDFT calculations at their respective optimized ground state geometry. It was found that the S0 → 1LMCT excitation is at 283 nm in toluene for both complexes, which is in agreement with the excitation λmax of [WO2X4]2− at ∼250–320 nm (X = Cl and F).17The intensity of an absorption band intimately depends on the transition dipole moment (M) between the ground state and the excited state (say, Sn),
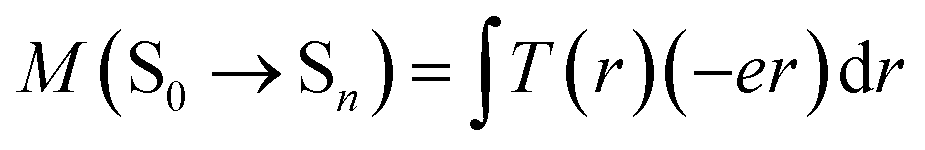
T(r) is the transition density of the S0 → Sn transition, r is a point in three-dimensional space, and e is the electronic charge. The transition density T(r) is related to the overlap density between the two MOs (φi and φa) involved in an excitation:
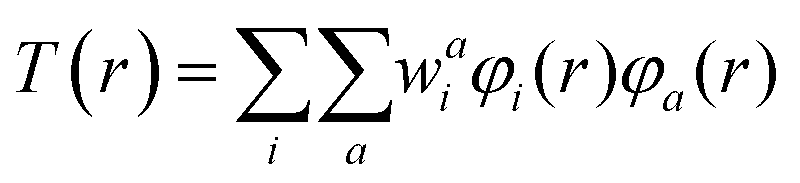
Since the transition dipole moment also depends on r, it may be more intuitive to compare the transition dipole moment density (TDMD), T(r) r and the fragment transition dipole moment (TDM) which is the spatial integrals of the TDMD of the fragment. Fig. 5 depicts the TDMD and fragment TDM along the z-direction of the S0 → S1 excitation. Attaching TPA to the Schiff base ligands extends the transition density distribution onto the TPA ligand, leading to an increase in the TDMD and a substantial fragment TDM at the TPA moieties (−0.086 and 0.916 a.u. for W1c at the Schiff base ligand and 0.516 and −3.23 a.u. at the TPA moieties for W1a). Similarly, the TDMD and the fragment TDM of the S0 → S2 and S0 → S3 excitations have substantial contributions at the TPA moieties of W1a. (Fig. S19 and S20†) Thus, increasing the donor–acceptor distance while maintaining significant transition density could lead to substantial enhancements of the S0 → Sn intensities, which accounts for the intense absorption of W1a.
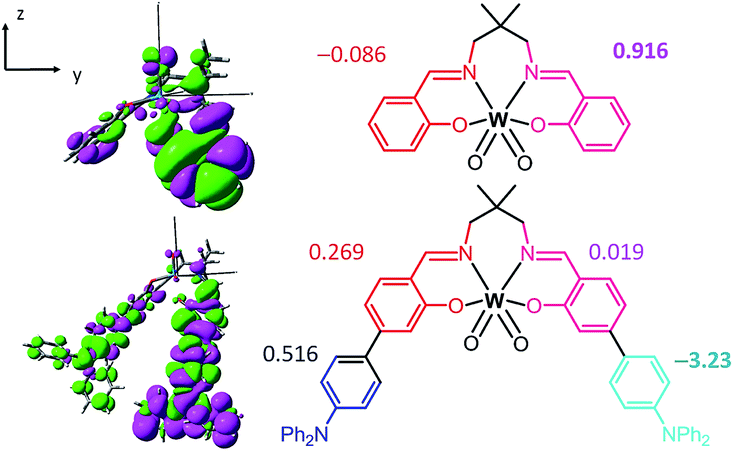 | ||
| Fig. 5 Transition dipole moment density (left) and the fragment transition dipole moment (right) along the z-direction of the S0 → S1 excitation in toluene solution at the respective optimized S0 geometries of W1c (top) and W1a (bottom). Colour code: magenta, negative; green, positive; isovalue = 0.001 a.u. | ||
Excited state dynamics
The early events of excited state dynamics have been probed by femtosecond time-resolved absorption (fs-ta) spectroscopy. As portrayed in Fig. 6a, the fs-ta spectrum obtained for W1a in CH2Cl2 at various time intervals after laser excitation (400 nm) evolved from having an absorption difference peak maximum at ∼480 nm and shoulder at 570 nm to a broad absorption profile spanning from 500 to 750 nm with a time constant of 370 ps, the latter of which is close to the fs-tre decay time constant (τ620nm = 324 ps). Excitation at 266 nm gave similar spectral evolution (Fig. S7†). For W1b, the fs-ta spectra recorded within 10 ps show a peak maximum at ∼470 nm and a broader absorption band covering 550–700 nm (Fig. 6b). Then, the absorption difference at ∼470 nm decreases while that at 500–700 nm grows with a time constant of 83 ps. For W1c, at 1.9 ps after laser excitation, the fs-ta spectrum shows a peak maximum at ∼480 nm and a broad, less intense absorption band beyond 650 nm (Fig. S8†). The spectrum then evolves with a time constant of 42 ps to generate a new profile with a broader absorption band with the peak maximum at ∼490 nm and a weaker, broad absorption band beginning at ∼570 nm. Since the final spectral profiles observed in these fs-ta spectra highly resemble those obtained in ns-ta spectra (Fig. S6†), these spectral transformations are ascribed to intersystem crossing (ISC) from S1 to the triplet manifold. The ISC time constants of 101–102 ps observed for these complexes are of the same order of magnitude as reported for the W(VI) Schiff base dioxo complexes.13,15 However, these ISC time constants are quite large when compared to those of Pt(II) and Au(III) complexes which usually lie in the range of 0.1 to a few ps.18 These slow ISC time constants also account for the observation of fluorescence bands of W1b and W1c in steady state emission measurements. For W1a, since the ILCT excited state would lead to a small energy difference between the S1 and T1 excited states, the emission originating from these two states have significant spectral overlap and the prompt fluorescence band (∼560–620 nm) could only be observed with femtosecond time-resolved emission spectroscopy (Fig. 4a).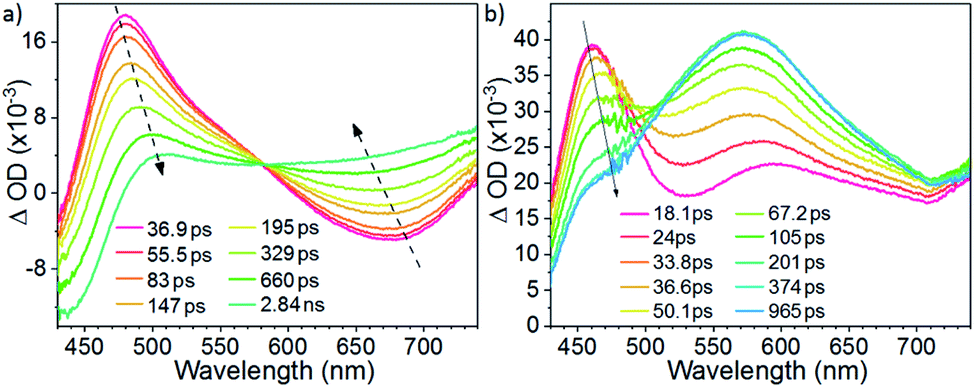 | ||
| Fig. 6 fs-TA spectra of (a) W1a and (b) W1b in CH2Cl2 at room temperature. | ||
Electrochemistry and excited state redox potentials
The electrochemical properties of these complexes have been examined by cyclic voltammetry in DMF containing 0.1 M nBu4NPF6 as the electrolyte. These complexes exhibit an irreversible ligand-centred reduction wave at −1.24 to −1.34 V vs. SCE and an irreversible ligand-centred oxidation wave at +1.10, +1.41 and +1.64 V vs. SCE for W1a, W1b and W1c, respectively. The reduction wave of W1a remains irreversible even at a scan speed of 1 V s−1, suggesting that the reduced form of W1a has limited stability under electrochemical conditions (Fig. S13†). The increasing ease of oxidation on moving from W1c to W1a is ascribed to the presence of electron-donating carbazole and diphenylamine groups on their respective phenoxide moieties. The E0–0 values of W1a, W1b and W1c are estimated to be 2.39, 2.70 and 2.59 eV, respectively, accordingly to the onset of their triplet emission in MeCN. By combining their spectroscopic and electrochemical data, the excited state reduction potentials [E(W*/W−)] of W1a, W1b and W1c were estimated to be +1.10, +1.46 and +1.25 V vs. SCE, respectively (Table 2), indicating that these tungsten complexes are good photo-oxidants. The redox potentials [E(W+/W*)] are estimated to be −1.29, −1.29 and −0.95 V vs. SCE, respectively, which denotes the reducing nature of their excited states.Photocatalysis
We first examined the feasibility of the WVI-oxo complex to perform photochemical dehydrogenation of alcohols by photolysis of W1a in the presence of secondary alcohol substrates. It was found that 1-indanol and 1-phenylethanol could be oxidized to the corresponding ketones in 52% and 16% yields, respectively, after 12 hours of light irradiation with LEDs (365 nm) (Scheme 1; Table S3†). No reaction was observed in the absence of oxygen. Changing the light source to 450 nm LEDs resulted in only 15% and 9% conversion of these alcohols to ketones, suggestive of the need for high-energy excitation for efficient dehydrogenation of benzyl alcohols. Cyclohexa-1,4-diene and Hantzsch ester, which have much weaker C–H bonds, were readily converted to benzene and the corresponding pyridine in almost quantitative yields (Scheme 1).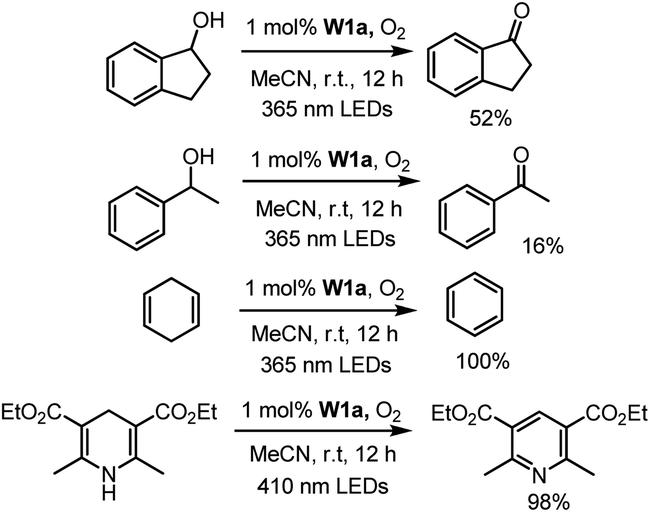 | ||
| Scheme 1 Photo-induced dehydrogenation reactions catalysed by W1a. | ||
Next, we examined the catalytic activity of these complexes in photo-redox reactions. Aryl boronic acid and esters are important building blocks for the formation of aryl carbon–carbon or carbon-heteroatom bonds.19 Recently, a number of strategies for photo-induced borylation of aryl halides via direct ultraviolet light irradiation,20 in the presence of a metal-based photocatalyst8i,21 or in situ generated electron donor21 have been developed. At the outset, visible-light-induced borylation of 1-iodo-3,5-dimethylbenzene with bis(pinacolato)-diboron (B2Pin2) was chosen as a model reaction with the most commonly used Hunig base (DIPEA) as a reductive quencher. To our delight, the borylation of aryl iodide proceeded smoothly and gave the desired product in 52% yield together with m-xylene derived from the hydrodehalogenation of aryl halide as the major by-product. Other reaction parameters such as the base, additive, and solvent were further optimized (see Table S4† for details). Meanwhile, we discovered that the reaction protocol was also applicable to aryl bromide and aryl chloride, the latter of which was recently reported to be an effective substrate in photo-redox reactions.8i,22 The reaction of ethyl 4-chlorobenzoate (1b) and B2Pin2 under the same conditions afforded the desired product 2b in 53% yield and the by-product ethyl benzoate in 41% yield (Table S5,† entry 1). Further screening of reaction conditions revealed that when tetrabutylammonium bromide (TBAB) or TBAB/KI was used, the formation of the dehalogenated by-product was suppressed efficiently (Table S5,† entry 15, 16) to ≤9%. In the absence of a photosensitizer, the product yield was greatly reduced to 20%, indicative of the role of W1a in increasing the efficiency of the reaction (Table S5,† entry 17). The reaction conducted in the absence of light did not show conversion of the substrate, confirming that visible light is critical to the reaction (Table S5,† entry 18). Under the same conditions, low product yields of 14–30% were obtained when using W1b and W1c as the catalyst.
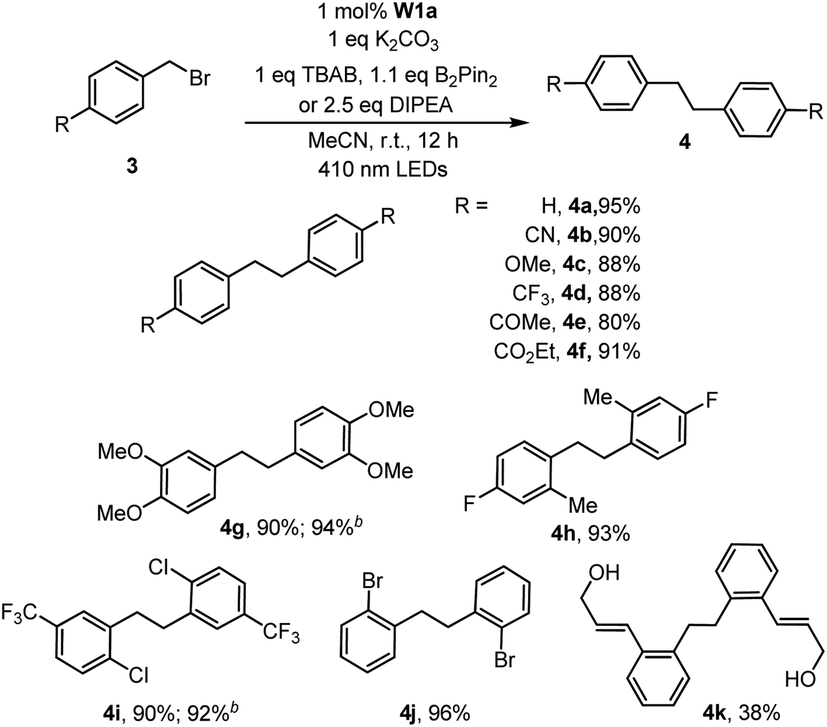
With the optimal conditions, we explored the substrate scope of the borylation reaction with aryl halides (Table 3). Reactions of aryl chlorides with an electron-withdrawing group such as ester, amide, ketone and nitrile proceeded smoothly to give the corresponding products in good to excellent yields (Table 3, 2b–c, 2f–h). This borylation reaction showed good tolerance to various functional groups such as free alcohol and alkyl bromide (Table 3, 2i, 2l). Sterically hindered aryl chloride could also give the corresponding product in 81% yield (Table 3,2j). However, substrates with methyl substituent(s) generally gave a moderate yield (50–66%) of products (Table 3, 2a, 2m–o). The reaction of alkyl halides under the same reaction conditions has also been examined. When 2-(bromomethyl)-1-chloro-4-(trifluoromethyl)benzene (3i) was subjected to the borylation reaction, the borylated product was not detected. Instead the 1,2-diphenylethane product was isolated in high yield (Table 4, 4i). The reductive homocoupling reaction of benzyl halides has been reported.2b,c,8a,23,24 In this work, reductive coupling of benzyl bromide occurred smoothly to give the dimerization product, 1,2-diphenylethane, under similar reaction conditions (Table 4). Interestingly, although no borylation product was found in the reaction mixture, the presence of B2Pin2 is critical for the photo-redox reaction, without which the yield of the desired coupling product decreased dramatically to 30%. After further screening, we discovered that the use of DIPEA instead of the combination of TBAB and B2Pin2 gave slightly higher product yields of 4g and 4i. Both methods are effective for the reductive coupling reaction of benzyl bromide. A variety of benzyl bromides with electron-withdrawing groups or electron-donating groups delivered the products in 80–95% yields (Table 4, 4a–g). As for the substrate having both benzylic and arylic bromide groups, benzylic bromide showed its priority in the reaction (Table 4, 4j).
| a Reaction conditions: 1 (0.5 mmol), B2Pin2 (0.55 mmol), W1a (1 mol%), K2CO3 (0.5 mmol), TBAB (0.5 mmol) in MeCN and irradiated using 410 nm LEDs at room temperature for 12 h. Isolated yields. |
|---|
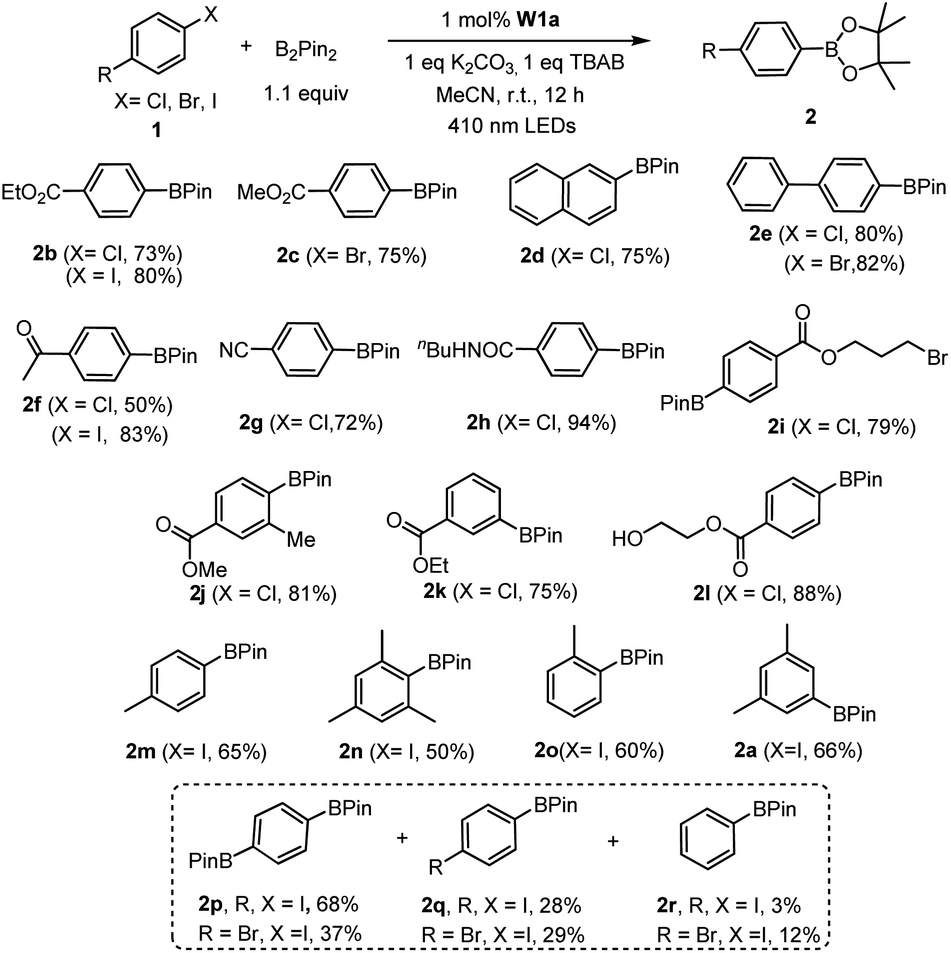
|
The capacity of W1a in catalysing the reductive dehalogenation of arylacyl bromide which has been reported by other groups25 was also examined (Table 5). 1,4-Dicarbonyl compounds were obtained in 74–94% yields, presumably due to the absence of a proton source in the reaction. When cinnamyl bromide was used, the dimerized product was obtained in 73% yield, with the ratio of isomer products being about 1:0.6:0.1.
| a Reaction conditions: 7 (0.5 mmol), B2Pin2(0.55 mmol), W1a (1 mol%), K2CO3 (0.5 mmol), TBAB (0.5 mmol) in MeCN and irradiated using 410 nm LEDs at room temperature for 12 h. Isolated yields. |
|---|
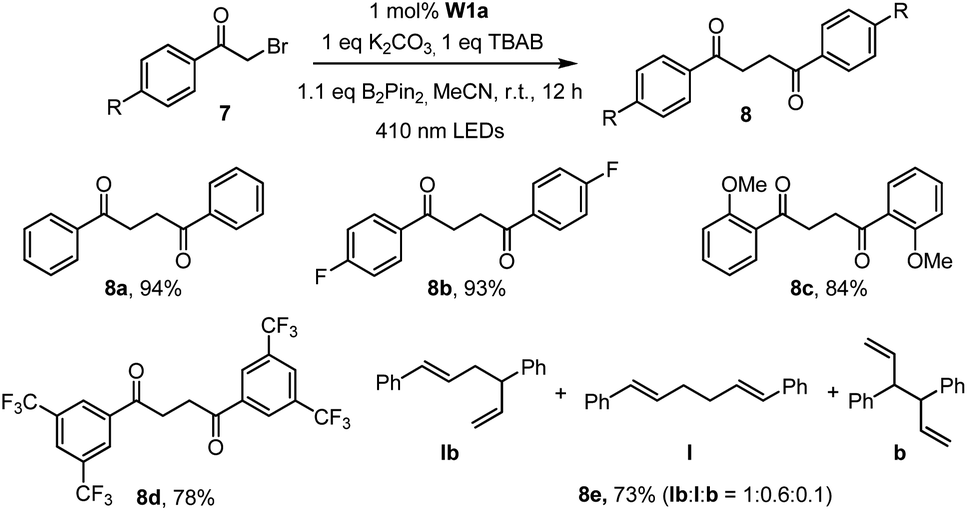
|
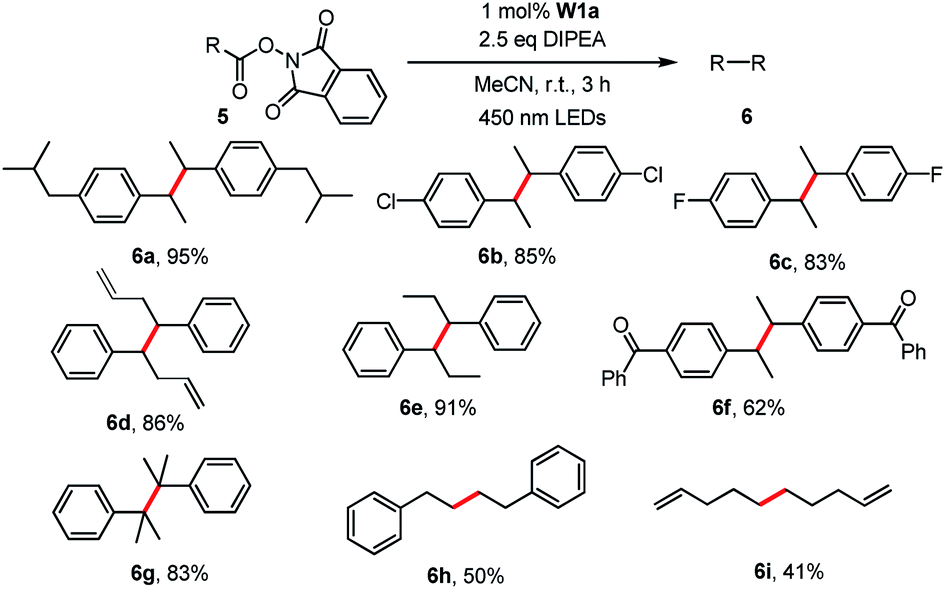
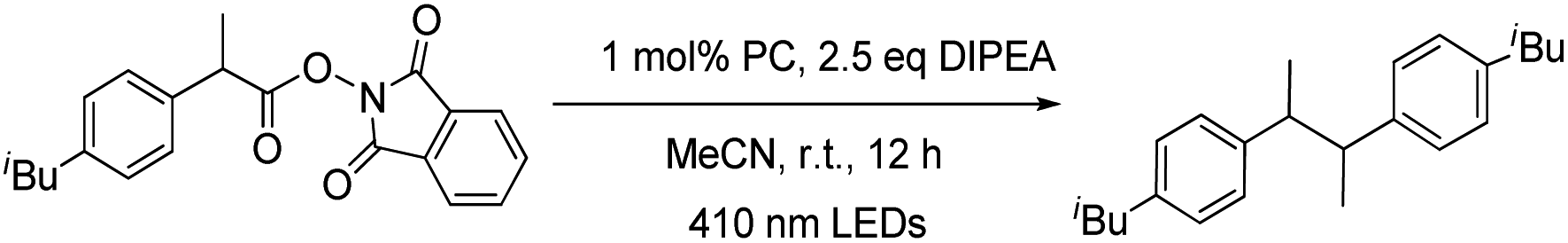
Redox-active esters26 (RAEs; e.g. N-hydroxyphthalimide ester 5) derived from alkyl carboxylic acid as convenient surrogates for alkyl halides were examined for the generation of alkyl radicals which could be trapped with various reagents (e.g. diselenide,27 Michael acceptors,26e,28 CCl4,29 1-bromocarbagermatranes30 and benzophenone imine31). In this work, the light-induced decarboxylative radical addition reaction of redox-active esters in the presence of styrene or acrylate ester was examined. No radical addition product was detected in the presence of W1a and DIPEA. Different from most related literature reports, we found that the light-induced decarboxylative coupling reaction occurred to give sterically hindered 1,2-diphenylethanes (Table 6).32 Decarboxylative coupling products, including those having Cl and F substituents, could be obtained in 50–95% yields. Alkene groups were tolerated albeit at low yield (41%) due to the formation of the decarboxylative hydrogenation product.
| a Reaction conditions: 5 (0.5 mmol), W1a (1 mol%), DIPEA (1.25 mmol) in MeCN and irradiated using 450 nm LEDs at room temperature for 3 h. Isolated yields. |
|---|
|
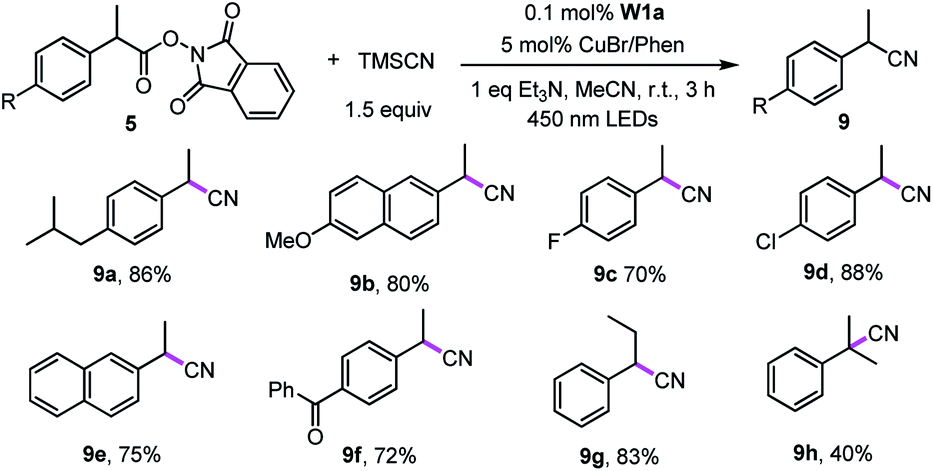
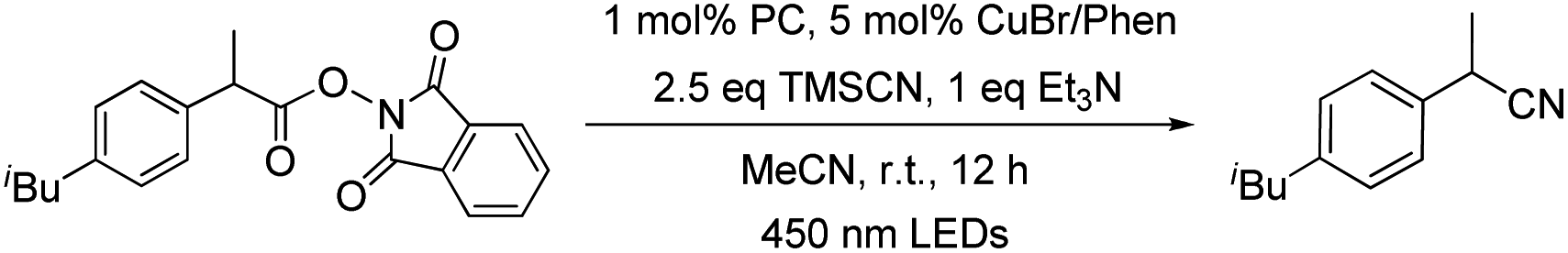
Recently, Liu group reported that benzyl radicals generated from N-hydroxylphthalimide ester can be trapped by a reactive copper(II) cyanide to deliver benzyl nitriles.33 Since our tungsten system could provide benzyl radicals via light irradiation, the decarboxylative cyanation of redox-active esters employing cooperative tungsten photocatalysis and copper catalysis was examined (Table 7). Thus, various redox-active-esters derived from simple carboxylic acids were found to provide the corresponding nitrile products in good yields (72–88%) in the presence of only 0.1 mol% W1a. Quaternary benzyl nitrile, which could not be obtained in Liu's catalytic system, was furnished using our protocol in 40% yields.
| a Reaction conditions: 5 (0.5 mmol) W1a (0.1 mol%), TMSCN (0.75 mmol), CuBr (5 mol%), Phen (5 mol%), Et3N (0.5 mmol) in MeCN and irradiated using 450 nm LEDs at room temperature for 3 h. Isolated yields. |
|---|
|
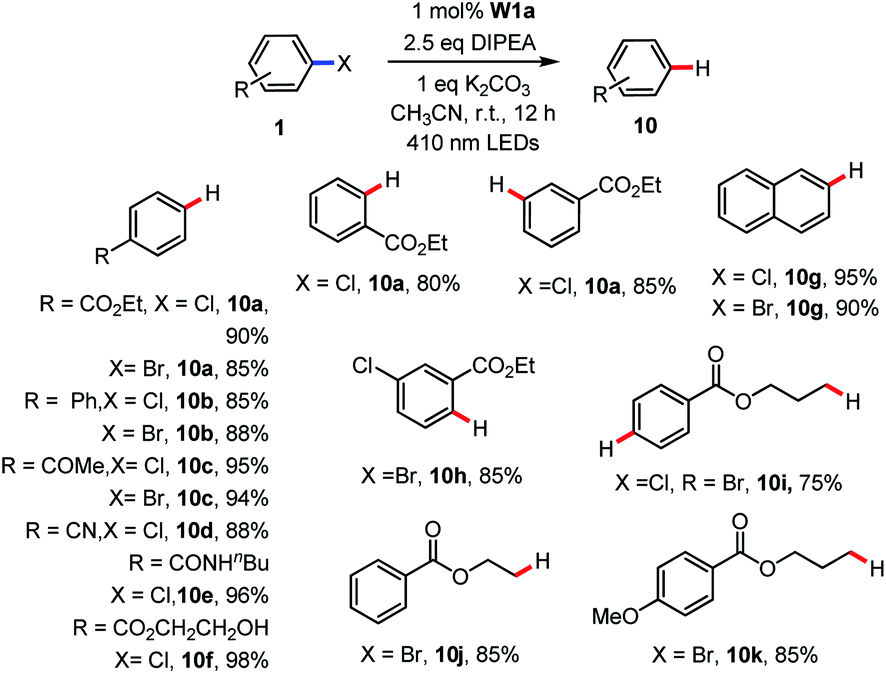
During the study on the C–B bond formation reaction (Table 3), we observed the formation of dehalogenated by-products. In the literature, there are a number of examples on light-induced hydrodehalogenation of aryl halides.14,34 Here, by simply removing B2Pin2 from the reaction conditions, the reductive dehalogenation of aryl halides occurred smoothly with good to excellent yields (Table 8). A panel of functional groups such as ester, ketone, cyano, amide and alcohol were tolerated. The reaction was not sensitive to steric hindrance. Ortho or meta substituted aryl chloride delivered the corresponding product in 80–85% yields. In the presence of aryl bromide and chloride, the aryl bromide was reduced preferentially with 85% yield while the chloride remained intact. In particular, alkyl bromides were reduced to afford the hydrodehalogenated product in 75–85% yields (10i–10k). When the substrate 3-bromopropyl 4-chlorobenzoate which contains aryl chloride and alkyl bromide in the same molecule was used, double reductive dehalogenation occurred to give propyl benzoate (10i) in 75% yield.
| a Reaction conditions: 1 (0.5 mmol), W1a (1 mol%), DIPEA (1.25 mmol), K2CO3 (0.5 mmol) in MeCN at room temperature and irradiated using 410 nm LEDs for 3 h. Isolated yields. |
|---|
|
Photo-induced decarboxylative alkylation of silyl enol ether was reported to give aryl alkyl ketones with N-(acyloxy)phthalimide serving as the alkyl radical source.35 Inspired by this work, we attempted tungsten catalysed alkylation of silyl enol ether with benzyl bromide. Instead of the expected product, bibenzyl and pinacol products (2,3-diphenylbutane-2,3-diol) were detected by GC-MS and 1H NMR spectroscopy. Interestingly, in the absence of benzyl bromide, pinacol product 12a was obtained in 94% yield (Table 9). Silyl enol ethers of acetophenone with methyl or trifluoromethyl groups afforded the corresponding products in 72% and 52% yields, respectively. While the mechanism of the homocoupling reaction of silyl enol ether to give pinacol is not clear, the ketyl radical36b–d is speculated to be involved in the reaction. It is noted that the light-induced reductive homocoupling reaction of benzaldehyde or acetophenone to synthesize pinacol has been reported in recent years.14,34i,36
| a Reaction conditions: 11 (0.5 mmol), W1a (1 mol%), DIPEA (1.25 mmol), K2CO3 (0.5 mmol) in MeCN at room temperature and irradiated using 410 nm LEDs for 12 h. Isolated yields. |
|---|

|
Mechanistic study
Several reactions shown in Fig. S10† were carried out to gain insights into the reaction mechanism. Firstly, when TEMPO (2,2,6,6-tetramethyl-1-piperidyloxy) as a radical scavenger was added to the reaction mixture for photo-induced homocoupling reaction of benzyl bromide (Table 4), the dimerization reaction was inhibited completely, and a new product, derived from trapping of the benzylic radical by TEMPO, was obtained in 73% yield (Fig. S10a†). Secondly, when the photo-induced homocoupling reaction of benzyl bromide was performed under an O2 atmosphere, benzyl benzoate 6 and benzaldehyde were detected by GC-MS as the major product, the formation of which indicated the generation of benzyl radicals during the reaction (Fig. S10b†).2b,37 Thirdly, a competitive reaction performed using an equivalent mixture of benzyl bromide with an electron-rich group (1-(bromomethyl)-4-methoxybenzene 3c) and benzyl bromide with electron-withdrawing group (ethyl 4-(bromomethyl)benzoate 3e) was found to give cross-over coupling product 4l in 10% yield (Fig. S10c†). When a mixture of two benzyl bromides with electron-donating groups such as 1-(bromomethyl)-4-methoxybenzene 3c and 4-(bromomethyl)-1,2-dimethoxybenzene 3g was subjected to the reaction, the cross-over product 4m was isolated in 40% yield (Fig. S10d†). The cross-over experiments indicated that the major product originated from a more stable radical intermediate. Fourthly, in the presence of styrene, the reductive coupling product 4f (Table 4) was also obtained in 46% yield (Fig. S10e†). However, in contrast to the traditional photo-induced reaction in the presence of alkene or alkynes, the expected radical addition product38 was not detected by GC-MS and 1H NMR spectroscopy. DFT calculations revealed that the homocoupling reaction between two benzyl radicals is barrierless with a large driving force (ΔG) of −42.3 kcal mol−1, whereas the radical addition to styrene is less prevalent with a barrier of 8.4 kcal mol−1 (Fig. S21†).For the role of the W(VI) complex in the catalysis, the Stern–Volmer emission quenching experiment demonstrated that the emission of W1a was quenched by DIPEA with a quenching rate constant (kq) of 8.2 × 107 dm3 mol−1 s−1, together with the formation of a new absorption profile with a lifetime of 10.1 μs in ns-ta measurement (Fig. 7), while emission quenching was not observed with benzyl bromide. Since the oxidation of DIPEA by triplet excited W1a is thermodynamically favourable (E(W1a*/W1a−) = +1.10 V vs. SCE; E(DIPEA+/0) = +0.81 V vs. SCE), the observed emission quenching is ascribed to the reductive quenching of W1a by DIPEA. With the addition of benzyl bromide, the new transient absorption profile was found to exhibit a faster decay lifetime of 5.1 μs (vs. 10.1 μs), suggesting that the proposed W1a˙− species generated via reductive quenching with DIPEA may react with benzyl bromide. Besides, electrochemical measurements revealed that an enhanced current was observed at the 1st reduction wave (Epc = −1.29 V vs. SCE) of W1a upon addition of benzyl bromide, supporting that the one-electron reduced W1a could react with benzyl bromide (Fig. S9†).
 | ||
| Fig. 7 Nanosecond time-resolved absorption difference spectra of (left) W1a only and (right) W1a in the presence of N,N-diisopropylethylamine (0.02 M) in degassed CH3CN at room temperature. | ||
Based on the aforementioned findings, a plausible mechanism was proposed. In the excited state, W1a* accepts an electron from an electron-donor (D) such as DIPEA to form a reduced species, W1a˙−. Spin density calculations for W1a˙− reveal that the unpaired electron mainly resides on the imine ligand rather than the W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moiety (Fig. S22†). The delocalized spin population also helps to hamper further interaction between W1a˙− and the benzyl radical to form the plausible tungsten-oxo adducts. Therefore, the one-electron reduced W1a˙− is proposed to serve as a reductant to react with substrates (benzyl halides) to generate the reactive radical and an anion (X−) accompanied by the regeneration of W1a. The reactive radical species could be trapped by terminal reagents to yield the coupling products.
O moiety (Fig. S22†). The delocalized spin population also helps to hamper further interaction between W1a˙− and the benzyl radical to form the plausible tungsten-oxo adducts. Therefore, the one-electron reduced W1a˙− is proposed to serve as a reductant to react with substrates (benzyl halides) to generate the reactive radical and an anion (X−) accompanied by the regeneration of W1a. The reactive radical species could be trapped by terminal reagents to yield the coupling products.
To gain insight into the photo-induced dehalogenation of aryl halides catalysed by W1a, the reduction potentials of several aryl chlorides have been measured and these values range from −1.99 to −2.28 V vs. SCE (Table S2†). By considering the excited state potential E(W1a+/W1a*) (−1.29 V) and also the ground state potential Epc(W1a/W1a−) (−1.29 V), both the triplet excited tungsten photo-catalyst and one-electron reduced photo-catalyst are unable to reduce these chloro-substituted benzene substrates by outer sphere electron transfer reactions. In a recent study by Connell, Polyzos and Francis and co-workers,39 it was found that the in situ reduction and hydrogenation of the ligand bound to the metal may furnish another species that is more reducing than the original one. To test if this may have occurred in our photochemical reactions, CD2Cl2 solutions containing both W1a and DIPEA were subjected to light irradiation and the reaction was monitored with 1H NMR spectroscopy. The 1H signals at δ 8.0–8.30 ppm assigned to the two non-equivalent hydrogen atoms on the imine bond (H–C(R)N) of W1a vanished after light irradiation in the presence of excess DIPEA, indicative of the generation of new species from W1a (Fig. S11 and S12†). Thus, it is possible that the photoreduction of the tungsten complex afforded new species which may function as another active photocatalyst in the reaction. However, attempts to isolate and characterize the corresponding intermediate/product have not yet been successful. Electron-primed photo-redox catalysis has recently been reported as a new strategy to enable photo-catalysts to catalyse thermodynamically challenging reactions such as reduction of aryl chlorides.40 This strategy involves the electrochemical reduction of the photo-catalyst (PC) at a constant potential to generate a one-electron reduced PC (PC˙−), which is then excited by light to generate a strong reductant (PC˙−*) with reducing power comparable to that of alkali metals. Since W1a could be reduced photo-chemically in the presence of DIPEA to give W1a˙− with a ΔO.D. at 446 nm decaying back to the baseline in ∼20 μs, it may be possible that W1a undergoes electron-primed photoredox catalysis, though the contribution of this reaction pathway to the product yield is difficult to estimate. To examine if the dehalogenation reaction proceeded via the radical chain pathway, we measured the reaction quantum yield of the dehalogenation of ethyl 4-chlorobenzoate with light excitation at 420 nm. After 2 hours of light irradiation, the amount of product formed corresponded to a quantum yield of 2.7%, suggesting that this photochemical reaction is less likely to involve a radical chain mechanism.41
As to the reaction mechanism for borylation of aryl halides, we found that the emission of W1a is quenched by ethyl 4-chlorobenzoate and TBAB with kq values of 2.3 × 105 M−1 s−1 and 5.7 × 106 M−1 s−1, respectively, while emission quenching is not observed with B2Pin2. Steady state photolysis of deaerated MeCN solutions containing (i) W1a (2.2 × 10−5 M) and TBAB (0.1 M) and (ii) W1a (2.2 × 10−5 M) and ethyl 4-chlorobenzoate (0.1 M) with a blue LED has been conducted. Upon light irradiation for 7 h, the solution containing TBAB showed isosbestic UV-vis absorption spectral changes with new absorption peak maxima at 292 and 362 nm, while the solution with ethyl 4-chlorobenzoate showed a minor decrease in the absorbance at 392 nm (Fig. S14†). As the triplet excited state of W1a should be capable of oxidizing Br− (E(W1a*/W1a−) = +1.1 V vs. SCE; E(Br˙/Br−) = +0.8 V vs. SCE42), W1a* may have been reduced to W1a˙− in the reaction, though the subsequent reactions leading to product formation may be complicated and may require further study. The emissive excited state of W1a is ineffectively quenched by ethyl 4-chlorobenzoate with a rate constant of 2.3 × 105 M−1 s−1 only, which is consistent with the fact that the triplet excited state of W1a is thermodynamically not capable of reducing aryl chlorides. Given that the kq of TBAB (5.7 × 106 M−1 s−1) is 20 times larger than that of ethyl 4-chlorobenzoate (2.3 × 105 M−1 s−1), under the stated reaction conditions (the ratio of TBAB to ethyl 4-chlorobenzoate is 1:1), reductive quenching of the triplet excited state of W1a by TBAB could occur upon light irradiation. The photo-stability of W1a in the presence of TBAB was also monitored by using 1H NMR spectroscopy. Upon blue LED irradiation, singlet signals at 11.1 and 9.9 ppm gradually emerged. The emergence of these signals, as well as the disappearance of 1H signals of the propylene linkage at 3.8–4.8 ppm, is indicative of W1a undergoing secondary reaction to give the diphenylamino-substituted salicylaldehyde. Recently other mechanisms such as two-photon-absorption,43in situ generated electron donor,22 and homolytic C–X cleavage of aryl halide by UV light irradiation to generate aryl radicals20 have also been proposed to be the mechanism of borylation of aryl chlorides. We found that in the absence of W1a, the borylated product could also be isolated in 20% yield after 12 hours (Table S5,† entry 17) and the yield increased to 68% by prolonging the reaction time to 2 days under 410 nm LED irradiation (Table S5,† entry 22). This indicates that while W1a indeed facilitated the borylation of aryl chloride upon light excitation, a photocatalyst-free reaction pathway (slow under the present reaction conditions) could also be involved.
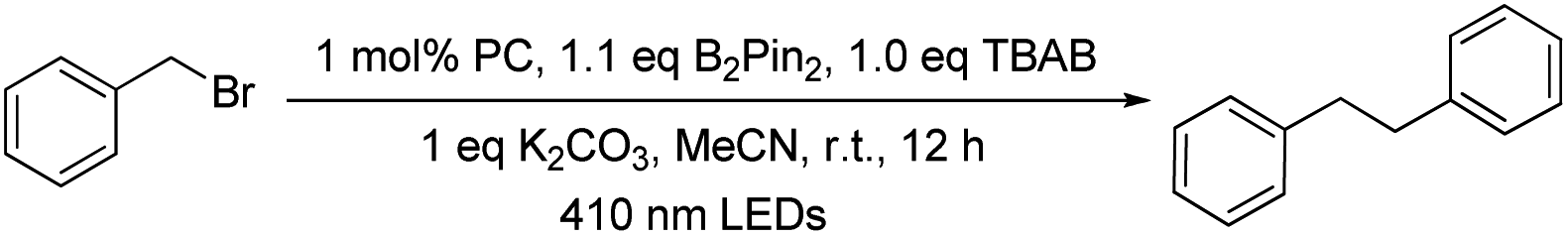
For the light-induced homocoupling of 2-bromoacetophenone (Table 5, 7a) and 1,3-dioxoisoindolin-2-yl 2-(4-isobutylphenyl)propanoate (Table 6 and 5a), as their reduction potentials (−1.18 V and −1.14 V vs. SCE, respectively) are less cathodic than E(W1a+/W1a*) and Epc(W1a/W1a−) (both values are −1.29 V vs. SCE; Table 2), these two light-induced homocoupling reactions may proceed via (i) the generation of W1a˙− by reductive quenching with Br−/DIPEA, followed by the reduction of the substrate with W1a˙−, and/or (ii) the direct reduction of the substrate by the triplet excited state of W1a. The efficiency of W1a in catalysing these photo-induced reactions has been evaluated by drawing a comparison with the yields of the product obtained with [Ir(ppy)3] and [Ru(bpy)3]2+ as photocatalysts (Table 10). It was found that in most cases, the performance of W1a was comparable to that of [Ir(ppy)3] and significantly better than that of [Ru(bpy)3]2+. For example, as shown in Table 4, W1a catalysed light-induced reductive homocoupling of benzyl bromide to give 1,2-diphenylethane in 95% yield, but the use of [Ir(ppy)3] and [Ru(bpy)3]2+ as the photocatalyst only gave the product in 61% and 5% yields, respectively, under the same reaction conditions. These results confirm the competitiveness of W1a as an earth-abundant photocatalyst for organic transformation reactions.
| Reaction | Yield | ||
|---|---|---|---|
| W1a | [Ir(ppy)3] | [Ru(bpy)3]2+ | |
| a Yields were determined by 1H NMR spectroscopy using dibromomethane as the internal standard. Anhydrous solvents were used. | |||
|
83 | 93 | 0 |
|
95 | 61 | 5 |
|
95 | 50 | 55 |
|
86 | 74 | 90 |
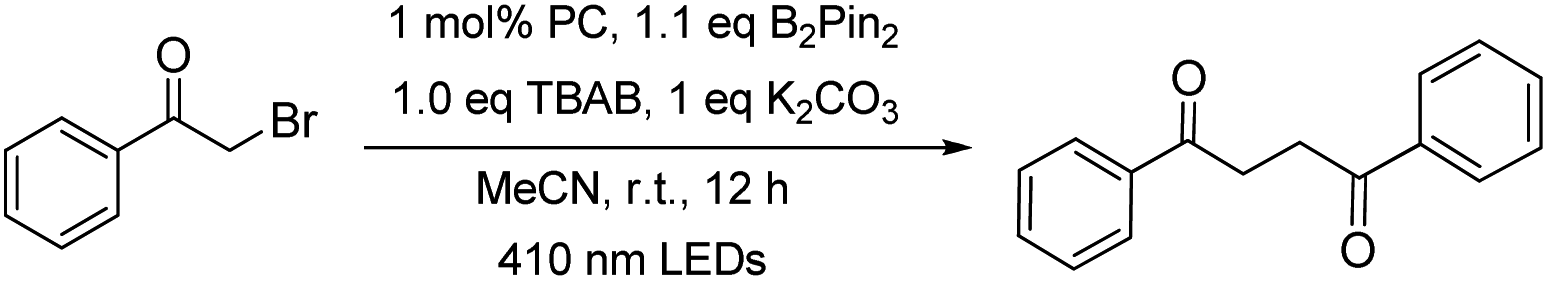
|
94 | 20 | 0 |
The LMCT excited state of decatungstate (W10O324−) is known to activate C–H bonds of aliphatic alcohols via hydrogen-atom abstraction (HAT) giving ketones as products.10d,j,n For the WVI-oxo complexes described herein, the LMCT transitions are high-lying in energy (∼300 nm) which results in fast internal conversion to low-lying IL/ILCT excited states as revealed by fs-ta measurements, hampering the manifestation of LMCT excited state chemistry of these complexes with visible light irradiation. Nevertheless, in this work, dehydrogenation of alcohols could be observed with the use of LEDs (365 nm) as the light source. The involvement of the high-energy 3LMCT excited state could not be completely excluded though we acknowledge that such an excited state is very short-lived and its formation is of low efficiency. Substrates with weaker C–H bonds, such as cyclohexa-1,4-diene and Hantzsch ester, were dehydrogenated in almost quantitative yields, suggesting that these reactions are dependent on the C–H bond strength which is consistent with the HAT pathway. However, since the reaction only occurred in the presence of oxygen, singlet oxygen and peroxy intermediates may also participate in the reaction giving the observed oxidized products. As tetraphenylporphyrin (H2TPP) is a known photosensitizer of singlet oxygen,44 we examined the light-induced dehydrogenation of 1-indanol by using H2TPP as the photocatalyst. Under similar reaction conditions, the dehydrogenated product 1-indanone was obtained in 15% yield (see Table S3,† entry 8). The capacity of W1a to sensitize the formation of singlet oxygen has been confirmed by measuring the emission intensity of singlet oxygen at ∼1270 nm in an aerated MeCN solution of W1a. The singlet oxygen quantum yield of W1a has been estimated to be 0.06. Also, we found that addition of one molar equivalent of hydrogen peroxide into an acetonitrile solution containing 1-indanol gave 1-indanone in 45% yield (Table S3,† entry 9), suggesting that reactive oxygen species which may be formed in the reaction between triplet excited W1a and dissolved oxygen could eventually lead to the oxidized product 1-indanone. The involvement of the superoxide radical anion in photo-catalysis has been reported in other reactions as well.45 It is possible that oxidation by reactive oxygen species generated from oxygen could be the major pathways for the formation of the dehydrogenated product. Regarding the mechanistic study on reductive homocoupling of benzyl bromides, DFT calculations revealed that the reduced W(VI) complex, W1a˙−, does not have any interaction/reaction with benzyl radicals.
Conclusion
In summary, we have developed air-stable, luminescent W(VI) complexes which could be obtained easily on the gram scale. The W(VI) complex having a diphenylamine substituent was found to catalyse visible-light-induced borylation of aryl halide, especially aryl chloride, the C–C reductive coupling reaction of benzyl bromide, the decarboxylative coupling reaction of redox-active esters of alkyl carboxylic acid, and the reductive coupling reaction of phenacyl bromide with high product yields and good tolerance to broad functional groups. It is envisioned that with further development, this class of photoluminescent W(VI) complexes could become an earth-abundant metal substitute for precious metal photosensitizers used in organic synthesis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the University Grants Committee Areas of Excellence Scheme (AoE/P-03/08), Shenzhen Science and Technology Innovation Committee (JCYJ20170412140251576 and JCYJ20180508162429786), Hong Kong Research Grants Council (HKU 17330416), China Postdoctoral Science Foundation (2018M633043) and CAS-Croucher Funding Scheme for Joint Laboratories.References
- (a) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898 CrossRef CAS PubMed; (b) Q. Liu and L.-Z. Wu, Natl. Sci. Rev., 2017, 4, 359 CrossRef CAS; (c) M. Parasram and V. Gevorgyan, Chem. Soc. Rev., 2017, 46, 6227 RSC; (d) J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS; (e) L. Fensterbank, J.-P. Goddard and C. Ollivier, in Visible Light Photocatalysis in Organic Chemistry, ed. C. Stephenson, T. Yoon and D. W. C. MacMillan, Wiley-VCH Verlag GmbH & Co.KGaA, Weinheim, 2018 Search PubMed; (f) C. B. Larsen and O. S. Wenger, Chem. – Eur. J., 2018, 24, 2039 CrossRef CAS PubMed; (g) L. Marzo, S. K. Pagire, O. Reiser and B. Konig, Angew. Chem., Int. Ed., 2018, 57, 10034 CrossRef CAS PubMed; (h) R. C. McAtee, E. J. McClain and C. R. J. Stephenson, Trends Chem., 2019, 1, 111 CrossRef; (i) F. Glaser and O. S. Wenger, Coord. Chem. Rev., 2020, 405, 213129 CrossRef CAS; (j) B. M. Hockin, C. Li, N. Robertson and E. Zysman-Colman, Catal. Sci. Technol., 2019, 9, 889 RSC; (k) C. Förster and K. Heinze, Chem. Soc. Rev., 2020, 49, 1057 RSC.
- (a) C. O. Dietrich-Buchecker, P. A. Marnot, J.-P. Sauvage, J. R. Kirchhoff and D. R. McMillin, J. Chem. Soc., Chem. Commun., 1983, 513 RSC; (b) J.-M. Kern and J.-P. Sauvage, J. Chem. Soc., Chem. Commun., 1987, 546 RSC; (c) D. Li, C.-M. Che, H.-L. Kwong and V. W.-W. Yam, J. Chem. Soc., Dalton Trans., 1992, 3325 RSC; (d) D. G. Cuttell, S.-M. Kuang, P. E. Fanwick, D. R. McMillin and R. A. Walton, J. Am. Chem. Soc., 2002, 124, 6 CrossRef CAS PubMed; (e) B. Wang, D. P. Shelar, X.-Z. Han, T.-T. Li, X. Guan, W. Lu, K. Liu, Y. Chen, W.-F. Fu and C.-M. Che, Chem. – Eur. J., 2015, 21, 1184 CrossRef CAS PubMed; (f) M. Knorn, T. Rawner, R. Czerwieniec and O. Reiser, ACS Catal., 2015, 5, 5186 CrossRef CAS; (g) M. M. Cetin, R. T. Hodson, C. R. Hart, D. B. Cordes, M. Findlater, D. J. Casadonte Jr, A. F. Cozzolino and M. F. Mayer, Dalton Trans., 2017, 46, 6553 RSC.
- (a) M. Grubel, I. Bosque, P. J. Altmann, T. Bach and C. R. Hess, Chem. Sci., 2018, 9, 3313 RSC; (b) L. A. Buldt, C. B. Larsen and O. S. Wenger, Chem. – Eur. J., 2017, 23, 8577 CrossRef PubMed; (c) S. Malzkuhn and O. S. Wenger, Coord. Chem. Rev., 2018, 359, 52 CrossRef CAS; (d) C.-H. Lim, M. Kudisch, B. Liu and G. M. Miyake, J. Am. Chem. Soc., 2018, 140, 7667 CrossRef CAS; (e) X. Shen, Y. Li, Z. Wen, S. Cao, X. Hou and L. Gong, Chem. Sci., 2018, 9, 4562 RSC; (f) B. J. Shields, B. Kudisch, G. D. Scholes and A. G. Doyle, J. Am. Chem. Soc., 2018, 140, 3035 CrossRef CAS PubMed; (g) R. C. G. Frem, A. C. Massabni, A. M. G. Massabni and A. E. Mauro, Inorg. Chim. Acta, 1997, 255, 53 CrossRef CAS; (h) H. Kunkely and A. Vogler, Inorg. Chem. Commun., 2000, 3, 143 CrossRef CAS.
- (a) Y. Zhang, J. L. Petersen and C. Milsmann, J. Am. Chem. Soc., 2016, 138, 13115 CrossRef CAS PubMed; (b) Y. Zhang, T. S. Lee, J. L. Petersen and C. Milsmann, J. Am. Chem. Soc., 2018, 140, 5934 CrossRef CAS PubMed.
- (a) L. A. Büldt, X. Guo, A. Prescimone and O. S. Wenger, Angew. Chem., Int. Ed., 2016, 55, 11247 CrossRef PubMed; (b) P. Herr, F. Glaser, L. A. Büldt, C. B. Larsen and O. S. Wenger, J. Am. Chem. Soc., 2019, 141, 14394 CrossRef CAS PubMed.
- (a) S. M. Stevenson, M. P. Shores and E. M. Ferreira, Angew. Chem., Int. Ed., 2015, 54, 6506 CrossRef CAS PubMed; (b) R. F. Higgins, S. M. Fatur, S. G. Shepard, S. M. Stevenson, D. J. Boston, E. M. Ferreira, N. H. Damrauer, A. K. Rappé and M. P. Shores, J. Am. Chem. Soc., 2016, 138, 5451 CrossRef CAS PubMed; (c) S. Otto, A. M. Nauth, E. Ermilov, N. Scholz, A. Friedrich, U. Resch-Genger, S. Lochbrunner, T. Opatz and K. Heinze, ChemPhotoChem, 2017, 1, 344 CrossRef CAS; (d) S. M. Stevenson, R. F. Higgins, M. P. Shores and E. M. Ferreira, Chem. Sci., 2017, 8, 654 RSC.
- (a) P. Chábera, Y. Z. Liu, O. Prakash, E. Thyrhaug, A. El Nahhas, A. Honarfar, S. Essen, L. A. Fredin, T. C. B. Harlang, K. S. Kjær, K. Handrup, F. Ericson, H. Tatsuno, K. Morgan, J. Schnadt, L. Häggström, T. Ericsson, A. Sobkowiak, S. Lidin, P. Huang, S. Styring, J. Uhlig, J. Bendix, R. Lomoth, V. Sundström, P. Persson and K. Wärnmark, Nature, 2017, 543, 695 CrossRef PubMed; (b) K. S. Kjær, N. Kaul, O. Prakash, P. Chábera, N. W. Rosemann, A. Honarfar, O. Gordivska, L. A. Fredin, K.-E. Bergquist, L. Häggström, T. Ericsson, L. Lindh, A. Yartsev, S. Styring, P. Huang, J. Uhlig, J. Bendix, D. Strand, V. Sundström, P. Persson, R. Lomoth and K. Wärnmark, Science, 2019, 363, 249 CrossRef PubMed; (c) A. Gualandi, M. Marchini, L. Mengozzi, M. Natali, M. Lucarini, P. Ceroni and P. G. Cozzi, ACS Catal., 2015, 5, 5927 CrossRef CAS; (d) J. Zhang, D. Campolo, F. Dumur, P. Xiao, J. P. Fouassier, D. Gigmes and J. Lalevée, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 42 CrossRef CAS; (e) J. Zhang, D. Campolo, F. Dumur, P. Xiao, J. P. Fouassier, D. Gigmes and J. Lalevée, ChemCatChem, 2016, 8, 2227 CrossRef CAS; (f) P. Zimmer, P. Müller, L. Burkhardt, R. Schepper, A. Neuba, J. Steube, F. Dietrich, U. Flörke, S. Mangold, M. Gerhards and M. Bauer, Eur. J. Inorg. Chem., 2017, 1504 CrossRef CAS.
- (a) H. Yin, P. J. Carroll, J. M. Anna and E. J. Schelter, J. Am. Chem. Soc., 2015, 137, 9234 CrossRef CAS PubMed; (b) J.-J. Guo, A. Hu, Y. Chen, J. Sun, H. Tang and Z. Zuo, Angew. Chem., Int. Ed., 2016, 55, 15319 CrossRef CAS PubMed; (c) H. Yin, P. J. Carroll, B. C. Manor, J. M. Anna and E. J. Schelter, J. Am. Chem. Soc., 2016, 138, 5984 CrossRef CAS PubMed; (d) H. Yin, Y. Jin, J. E. Hertzog, K. C. Mullane, P. J. Carroll, B. C. Manor, J. M. Anna and E. J. Schelter, J. Am. Chem. Soc., 2016, 138, 16266 CrossRef CAS PubMed; (e) A. Hu, Y. Chen, J.-J. Guo, N. Yu, Q. An and Z. Zuo, J. Am. Chem. Soc., 2018, 140, 13580 CrossRef CAS PubMed; (f) A. Hu, J.-J. Guo, H. Pan, H. Tang, Z. Gao and Z. Zuo, J. Am. Chem. Soc., 2018, 140, 1612 CrossRef CAS PubMed; (g) A. Hu, J.-J. Guo, H. Pan and Z. Zuo, Science, 2018, 361, 668 CrossRef CAS PubMed; (h) T. C. Jenks, M. D. Bailey, J. L. Hovey, S. Fernando, G. Basnayake, M. E. Cross, W. Li and M. J. Allen, Chem. Sci., 2018, 9, 1273 RSC; (i) Y. Qiao, Q. Yang and E. J. Schelter, Angew. Chem., Int. Ed., 2018, 57, 10999 CrossRef CAS.
- (a) Y.-Q. Wu, G.-X. Lu and S.-B. Li, Acta Chim. Sin., 2004, 62, 1134 CAS; (b) Z. Chen and J. Su, J. Jiangsu Univ., 2005, 26, 57 CAS; (c) Y. Cui, H. Li and K. Zhang, Preparation of tungsten trioxide photocatalyst and its Application, Chemical Industry Press. Co. Ltd., Beijing, 2012 Search PubMed; (d) J. G. West and E. J. Sorensen, Isr. J. Chem., 2017, 57, 259 CrossRef CAS; (e) K. Suzuki, N. Mizuno and K. Yamaguchi, ACS Catal., 2018, 8, 10809 CrossRef CAS; (f) Z. Yuan, H. Yang, N. Malik, M. Čolović, D. S. Weber, D. Wilson, F. Bénard, R. E. Martin, J. J. Warren, P. Schaffer and R. Britton, ACS Catal., 2019, 9, 8276 CrossRef CAS.
- (a) C. L. Hill, Synlett, 1995, 127 CrossRef CAS; (b) A. Maldotti, R. Amadelli, V. Carassiti and A. Molinari, Inorg. Chim. Acta, 1997, 256, 309 CrossRef CAS; (c) D. Dondi, M. Fagnoni, A. Molinari, A. Maldotti and A. Albini, Chem. – Eur. J., 2004, 10, 142 CrossRef CAS; (d) A. Maldotti, A. Molinari and F. Bigi, J. Catal., 2008, 253, 312 CrossRef CAS; (e) M. D. Tzirakis and M. Orfanopoulos, J. Am. Chem. Soc., 2009, 131, 4063 CrossRef CAS PubMed; (f) M. D. Tzirakis and M. Orfanopoulos, Angew. Chem., Int. Ed., 2010, 49, 5891 CrossRef CAS PubMed; (g) S. D. Halperin, H. Fan, S. Chang, R. E. Martin and R. Britton, Angew. Chem., Int. Ed., 2014, 53, 4690 CrossRef CAS PubMed; (h) M. Okada, T. Fukuyama, K. Yamada, I. Ryu, D. Ravelli and M. Fagnoni, Chem. Sci., 2014, 5, 2893 RSC; (i) S. D. Halperin, D. Kwon, M. Holmes, E. L. Regalado, L.-C. Campeau, D. A. DiRocco and R. Britton, Org. Lett., 2015, 17, 5200 CrossRef CAS PubMed; (j) J. G. West, D. Huang and E. J. Sorensen, Nat. Commun., 2015, 6, 10093 CrossRef PubMed; (k) K. Yamada, M. Okada, T. Fukuyama, D. Ravelli, M. Fagnoni and I. Ryu, Org. Lett., 2015, 17, 1292 CrossRef CAS PubMed; (l) M. Meanwell, M. B. Nodwell, R. E. Martin and R. Britton, Angew. Chem., Int. Ed., 2016, 55, 13244 CrossRef CAS PubMed; (m) J. J. Murphy, D. Bastida, S. Paria, M. Fagnoni and P. Melchiorre, Nature, 2016, 532, 218 CrossRef CAS PubMed; (n) D. J. Abrams, J. G. West and E. J. Sorensen, Chem. Sci., 2017, 8, 1954 RSC; (o) M. C. Quattrini, S. Fujii, K. Yamada, T. Fukuyama, D. Ravelli, M. Fagnoni and I. Ryu, Chem. Commun., 2017, 53, 2335 RSC; (p) D. M. Schultz, F. Lévesque, D. A. DiRocco, M. Reibarkh, Y. Ji, L. A. Joyce, J. F. Dropinski, H. Sheng, B. D. Sherry and I. W. Davies, Angew. Chem., Int. Ed., 2017, 56, 15274 CrossRef CAS; (q) G. Laudadio, S. Govaerts, Y. Wang, D. Ravelli, H. F. Koolman, M. Fagnoni, S. W. Djuric and T. Noël, Angew. Chem., Int. Ed., 2018, 57, 4078 CrossRef CAS; (r) M. Meanwell, J. Lehmann, M. Eichenberger, R. E. Martin and R. Britton, Chem. Commun., 2018, 54, 9985 RSC; (s) I. B. Perry, T. F. Brewer, P. J. Sarver, D. M. Schultz, D. A. DiRocco and D. W. C. MacMillan, Nature, 2018, 560, 70 CrossRef CAS PubMed; (t) Z. Yuan, M. B. Nodwell, H. Yang, N. Malik, H. Merkens, F. Bénard, R. E. Martin, P. Schaffer and R. Britton, Angew. Chem., Int. Ed., 2018, 57, 12733 CrossRef CAS PubMed; (u) V. I. Supranovich, V. V. Levin and A. D. Dilman, Org. Lett., 2019, 21, 4271 CrossRef CAS PubMed; (v) H.-B. Yang, A. Feceu and D. B. C. Martin, ACS Catal., 2019, 9, 5708 CrossRef CAS.
- D. C. Duncan, T. L. Netzel and C. L. Hill, Inorg. Chem., 1995, 34, 4640 CrossRef CAS.
- (a) K. R. Mann, M. Cimolino, G. L. Geoffroy, G. S. Hammond, A. A. Orio, G. Albertin and H. B. Gray, Inorg. Chim. Acta, 1976, 16, 97 CrossRef CAS; (b) K. R. Mann, H. B. Gray and G. S. Hammond, J. Am. Chem. Soc., 1977, 99, 306 CrossRef CAS; (c) W. Sattler, L. M. Henling, J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 2015, 137, 1198 CrossRef CAS PubMed; (d) K. Takematsu, S. A. M. Wehlin, W. Sattler, J. R. Winkler and H. B. Gray, Dalton Trans., 2017, 46, 13188 RSC.
- K.-T. Yeung, W.-P. To, C. Sun, G. Cheng, C. Ma, G. S. M. Tong, C. Yang and C.-M. Che, Angew. Chem., Int. Ed., 2017, 56, 133 CrossRef CAS PubMed.
- K. Li, Q. Wan, C. Yang, X.-Y. Chang, K.-H. Low and C.-M. Che, Angew. Chem., Int. Ed., 2018, 57, 14129 CrossRef CAS PubMed.
- K.-T. Chan, T.-L. Lam, D. Yu, L. Du, D. L. Phillips, C.-L. Kwong, G. S. M. Tong, G. Cheng and C.-M. Che, Angew. Chem., Int. Ed., 2019, 58, 14896 CrossRef CAS PubMed.
- W.-P. To, D. Zhou, G. S. M. Tong, G. Cheng, C. Yang and C.-M. Che, Angew. Chem., Int. Ed., 2017, 56, 14036 CrossRef CAS PubMed.
- A. M. Srivastava and J. F. Ackerman, Mater. Res. Bull., 1999, 34, 1397 CrossRef CAS.
- (a) W.-P. To, K. T. Chan, G. S. M. Tong, C. Ma, W.-M. Kwok, X. Guan, K.-H. Low and C.-M. Che, Angew. Chem., Int. Ed., 2013, 52, 6648 CrossRef CAS PubMed; (b) G. Cheng, S. C. F. Kui, W.-H. Ang, M.-Y. Ko, P.-K. Chow, C.-L. Kwong, C.-C. Kwok, C. Ma, X. Guan, K.-H. Low, S.-J. Su and C.-M. Che, Chem. Sci., 2014, 5, 4819 RSC; (c) P.-K. Chow, G. Cheng, G. S. M. Tong, W.-P. To, W.-L. Kwong, K.-H. Low, C.-C. Kwok, C. Ma and C.-M. Che, Angew. Chem., Int. Ed., 2015, 54, 2084 CrossRef CAS PubMed; (d) K. T. Chan, G. S. M. Tong, W.-P. To, C. Yang, L. Du, D. L. Phillips and C.-M. Che, Chem. Sci., 2017, 8, 2352 RSC.
- (a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS; (b) A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722 CrossRef CAS PubMed; (c) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed; (d) L. Xu, S. Zhang and P. Li, Chem. Soc. Rev., 2015, 44, 8848 RSC.
- (a) K. Chen, M. S. Cheung, Z. Lin and P. Li, Org. Chem. Front., 2016, 3, 875 RSC; (b) K. Chen, S. Zhang, P. He and P. Li, Chem. Sci., 2016, 7, 3676 RSC; (c) A. M. Mfuh, J. D. Doyle, B. Chhetri, H. D. Arman and O. V. Larionov, J. Am. Chem. Soc., 2016, 138, 2985 CrossRef CAS PubMed; (d) A. M. Mfuh, V. T. Nguyen, B. Chhetri, J. E. Burch, J. D. Doyle, V. N. Nesterov, H. D. Arman and O. V. Larionov, J. Am. Chem. Soc., 2016, 138, 8408 CrossRef CAS PubMed; (e) W. Liu, X. Yang, Y. Gao and C.-J. Li, J. Am. Chem. Soc., 2017, 139, 8621 CrossRef CAS PubMed.
- (a) M. Jiang, H. Yang and H. Fu, Org. Lett., 2016, 18, 5248 CrossRef CAS PubMed; (b) A. Nitelet, D. Thevenet, B. Schiavi, C. Hardouin, J. Fournier, R. Tamion, X. Pannecoucke, P. Jubault and T. Poisson, Chem. – Eur. J., 2019, 25, 3262 CAS.
- (a) L. Zhang and L. Jiao, J. Am. Chem. Soc., 2017, 139, 607 CrossRef CAS PubMed; (b) L. Zhang and L. Jiao, J. Am. Chem. Soc., 2019, 141, 9124 CrossRef PubMed.
- K. Hironaka, S. Fukuzumi and T. Tanaka, J. Chem. Soc., Perkin Trans. 2, 1984, 1705 RSC.
- G. Park, S. Y. Yi, J. Jung, E. J. Cho and Y. You, Chem. – Eur. J., 2016, 22, 17790 CrossRef CAS PubMed.
- (a) S. Fukuzumi, S. Mochizuki and T. Tanaka, J. Phys. Chem., 1990, 94, 722 CrossRef CAS; (b) J. Jung, J. Kim, G. Park, Y. You and E. J. Cho, Adv. Synth. Catal., 2016, 358, 74 CrossRef CAS; (c) M. Hou, L. Lin, X. Chai, X. Zhao, B. Qiao and Z. Jiang, Chem. Sci., 2019, 10, 6629 RSC; (d) X. Zhu, Y. Lin, Y. Sun, M. C. Beard and Y. Yan, J. Am. Chem. Soc., 2019, 141, 733 CrossRef CAS PubMed.
- (a) K. Okada, K. Okamoto and M. Oda, J. Am. Chem. Soc., 1988, 110, 8736 CrossRef CAS; (b) K. Okada, K. Okamoto, N. Morita, K. Okubo and M. Oda, J. Am. Chem. Soc., 1991, 113, 9401 CrossRef CAS; (c) M. Zlotorzynska and G. M. Sammis, Org. Lett., 2011, 13, 6264 CrossRef CAS PubMed; (d) C. R. Jamison and L. E. Overman, Acc. Chem. Res., 2016, 49, 1578 CrossRef CAS PubMed; (e) J. Schwarz and B. König, Green Chem., 2016, 18, 4743 RSC; (f) S. Murarka, Adv. Synth. Catal., 2018, 360, 1735 CrossRef CAS.
- K. Okada, K. Okubo, N. Morita and M. Oda, Chem. Lett., 1993, 22, 2021 CrossRef.
- W. Sha, L. Deng, S. Ni, H. Mei, J. Han and Y. Pan, ACS Catal., 2018, 8, 7489 CrossRef CAS.
- K. Okada, K. Okamoto and M. Oda, J. Chem. Soc., Chem. Commun., 1989, 1636 RSC.
- W.-T. Jiang, S. Yang, M.-Y. Xu, X.-Y. Xie and B. Xiao, Chem. Sci., 2020, 11, 488 RSC.
- R. Mao, J. Balon and X. Hu, Angew. Chem., Int. Ed., 2018, 57, 9501 CrossRef CAS PubMed.
- (a) Z.-J. Wang, J.-J. Lv, R.-N. Yi, M. Xiao, J.-J. Feng, Z.-W. Liang, A.-J. Wang and X. Xu, Adv. Synth. Catal., 2018, 360, 932 CrossRef CAS; (b) Y. Liu, S. Xiao, Y. Qi and F. Du, Chem.–Asian J., 2017, 12, 673 CrossRef CAS PubMed; (c) B. J. Fallon, V. Corcé, M. Amatore, C. Aubert, F. Chemla, F. Ferreira, A. Perez-Luna and M. Petit, New J. Chem., 2016, 40, 9912 RSC; (d) K. Sato, Y. Inoue, T. Mori, A. Sakaue, A. Tarui, M. Omote, I. Kumadaki and A. Ando, Org. Lett., 2014, 16, 3756 CrossRef CAS PubMed; (e) S.-H. Kim and R. D. Rieke, J. Org. Chem., 2000, 65, 2322 CrossRef CAS PubMed.
- D. Wang, N. Zhu, P. Chen, Z. Lin and G. Liu, J. Am. Chem. Soc., 2017, 139, 15632 CrossRef CAS PubMed.
- (a) H. Kim and C. Lee, Angew. Chem., Int. Ed., 2012, 51, 12303 CrossRef CAS PubMed; (b) G. Revol, T. McCallum, M. Morin, F. Gagosz and L. Barriault, Angew. Chem., Int. Ed., 2013, 52, 13342 CrossRef CAS PubMed; (c) S. M. Senaweera, A. Singh and J. D. Weaver, J. Am. Chem. Soc., 2014, 136, 3002 CrossRef CAS PubMed; (d) M. Häring, R. Pérez-Ruiz, A. Jacobi von Wangelin and D. D. Díaz, Chem. Commun., 2015, 51, 16848 RSC; (e) S. O. Poelma, G. L. Burnett, E. H. Discekici, K. M. Mattson, N. J. Treat, Y. Luo, Z. M. Hudson, S. L. Shankel, P. G. Clark, J. W. Kramer, C. J. Hawker and J. Read de Alaniz, J. Org. Chem., 2016, 81, 7155 CrossRef CAS PubMed; (f) B. Michelet, C. Deldaele, S. Kajouj, C. Moucheron and G. Evano, Org. Lett., 2017, 19, 3576 CrossRef CAS PubMed; (g) J. I. Bardagi, I. Ghosh, M. Schmalzbauer, T. Ghosh and B. König, Eur. J. Org. Chem., 2018, 34 CrossRef CAS; (h) T. U. Connell, C. L. Fraser, M. L. Czyz, Z. M. Smith, D. J. Hayne, E. H. Doeven, J. Agugiaro, D. J. D. Wilson, J. L. Adcock, A. D. Scully, D. E. Gómez, N. W. Barnett, A. Polyzos and P. S. Francis, J. Am. Chem. Soc., 2019, 141, 17646 CrossRef CAS PubMed; (i) A. Steiner, J. D. Williams, J. A. Rincón, O. de Frutos, C. Mateos and C. O. Kappe, Eur. J. Org. Chem., 2019, 5807 CrossRef CAS.
- W. Kong, C. Yu, H. An and Q. Song, Org. Lett., 2018, 20, 349 CrossRef CAS PubMed.
- (a) N. Ma, W. Shi, R. Zhang, Z. Zhu and Z. Jiang, Tetrahedron Lett., 2011, 52, 718 CrossRef CAS; (b) M. Nakajima, E. Fava, S. Loescher, Z. Jiang and M. Rueping, Angew. Chem., Int. Ed., 2015, 54, 8828 CrossRef CAS PubMed; (c) S. Okamoto, K. Kojiyama, H. Tsujioka and A. Sudo, Chem. Commun., 2016, 52, 11339 RSC; (d) A. Gualandi, G. Rodeghiero, E. Della Rocca, F. Bertoni, M. Marchini, R. Perciaccante, T. P. Jansen, P. Ceroni and P. G. Cozzi, Chem. Commun., 2018, 54, 10044 RSC; (e) T. Kohlmann, C. Kerzig and M. Goez, Chem. – Eur. J., 2019, 25, 9991 CrossRef CAS PubMed.
- D. Saberi and H. Hashemi, Catal. Commun., 2018, 106, 50 CrossRef CAS.
- (a) J. D. Nguyen, J. W. Tucker, M. D. Konieczynska and C. R. J. Stephenson, J. Am. Chem. Soc., 2011, 133, 4160 CrossRef CAS PubMed; (b) C.-J. Wallentin, J. D. Nguyen, P. Finkbeiner and C. R. J. Stephenson, J. Am. Chem. Soc., 2012, 134, 8875 CrossRef CAS PubMed.
- T. U. Connell, C. L. Fraser, M. L. Czyz, Z. M. Smith, D. J. Hayne, E. H. Doeven, J. Agugiaro, D. J. D. Wilson, J. L. Adcock, A. D. Scully, D. E. Gómes, N. W. Barnett, A. Polyzos and P. S. Francis, J. Am. Chem. Soc., 2019, 141, 17646 CrossRef CAS PubMed.
- (a) L. Capaldo, L. L. Quadri and D. Ravelli, Angew. Chem., Int. Ed., 2019, 58, 17508 CrossRef CAS PubMed; (b) H. Huang, Z. M. Strater, M. Rauch, J. Shee, T. J. Sisto, C. Nuckolls and T. H. Lambert, Angew. Chem., Int. Ed., 2019, 58, 13318 CrossRef CAS PubMed; (c) H. Yan, Z.-W. Hou and H.-C. Xu, Angew. Chem., Int. Ed., 2019, 58, 4592 CrossRef CAS PubMed; (d) N. G. W. Cowper, C. P. Chernowsky, O. P. Williams and Z. K. Wickens, J. Am. Chem. Soc., 2020, 142, 2093 CrossRef CAS PubMed; (e) H. Kim, H. Kim, T. H. Lambert and S. Lin, J. Am. Chem. Soc. Chem, 2020, 142, 2087 CrossRef CAS PubMed.
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426 RSC.
- Z. Wang, Q. Liu, X. Ji, G.-J. Deng and H. Huang, ACS Catal., 2020, 10, 154 CrossRef CAS.
- (a) I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725 CrossRef CAS PubMed; (b) C. Kerzig and O. S. Wenger, Chem. Sci., 2019, 10, 11023 RSC; (c) F. Glaser, C. Kerzig and O. S. Wenger, Angew. Chem., Int. Ed., 2020, 59 DOI:10.1002/anie.201915762.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233–234, 351 CrossRef CAS.
- (a) Y.-Q. Zou, L.-Q. Lu, L. Fu, N.-J. Chang, J. Rong, J.-R. Chen and W.-J. Xiao, Angew. Chem., Int. Ed., 2011, 50, 7171 CrossRef CAS PubMed; (b) Y.-Q. Zou, J.-R. Chen, X.-P. Liu, L.-Q. Lu, R. L. Davis, K. A. Jørgensen and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 784 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, photophysical data, DFT calculations and NMR spectral data. See DOI: 10.1039/d0sc01340d |
| This journal is © The Royal Society of Chemistry 2020 |