
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bimetallic metal–organic frameworks and their derivatives
Liyu
Chen
 a,
Hao-Fan
Wang
a,
Caixia
Li
a and
Qiang
Xu
*ab
a,
Hao-Fan
Wang
a,
Caixia
Li
a and
Qiang
Xu
*ab
aAIST-Kyoto University Chemical Energy Materials Open Innovation Laboratory (ChEM-OIL), National Institute of Advanced Industrial Science and Technology (AIST), Yoshida, Sakyo-ku, Kyoto 606-8501, Japan. E-mail: q.xu@aist.go.jp
bSchool of Chemistry and Chemical Engineering, Yangzhou University, Yangzhou 225002, China. E-mail: qxuchem@yzu.edu.cn
First published on 28th April 2020
Abstract
Bimetallic metal–organic frameworks (MOFs) have two different metal ions in the inorganic nodes. According to the metal distribution, the architecture of bimetallic MOFs can be classified into two main categories namely solid solution and core–shell structures. Various strategies have been developed to prepare bimetallic MOFs with controlled compositions and structures. Bimetallic MOFs show a synergistic effect and enhanced properties compared to their monometallic counterparts and have found many applications in the fields of gas adsorption, catalysis, energy storage and conversion, and luminescence sensing. Moreover, bimetallic MOFs can serve as excellent precursors/templates for the synthesis of functional nanomaterials with controlled sizes, compositions, and structures. Bimetallic MOF derivatives show exposed active sites, good stability and conductivity, enabling them to extend their applications to the catalysis of more challenging reactions and electrochemical energy storage and conversion. This review provides an overview of the significant advances in the development of bimetallic MOFs and their derivatives with special emphases on their preparation and applications.
 Liyu Chen | Liyu Chen received his BS degree in 2013 and his PhD in 2017, both from the South China University of Technology. In August 2017, he joined Prof. Qiang Xu's group in the AIST-Kyoto University Chemical Energy Materials Open Innovation Laboratory (ChEM-OIL) as a postdoctoral researcher. His research is centered on the design and preparation of MOF-based materials and their applications in heterogeneous catalysis. |
 Hao-Fan Wang | Hao-Fan Wang received his BS and PhD degrees from the Department of Chemical Engineering, Tsinghua University in 2013 and 2018, respectively. He is currently a postdoctoral researcher in the AIST-Kyoto University Chemical Energy Materials Open Innovation Laboratory (ChEM-OIL). His research interests include carbon materials, MOF-derived materials, and their applications in electrocatalysis and metal–air batteries. |
 Caixia Li | Caixia Li received her BS degree in 2014 and her PhD in 2019, both from Shandong University. In August 2019, she joined Prof. Qiang Xu's group in the AIST-Kyoto University Chemical Energy Materials Open Innovation Laboratory (ChEM-OIL) as a postdoctoral research fellow. Her research is focused on the design and preparation of MOF-derived materials and their applications in the energy storage area. |
 Qiang Xu | Qiang Xu received his PhD in 1994 from Osaka University. He is the director of AIST-Kyoto University ChEM-OIL, adjunct Professor of Kobe University/Kyoto University and specially-appointed professor of Yangzhou University. He received the Thomson Reuters Research Front Award in 2012 and Humboldt Research Award in 2019 and was recognized as a highly cited researcher (2014–2019) in Chemistry and Engineering/Materials Science by Thomson Reuters/Clarivate Analytics. His research interests include the chemistry of nanostructured materials and their applications, especially for catalysis and energy. He is a fellow of the Engineering Academy of Japan (EAJ), European Academy of Sciences (EURASC) and National Academy of Sciences, India (NASI). |
1. Introduction
Metal–organic frameworks (MOFs), or porous coordination polymers (PCPs), constructed from inorganic nodes with organic linkers, have emerged as a promising class of materials with high porosity, diverse composition and tuneable pore structures.1,2 These characteristics have attracted significant research interest in a variety of fields, such as gas adsorption and separation,3–5 catalysis,6–8 luminescence,9 sensing,10 biomedicine,11 and energy.12–14In order to enhance the catalytic, electronic, and luminescence properties of MOFs, it has been proposed to incorporate second metal ions into the nodes of frameworks for the preparation of bimetallic MOFs.15,16 The partial substitution by second metal ions in the inorganic nodes or secondary-building units (SBUs) in the framework will allow the bimetallic system to show synergistic effects. In bimetallic MOFs, the proportions of the metals can be adjusted or even controlled, offering the possibility to tune the physicochemical properties of bimetallic MOFs.17,18
According to the distribution of metal ions, bimetal MOFs can adopt “solid solution” or “core–shell” structures (Fig. 1).19 In solid solution bimetallic MOFs, the metals show delocalized or even homogeneous distributions through the whole crystal. Solid solution bimetallic MOFs can be synthesized by direct synthesis, post-synthetic modification and template synthesis. In core–shell bimetallic MOFs, the MOF shell is chemically different from the MOF core while they are integrated into a single architecture. Core–shell bimetallic MOFs can be synthesized through seed-induced growth of a MOF onto the surface of another, post-synthetic exchange of metal ions in the MOF surface, or one-pot synthesis. In this review, we mainly discuss the bimetallic MOFs in which both of metal ions serve as SBUs of MOFs. Bimetallic systems of immobilization of metal ions, metal nanoparticles (NPs), and metal complexes on the inorganic nodes,20,21 ligands,22–24 or inside MOF cavities25–27 as guests are outside of the scope of this review.28,29
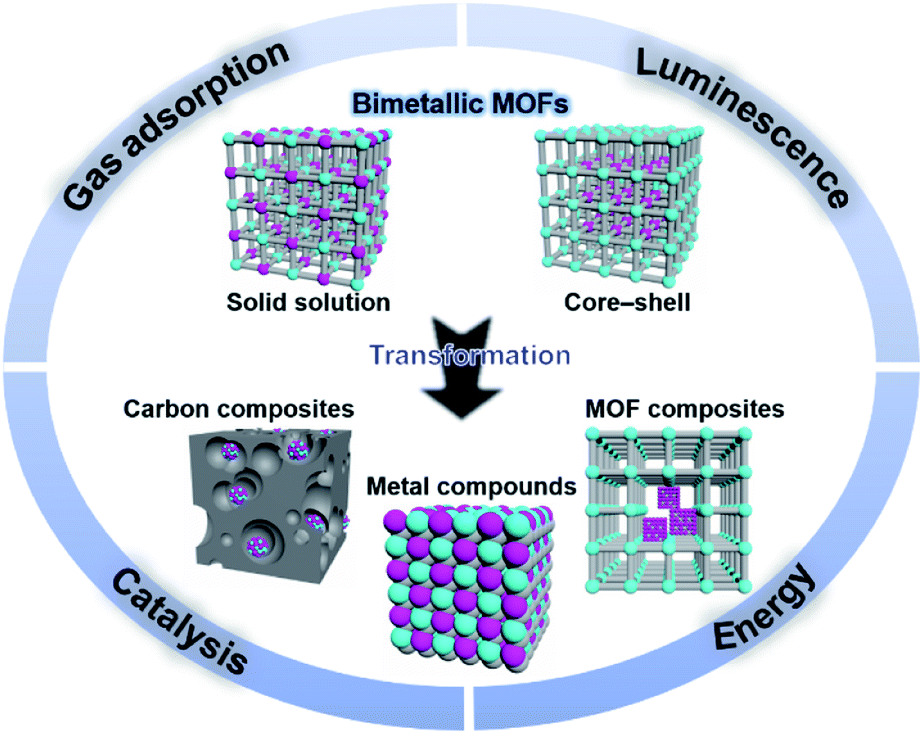 | ||
| Fig. 1 Schematic organization of the main contents including bimetallic MOFs and their derivatives, and some major applications. | ||
Bimetallic MOFs with tuneable compositions and structures have shown enhanced properties compared to their monometallic counterparts, exhibiting superior performance in many applications, including gas adsorption, catalysis, energy storage and conversion, and luminescence sensing.15,30–32
Moreover, bimetallic MOFs can be used as precursors/templates for the synthesis of a variety of nanostructured materials, including carbon composites, metal compounds, and MOF composites (Fig. 1).33,34 By selecting appropriate bimetallic MOFs and controlling the post-treatment process (e.g., pyrolysis, hydrolysis, and hydrothermal treatment), the compositions and structures of the afforded nanomaterials can be controlled. Bimetallic MOF-derived nanomaterials exhibit exposed active sites and high stability and conductivity, which can benefit their applications in the catalysis of more challenging reactions under harsh conditions and electrochemical energy storage and conversion.
This review provides an overall picture of the significant advances of bimetallic MOFs (Fig. 2). Synthetic strategies of bimetallic MOFs and their derivatives are summarized. The applications of bimetallic MOFs and their derivatives in various fields including gas adsorption, catalysis, energy storage and conversion, and luminescence sensing are also presented. This review is expected to inspire more efforts in the development of bimetallic MOFs and their derivatives for a variety of applications.
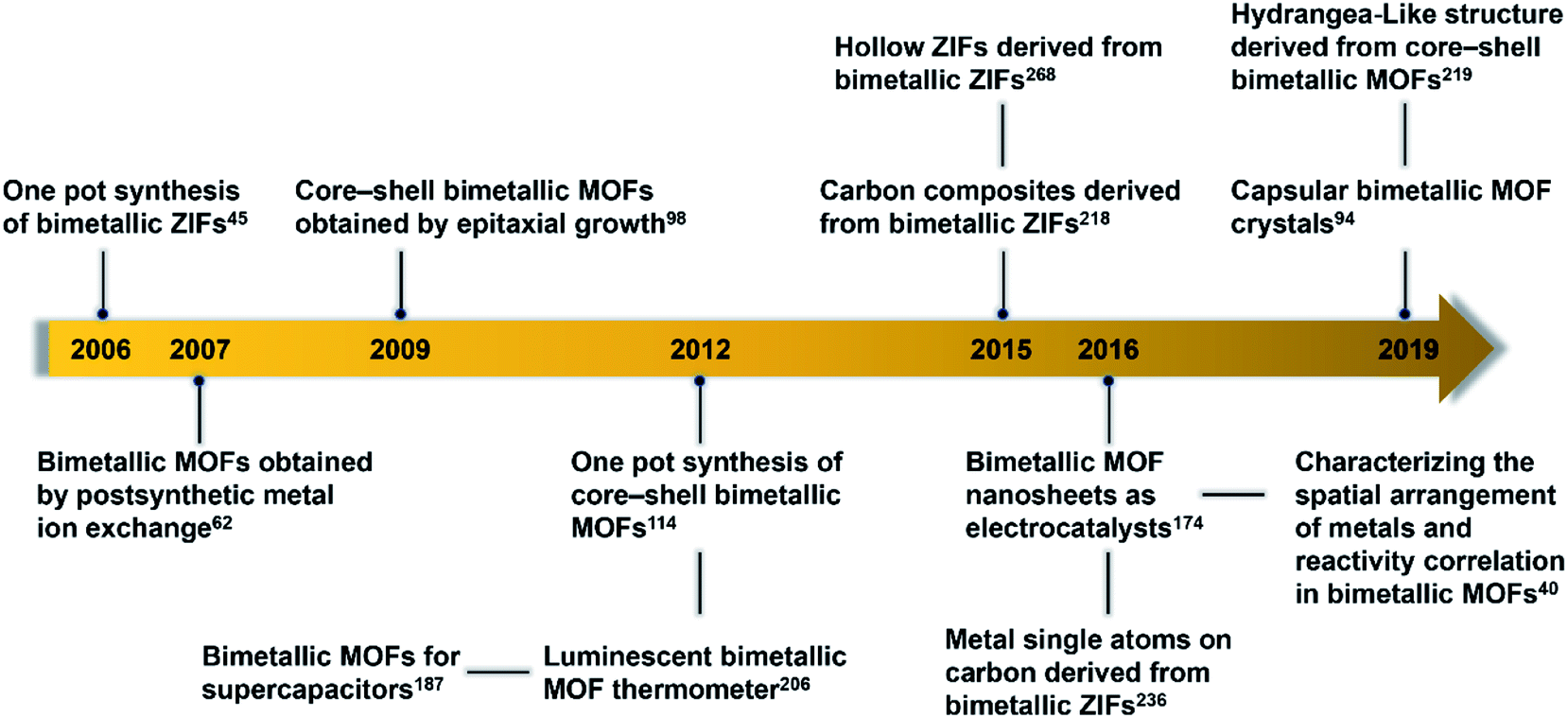 | ||
| Fig. 2 Timeline of important breakthroughs regarding bimetallic MOFs and their derivatives. | ||
2. Synthesis of bimetallic MOFs
Many strategies have been developed for the synthesis of bimetallic MOFs. This section focuses on the synthetic strategies of both solid solution and core–shell bimetallic MOFs. For the obtained bimetallic MOFs, using appropriate physicochemical techniques is essential for characterizing the compositions, distributions, and structures of the bimetallic MOFs. X-ray diffraction (XRD) and energy-dispersive X-ray (EDX) mapping can be combined to identify the crystalline phases of MOFs. In accordance with Vegard's law, bimetallic MOFs show diffraction peak positions between those of monometallic MOFs.35,36 Atomic absorption spectroscopy (AAS), inductively coupled plasma (ICP), EDX mapping and aerosol time-of-flight mass spectrometry (ATOFMS) can be coupled to calculate the global and local metal concentrations and distributions.37,38 X-ray absorption techniques, such as X-ray photoelectron spectroscopy (XPS) and X-ray absorption fine structure (XAFS), can determine the location of metals (present as nodes, bonded to the nodes, or present in the pores) in bimetallic MOFs.39 Extended X-ray absorption fine structure spectroscopy (EXAFS), neutron powder diffraction (NPD) and XPS can determine the spatial arrangements of metals in the SBUs of bimetallic MOFs.40–42 Different techniques should be combined for the accurate characterization of the synthesized bimetallic MOFs.2.1 Solid solution bimetallic MOFs
The term “solid solution” is used somewhat lightly, as in some cases a completely homogeneous distribution of metals may not be present.16,35,43 In this review, we divide solid solution bimetallic MOFs into two categories based on the spatial arrangements of metals: (1) two different metals are in the same SBU and the mixed metal SBUs are present throughout the MOF structure (Fig. 3A) and (2) each SBU consists of the same kind of metal and the two different SBUs are well mixed in the MOF structure (Fig. 3B). Two metals with a similar ionic radius and coordination sphere are more likely to exist in the same SBUs.44 It has been demonstrated that metals mixed in the same SBU can show stronger synergistic interactions compared to metals in different SBUs.40 However, in many bimetallic MOFs, the specific arrangement of metals in the SBUs throughout the crystal structure remains undetermined. Nevertheless, we generally define that in solid solution bimetallic MOFs the two metals are well dispersed throughout the MOF crystal. In this section, we focus on the discussion of methodologies to prepare solid solution bimetallic MOFs.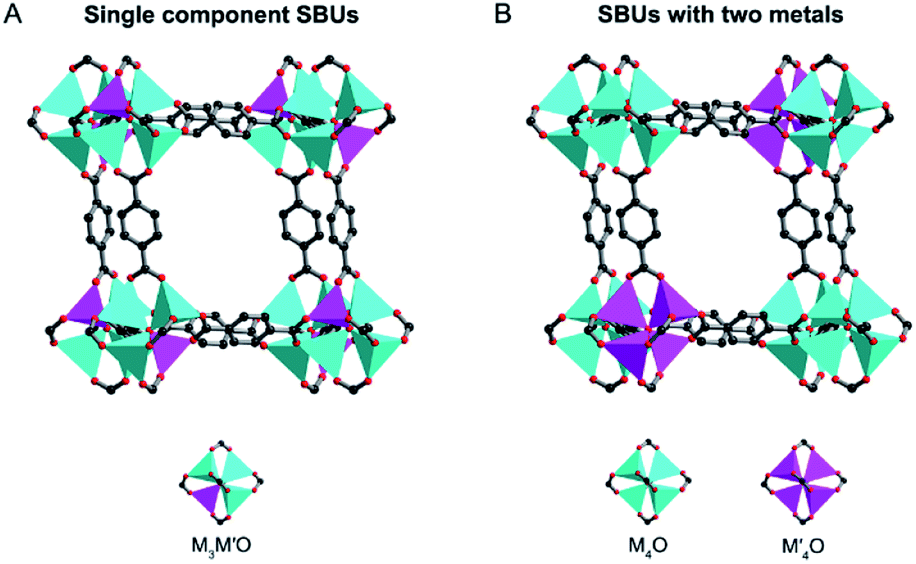 | ||
| Fig. 3 Two spatial arrangements of metals in solid solution bimetallic MOFs and their SBUs. | ||
A variety of bimetallic MOFs have been successfully synthesized through adding second metal precursors in the recipe of monometallic MOFs, such as Co2+ doping into MOF-5 (IRMOF-1, Zn4O(BDC)3, BDC = terephthalate) for Co/MOF-5,49 Zn2+ doping into HKUST-1 (Cu3(BTC)2, BTC = 1,3,5-benzenetricarboxylate) for Zn/Cu-BTC,50 Co doping into MOF-74 (Zn2(DOT), DOT = dioxidoterephthalate) for Co/MOF-74,51 Cu2+ doping into ZIF-67 (Co(2-MeIm)2, 2-MeIm = 2-methylimidazole, ZIF stands for the zeolitic imidazolate framework) for Cu/ZIF-67,52 Cu into ZIF-8 (Zn(2-MeIm)2) for Cu/ZIF-8 (Fig. 4A),53 Mg2+ doping into MIL-101(Cr) (MIL stands for Materials Institute Lavoisier),54 MIL-53(Cr–Fe) [(MIIIOH)(BDC)(H2BDC)x],55 and Ce doping into UiO-66(Zr) (UiO stands for Universitetet i Oslo).56
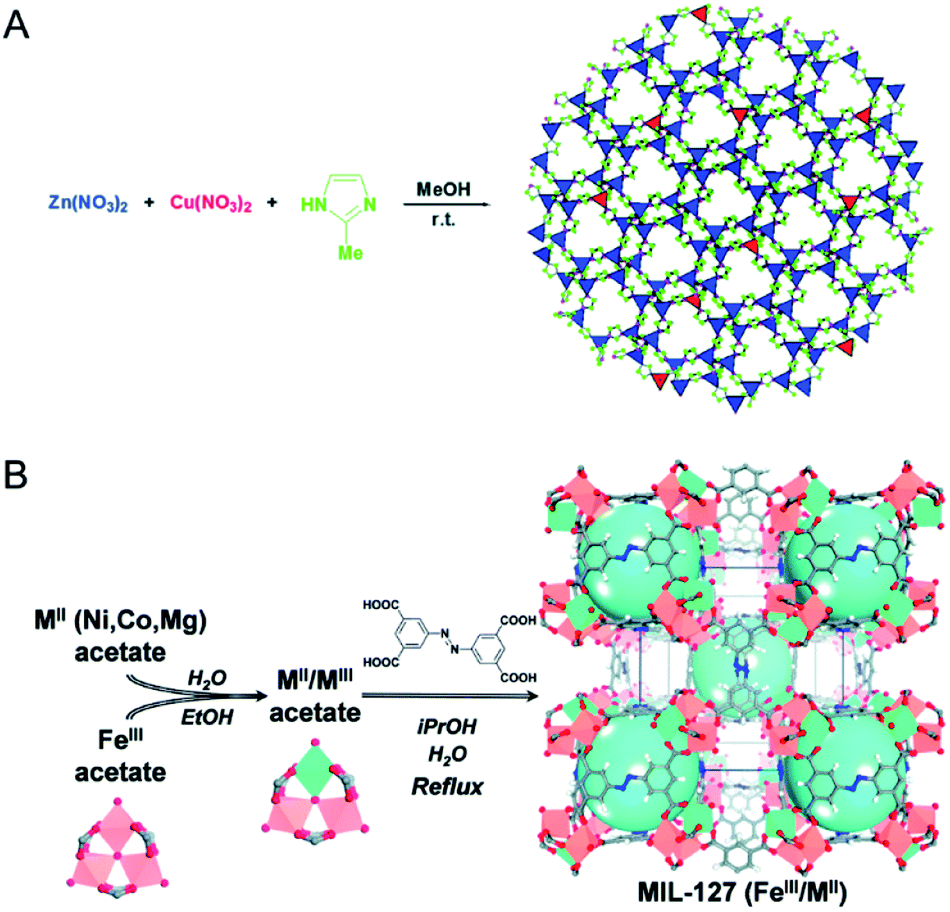 | ||
| Fig. 4 (A) Schematic representation of the synthesis of Cu-doped ZIF-8 crystals. Reproduced with permission from ref. 53. Copyright 2015, Royal Society of Chemistry. (B) Schematic representation of the building block approach to produce mixed-metal carboxylate MOFs such as MIL-127. From left to right, mixing iron acetate trimeric building blocks with MII acetate compounds leads to neutral mixed-metal trimeric building blocks that are used to produce the titled MOF via acetate/ligand exchange. Reproduced with permission from ref. 60. Copyright 2015, Royal Society of Chemistry. | ||
Mixed-metal MOFs containing more than two kinds of metal ions have been achieved. Yaghi and co-workers use a one-pot reaction to make crystalline mixed-metal MOF-74 [M2(DOT)] of varying metal ions (up to 10 different metal ions) in the nodes.57 H4DOT and selected metal salts were dissolved in a solvent mixture of N,N-dimethylformamide (DMF), ethanol, and water, heated at 120 °C for 20–24 h, affording mixed-metal MOF-74 (MM-MOF-74) materials. It is worth mentioning that this approach allowed the incorporation of typical metal ions (i.e., Ca, Sr, Ba, and Cd) in MM-MOF-74 from which a single-metal-containing MOF structure could not be made. XRD and SEM confirmed the pure phase of MM-MOF-74. Inductively coupled plasma-optical emission spectrometry (ICP-OES) demonstrated that MM-MOF-74 contained all metal ions added to the reaction mixture, while Ca, Sr, and Ba ions were less present because of their higher coordination number (8) than that of Mg, Mn, Fe, Co, Ni, Zn and Cd (6). EDX indicated that MM-MOF-74 crystals contained all the metals but had different distributions of the metals in each crystal.
To reach a higher degree of control over the synthesis of mixed-metal MOFs, the use of pre-defined SBUs is a promising approach to control metal stoichiometry.58–61 Feng, Bu and co-workers created a variety of In-M clusters as inorganic building blocks for fabricating five series of MOFs, CPM-18 M (M = Nd, Sm), CPM-19 M (M = Nd, Pr), CPM-20 (Co), CPM-21 M (M = Mn, Co, Cu), and CPM-23 (Mg).58 This work demonstrated the successful synthesis of bimetallic MOFs consisting of a p-block element (indium) and s-, d-, and f-block elements with differing nuclearities, metal-to-metal ratios, geometries, and charges. Serre and co-workers used oxocentered trimeric mixed iron(III)/metal(II) acetates (M = Co, Ni, Mg) as building blocks for the scalable and green preparation of mixed-metal MIL-127 (Fig. 4B).60 They first synthesized neutral mixed metal building blocks with the general formula FeIII2MIIO(H2O)2[O2C-CH3]6·nH2O (with M = Co or Ni), which were then used as precursors for the synthesis of Fe/Co, Fe/Ni and Fe/Mg MIL-127 MOFs. EDX analysis indicated that the ratio of Fe(III) to metal(II) remained at ca. 2 to 1 in the obtained MOFs. A control experiment using a 2 to 1 ratio of Fe(III) and metal(II) salts under similar synthesis conditions led to either amorphous or recrystallized linkers due to the different acidity and metal–ligand bond lability between metal(III) and metal(II) cations.
Long and co-workers demonstrated the concept of cation exchange of MOFs by partially exchanging Mn2+ in Mn3[(Mn4Cl)3(BTT)8(CH3OH)10]2 (1-Mn2+; BTT = 1,3,5-benzenetristetrazolate) with selected cations.62 The MOFs were soaked in methanolic solutions of Fe2+, Co2+, Ni2+, Cu2+, Zn2+, Li+, and Cu+ salts at room temperature. The degree of cation exchange in 1-Mn2+ was determined by inductively coupled plasma-atomic absorption (ICP-AA). The 3 equiv. of Mn2+ in 1-Mn2+ could be replaced with Fe2+, Co2+, Ni2+, Cu2+, and Zn2+. However, the metathesis of Mn2+ with monovalent cations (Li+ or Cu+) showed incomplete exchange with Li+ or almost negligible substitution with Cu+ due to charge repulsion effects.
Kim and co-workers reported the complete and reversible exchange of metal ions in the SBUs of a microporous framework while maintaining the structural integrity of the framework and single crystallinity.75 The Cd2+ ions in MOF 1, Cd1.5(H3O)3[(Cd4O)3(hett)8]·6H2O (hett = ethyl substituted truxene tricarboxylate), were exchanged with Pb2+ by immersing MOF 1 in aqueous Pb(NO3)2 solution. The monitoring of the ion exchange process by inductively coupled plasma atomic emission spectroscopy (ICP-AES) showed that 98% of Cd2+ was replaced by Pb2+ within 2 h and a complete exchange of Cd2+ with Pb2+ occurred in 2 days. A reverse exchange of Pb2+ with Cd2+ took a longer time with ∼50% exchange of Pb2+ in 1 day and a complete exchange in almost 3 weeks. Moreover, the Cd2+ ions of MOF 1 could be exchanged with trivalent lanthanide ions (Dy3+ and Nd3+) balanced by NO3− ions without losing crystallinity.
Metal ion exchange has focused largely on MOFs that are not considered highly robust,63,76 while it is also observed on MOFs that are considered to possess high structural stability, such as ZIFs,38 UiO-66(Zr),37,80 and MILs.81
Cohen and co-workers reported the synthesis of bimetallic ZIFs (Mn(II)-based ZIFs) through a postsynthetic exchange approach (Fig. 5A).38 ZIF-71(Zn) was incubated in a solution of Mn(acetylacetonate)2 in MeOH to afford ZIF-71(Zn/Mn). Results showed that all of the ZIF-71 particles participated in the exchanged process with 12% of the tetrahedral Zn2+ centres exchanged for Mn2+. ZIF-8 could also exchange ∼10% of Zn(II) centres for Mn(II). Cohen and co-workers also demonstrated the metal exchange of Zr4+ in UiO-66(Zr) with Ti4+ and Hf4+.37 UiO-66(Zr) was exposed to DMF solutions of different Ti4+ salts, such as TiCp2Cl2, TiCl4(THF)2, or TiBr4 (Cp = η5-cyclopentadienyl, THF = tetrahydrofuran) for 5 days at 85 °C. The amount of exchanged Ti4+ depended on the metal salt used. TiCl4(THF)2 showed the highest substitution level (38 wt%), while TiBr4 showed the lowest level due to its reactivity and instability. UiO-66(Zr) allowed very little metal ion exchange with HfCl4 at room temperature. Even at elevated temperatures, only ∼20% of the UiO-66(Zr) particles incorporated Hf4+.
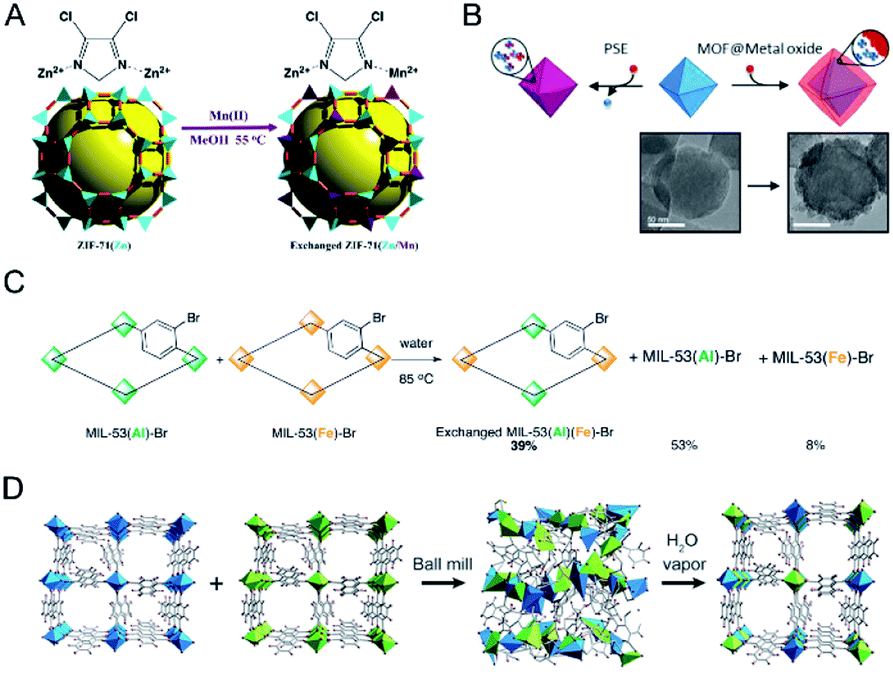 | ||
| Fig. 5 (A) Postsynthetic Mn(II) exchange of ZIF-71. Reproduced with permission from ref. 38. Copyright 2013, American Chemical Society. (B) The addition of exogenous Ti4+ and Hf4+ to UiO-66 resulted in the formation of metal oxides on the surface of UiO-66 particles, giving a core–shell, MOF@metal oxide structure. Reproduced with permission from ref. 83. Copyright 2018, American Chemical Society. (C) Solid–solid postsynthetic cation exchange in MIL-53. Reproduced with permission from ref. 37. Copyright 2012, American Chemical Society. (D) A possible formation mechanism for the solid solution of Al-ndc and Ga-ndc by ball milling and vapor exposure processes. Reproduced with permission from ref. 85. Copyright 2017, Wiley-VCH. | ||
One should keep in mind that the postsynthetic exchange of metal ions cannot always allow the replacement of metal ions in the SBUs. The metal ions may graft onto the surface of the inorganic node at a linker vacancy site or deposit on the surface of MOFs in the form of nanoscale metal oxides (Fig. 5B).82,83 Different analytical methods should be combined to confirm the successful exchange of metal ions in the SBUs of MOFs after synthesis.
Moreover, metal exchange can also occur between two robust MOF solids. Cohen and co-workers demonstrated that robust MOFs would readily exchange their structural metal ions in a solid–solid postsynthetic exchange process (Fig. 5C).37 MIL-53(Al)–Br and MIL-53(Fe)–Br as dry solids were mixed and then incubated in water for 5 days at 85 °C. ATOFMS showed that 40% of the particles contained both metal ions [i.e., MIL-53(Al/Fe)].
In addition, mechanical milling has also been demonstrated to be a powerful tool for metal ion exchange between two solid MOFs for the synthesis of solid solution bimetallic MOFs.84–86 Horike, Kitagawa and co-workers demonstrated that solvent-free mechanical milling of two MOF crystals could induce the dissolution of each metal ion into the amorphous solid state, which subsequently reconstructed to crystalline structures via vapor treatment (Fig. 5D).85 The authors prepared nine powder mixtures of Al-ndc and Ga-ndc (ndc = 1,4-naphthalenedicarboxylate) with different molar ratios from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 to 9:1. The mixtures were treated by ball-milling, followed by exposure to saturated water for 3 days to form solid solution Al/Ga-ndc materials, as determined by XRD. This method could also be applied for the preparation of other solid solution MOFs, such as Zn/Mg-MOF-74, Zn/Co-ZIF-8, and Zn/Cd-ZIF-8.
:9 to 9:1. The mixtures were treated by ball-milling, followed by exposure to saturated water for 3 days to form solid solution Al/Ga-ndc materials, as determined by XRD. This method could also be applied for the preparation of other solid solution MOFs, such as Zn/Mg-MOF-74, Zn/Co-ZIF-8, and Zn/Cd-ZIF-8.
:1 regardless of the initial metal concentrations in the reaction mixture.
 | ||
| Fig. 6 (A) Synthetic strategy for the heterogeneous phase preparation of bimetallic MOF-74 structures via a template-directed approach, in which a 1D metal–organic polymer with well-defined binding pockets was used as a structural template for the preparation of well-ordered bimetallic MOF-74s. Reproduced with permission from ref. 87. Copyright 2017, Wiley-VCH. (B) Schematic illustration of crystal-structure transformation of a MOF combined with its shape evolution. Reproduced with permission from ref. 93. Copyright 2014, Wiley-VCH. (C) Schematic illustration for the formation of capsular-MOF from FeNi-MIL-88B. Reproduced with permission from ref. 94. Copyright 2019, American Chemical Society. | ||
In addition to achieving a better control of the metal arrangement in bimetallic MOFs, templated synthesis can also allow the construction of hollow bimetallic MOFs. Hollow MOFs can not only inherit the merits of MOFs, but also have additional advantages such as facile mass transport and rich active sites.88 Templated synthesis methods for the synthesis of hollow MOFs can be divided into the exterior-template method and self-template method. Exterior-template synthesis of hollow MOFs usually uses a sacrificial template to prefabricate a core–shell intermediate and subsequently removes the template.89,90 This exterior-template method has been applied for the synthesis of hollow bimetallic MOFs.91 The self-template method has also been reported for the synthesis of hollow bimetallic MOFs.92–95 The self-template method usually goes through a dissolution–regrowth process, leading to the formation of bimetallic MOFs with hollow structures. The self-template method does not require a further step to remove the templates, which is simple and facile. For example, Wang and co-workers reported the synthesis of hierarchical Zn/Ni-MOF-2 nanosheet assembled hollow nanocubes (NAHNs) transformed from Zn/Ni-MOF-5 via a dissolution and recrystallization process (Fig. 6B).93 H2BDC, Zn2+ and Ni2+ ions were dissolved in a mixed solvent of N,N-dimethylacetamide (DMAC) and ethanol. During the synthesis process, a high concentration of precursors led to a fast formation of cubic crystalline Zn/Ni-MOF-5, which was gradually etched and acted as the template for the growth of thermodynamically favoured Zn/Ni-MOF-2 nanosheets. Xu and co-workers constructed an iron-nickel-based single-crystal open capsular-MOF through a crystal-structure transformation (Fig. 6C).94 Nonhollow crystals of FeNi-MIL-88B were dispersed in a hot DMF solution of 2,4,6-tris(4-pyridyl)pyridine (tpy), affording uniform capsular-MOF nanocrystals. A detailed study of the growth mechanism revealed that the parent FeNi-MIL-88B with plenty of concave defects dissolved and released Fe3+, Ni2+, and BDC-NH22− under solvothermal conditions, followed by the construction of FeNi-MIL-88B-tpy on the surface of FeNi-MIL-88B and final formation of capsular FeNi-MIL-88B-tpy with openings in the nanocrystal walls. FeNi-MIL-88B-tpy with additional size-matching ligands (tpy) in the structure showed enhanced structural stabilities compared to FeNi-MIL-88B, thus driving the dissolution–regrowth process in hot DMF.
2.2 Core–shell bimetallic MOFs
In core–shell bimetallic MOFs, the shell and core MOFs are formed from different metal centres. Three well-established strategies have been exploited for fabricating core–shell bimetallic MOFs, namely, seed-induced growth, post-synthetic exchange, and one-pot synthesis. | ||
| Fig. 7 (A) Synthetic scheme for the preparation of core–shell ZIF-8@ZIF-67 crystals. Reproduced with permission from ref. 99. Copyright 2015, American Chemical Society. (B) Schematic representation of the preparation of two types of heterocompositional CPPs, Hybrid CPP I (A@B) and Hybrid CPP II (C/A/C), through the isotropic and anisotropic nanoscale growth of secondary CPs on CPP-15. Here, A = Fe-MIL-88B, B = Ga-MIL-88B, and C = In-MIL-88B. Reproduced with permission from ref. 102. Copyright 2013, American Chemical Society. (C) TEM image of UiO-66@ZIF-8. Reproduced with permission from ref. 103. Copyright 2015, Wiley-VCH. | ||
Oh and co-workers further demonstrated that lattice parameter variation of isostructural MOFs could lead to different growth behaviors in an isotropic or anisotropic fashion (Fig. 7B).102 They employed Fe-MIL-88B as the seed to direct the formation of Ga-MIL-88B and In-MIL-88B with analogous structures but different sizes of metal ions. Ga-MIL-88B isotropically grew on the entire surface of Fe-MIL-88B, while In-MIL-88B anisotropically grew in the c-direction at both tips of Fe-MIL-88B. The different behaviors are due to the size variation of the metal ions. The ion size of Ga(III) is quite similar to that of Fe(III), resulting in a good similarity in the cell parameters for growth in an isotropic manner. However, the ion size of In(III) is larger than that of Fe(III), leading to a significant increase in the c cell parameters of In-MIL-88B and thus the anisotropic growth of In-MIL-88B in the c-direction.
The epitaxial growth method is usually limited to the synthesis of core–shell MOFs with similar crystallographic parameters. The assembly of two MOFs with different crystal structures would inevitably suffer from the separate growth of MOFs in solution. To address this issue, it has been demonstrated that the use of capping agents can promote a conformal and oriented overgrowth of one MOF on another MOF with different crystal structures.103–105 Tsung and co-workers overgrew uniform ZIF-8 shells on monodisperse UiO-66 microcrystal cores to form UiO-66@ZIF-8 with the assistance of cetyltrimethylammonium bromide (CTAB).103 The authors proposed that CTAB could adsorb small and uniform ZIF-8 nuclei under sonication and then orient the nuclei to grow into a conformal shell (Fig. 7C). Li, Kitagawa and co-workers developed an internal extended growth method (IEGM) for the synthesis of a MOF-on-MOF structure [NH2-UiO-66(Zr) on NH2-MIL-125(Ti)], in which the two MOFs have distinct morphologies and crystal structures.106 NH2-UiO-66(Zr) was exposed to NH2-MIL-125(Ti) precursors with polyvinylpyrrolidone (PVP) as the structure-directing agent through microwave heating, resulting in the interspersion of NH2-UiO-66(Zr) particles on the NH2-MIL-125(Ti) nanotablet. During the synthesis, PVP covered NH2-UiO-66(Zr) NPs to coordinate Ti4+, thus allowing the nucleation and growth of NH2-MIL-125(Ti) around NH2-UiO-66(Zr) NPs. Various MOF-on-MOF materials, such as MIL-101(Cr) on NH2-MIL-125(Ti), MOF-76 (Tb) on NH2-MIL-125, could be successfully fabricated by this method.
Although capping agents can facilitate the integration of core MOFs and shell MOFs with distinct crystallographic parameters, the capping agent can potentially make the MOF–MOF interface complex and ill defined. In this regard, Zhou and co-workers reported the fabrication of MOF-on-MOF structures with mismatched lattices through retrosynthetic design without the use of capping agents.107 The authors chose stable PCN-222(Zr) (also known as MOF-545) as the core MOF, which was pretreated with linkers (H2BDC) of the shell MOF (MOF-5), resulting in the coordination of H2BDC on the surface of PCN-222(Zr). Further adding metal precursors (Zn(NO3)2) allowed the gradual formation of MOF-5 on the surface of PCN-222(Zr). This retrosynthetic method could be successfully applied to the synthesis of other MOF-on-MOF materials with stable MOFs (UiO-66 (Zr), UiO-67 (Zr), PCN-160 (Zr), MOF-808 (Zr), PCN-250 (Fe), and MIL-125(Ti)) as core MOFs and M(II) based carboxylate MOFs as the shell MOFs [MOF-177 (Zn), HKUST-1 (Cu), and MOF-1114 (Yb)] due to their mild synthetic conditions and relatively large crystal sizes.
 | ||
| Fig. 8 (A) Synthetic scheme for the preparation of core–shell heterostructures via selective transmetalations. Reproduced with permission from ref. 78. Copyright 2012, American Chemical Society. (B) Transmetalations in two MOFs (Zn-HKUST-1 and Zn-PMOF-2) with different framework flexibilities. Reproduced with permission from ref. 108. Copyright 2012, Royal Society of Chemistry. | ||
In the subsequent work, Lah and co-workers further demonstrated the concept of selective transmetalation using two Zn-based MOFs with different ligand lengths, Zn-HKUST-1 and Zn-PMOF-2 (Zn24L8(H2O)12, L = 1,3,5-tris(3,5-dicarboxylphenylethynyl)benzene).108 In the methanol solution of Cu(II), the transmetalation of Zn-HKUST-1 was incomplete even after three months. Zn-PMOF-2 showed a higher rate and extent in the transmetalation process due to its more flexible structure compared with Zn-HKUST-1 (Fig. 8B). For both MOFs, the exchange of the metal ions occurred selectively at the external shell, leading to a core–shell heterostructure.
Zou and co-workers investigated the reaction kinetics of Co2+ and Zn2+ with 2-MeIM for bimetallic Co/Zn ZIFs (Fig. 9A).112 The authors found that a low Co/Zn ratio resulted in Co-rich cores and Zn-rich shells. The growth went through a nucleation-growth separated process; Co2+ with higher reaction kinetics formed the cores first, followed by the growth of zinc shells. At a high Co/Zn ratio, the nucleation and growth processes were not separated, leading to a quick formation of solid solution Co/Zn ZIFs. Moreover, when adding Co2+ first, ZIF-67@ZIF-8/67 core–shell nanocrystals were obtained with tuneable core/shell thickness ratios. However, exchanging the sequence by adding Zn2+ first only formed agglomerates with irregular shapes.
 | ||
| Fig. 9 (A) Schematic illustration of Co20Zn80-ZIF, Co50Zn50-ZIF, and Co80Zn20-ZIF. Reproduced with permission from ref. 112. Copyright 2017, Wiley-VCH. (B) A one pot microwave procedure for creating mixed-metal “egg yolk” MOFs with a core of (Cr/V)-MIL-53 and a shell of Cr-MIL-53 (top). A solvothermal method for producing homogeneous mixed-metal MOFs (bottom). Reproduced with permission from ref. 113. Copyright 2017, Royal Society of Chemistry. (C) Schematic illustration of the construction of PCP crystals from binary crystal domain modules. Reproduced with permission from ref. 114. Copyright 2012, American Chemical Society. | ||
Van Der Voort and co-workers reported egg-yolk bimetallic MOFs with a core of (Cr/V)-MIL-53 and a shell of Cr-MIL-53 using a one-pot microwave (MW) procedure.113 V3+ and Cr3+ salts were mixed with H2BDC in demineralized water in a MW for 3 h, resulting in the formation of (Cr/V)-MIL-53@ Cr-MIL-53 (Fig. 9B). For comparison, using a solvothermal method for 96 h resulted in one homogeneous phase of (Cr/V)-MIL-53 with uniform dispersion of V3+ and Cr3+. This work demonstrated that the synthesis time and method had a crucial influence on the metal dispersion in the crystals.
Kitagawa and co-workers controlled the kinetics of the crystal formation process for the preparation of core–shell MOFs in one pot by using precursors of distinct chemical reactivities (Fig. 9C).114 The authors employed [M(NO2-ip) (bpy)]0.5DMF·0.5MeOH (MCID-5⊃G, M = Zn, Mn, and Ni, NO2-ip = 5-nitroisophthalate, and bpy = 4,4′-bipyridyl) for the study. When using 5-nitroisophthalic acid (H2NO2-ip) as a precursor, the crystal formation rate of ZnCID-5⊃G was much quicker than that of MnCID-5⊃G. The one-pot reaction of H2NO2-ip and bpy with zinc and manganese perchlorate salts was conducted in a DMF/MeOH solution at 70 °C for 3 days, yielding crystals with a Zn-rich inner core and a Mn-rich outer shell. During the synthesis of Zn/MnCID-5⊃G, the nucleation process was dominated by the Zn rich crystals, which could facilitate the heterogeneous nucleation and growth of Mn-rich crystals. Moreover, additionally adding the precursors of NiCID-5⊃G with a moderate formation rate could yield Zn/Ni/MnCID-5⊃G crystals with a sandwich structure.
3. Applications of bimetallic MOFs
The mixing of metals in the SBUs of a MOF structure is widely recognized as an effective way to achieving optimal catalytic, optical, and electronic properties. A number of composition–property relationships have been established for many different MOF families.The incorporation of second metal ions in the unstable SBUs of MOFs may enhance the stability of MOFs. For example, MOFs constructed from zinc(II) paddlewheel [Zn2(OCO)4] SBUs frequently with low stability can show enhanced stability when exchanging the ZnII ion for other metal ions (such as Cu2+ and Ni2+).64,115–117 Mg-MOF-74 can gain improved water stability by the incorporation of Ni2+ or Co2+ ions that are less likely to hydrolyze than Mg2+.118
The flexibility of MOFs can be modulated by adopting different metal ions in the SBUs to meet the requirements of desired applications, such as storage, separation, and sensing.119 Yeung, Goodwin and co-workers studied the thermal expansion in Zn/Cd ZIF-8.35 The coefficient of thermal expansion showed a continuous and monotonic decrease with the increase of Cd content in Zn/Cd ZIF-8. These results demonstrated that Cd substitution in ZIF-8 made the framework structure increasingly flexible, due to a weaker bonding of Cd2+ with 2-MeIM compared with Zn2+. Serre and co-workers reported the tuning of the breathing behaviour of MIL-53 by cation mixing.55 MIL-53(Cr–Fe) adopted a hydrated narrow pore (np) form below 343 K, and then changed to a closed pore (cp) form at 343 K and a large pore (lp) form upon further heating above 463 K. This breathing behaviour is different from that of MIL-53(Cr) and MIL-53(Fe), which showed a direct np to lp transformation and a two-step np to cp transformation, respectively.
The electronic properties of MOFs can be tailored through metal node engineering.120,121 Mixing different metals in the SBUs of MOFs may provide MOFs with excellent electrical conductivity for a variety of applications, such as semiconductors, supercapacitors, thermoelectrics, and resistive sensing. Chen, Shustova and co-workers prepared bimetallic MOFs, Cu3−yMy(BTC)2 (M = Co, Zn), and studied how the substitution of a secondary metal would affect the electronic properties of MOFs.122 Monometallic M-MOFs (M = Cu and Zn) and bimetallic CuZn-MOFs exhibited zero intensity near the Fermi level which were characteristic of insulators, while CuCo-MOFs exhibited semiconductor behaviour. Microwave conductivity measurements showed that effective conductivities were 0.1 × 10−4 and 3.5 × 10−4 S cm−1 for Cu3(BTC)2 and Cu2.4Co0.6(BTC)2, respectively. Density functional theory (DFT) calculations revealed that the Co incorporation into the Cu-BTC matrix resulted in a decrease in the band gap and thus a higher conductivity.
Bimetallic MOFs show enhanced physical and chemical properties due to the synergistic effect of the two metals. The remarkable features of bimetallic MOFs make them suitable for applications in a variety of fields, including gas adsorption, catalysis, energy storage and conversion, and luminescence sensing.
3.1 Gas adsorption
Developing efficient porous materials for gas adsorption is fundamentally and industrially important. Bimetallic MOFs with large surface areas, adjustable pore sizes, and open metal sites are promising candidates as adsorbents for gas adsorption. Moreover, the adsorption strength of a specific adsorbate in bimetallic MOFs can be optimized through tuning the compositions of bimetallic MOFs. Many bimetallic MOFs have been designed as excellent adsorbents for gas adsorption.Singh and co-workers prepared CoxZn100−x-ZIF-8 with varying Co contents (x = 0, 25, 50, 75, 90 and 100).123 CoZn-ZIF-8 showed an enhancement of the surface area by ∼40% and the pore volume by ∼33% as compared to monometallic Zn-ZIF-8. At 77 K and 1 bar, Co75Zn25-ZIF-8 showed an enhancement of ∼23% in the H2 uptakes as compared to Zn-ZIF-8 (Fig. 10A). The enhanced H2 uptake capacities of CoZn-ZIF-8 could be attributed to the enhanced pore volume, surface area, microporosity, and heterogeneity in the pores.
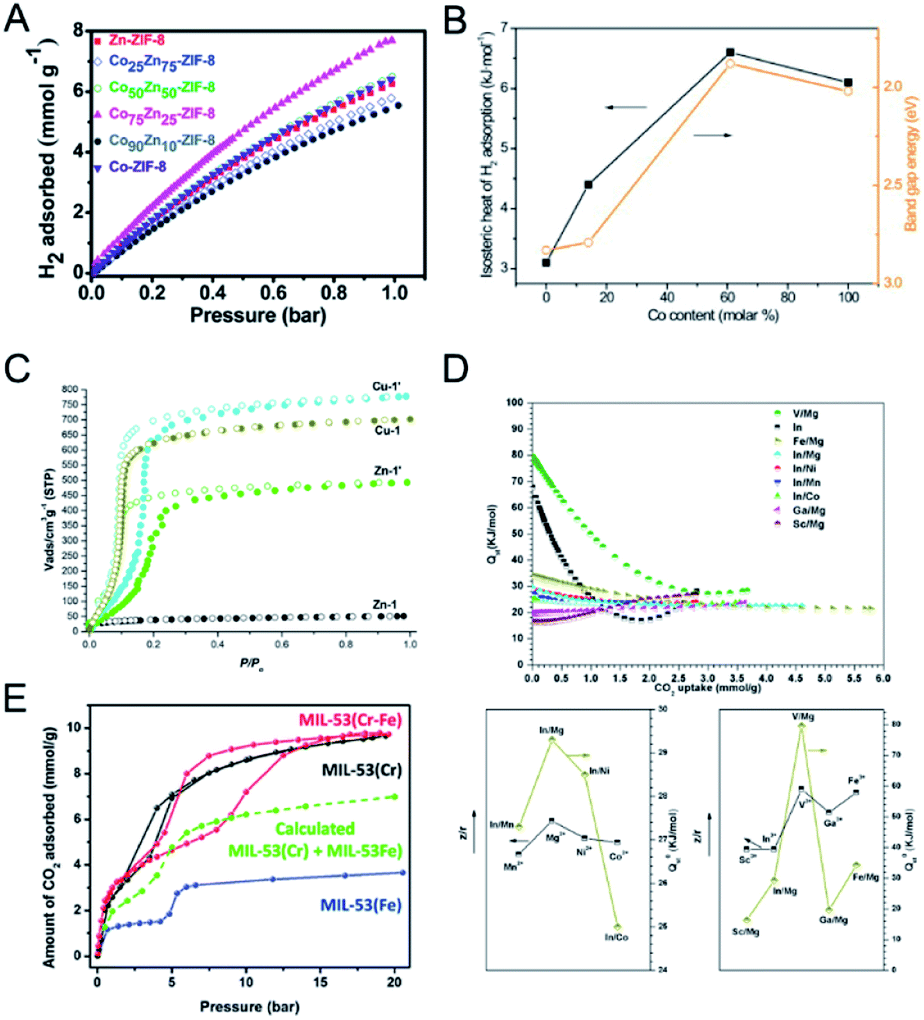 | ||
| Fig. 10 (A) H2 uptake isotherms of Zn-ZIF-8, Co-ZIF-8 and CoZn-ZIF-8 at 77 K. Reproduced with permission from ref. 123. Copyright 2016, Royal Society of Chemistry. (B) Hydrogen isosteric heat of adsorption, Qst and band gap energy at 1.5% H2 coverage versus Co content (x) in Zn1−xCoxMOF-74. Reproduced with permission from ref. 51. Copyright 2011, Elsevier. (C) CO2 (195 K) adsorption isotherms of activated Zn-1, Zn-1′, Cu-1, and Cu-1′. Reproduced with permission from ref. 125. Copyright 2017, American Chemical Society. (D) Isosteric heat for CO2 for CPM-200s (top). Correlation between the isosteric heat at zero loading of CO2 (Qst0) and the charge-to-radius ratio (z/r) of metal ions for CPM-200 s (bottom). Reproduced with permission from ref. 126. Copyright 2016, American Chemical Society. (E) CO2 sorption isotherms at 283 K for the MIL-53 materials. Reproduced with permission from ref. 55. Copyright 2012, Royal Society of Chemistry. | ||
Botas and co-workers prepared MOF-74 with different Co contents in the range of 0–100% for H2 adsorption at 0 °C and 10 bar.51 The H2 adsorption capacity of ZnCo-MOF-74 generally increased with the increase of Co content. The authors found a correlation between the isosteric heat of adsorption (Qst0) for H2 and the band gap energy values of Zn1−xCoxMOF-74 (Fig. 10B), implying that H2-metal cluster interactions have a more decisive role in H2 adsorption.
Long and co-workers exchanged Mn2+ ions in Mn3[(Mn4Cl)3(BTT)8(CH3OH)10]2 with selected cations (M = Li+, Cu+, Fe2+, Co2+, Ni2+, Cu2+, Zn2+) to tune the interaction strength between H2 and unsaturated metal centres in the frameworks.62 All the materials showed similar H2 uptakes ranging from 2.00 wt% to 2.29 wt% at 77 K and 900 torr. However, the materials showed significantly different uptake slopes at very low pressure, indicating different metal–hydrogen binding strengths in the materials. The results showed that Mn2+, Fe2+, and Co2+ ions yielded the strongest H2 binding among the cations studied.
The incorporation of a second metal in MOFs can improve the stability of MOFs and provide stronger interaction with CO2. Sun and co-workers used the metal ion exchange strategy to fabricate bimetallic MOFs with improved stability and CO2 adsorption capacity.125 ZnII ions in Zn-1 ([Zn3(L)2(dabco)(H2O)]·9DMF, H3L = [1,1′:3′,1′′-terphenyl]-4,4′′,5′-tricarboxylic acid) were partially replaced by CuII ions to fabricate bimetallic Zn-1′. The copper(II) analogue, Cu-1, was exchanged with ZnII ions to fabricate Cu-1′. Zn-1′ showed an enhanced N2 adsorption amount (2063 m2 g−1) than Zn-1 (59 m2 g−1), due to the high stability of the hybrid framework. Cu-1 and Cu-1′ showed similar N2 adsorption amounts. At 195 K and 1 bar, the CO2 uptakes of Zn-1, Zn-1′, Cu-1, and Cu-1′ were 52, 490, 703, and 770 m3 g−1, respectively. Both bimetallic Zn-1′ and Cu-1′ exhibited improved adsorption capacities than their pristine counterparts. Moreover, bimetallic Zn-1′ and Cu-1′ showed large hysteresis in the CO2 adsorption (Fig. 10C), implying enhanced interactions between the frameworks and CO2 molecules. Xiao, Li and co-workers doped Mg in MIL-101(Cr) to synthesize bimetallic MIL-101(Cr, Mg), showing improved CO2 adsorption capacity.54 The CO2 adsorption capacity of MIL-101(Cr, Mg) reached 3.28 mmol g−1 at 298 K and 1 bar, with an increase of 40% in comparison with MIL-101(Cr). The enhanced CO2 adsorption of MIL-101(Cr, Mg) was attributed to the higher surface area and stronger adsorptive sites for CO2 due to the doping of Mg.
Bu, Feng and co-workers reported the cooperative effect of dissimilar metals in the SBUs of MOFs for CO2 adsorption.126 The authors prepared a family of isostructural heterometallic MOFs (MII2MIII(μ3-OH)(CO2)6, CPM-200 series, CPM = crystalline porous materials) with combinations of trivalent (In3+, Ga3+, Fe3+, V3+, and Sc3+) and divalent metals (Mg2+, Mn2+, Co2+, and Ni2+) in the SBUs.126 At 273 K and 1 bar, the CO2 uptakes of the CPM-200 series, in cm3 g−1, were in the order of –Fe/Mg (207.6) > –In/Mg (190.8) > –V/Mg (155.4) > –In/Co (136.9) > –Ga/Mg (136.2) > –In/Mn (126.5) > –Sc/Mg (122.4) > –In/Ni (100.4). Among the CPM-200 series, CPM-200-Sc/Mg showed the highest Qst0 for CO2 (−79.6 kJ mol−1). The authors demonstrated a strong correlation between the charge-to-radius ratio (z/r) of metal ions in MOFs and Qst0 for CO2 (Fig. 10D).
In addition to the open metal sites, the pore architecture is of vital importance for CO2 capture performance. Hill and co-workers reported the exchange of Zr in UiO-66 for Ti to yield bimetallic UiO-66 with a smaller pore size and higher adsorption enthalpy for an enhanced CO2 uptake.80 With the increase of Ti substitution, the octahedral cages shrank by ∼1 Å and became increasingly broader. The CO2 uptake at 273 K of UiO-66(Zr100), UiO-66(Ti32) and UiO-66(Ti56) was 2.2, 2.3 and 4 mmol g−1, respectively. Considering that theoretical enhancement in the CO2 gravimetric uptake of UiO-66(Ti100) is ∼19% while UiO-66(Ti56) showed an enhancement of 81% compared with UiO-66(Zr100), the authors proposed that both the decrease of pore sizes and stronger adsorption characteristics of Ti(IV) contributed to the enhanced CO2 uptake in Ti-exchanged UiO-66.
Moreover, the structural flexibility change of bimetallic MOFs will lead to a different CO2 sorption behaviour compared with their single cation analogues. Serre and co-workers reported different CO2 sorption isotherms of bimetallic MIL-53(Cr–Fe) compared to monometallic MOFs (Fig. 10E).55 CO2 sorption tests at 283 K showed that the np to lp transition of MIL-53(Cr–Fe) occurred at a pressure of ∼10 bar, which was an intermediate between those of MIL-53(Cr) (3 bar) and MIL-53(Fe) (20 bar). This result demonstrated that MIL-53(Cr–Fe) was an intermediate between MIL-53(Cr) (‘easy’ to open) and MIL-53(Fe) (‘hard’ to open) in terms of ease of pore opening.
3.2 Catalysis
Although catalytically active metal ions can be introduced through immobilization on the SBUs or organic linkers, the incorporation of a second metal on the SBUs of MOFs for bimetallic MOFs results in distinct catalytic properties compared with other ways. In this regard, Farha, Hupp, Nguyen and co-workers compared the catalytic activity of UiO-66(Zr) functionalized with TiIV ions as part of the node (Ti-UiO-66), attached to the node (UiO-66-Tiex), and on the organic linker (UiO-66-Cat-Ti) in the oxidation of cyclohexene.137 These three materials had TiIV ions with different coordination environments at different sites of the UiO-66 support (Fig. 11A). Three UiO-66-based catalysts showed the catalytic activity in the order of Ti-UiO-66 ≫ UiO-66-Cat-Ti > UiO-66-Tiex, indicating that tetrahedral TiIV ions were more active than the more saturated octahedral TiIV ions.
 | ||
| Fig. 11 (A) Proposed structures for TiIV ions supported on the nodes (a), as the nodes (b), and on the struts (c) of UiO-66. Reproduced with permission from ref. 137. Copyright 2015, Royal Society of Chemistry. (B) MOF-74(Cu/Co) with different Cu/Co ratios as catalysts in the oxidation of styrene with O2 under solvent-free and mild reaction conditions. Reproduced with permission from ref. 139. Copyright 2016, Elsevier. (C) Time conversion plot of β-pinene, using as a catalyst either the mixed-metal MIL-100(Fe, Ni) 1–3 or homometallic MIL-100(Fe). Reproduced with permission from ref. 144. Copyright 2019, Royal Society of Chemistry. (D) Different behaviours of GaPF-1, InPF-11β and InxGa1−xMOF as heterogeneous catalysts in the three-component, one pot Strecker reaction. Reproduced with permission from ref. 149. Copyright 2015, American Chemical Society. (E) Bimetallic MOFs with two different metals catalyzing two kinds of reactions, viz., olefin to its epoxide followed by epoxide ring opening to afford the corresponding vicinal diol in a sequential manner. Reproduced with permission from ref. 150. Copyright 2016, American Chemical Society. | ||
Monometallic MOF catalysts usually exhibit either unsatisfactory activity or selectivity. Bimetallic MOFs can show a synergistic effect between the two metal ions to activate the reactants and reduce the reaction energy barrier in heterogeneous catalysis.31 Bimetallic MOFs have shown promising catalytic performances in a variety of reactions, including oxidation reactions,56,76,138–141 reduction reactions,142,143 condensation reactions,144–146 addition reactions,53,147 and substitution reactions.148
Zhu, Fan and co-workers applied bimetallic MOF-74(Cu/Co) with different Cu/Co ratios in styrene oxidation with O2 (Fig. 11B).139 MOF-74(Cu) showed a low catalytic activity (0.6% conversion) but an absolute selectivity for benzaldehyde. MOF-74(Co) possessed a high catalytic activity (47.3% conversion) but a low selectivity (35%) for benzaldehyde. The incorporation of Co2+ in MOF-74 (Cu) could significantly enhance the conversion of styrene. With the increase of Co2+ content in MOF-74(Cu/Co), the conversion of styrene increased while the selectivity for benzaldehyde decreased. A physical mixture of MOF-74(Cu) and MOF-74(Co) exhibited a lower activity than MOF-74(Cu/Co), indicating the synergistic effect of Cu2+ and Co2+ in the same framework.
Garcia, Serre and co-workers synthesized a series of MIL-100(Fe, Ni) materials with different metal ratios as catalysts for the Prins reaction.144 MIL-100(Fe, Ni) (1–3) samples contained a Ni amount of 1%, 3%, and 5%, respectively. In the Prins reaction between β-pinene and paraformaldehyde, monometallic MIL-100(Fe) only showed a low conversion (<20%). Bimetallic MIL-100(Fe, Ni) afforded higher conversions between 70 and 100%. MIL-100(Fe, Ni) 3 with the highest Ni percentage showed the highest activity, with a total conversion to nopol in 10 h (Fig. 11C). The authors proposed that the incorporation of NiII could cause the distortion of the framework to facilitate the access to the FeIII sites, thus enhancing the catalytic activity of the catalyst.
Park and co-workers synthesized a solid solution Zn–Co ZIF (CZ-ZIF) for chemical fixation of CO2.147 In the cycloaddition reaction of CO2 with epichlorohydrin (ECH), ZIF-8 showed an excellent ECH conversion of 98% but a low selectivity of 33% towards epichlorohydrin carbonate (ECC). ZIF-67 exhibited high selectivity to ECC (98%) but an inferior conversion of 66%. Solid solution CZ-ZIF afforded both high conversion of ECH (94%) and high selectivity (98%) to the desired ECC. The bimetallic CZ-ZIF could combine the advantageous properties of Zn (high activity) and Co (high selectivity and low leaching), achieving enhanced catalytic performance compared to its monometallic counterparts.
Multicomponent reactions and cascade reactions which require the cooperation of different active sites can be accomplished in bimetallic MOFs.149–152 Monge and co-workers demonstrated the control of the catalytic activity and selectivity of bimetallic MOFs in a multicomponent one pot Strecker reaction by modulating specifically selected metal ratios (Fig. 11D).149 In the A3 reaction between benzaldehyde, trimethylsilyl cyanide (TMSCN), and aniline, monometallic GaPF-1 [Ga(OH)(hfipbb)] and InPF-11β [In(O2C2H4)0.5(hfipbb)] (H2hfipbb = 4,4′-(hexafluoroisopropylidene) bis(benzoic acid)) gave only aldehyde cyanosilylation and imine as the main product, respectively. Solid-solution bimetallic MOFs [InxGa1−x(O2C4H4)0.5(hfipbb), x = 0.72, 0.55, and 0.28] could selectively yield the final Strecker product (α-aminonitrile) with different reaction rates. InGaPF-3 with the least amount of In showed the highest reaction rate, with 96% conversion in 0.33 h. Koner and co-workers reported a series of bimetallic MOFs based on copper and alkaline-earth metals (Mg, Ca, Sr, and Ba) to effectively catalyse olefin epoxidation and subsequent epoxide ring opening (Fig. 11E).150 In the tandem reaction, CuII acted as an active centre in epoxidation reactions and alkaline-earth metal ions served as Lewis acidic sites for epoxide ring opening subsequently. These MOFs with different alkaline-earth metals showed different reaction rates, increasing in the order of Mg < Ca < Sr ≈ Ba. The authors proposed that the increase of the size of the alkaline-earth metals led to a greater number of ligands accommodated in the coordination sphere and thus formed more open sites to enhance the catalytic activity.
Ti-based MOFs have shown great promise in photocatalysis. The incorporation of second metal ions in the SBUs of Ti-based MOFs can greatly enhance the photocatalytic activity. Martí-Gastaldo and co-workers reported two Ti-based MOFs (MUV-10, TiIV3MII3(μ3-O)2(btc)4-(H2O)6, M = CaII, MgII) by metal doping for photocatalytic H2 production (Fig. 12A).153 The authors found that heterometallic clusters served as excellent platforms to manipulate the electronic structure and in turn regulate the photoactivity. MUV-10(Mn) could produce 6500 μmol g−1 of H2 after 24 hours under the irradiation of a Xe lamp (300 W), showing two times higher activity than that of MUV-10(Ca). However, few Ti-based MOFs have been reported due to the high reactivity of Ti precursors. Alternatively, Ti can be incorporated into MOFs through post-synthetic exchange methods. For example, Ti-substituted UiO-66 (Zr/Ti) materials have shown enhanced photocatalytic performance compared to monometallic UiO-66(Zr).154–157 Zhou and co-workers synthesized four Ti based-MOFs, PCN-333(Sc)–Ti, MIL-100(Sc)–Ti, MOF-74(Zn)–Ti and MOF-74(Mg)–Ti, using a high valence metathesis and oxidation (HVMO) method for photodegradation of methylene blue (MB).158 The metal exchange ratios of PCN-333(Sc), MIL-100(Sc), MOF-74(Zn) and MOF-74(Mg) were 85.9%, 52.0%, 100%, and 35.1%, respectively (Fig. 12B). PCN-333(Sc)–Ti and MIL-100(Sc)–Ti showed 35% and 64% degradation of MB under a 300 W Xe lamp for nine minutes, respectively. MOF-74(Zn)–Ti and MOF-74(Mg)–Ti both showed conversions up to 98% in three minutes. All the synthesized Ti based-MOFs showed enhanced activities compared to TiO2 and monometallic MOFs.
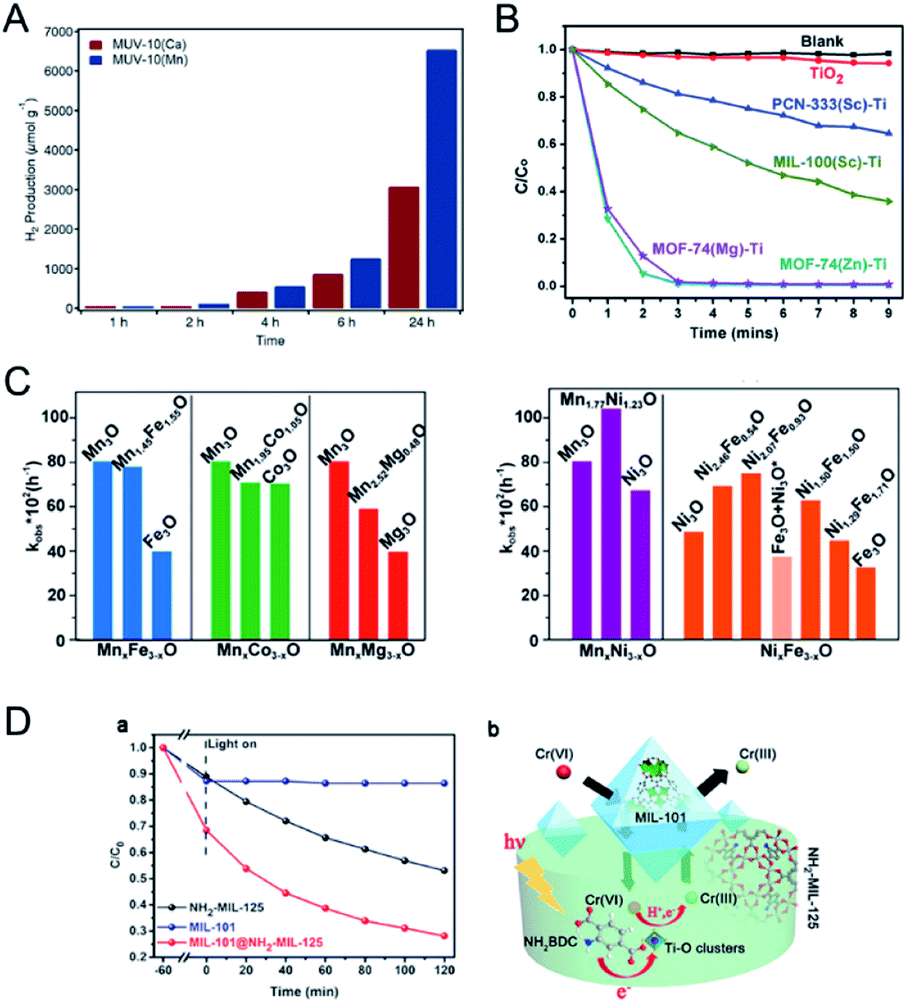 | ||
| Fig. 12 (A) Photocatalytic generation of hydrogen after 24 hours of irradiation using MUV-10(Ca) and MUV-10(Mg). Reproduced with permission from ref. 153. Copyright 2018, Wiley-VCH. (B) Photodegradation of MB using no catalyst (blank), TiO2, PCN-333(Sc)–Ti, MIL-100(Sc)–Ti, MOF-74(Zn)–Ti and MOF-74(Mg)–Ti with 300 W xenon light irradiation. Reproduced with permission from ref. 158. Copyright 2016, Royal Society of Chemistry. (C) Comparison between kobs of MTV-MOFs in a domain metal arrangement (left) and well-mixed metal arrangement (right) and those of their corresponding single component MOFs. Reproduced with permission from ref. 40. Copyright 2016, American Chemical Society. (D) Adsorption and photocatalytic degradation toward CrVI with MIL-101, NH2-MIL-125, and MIL-101@NH2-MIL-125 (a). Illustration of the enhanced photocatalytic degradation process towards CrVI with micro/mesoporous MIL-101@NH2-MIL-125 materials (b). Reproduced with permission from ref. 106. Copyright 2017, Wiley-VCH. | ||
Deng and co-workers demonstrated the synergistic effect of two different metals within the SBUs of MOFs for the photo-oxidation of 1,5-dihydroxynaphthalene (DHN).40 The authors synthesized a series of multivariate MOFs (MTV-MOFs) with a formula of (M3O)2(TCPP-M)3 (TCPP = tetrakis (4-carboxyphenyl) porphyrin). MTV-MOFs composed of MnxNi3−xO and NixFe3−xO SBUs showed a well-mixed spatial arrangement of metals, which exhibited enhanced conversion rates compared to their corresponding single component MOFs. Detailed characterization demonstrated that band gaps changed dramatically when different metals were present in the same SBU with a synergistic effect, leading to effective electron/hole transfer to oxygen for a high reaction rate. For comparison, in the case of MTV-MOFs composed of MnxFe3−xO, MnxCo3−xO and MnxMg3−xO SBUs, the two kinds of metals were in different SBUs and thus showed negligible interactions due to the separation by organic linkers (Fig. 12C). Therefore, these bimetallic MOFs exhibited the highest catalytic rates among their corresponding single component MOFs.
Core–shell MOFs have been successfully applied in photocatalysis, in which the shell MOF can accumulate/recognize reactants and the core MOF with catalytically active sites can transform the reactants to products.106,107,159 Kitagawa and co-workers applied MIL-101(Cr)@NH2-MIL-125(Ti) in the adsorption-photocatalytic removal of CrVI (Fig. 12D).106 MIL-101(Cr)@NH2-MIL-125(Ti) showed enhanced adsorption capacity on CrVI (3.16 mg g−1) compared to those of NH2-MIL-125(Ti) (1.12 mg g−1) and MIL-101(Cr) (1.28 mg g−1). MIL-101(Cr)@NH2-MIL-125(Ti) removed 72% of CrVI from the solution under visible light irradiation. However, MIL-101(Cr) showed no activity and NH2-MIL-125(Ti) removed only 47% of CrVI under similar conditions. The MOF on MOF heteroarchitecture could combine the high adsorption capacity of MIL-101(Cr) and photocatalytic activity of NH2-MIL-125(Ti) to exhibit high efficiency in photocatalysis.
In bimetallic MOFs, the electron configurations and d-band centres can be tailored through the mix of different metals in the SBUs, offering opportunities to enhance the electrocatalytic performance.122,171 Li, Lan and co-workers prepared four isostructural MOFs (NNU-21–24) based on Fe2M (M = Fe, Co, Ni, Zn) clusters bridged with biphenyl-3,4′,5-tricarboxylic acid (BPTC) ligands for the electrocatalysis of the OER.163 Among the four synthesized MOFs, monometallic NNU-21 (Fe3-BPTC) showed lower OER performance than bimetallic MOFs (NNU-22 (Fe2Co-BPTC), NNU-23 (Fe2Ni-BPTC) and NNU-24 (Fe2Zn-BPTC)) (Fig. 13A). NNU-23 exhibited the best OER performance with an overpotential of 365 mV at a current density of 10 mA cm−2 in 0.1 M KOH. Moreover, all the catalysts exhibited high stability with almost no decrease of activity after 2000 cycles. DFT calculation revealed that the incorporation of a second metal (Co, Ni or Zn) into the Fe cluster could induce the d-band centre to be close to the Fermi level, leading to a stronger binding interaction between the O* intermediate and catalysts, thus improving the OER performance. In addition to solid solution bimetallic MOFs, MOF@MOF structures can also show a synergistic effect in electrocatalysis.172 Zhu, Sun and co-workers grew Fe-MOF NPs onto Ni-MOF nanosheets (Ni-MOF@Fe-MOF) for the OER.173 The deposition of catalytically inert Fe-MOF on Ni-MOF could enhance the electrocatalytic performance. Ni-MOF@Fe-MOF exhibited an overpotential of 265 mV to reach a current density of 10 mA cm−2 in 1.0 M KOH, which was lower than that of Ni-MOF (370 mV). The enhanced performance of Ni-MOF@Fe-MOF could be attributed to the synergistic effect between Ni active centres and Fe species.
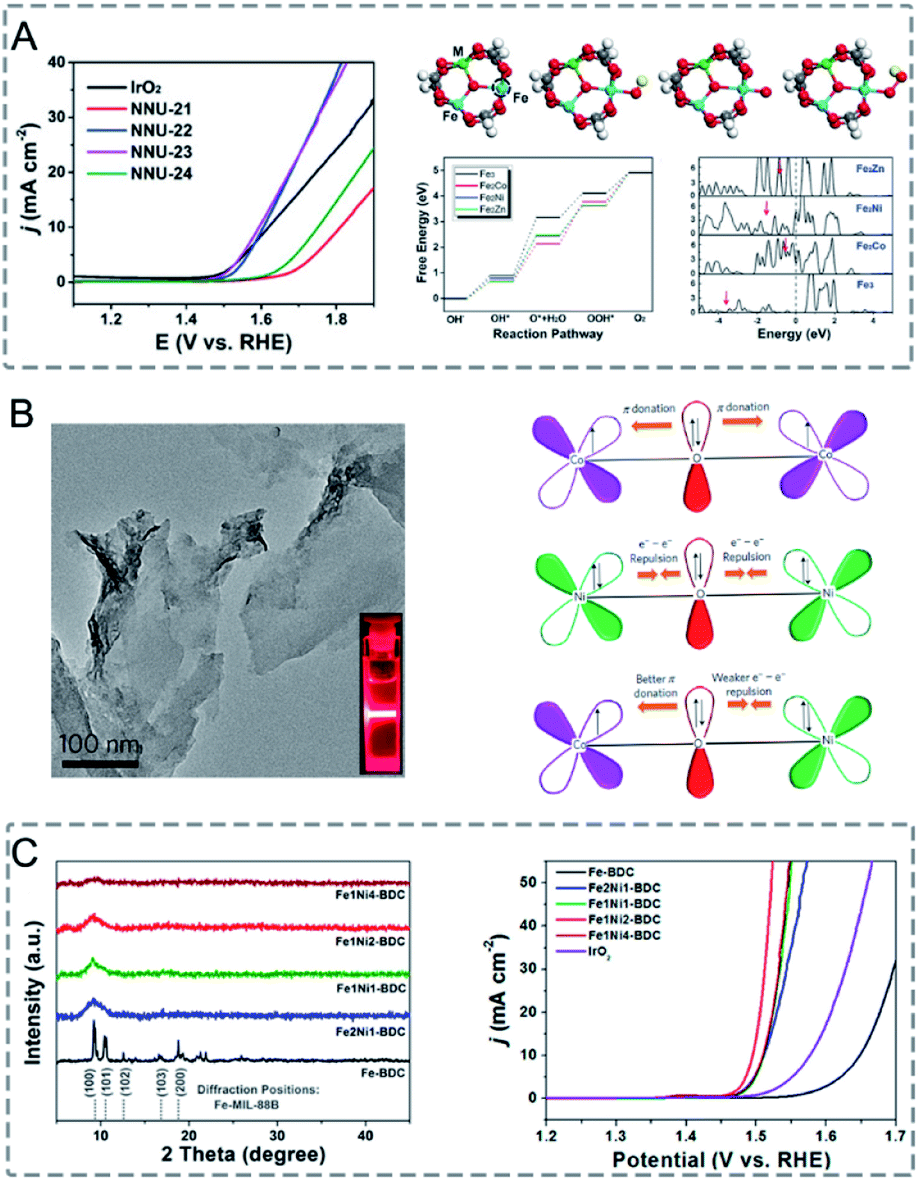 | ||
| Fig. 13 (A) LSV curves of IrO2 and NNU-21–24 for the OER in 0.1 m KOH (left). Initial structure of the Fe2M cluster, followed by the adsorption of OH*, O*, and OOH* intermediates on the Fe site. The active site is marked by a dashed circle. The free energy profile for the OER pathway and projected density of states of the Fe2M-cluster (right). Red arrows denote the d-band center. Reproduced with permission from ref. 163. Copyright 2018, Wiley-VCH. (B) TEM image of NiCo-UMOFNs (left). Schematic representation of the electronic coupling between Co and Ni in UMOFNs (right). Reproduced with permission from ref. 174. Copyright 2016, Nature Publishing Group. (C) XRD patterns of Fe-BDC, Fe2Ni1-BDC, Fe1Ni1-BDC, Fe1Ni2-BDC, and Fe1Ni4-BDC (diffraction positions of Fe-MIL-88B are shown in the bottom) (left). LSV curves of Fe-BDC, Fe2Ni1-BDC, Fe1Ni1-BDC, Fe1Ni2-BDC, Fe1Ni4-BDC, and IrO2 in 1 M KOH at a scan rate of 5 mV s−1 (right). Reproduced with permission from ref. 164. Copyright 2019, American Chemical Society. | ||
However, bimetallic MOFs usually suffer from poor conductivity, blockage of active metal centres by organic linkers, and low stability, which dramatically limit their utilization as electrocatalysts. To address these challenges, thinning bimetallic MOFs into nanosheets, fabricating an amorphous structure with plenty of defects, and integrating bimetallic MOFs with conductive supports have been proposed to enhance the electrocatalytic performance of bimetallic MOFs.
Thinning bimetallic MOFs into two-dimensional (2D) nanosheets can enhance mass transport and electron transfer and maximize unsaturated metal sites on the surfaces to improve the electrocatalytic performance.8,160,174–177 Liu, Zhao, Tang and co-workers reported ultrathin NiCo bimetallic MOF nanosheets (NiCo-UMOFNs) as effective electrocatalysts for the OER (Fig. 13B).174 NiCo-UMOFNs on a glassy-carbon electrode showed an overpotential of 250 mV at a current density of 10 mA cm−2 in 1.0 M KOH solution, smaller than those of Ni-UMOFNs (321 mV), Co-UMOFNs (371 mV), bulk NiCo-MOFs (317 mV) and commercial RuO2 (279 mV). XAFS analysis and DFT calculations demonstrated that the coordinatively unsaturated metal atoms were the dominating active centres and the synergistic effect between Co and Ni further improved the electrocatalytic OER activity. Electron transfer from the Ni2+ sites to the Co2+ sites could enhance the interaction between Ni2+ sites and water, thus offering a lower energy barrier for water oxidation. Oh and co-workers prepared bimetallic conductive 2D MOFs (CoxNiy-CATs) with varied ratios of Co2+ to Ni2+ for the ORR in 0.1 M NaClO4 and 0.02 M PBS electrolytes.160 Ni-CAT showed a high onset potential of 0.47 V (vs. the reversible hydrogen electrode, RHE, the same below if not mentioned), but a low diffusion limiting current density at 0.0 V (−3.62 mA cm−2). However, Co-CAT displayed a high diffusion limiting current density (−5.59 mA cm−2) but a low onset potential (0.42 V). Bimetallic Co0.27Ni0.73-CAT combined the advantages of Co-CAT (effective adsorption for O2) and Ni-CAT (high conductivity), showing a diffusion-limiting current density (−5.68 mA cm−2) and an onset potential (0.46 V). With the increase of the proportion of Ni from 50% to 73%, the onset potential of CoxNiy-CATs showed a slight increase. Further increasing the proportion of Ni to 86% resulted in a decrease in diffusion-limiting current density. Rotating ring disk electrode (RRDE) investigation showed that the electron transfer number of Co-CAT, Ni-CAT, and Co0.27Ni0.73-CAT was 3.91, 3.31, and 3.95, respectively. The bimetallic Co0.27Ni0.73-CAT was more preferable for a four-electron process.
In MOFs with good crystallinity, the metal sites are confined in the crystalline frameworks and blocked by the organic linkers, which is not good for electrocatalysis. In this regard, low-crystalline bimetallic MOFs with highly exposed active sites and porous structures hold great promise for electrocatalysis. Mai and co-workers reported the synthesis of low-crystalline bimetallic Fe/Ni-MOF NPs (FexNiy-BDC) for the OER.164 The addition of Ni precursors in the synthesis solution of Fe-BDC (Fe-MIL-88B) caused mismatched coordination with organic linkers, thus resulting in the loss of the long-range ordered structure and formation of rich defects (Fig. 13C). Fe1Ni2-BDC showed a small overpotential of 260 mV at a current density of 10 mA cm−2 and a high Faradaic efficiency (FE) of 99.5% for the OER in 1.0 M KOH. The superior catalytic performance of Fe1Ni2-BDC could be attributed to the exposed active sites, synergistic effect between Fe and Ni, and fast charge transfer in the amorphous structure.
The integration of bimetallic MOFs with functional materials can enhance the characteristics of MOFs with improved electrical conductivity, chemical stability and mechanical strength for electrocatalytic applications.6,167,178–180 Lu and co-workers reported the in situ growth of amino-functionalized bimetallic NH2-MIL-88B(Fe2Ni) (NFN-MOF) on 3D microporous nickel foam (NF) as an efficient bifunctional electrocatalyst for overall water splitting.181 In a 1.0 M KOH electrolyte, the synthesized NFN-MOF/NF exhibited overpotentials of 240 and 87 mV at a current density of 10 mA cm−2 for the OER and HER, respectively. For the overall water splitting with NFN-MOF/NF as both the anode and cathode, a cell voltage of only 1.56 V was required to achieve a current density of 10 mA cm−2. The excellent catalytic performance of NFN-MOF/NF can be attributed to the synergistic effect between Ni and Fe metal ions in the MOF, and a positive coupling effect between the MOF and NF.
3.3 Energy storage and conversion
MOFs with large surface areas, adjustable pore structures, and redox metal centres are promising electrode materials for electrochemical capacitors and rechargeable batteries.14,182–186 However, the inherent poor electrical conductivity of MOFs is one of the greatest obstacles to achieve high performance in capacitors. In bimetallic MOFs, the electrochemical properties can be tuned by mixing two different metals in the SBUs. Bimetallic MOFs with enhanced electrical conductivity have been applied as the electrode materials for supercapacitors.187–193 Pei, Chen and co-workers improved the electrical conductivity of MOFs by partially exchanging Ni2+ in the Ni-MOF ([Ni3(OH)2(tp)2(H2O)4]·2H2O, tp = C8H4O42−) with Co2+ or Zn2+ for hybrid supercapacitors.190 The specific capacities of Co/Ni-MOF, Zn/Ni-MOF and Ni-MOF were 236.1, 161.5 and 142.3 mA h g−1 at 1 A g−1, with a capacity retention of 82.8%, 56% and 37% at 10 A g−1, respectively (Fig. 14A). The assembled hybrid supercapacitors (Co/Ni-MOF//CNTs–COOH) exhibited an energy density of 49.5 W h kg−1 and a power density of 1450 W kg−1. The metal ion exchange resulted in an increase of free holes and the interlayer distance and in turn led to an enhancement of the electrical conductivity and surface area, explaining the enhanced performance of bimetallic MOFs for capacitors.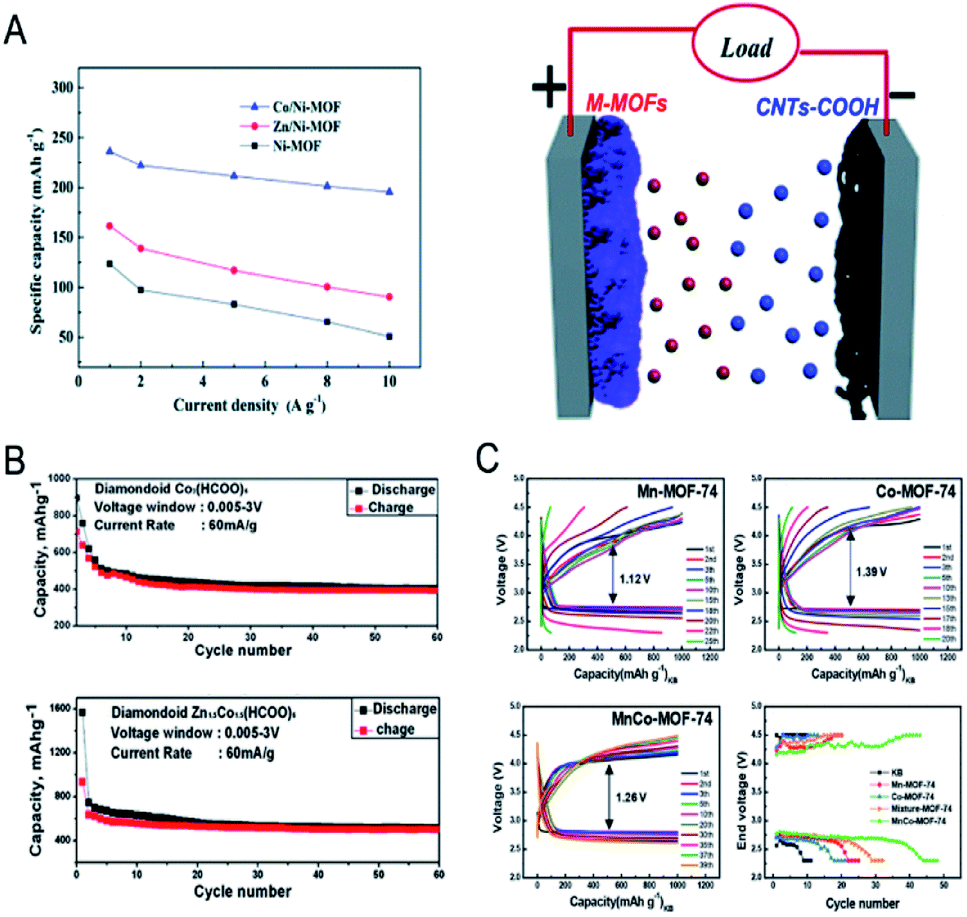 | ||
| Fig. 14 (A) Specific capacity of pristine Ni-MOF, Zn/Ni-MOF and Co/Ni-MOF at various current densities (left). An abstract illustration of hybrid supercapacitors containing M-MOFs and CNTs–COOHs as a positive and negative electrode, respectively (right). Reproduced with permission from ref. 190. Copyright 2017, Royal Society of Chemistry. (B) Capacity vs. cycle number plot for FOR3 (top) and FOR4 (bottom) (current density of 60 mA g−1, potential window 0.005–3 V, recorded at room temperature). Reproduced with permission from ref. 195. Copyright 2010, Royal Society of Chemistry. (C) Galvanostatic cycling performance of Li–O2 batteries using Mn-MOF-74, Co-MOF-74, and MnCo-MOF-74 electrodes at a limited capacity of 1000 mA h g−1 with a current density of 200 mA g−1. End voltage vs. cycle number of the electrodes. Reproduced with permission from ref. 198. Copyright 2018, American Chemical Society. | ||
Bimetallic MOFs have been investigated as anode and cathode materials for lithium-ion batteries (LIBs). When using bimetallic MOFs as anode materials, the adjustable pores can allow Li ions to be stored and reversibly inserted/extracted, leading to insertion-type electrodes.194 Some bimetallic MOFs enabling reversible transformation/regeneration can be employed as conversion-type electrodes.195 For example, Vittal et al. investigated the electrochemical performance of FOR4 (Zn1.5Co1.5(HCOO)6).195 The formate-bridged MOFs were converted to lithium formate MOFs upon lithiation and regenerated upon de-lithiation. The reversible conversion reaction led to good cycling stability of FOR4 (Fig. 14B), with a high reversible capacity of 510 mA h g−1 up to 60 cycles at 60 mA g−1, exceeding that of monometallic CoMOF (FOR3). As cathodes in LIBs, bimetallic MOFs with redox metal centres and good Li-ion mobility within the solid are good candidates, such as K0.14Mn1.43[Fe(CN)6]·6H2O196 and K0.1Ni[Fe(CN)6]0.7 [Fe(CN)6]0.3·4.7H2O.197
Bimetallic MOFs can be applied as cathode materials for lithium–oxygen batteries (LOBs). The porous structures and tuneable bimetallic active sites endow bimetallic MOFs with the capabilities to optimize the performance. Lee and co-workers used bimetallic MnCo-MOF-74 materials as cathode catalysts for Li–O2 batteries.198 MnCo-MOF-74 delivered a discharge capacity of 11150 mA h g−1 and excellent cyclability (44 cycles) with a low overpotential at a limited capacity of 1000 mA h g−1, outperforming the monometallic counterparts, Mn- and Co-MOF-74 (Fig. 14C). The synergistic integration of Mn- and Co-metal clusters contributed to the improved efficiency and reversibility.
3.4 Luminescence sensing
Luminescent MOFs (LMOFs) are an important sub-category of MOFs.199 Lanthanide (Ln) cations are particularly attractive for the construction of LMOFs because of their characteristic sharp emission bands and high quantum yields in the near-infrared and visible regions. Typically, Eu3+ and Tb3+ are usually used in LnMOFs because of their strong characteristic red emission at 614 nm (5D0 → 7F2) and green emission at 541 nm (5D4 → 7F5), respectively.Mixed Ln3+ ions with different emissions can be integrated into a MOF framework to tune the luminescence properties.200 Moreover, the metal distribution (solid solution or core–shell) in mixed LnMOFs influences the luminescence properties. Mahon, Burrows and co-workers studied the effect of metal distribution on the luminescence properties of mixed LnMOFs.201 Solid solution mixed LnMOFs, [Gd0.17Tb0.19Eu0.64(Hodip) (H2O)]·nH2O (H4odip = 5,5′-oxydiisophthalic acid), showed an emission spectrum with strong bands of Eu3+ and weak bands of Tb3+ and H4odip. This is due to the energy transfer from Tb3+ to Eu3+, resulting in a quenching of Tb3+ transitions. Core–shell mixed LnMOFs displayed different emission spectra because the metal energy transfer between Ln3+ ions was prevented. Gd@Tb@Eu and Tb@Eu@Gd showed emission spectra dominated by Tb3+ emissions with lower intensity Eu3+ and ligand transitions. Eu@Gd@Tb exhibited an emission spectrum with only Eu3+ and ligand transitions present. In all the core–shell structures, the core Ln3+ emissions were not observable. Therefore, alteration of shell ordering and thickness could tailor the luminescence properties of core–shell mixed Ln-MOFs.
A variety of bimetallic LnMOFs have acted as ratiometric sensors with high sensitivity and high selectivity toward ionic species, pH, temperature, environmental toxins, explosives, biomolecules, etc.202–204
Bimetallic LnMOFs with dual emissions can be employed as self-referencing luminescence thermometers based on the intensity ratios of two separate transitions.205 Qian, Chen and co-workers reported luminescent mixed Ln-MOFs, (EuxTb1−x)2(DMBDC)3(H2O)4·DMF·H2O (EuxTb1−x–DMBDC, DMBDC = 2,5-dimethoxy-1,4-benzenedicarboxylate, x = 0.0011, 0.0046, and 0.0069) as luminescent thermometers.206 From 10 to 300 K, the luminescence intensity of Tb3+ at 545 nm in Tb-DMBDC and Eu3+ at 613 nm in Eu-DMBDC both decreased gradually as the temperature increases (Fig. 15A and B). The mixed lanthanide MOFs exhibited a significantly different temperature-dependent luminescence behaviour. In Eu0.0069Tb0.9931–DMBDC, the emission intensity of Tb3+ decreased, while that of Eu3+ increased with the increase of temperature (Fig. 15C). The emission intensity ratio (ITb/IEu) correlated well with the temperature in the range of 50 to 200 K (Fig. 15D). Moreover, Eu0.0069Tb0.9931–DMBDC emitted temperature dependent luminescence colours from green-yellow to red at temperatures ranging from 10 to 300 K. The exhibited temperature-dependent emissions and luminescence colours in Eu/Tb-DMBDC may be attributed to the temperature-dependent energy transfer from Tb3+ to Eu3+ ions.
 | ||
| Fig. 15 (A–C) Emission spectra of (A) Tb-DMBDC, (B) Eu-DMBDC, and (C) Eu0.0069Tb0.9931-DMBDC recorded between 10 and 300 K (excited at 355 nm from the third harmonic of a Nd:YAG laser). (D) Temperature dependence of the integrated intensity ratio of Tb3+ (545 nm) to Eu3+ (613 nm) for Eu0.0069Tb0.9931-DMBDC (black squares), and temperature dependence of the integrated intensity of Tb3+ (545 nm) for Tb-DMBDC (red triangles). (Inset) Fitted curves of the integrated intensity ratio for Eu0.0069Tb0.9931-DMBDC from 50 to 200 K. Reproduced with permission from ref. 206. Copyright 2012, American Chemical Society. (E–G) Relative luminescence intensity changes of (E) Tb-ZMOF, (F) Eu-ZMOF, and (G) MZMOF-3 in the presence of different analytes. The intensity change is indicated by the colormap from −0.5 to 0.9. (H) Integrated luminescence intensity changes of MZMOF-3 suspended in MeOH toward 0.03, 0.10, and 0.13 mM LPA. Reproduced with permission from ref. 207. Copyright 2015, American Chemical Society. (I) Chemical Structure of PCM-22. (J) Relative photoemission response ratios obtained upon addition of trace H2O to Eu1:Tb5-PCM-22 pre-soaked in D2O showing a linear response. Error bars were obtained from three separate experiments. Reproduced with permission from ref. 208. Copyright 2017, Elsevier. | ||
Shi, Zaworotko and co-workers synthesized mixed Ln zeolite-like MOFs (Ln-ZMOFs), EuxTb1−x-ZMOF, as a fluorescent indicator for lysophosphatidic acid (LPA), a cancer biomarker.207 In Eu0.6059Tb0.3941-ZMOF (MZMOF-3), the integrated intensity ratio of Eu3+ and Tb3+ (IEu/ITb) correlated well with the concentration of LPA in MeOH (Fig. 15H). Moreover, MZMOF-3 exhibited selective detection for LPA over additives (water, glucose, Na+, Cl−, proline, and urea). For MZMOF-3, the luminescence intensity of LPA was 10–80 times higher than those of the additives (Fig. 15G). However, Tb-ZMOF and Eu-ZMOF exhibited similar fluorescence emission signals towards additives and LPA (Fig. 15E and F).
Mixed Ln-MOFs have also been studied as sensors for quantitative detection of water and identification of solvents. Humphrey and co-workers synthesized mixed-metal PCM-22 with controlled amounts of Eu3+, Gd3+ and Tb3+ as sensors for the identification of a wide range of solvents, and especially for trace H2O detection in D2O (Fig. 15I and J).208 PCM-22, with a formula of [Ln(tctp) (OH2)3]·3(1,4-dioxane), was synthesized by the reaction of Ln(NO3)3 with P(C6H4-p-CO2H)3 (tctpH3). For binary Eu1:Tb1-PCM-22, based on the emissions of Tb3+ (616 nm) and Eu3+ (543 nm), the ratio of I543/I616 showed a linear relationship with H2O concentration in D2O over the range of 10–120000 ppm. For ternary PCM-22 with four Eu:Gd:Tb compositions, they provided a unique “eight-factor” fingerprint for rapid identification of 18 solvents.
In conclusion, due to the synergistic effect between the two metals, bimetallic MOFs can exhibit significantly enhanced physical and chemical features compared with their monometallic counterparts. Therefore, through the rational design of bimetallic MOFs, they can afford excellent performance in gas adsorption, catalysis, energy storage and conversion, luminescence sensing, and so on.
4. Synthesis of bimetallic MOF derivatives
Since the MOF pyrolysis was reported by Xu and co-workers for porous carbon synthesis in 2008209 and for metal oxide synthesis in 2010,210 MOF derivatives have been intensively designed and synthesized.33,211 Especially, bimetallic MOFs with controllable compositions in the SBUs provide a promising platform for the preparation of a variety of functional materials, such as carbon composites (carbon supported atomically dispersed metals, metal NPs, alloy, oxides, carbides, sulphides, and phosphides), metal compounds (oxides, hydroxides, nitrides, sulphides, and phosphides), and MOF composites, which have a wide variety of physicochemical properties and applications.4.1 Carbon composites
The pyrolysis of MOFs has been adopted as an effective way to obtain carbon composites with various morphologies and compositions. The texture and graphitization degree of carbon supports have a great influence on the performance of the composites. Carbon supports with a high surface area and hierarchical pores can guarantee the free diffusion of substrates.212 Graphitized carbon can enhance electrical conductivity and chemical and thermal stability.ZIFs have been widely investigated as precursors to synthesize carbon-based materials. The pyrolysis of bimetallic ZIFs can realize well-graphitized carbons, high-surface-area carbons, and active metal–Nx–C sites at the same time, which cannot be achieved by monometallic ZIFs. For example, the pyrolysis of Zn-based ZIF-8 can afford high-surface-area carbon with high N contents but fail to provide active metal–Nx–C sites and well-graphitized carbon.213,214 On the other hand, the pyrolysis of Co-based ZIF-67 can give well-graphitized carbon and highly active Co–Nx–C sites but only offer carbon with a low surface area and porosity.215–217 In this regard, Xiong, Jiang and co-workers employed a series of bimetallic ZIFs (BMZIFs-n, n represents the ratio of Zn/Co) as precursors to afford Co, N-doped porous carbons (CNCo-n) with a large surface area, high graphitization degree, and highly dispersed N and CoNx active species (Fig. 16A).218 BMZIFs-n were pyrolyzed at 900 °C under N2, during which the generated ZnO was reduced by carbon and then evaporated. After treating the calcined products in H2SO4 at 80 °C to remove the inactive and unstable Co species, CNCo-n could be afforded. The BET surface areas of CNCo-n (n = 0, 0.2, 5, 10, 20, 40, and 100) were 270, 362, 550, 1090, 1225, 1480, and 2184 m2 g−1, respectively. Besides, Yamauchi and co-workers thermally transformed core–shell ZIF-8@ZIF-67 into nanoporous nitrogen-doped carbon@graphitic carbon (NC@GC) materials.99 ZIF-8@ZIF-67(x) (x represents the molar ratio of Co2+/Zn2+) materials were thermally treated at 800 °C under a N2 flow, followed by washing with HF solution to remove the residual Zn and unstable Co species. The obtained NC@GC samples showed intermediate surface areas between those of NC and GC derived from ZIF-8 and ZIF-67, respectively. In addition, NC@GC showed mesopores on the particle surface and abundant layered graphitic carbon structures, which were similar to those of GC.
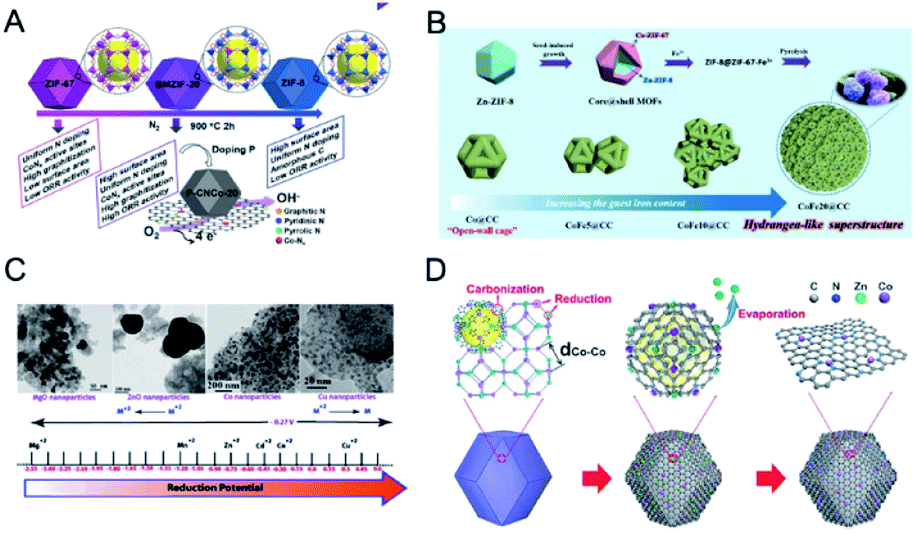 | ||
| Fig. 16 (A) Schematic illustration for the preparation of porous carbons from BMZIFs. Reproduced with permission from ref. 218. Copyright 2015, Wiley-VCH. (B) Schematic illustration of the construction of the nature-inspired hydrangea-like superstructure of open carbon cages via morphology-controlled thermal transformation of core@shell MOFs. Reproduced with permission from ref. 219. Copyright 2019, Wiley-VCH. (C) The effect of reduction potential of metal atoms present in the MOFs on the formation of metal/metal oxide nanoparticles. Metals having a reduction potential above −0.27 V undergo thermolysis in an N2 atmosphere to give pure metal nanoparticles, whereas metals with a reduction potential less than −0.27 V, even in an N2 atmosphere, produce metal oxides only. Reproduced with permission from ref. 223. Copyright 2012, Royal Society of Chemistry. (D) The formation of Co SAs/N-C. Reproduced with permission from ref. 236. Copyright 2016, Wiley-VCH. | ||
Recently, Xu and co-workers reported that introducing guest Fe ions into a ZIF-8@ZIF-67 precursor led to the self-assembly of open carbon cages into a hydrangea-like 3D superstructure (Fig. 16B).219 Direct pyrolysis of core–shell ZIF-8@ZIF-67 produced isolated open-wall N-doped carbon cages with insufficient contact and total conductivity. The introduction of guest Fe ions into the MOF precursor formed FeCo alloy NPs during the pyrolysis, which could catalyse the growth of carbon nanotubes and thus interconnect neighbouring cages to form a hydrangea-like 3D superstructure.
The employment of bimetallic MOFs as precursors can afford precise size, composition, and structure control of the supported metal/oxide NPs.220–222 This is benefited from the ability to control the compositions and distributions of metal ions in MOFs, which are not achievable by other synthetic methods. The calcination of bimetallic MOFs under an inert atmosphere can afford metal alloy or oxide NPs depending on the reduction potential of the metal ions.223 Metal ions (e.g., Cu2+ and Co2+) with a reduction potential of −0.27 V or higher usually form metal NPs after calcination under an inert atmosphere, whereas metal ions (e.g., Cd2+ and Mg2+) with reduction potentials lower than −0.27 V form metal oxide NPs (Fig. 16C). For example, Chen and co-workers calcined bimetallic MOFs (Fe3[Co(CN)6]2) at 600 °C in N2, affording FeCo alloy NPs encapsulated in N-doped graphene layers.220 Gao, Lou and co-workers pyrolyzed Co/Mn-MIL-100 at 800 °C in a 5% H2/Ar atmosphere, affording a MnO/Co hybrid supported by porous graphitic carbon.224
The type of metal compound generated from the pyrolysis of bimetallic MOFs can be affected by the elements in the ligands. Carbon atoms can diffuse into the interstices of metal atoms to form bimetallic carbides.225–227 For example, Su and co-workers reported the synthesis of Co/Mo2C/Mo3Co3C@C and Ni/Mo2C@C derived from two bimetallic MOFs (CoMo-MOF and NiMo-MOF) annealed at 900 °C in N2, respectively.226 Other metal compounds (metal nitrides, phosphides and sulphides) supported on carbon materials can also be synthesized from bimetallic MOFs if suitable ligands with heteroatoms are employed.
Bimetal nitrides, phosphides and sulphides embedded in carbon can also be obtained through introducing additional precursors (N/P/S-containing regents) before228–231 or after the pyrolysis process.94,232–235 Moreover, additional metal precursors can be introduced into MOFs before calcination to alter the composition of metal compounds.
The pyrolysis of MOFs serves as an ideal route for preparing various metal single atoms (SAs) on porous carbon (SAs/C). Delicate control is required to avoid the conversion of metal ions into aggregated NPs at high temperature. Bimetallic MOFs offer a promising platform for the synthesis of SAs/C based on the idea of using a second metal that is easily removed after pyrolysis to extend the distance between adjacent targeted metals to avoid aggregation during pyrolysis.236–240 Wu, Li and co-workers demonstrated the concept for the synthesis of Co SAs on N-doped porous carbon (Co SAs/N–C) through the pyrolysis of a bimetallic Zn/Co MOF (Zn/Co-BMOF).236 The addition of Zn2+ could dilute the concentration of Co2+ and extend the adjacent distances between Co atoms. During the pyrolysis process, Zn was evaporated at high temperature, leaving abundant N sites to anchor and stabilize the isolated Co atoms (Fig. 16D). Co SAs/N-C obtained by calcination at 800 and 900 °C showed a 4 and 2 metal coordination number with surrounding N atoms, respectively. Further increasing the calcination temperature to 1000 °C broke the Co–N bonds and caused the formation of Co NPs. In the sequent work, Wu, Li and co-workers synthesized Fe–Co dual sites embedded on N-doped porous carbon [(Fe,Co)/N–C] derived from Zn/Co-BMOF encapsulated FeCl3.241 FeCl3 was immobilized within the pores of Zn/Co-BMOF by a double-solvent method. During the calcination, Fe salts were reduced by carbon and bonded with neighbouring Co atoms to form Fe–Co dual-sites. The Fe species would accelerate the decomposition of MOFs and force the generation of voids inside the N-C support.
4.2 Metal compounds
The thermal calcination of bimetallic MOFs in air can lead to their decomposition into their corresponding bimetallic oxides. To achieve a higher surface area and porosity and preserve the original structure of parent MOFs to a certain extent, a two-step annealing (first in N2 and then in air) can be applied to synthesize metal oxide nanostructures. The use of bimetallic MOFs as precursors to obtain bimetallic oxides has emerged as a powerful synthetic route, offering the possibility to precisely control the compositions and structures of metal oxides.The achieved success in the control of metal distribution in MOFs can allow the tuning of the composition of the obtained metal oxides.242–245 Zhou and co-workers synthesized ZnxCo3−xO4 (0 < x ≤ 1) hollow polyhedra through calcination of bimetallic Zn-Co-ZIFs.242 Bimetallic Zn–Co-ZIFs were first heated in N2 and then in air at 400 °C. The first thermal treatment in N2 was of vital importance to partially preserve the porous structure of ZIFs. Monge, Gándara and co-workers demonstrated the control of the composition of multimetal oxides through the use of MOFs constructed from up to four different metal elements (Fig. 17A).244 Multimetallic MOFs, constructed from a helical inorganic SBU with different cations (Zn2+, Co2+, Mn2+, and Ca2+) and an organic linker H2hfipbb [4,4′-(hexafluoroisopropylidene)bis-(benzoic acid)], were subjected to a thermal treatment in air at 800 °C to obtain the corresponding multimetal oxides. The metal arrangements and ratios of MOFs could be translated to metal oxides after calcination.
 | ||
| Fig. 17 (A) Thermal decomposition of multimetal MOFs allows the obtaining of multication oxides with compositions not attainable with conventional oxide synthetic routes. The metal ratios in the resulting oxides are transferred from their corresponding parent MOFs. Reproduced with permission from ref. 244. Copyright 2019, American Chemical Society. (B) Schematic illustration of the formation process of Co3O4/Co–Fe oxide DSNBs: (I) ion–exchange reaction between ZIF-67 NCs and [Fe(CN)6]3− ions to form ZIF-67/Co–Fe PBA YSNCs and (II) subsequent conversion to Co3O4/Co–Fe oxide DSNBs by thermal annealing (top). (a, c) FESEM and (b, d–f) TEM images of (a, b) ZIF-67 NCs and (c–f) ZIF-67/Co–Fe PBA YSNCs. Reproduced with permission from ref. 248. Copyright 2018, Wiley-VCH. (C) Illustration of the synthesis process from MOF-74 to Ni–Co MDH. Reproduced with permission from ref. 250. Copyright 2017, American Chemical Society. (D) Schematic diagram of synthesis for porous CoxNiyN composites. Reproduced with permission from ref. 253. Copyright 2019, American Chemical Society. (E) Schematic illustration of fabrication of hollow Co-based bimetallic sulphides. Reproduced with permission from ref. 254. Copyright 2016, American Chemical Society. | ||
Metal oxides with complex structures can be fabricated from rationally designed bimetallic MOF precursors.246,247 Song, Lou and co-workers reported the preparation of Co3O4/Co–Fe oxide double-shelled nanoboxes (DSNBs) derived from ZIF-67/Co–Fe PBA yolk–shell nanocubes (YSNCs).248 ZIF-67/Co–Fe PBA YSNCs were fabricated through an anion-exchange reaction between ZIF-67 and [Fe(CN)6]3− ions. The ZIF-67/Co–Fe PBA YSNCs were further converted to Co3O4/Co–Fe oxide DSNBs by calcination in air (Fig. 17B).
Metal hydroxides can be fabricated from bimetallic MOF precursors through alkali hydrolysis.249–252 Zou, Liu and co-workers reported the synthesis of bimetallic double hydroxides (MDHs) derived from bimetallic NiCo-MOF-74 through an alkaline treatment (Fig. 17C).250 Ni/Co MOF-74 samples were dispersed in a 2 M KOH aqueous solution under stirring at room temperature, followed by hydrothermal treatment at 120 °C for 2 h. The hydrothermal treatment with KOH resulted in the destruction of micron-sized MOF-74 and the formation of nanoscale MDH.
Bimetallic nitrides can be prepared from bimetallic MOFs through nitridation under an NH3 atmosphere. Guo and co-workers synthesized bimetallic CoNi nitrides derived from bimetallic CoNi-MOF-74.253 CoNi-MOF-74 was first converted to Ni1Co2O4 through calcination at 400 °C in air, and then transformed into Co2Ni1N by nitridation under an NH3 atmosphere at 500 °C (Fig. 17D). The resulting metal nitrides inherited the morphology from their corresponding precursors.
Bimetallic sulphides can be synthesized using bimetallic MOFs as templates via a hydrothermal sulfidation reaction.193,254–256 Zou and co-workers reported the synthesis of hollow bimetallic sulphides (MxCo3−xS4, M = Zn, Ni, and Cu) by solvothermal sulfidation and thermal annealing from Co-based bimetallic MOFs.254 MCo-MOFs (M = Zn, Ni, and Cu) were treated in an ethanol solution of thioacetamide (TAA) at 120 °C for 4 h, followed by calcination under a N2 atmosphere at 350 °C for 2 h (Fig. 17E). The obtained MxCo3−xS4 preserved the rhombic dodecahedral morphology of MOF precursors, while formed a hollow structure after the sulfidation procedure. S2− ions released from TAA first reacted with metal ions on the MOF surface to form a thin layer of sulphides, which could promote the outward diffusion of metal ions against the inward diffusion of S2− ions. Then the inner framework was gradually dissolved and released metal ions to react with S2− ions on the outer surface, which finally led to the generation of a hollow void inside the shell.
Transition metal phosphides (TMPs) show extremely low cost and expected long-term stability in both acidic and alkaline operating environments. Bimetallic MOFs have been applied for the synthesis of TMPs.257–262 Zhao and co-workers synthesized nickel–cobalt mixed metal phosphide nanotubes (CoxNiyP, x and y represent the molar ratios of Co and Ni in the MOF precursor, respectively) derived from bimetallic CoNi-MOF-74.258 Bimetallic MOF-74 materials with different Co/Ni ratios were first calcined at 350 °C in air to afford CoxNiyO, followed by phosphorization with NaH2PO2 at 300 °C in N2 to afford CoxNiyP.
As summarized above, various types of metal compounds can be obtained through the treatment of bimetallic MOFs.
4.3 MOF composites
Complete decomposition of MOFs usually leads to a dramatic decrease of the surface area and loss of well-defined pore structures.209,213 Recently, Xu and co-workers have demonstrated a new concept of “quasi-MOFs” fabricated through controlled deligandation of MOFs, which can realize both a porous structure and exposed inorganic nodes.263 This controlled thermal transformation method has been successfully applied on solid solution bi- and multi-metallic MOFs for the synthesis of MOF immobilized metal NPs or oxides.264,265 This design takes advantage of different coordination abilities of the metal ions with the organic linkers. The metal ions with weaker coordination with the organic linkers transform into metal NPs/compounds under certain conditions while the other metal ions remain coordinated with the organic linkers to preserve the framework. Hu and co-workers reported a thermal transformation of a benzimidazole-modified Cu/Co bimetal-MOF into a Cu-nanowire@quasi-MOF composite (Fig. 18A).264 The Cu/Co bimetal-MOF was calcined at 300 °C for 30 min under a N2 atmosphere to afford a quasi-MOF matrix with a partially preserved MOF structure embedded with Cu nanowires. Under similar calcination conditions, monometallic Cu-MOF transformed into core–shell Cu2O@N doped carbon composites. During the calcination process, Cu-ligand bonds with weaker coordination would break and form metastable Cu NPs, while the Co-MOF part with higher stability would serve as a morphology retainer and a nano-channel template to reassemble Cu NPs into Cu nanowires. | ||
| Fig. 18 (A) Scheme and TEM images of Cu MOF(300), Cu/Ni bimetal-MOF(300), and Cu-nanowire@Quasi-MOF. Reproduced with permission from ref. 264. Copyright 2019, Royal Society of Chemistry. (B) Synthetic procedure of the Zn1−xCox-ZIF@Zn1−xCoxO hybrid photocatalyst. Reproduced with permission from ref. 266. Copyright 2018, Royal Society of Chemistry. (C) In situ self-assembly of MOA through the reaction between a metal ion and organic ligand. MOA@H-MOF composite formed by a one-pot self-assembly reaction. H-MOF formed through removing the MOA template. Reproduced with permission from ref. 267. Copyright 2015, Nature Publishing Group. (D) Ilustration of the structural evolution (top). TEM image, SEM image, and EDX mappings of H-Zn/Co-ZIF (bottom). Reproduced with permission from ref. 268. Copyright 2015, Wiley-VCH. | ||
Besides thermal transformation, solvent induced transformation is also a powerful strategy for the conversion of bimetallic MOFs to composites of MOFs and metal compounds. Luo and co-workers obtained Zn1−xCox-ZIF@Zn1−xCoxO through the transformation of Zn1−xCox-ZIF with AgNO3 (Fig. 18B).266 Zn1−xCox-ZIF was dispersed in an aqueous solution of silver nitrate (AgNO3) and stirred for 1 h to obtain Zn1−xCox-ZIF@Zn1−xCoxO. AgNO3 acted as the catalyst to break the coordinative bonds of MOFs and created a hydroxyl rich environment for the formation of Zn1−xCoxO on the surface of Zn1−xCox-ZIF.
Selectively removing the core MOFs in single- or multi-core–shell bimetallic MOFs can obtain hierarchical-pore or hollow MOF immobilized metal compound composites. In this design, less stable MOFs act as seeds to grow stable MOFs as the shell, followed by selective removal of the core MOFs to generate pores. The generated mesopores/macropores can facilitate the diffusion of substrates and the desorption of products.
For example, Li, Zhong and co-workers used MOF-5 as a template to grow UiO-66(Zr), followed by the removal of the cores to synthesize hierarchical UiO-66(Zr) (H-UiO-66).267 MOF-5 is stable in certain solvents but sensitive towards moisture and acid, while UiO-66(Zr) is stable even in acidic solutions. Nano-sized MOF-5 particles were first synthesized, followed by the growth of UiO-66(Zr). The obtained material was washed with acid aqueous solution to get the targeted material H-UiO-66(Zr) with Zr species anchoring in the mesopore surface (Fig. 18C). In another system, Wu, Li, and co-workers reported a mild phase transformation of core–shell ZIF-67@ZIF-8 structures into hollow Zn/Co ZIF particles.268 They treated core–shell ZIF-67@ZIF-8 with Co2+ in methanol at 120 °C for 4 h, resulting in the dissolution of the solid interior and then the formation of a hollow structure with interlaced nanoplates (Fig. 18D). The authors proposed that methanol molecules formed H-bonds in the presence of Co2+ to break the coordination bonds between Co2+ and 2-MeIm, driving the structural evolution of ZIF-67. Muhler, Fischer and co-workers reported similar results in parallel.269 During the epitaxial growth of ZIF-8 on preformed ZIF-67 under solvothermal conditions, ZIF-67 partially dissolved to release cobalt ions to form cobalt hydroxide with a sheet-like structure.
Carbon composites, metal compounds and MOF composites can be derived from bimetallic MOFs through pyrolysis, hydrothermal treatment, and so on. This wide variety of MOF-derived materials can be applied in many applications, which will be introduced in the following section.
5. Applications of bimetallic MOF derivatives
The applications of the majority of bimetallic MOFs are restricted by their low stability, poor conductivity and blockage of active sites. Bimetallic MOF-derived materials show high stability, high conductivity, and exposed active sites. These properties provide bimetallic MOF derivatives with more opportunities for applications in catalysis under harsh conditions and electrochemical energy storage and conversion.5.1 Catalysis
MOF-derived materials are active for many reduction reactions, such as the hydrogenation of phenol,270 nitro compounds271,272 and nitriles,221 and the reduction of NO273,274 and CO2.275 As an example, Li and co-workers applied bimetallic alloy NPs embedded in an N-doped carbon matrix (M–M′@C–N, M/M′ = Co, Ni, Cu) derived from bimetallic MOFs (M–M′(1,4-bdc)2(dabco)·4DMF·1/2H2O, M/M′ = Co, Ni, Cu) for transfer hydrogenation of nitriles to imines.221 In the hydrogenation of benzonitrile to the corresponding imine, Co–Ni(3:1)@C–N exhibited remarkable activity with over 98% yield for the desired product after 15 h, which was 5 times and 7 times as high as that of Ni@C–N and Co@C–N, respectively (Fig. 19A). Co–Ni@C–N with a Co–Ni molar ratio of 3:1 showed the highest activity; further decreasing or increasing the proportion of Co resulted in a decrease of activity.
 | ||
| Fig. 19 (A) The conversion of benzonitrile and selectivity of the corresponding imine with various catalysts for the transfer hydrogenation of benzonitrile. Reaction conditions: 0.5 mmol nitriles, 4 mL i-PrOH, catalyst (metal 10 mol%), 80 °C, and 15 h. Reproduced with permission from ref. 221. Copyright 2016, Royal Society of Chemistry. (B) Schematic diagram of the synthesis procedure of the bimetallic MOF-derived CeCuOx catalyst (top). Toluene conversion and CO2 yield versus temperature curves over CeO2, CuO, and CeCuOx catalysts (left bottom). Conversion versus temperature curves for complete oxidation of methanol, acetone, toluene, and o-xylene over the CeCuOx catalyst. (Note: [VOC] = 500 ppm, mcatalyst = 0.15 g, and gas hourly space velocity (GHSV) = 23000 h−1) (right bottom). Reproduced with permission from ref. 278. Copyright 2019, Wiley-VCH. (C) Products of HMF oxidation (top). DFF yield as a function of reaction time over the FeCo/C catalysts. Reaction conditions: 1 mmol HMF, catalyst (metal 20 mol%), Na2CO3 1 mmol, 2 mL toluene, 1 MPa O2, 100 °C, and 6 h (bottom). Reproduced with permission from ref. 290. Copyright 2016, Royal Society of Chemistry. (D) Depolymerization of renewable lignin extracted from poplar to monophenolic compounds with the Ni–Co@C catalyst was prepared from MOFs (top). Monophenol yield on the NixCo1−x/C catalyst (bottom). Reproduced with permission from ref. 294. Copyright 2019, American Chemical Society. | ||
Bimetallic MOF-derived porous catalysts have also been developed for a variety of oxidation reactions, such as the oxidation of alcohols,271,276,277 acetone, olefin and toluene,278 and organic pollutant degradation.279,280 As an example, Han, Yeung and co-workers prepared a binary metal oxide (CeCuOx) derived from bimetallic MOFs (CeCuBDC) as the catalyst for the oxidation of organic compounds.278 In the oxidation of toluene to CO2, CeCuOx exhibited high catalytic activity, with a T50 (temperature required for 50% conversion) and T90 of 150 and 186 °C, respectively (Fig. 19B). CeCuOx showed lower activation energy (52.4 kJ mol−1) than CeO2 (57.5 kJ mol−1) and CuO (70.5 kJ mol−1). The CeCuOx catalyst was also highly active for the oxidation of methanol, acetone and o-xylene, with a T50 of 80, 87 and 142 °C, respectively.
Controlled generation of hydrogen from chemical hydrogen storage materials is of vital importance to the hydrogen-economy paradigm in the future.281–284 Bimetallic MOF-derived nanomaterials exhibit high catalytic performance for the release of hydrogen from chemical hydrogen storage materials such as ammonia borane (NH3BH3, AB) and lithium borohydride (LiBH4).285–288 Chen and co-workers fabricated porous cobalt phosphide supported by carbon-based nanoframeworks (CoP@CNFs), which was derived from Zn/Co-ZIF and was applied for hydrogen generation from AB.286 Zn/Co-ZIF was calcined in air to get Zn–Co–O@CNF, followed by phosphidation using Na2H2PO2 and further acid treatment to get CoP@CNFs. CoP@CNFs gave an initial TOF value of 165.5 molH2 molmetal−1 min−1 at 298 K, maintaining 90% of its initial activity after 4 cycles.
Biomass has been an important resource for the production of chemicals, fuels, and energy.289 Bimetallic MOF-derived nanocatalysts have been applied in effective biomass transformation.290–295 Li, Luque and co-workers applied hollow Fe–Co supported on carbon (FeCo/C) derived from bimetallic Fe/Co-MIL-45b for chemical conversions of biomass-derived platform chemicals.290 In the aerobic oxidation of biomass-derived 5-hydroxymethylfurfural (HMF) to 2,5-diformylfuran (DFF), FeCo/C(T) prepared by calcination at different temperatures (T = 500, 600, 700 or 800 °C) showed distinct activities. FeCo/C(500) exhibited the highest activity, with a complete conversion and >99% selectivity to DFF at 100 °C after 6 h (Fig. 19C). Jones and co-workers prepared Co/Cu–carbon (Co/Cu–C) derived from Cu/Co MOF-74 for furfural hydrogenation.292 The catalyst performance was investigated at 180 °C and a weight/volume flow rate (W/F) of 3.63 gcat h mol−1, and monometallic Co–C showed high conversion (50%) and high selectivity for 2-methylfuran (49%), while Cu–C had low conversion but high selectivity toward furfuryl alcohol. CoCu–C-400 (pyrolyzed at a temperature of 400 °C) with a partial Cu shell showed reactivities characteristic of Co–C. CoCu–C-600 with a full Cu shell had reactivities similar to those of Cu–C. Although bimetallic CoCu catalysts did not show activity/selectivity improvements compared to those of monometallic catalysts for furfural conversion, these results have demonstrated synthesis–structure–property correlations, which would guide the future development of better catalysts. The conversion of lignin (10–35% by weight in biomass) to value-added chemicals, alternative fuels, and platform compounds is attractive. Bao, Ren and co-workers reported the use of bimetallic-MOF-derived Ni–Co/C materials with varying Ni/Co ratios as catalysts for lignin conversion into monophenols.294 In the hydrogenolysis of poplar lignin, Ni0.5Co0.5/C showed superior catalytic efficiency to Ni/C, with a yield of 55.2% to monophenols and a selectivity of 70.3% to guaiacol (Fig. 19D).
Photocatalytic degradation of organic pollutants from wastewater is of ecological and environmental importance. Nanomaterials derived from bimetallic MOFs have shown high activity and stability in the degradation of organic pollutants, such as organic dyes and antibiotics.266,279,296–299 Li and co-workers designed ZnO@C–N–Co core–shell nanocomposites derived from Zn/Co ZIF for the degradation of methyl orange (MO).297 When calcining Zn/Co ZIF at 600 °C, ZnO NPs generated from the ZIF-8 shell would aggregate and move to the hollow cavity while the internal Co NPs transferred inversely to the N–C shell, resulting in the unique ZnO@C–N–Co core–shell structure. Under the irradiation of a Xe lamp (300 W) for 2.5 h, ZnO@C–N–Co exhibited a high degradation percentage of MO (99.5%), much higher than that of ZIF-67-600 (29.4%) and ZIF-8-600 (41.9%). The superior performance of ZnO@C–N–Co could be attributed to the synergistic effect between the components, in which the porous carbon shell contributed to the stabilization of ZnO and the adsorption of reactants and Co NPs inhibited the recombination of electrons and holes (Fig. 20A).
 | ||
| Fig. 20 (A) Proposed mechanism of the photodegradation of organic pollutants using the as-prepared core–shell ZnO@C–N–Co as the catalyst (left). Photodegradation curves of MO over various catalysts under irradiation with UV-vis light (right). Reproduced with permission from ref. 297. Copyright 2017, Royal Society of Chemistry. (B) Schematic illustration of the synthetic procedure for NiS/ZnxCd1−xS (left). Comparison of the photocatalytic HER rates of NiS/ZnxCd1−xS under visible-light irradiation (right). Reproduced with permission from ref. 306. Copyright 2018, Wiley-VCH. (C) Comparison of the photocatalytic activities of the photocatalysts under UV-vis irradiation, calculated according to how each compound evolved after 6 h. Schematic illustration of the photocatalytic CO2 reduction with ZnO (middle) and ZnO@Co3O4 (right). Reproduced with permission from ref. 309. Copyright 2016, Royal Society of Chemistry. | ||
Photocatalytic water splitting for hydrogen production is a promising route to convert solar energy into renewable hydrogen energy.300,301 Bimetallic MOF-derived nanomaterials, such as Fe–Ni–P,302 CuS/ZnS,303 Co-doped Zn1−xCdxS,304 yolk–shell CdS microcubes,305 and NiS/ZnxCd1−xS,306 have been applied in photocatalytic water splitting for hydrogen production. As an example, Shi, Cheng and co-workers constructed non-noble metal co-catalyst/solid solution heterojunction NiS/ZnxCd1−xS derived from Ni-ZnxCd1−x-MOF for the production of hydrogen (Fig. 20B).306 Under visible-light irradiation (λ > 420 nm), NiS/Zn0.5Cd0.5S exhibited a high HER rate of 16.78 mmol g−1 h−1, much higher than those of NiS/ZnS (0.09 mmol g−1 h−1), NiS/ZnS (0.09 mmol g−1 h−1) and NiS/CdS (3.52 mmol g−1 h−1). The tuning of the Zn/Cd ratio in heterojunction ZnxCd1−xS pointed to an optimized x of 0.5 to achieve a good balance between the light absorption capacity of the catalyst and edge of the conduction band. The co-catalyst NiS could further accelerate the water dissociation kinetics to improve the photocatalytic hydrogen evolution activity. Furthermore, constructing composites of co-catalysts and bimetallic MOF-derived photocatalysts, such as Cu0.9Co2.1S4@MoS2,307 CdS/ZnxCo3−xO4,308 Pt–ZnO–Co3O4,262 Pt–ZnS–CoS,262 Pt–Zn3P2–CoP,262 and MoS2@ZnCoS,256 can greatly enhance the activity for photocatalytic hydrogen evolution.
Photocatalytic reduction of CO2 is considered as an attractive approach to address fossil fuel shortage and carbon emission problems. Ye and co-workers prepared a ZnO@Co3O4 composite derived from ZIF-8@ZIF-67 for CO2 photoreduction to CH4 and CO.309 Under UV-vis irradiation, ZnO@Co3O4 gave a CH4 generation rate of 0.99 μmol g−1 h−1, higher than those of ZnO derived from ZIF-8 and Co3O4 derived from ZIF-67 (Fig. 20C). In ZnO@Co3O4, ZnO played a main role in the catalytic transformation, while Co3O4 effectively protected ZnO from photocorrosion to enhance the photocatalytic stability. Deng, Peng and co-workers used carbonized cobalt composites (C-BMZIFs) derived from bimetallic Zn/Co-ZIF as co-catalysts for photocatalytic CO2 reduction.310 Under visible light irradiation with [Ru(bpy)3]2+ as the photosensitizer and triethanolamine (TEOA) as the electron donor, C-BMZIFs (Zn/Co = 3/1) delivered a high CO yield of 1.1 × 104 mmol g−1 h−1. Interestingly, decreasing the Zn/Co ratio in the Zn/Co-ZIF precursors could increase the size of Co particles in C-BMZIFs, which could favor the evolution of H2 against CO, resulting in the decrease of the CO/H2 ratio in the produced syngas. Other nanomaterials derived from bimetallic MOFs, such as CuNi/C311 and ZnO/NiO,312 have also been designed for photocatalytic reduction of CO2.
The ORR is the key reaction on the cathode of fuel cells and metal–air batteries. Bimetallic MOF derivatives,244 especially Fe and/or Co in N-doped carbon,218,236,237,241,316 have been investigated for ORR electrocatalysis.317 These supported metal catalysts, in the form of atomically dispersed metal sites, metal clusters, and metal NPs have demonstrated activities for the ORR. Deng and co-workers reported the controlled synthesis of NPs, atomic clusters (ACs), and SAs of Co catalysts on N-doped porous carbon derived from ZnCo-ZIFs for the ORR.318 Three samples with Zn/Co molar ratios of 0:1, 2:1, and 8:1 were synthesized, denoted as Co-ZIF, ZnCo-ZIF-2, and ZnCo-ZIF-8, respectively. Upon pyrolysis at 1173 K in Ar and subsequent reflux treatment in H2SO4 solution, Co-ZIF, ZnCo-ZIF-2, and ZnCo-ZIF-8 transformed into Co-NPs@NC, Co-ACs@NC, and Co–SAs@NC nanocomposites, respectively (Fig. 21A). These catalysts showed size-dependent ORR activity in a 0.1 M KOH electrolyte. Co–SAs@NC exhibited a superior activity with a half-wave potential (E1/2) of 0.82 V, outperforming those of Co-ACs@NC (0.81 V) and Co-NPs@NC (0.80 V). The determined electron transfer number of Co–SAs@NC is above 3.9, higher than those of Co-ACs@NC and Co-NPs@NC. These results demonstrated that isolated single Co atoms could provide maximum atom-utilization and be well stabilized by an N-doped carbon support to show superior reactivity and stability. Besides Fe and Co, Mn-based catalysts are promising alternatives to address the insufficient durability of ORR catalysts. Wang, Wu and co-workers synthesized an atomically dispersed Mn–N–C catalyst for the ORR, obtained by calcination of MnZn-ZIF-8 followed by adsorption and thermal activation processes to increase the density of MnN4 active sites (Fig. 21B).239 The 20Mn-NC-second catalyst (where 20 represents the molar percentage of Mn against the total metals for the synthesis of MnZn-ZIF-8; second refers to the second adsorption step) exhibited the most positive E1/2 of 0.8 V. Mn-NC-first showed poor ORR activity due to insufficient density of active sites. The 20Mn-NC-second catalyst showed a H2O2 yield of less than 2%, indicating a four-electron reduction pathway. The 20Mn-NC-second catalyst as a cathode in membrane electrode assemblies for fuel cells showed a power density of up to 0.46 W cm−2.
 | ||
| Fig. 21 (A) TEM images of Co-NPs@NC (left top), HAADF-STEM images of Co-ACs@NC (middle top) and Co–SAs@NC (right top). ORR polarization curves obtained using RRDEs at 1600 rpm and Jk at 0.6 V and E1/2 for synthesized Co-NPs@NC, Co-ACs@NC, Co–SAs@NC and Pt/C catalysts (bottom). Reproduced with permission from ref. 318. Copyright 2019, Wiley-VCH. (B) Schematic of atomically dispersed MnN4 site catalyst synthesis (top). Steady-state ORR polarization plots in 0.5 M H2SO4 electrolytes (Pt/C catalyst reference was studied in 0.1 M HClO4) to study the effect of synthesis steps (that is, first doping and second adsorption) on Mn–N–C catalyst activity (left bottom). Fuel cell performance of the best-performing 20Mn-NC-second and 20Fe-NC-second catalysts under both H2/O2 and H2/air conditions. Error bars represent the standard deviation from at least three independent measurements (right bottom). Reproduced with permission from ref. 239. Copyright 2018, Nature Publishing Group. (C) Scheme of the fabrication of trimetal NiCoFe-MOF-74 and the partial pyrolysis of NiCoFe-MOF-74 to NiCo/Fe3O4/MOF-74 (top). OER polarization curves of NiCoFe-MOF-74 and the derived partial pyrolyzed and completely pyrolyzed samples at 1600 rpm (left bottom). Relative current of initial current density at a constant potential of 1.47 V as a function of test time (chronoamperometric method) (right bottom). NiCoFe-MOF-74-400 °C-1 h (NiCo/Fe3O4/MOF-74) (red curve), MOF-74 (blue curve), and commercial RuO2 (green curve). Reproduced with permission from ref. 265. Copyright 2018, American Chemical Society. (D) LSV curves of FeP/C and NFP/C-3 in 0.5 M H2SO4 and 1.0 M PBS and 1.0 M KOH solutions. Chronopotentiometry curves at a constant current density of 10 mA cm−2 for 12 hours of NFP/C-3 in 0.5 M H2SO4, 1.0 M PBS, and 1.0 M KOH solutions. Reproduced with permission from ref. 228. Copyright 2019, American Association for the Advancement of Science. | ||
Efficient OER electrocatalysis is important for various energy-related processes, such as electrochemical water splitting and rechargeable metal–air batteries. Bimetallic MOF-derived metals/alloys embedded in carbon,319 metal compounds248,261 and their carbon composites233,320–322 have demonstrated OER activity.323,324 Xu and co-workers applied FeCo–P/C nanocomposites derived from bimetallic MOFs for the OER.232 FexCoy bimetallic MOFs with various Fe/Co ratios (x = 1, 2 and y = 1, 2) were employed as precursors to synthesize FexCoy-P/C. The overpotentials of FexCoy-P/C in 1.0 M KOH at a current density of 10 mA cm−2 showed a trend of Fe1Co1–P/C (360 mV) < Fe1Co2–P/C (362 mV) < Fe2Co1–P/C (368 mV). All the FexCoy-P/C materials exhibited a better OER activity than their FexCoy/C without P-doping. The authors demonstrated that both P- and Fe-doping could reduce the resistance of charge transfer to accelerate the electron transfer and thus improve the OER activity. Considering that the complete decomposition of MOFs usually leads to a dramatic decrease of the surface area and pore structure, partial decomposition of bimetallic MOFs can preserve the porous structure to a certain extent for effective substrate diffusion while producing active metal NPs. Li and co-workers reported the synthesis of NiCo/Fe3O4/MOF-74 through controlled pyrolysis of trimetallic NiCoFe-MOF-74 for efficient OER electrocatalysis (Fig. 21C).265 The obtained NiCo/Fe3O4/MOF-74 retained 68% of the specific surface area of the pristine NiCoFe-MOF-74 (820 m2 g−1). In NiCo/Fe3O4/MOF-74, NiCo/Fe3O4 heterostructures with a NiCo alloy enriched in the core and Fe3O4 distributed on the shell were well dispersed in the partially retained MOF-74 structure. In a 1.0 M KOH electrolyte, NiCo/Fe3O4/MOF-74 exhibited an overpotential of 238 mV at 10 mA cm−2 on a glassy carbon electrode, surpassing those of NiCoFe-MOF-74 (270 mV), the totally decomposed NiCoFe-MOF-74 derivative (366 mV) and the commercial RuO2 catalyst (310 mV). NiCo/Fe3O4/MOF-74 could retain 99.1% of its initial OER activity after 36 h. The excellent activity and stability of NiCoFe-MOF-74 for the OER could be attributed to the porous structures of MOF-74 and the synergistic effect between NiCo and Fe3O4.
Efficient and inexpensive HER electrocatalysis to produce hydrogen from water splitting is of significant importance. Bimetallic MOF-derived materials, especially Mo-based materials325 and metal nitrides/phosphides/sulphides,253,254,258,259,326 have been intensively investigated due to their favourable hydrogen adsorption energy toward the HER.327 Lou and co-workers synthesized carbon-supported Ni-doped FeP nanocrystals from Ni-doped MIL-88A as HER electrocatalysts.228 Ni-doped MIL-88A was treated with phytic acid and subsequent pyrolysis under Ar and H2 to obtain Ni-doped FeP/C. The overpotential at 10 mA cm−2 of NFP/C-3 (Ni/Fe atomic ratio of 3) was 72, 117, and 95 mV in 0.5 M H2SO4 (pH = 0), 1.0 M phosphate-buffered saline (pH ≈ 7) and 1.0 M KOH (pH = 14), respectively (Fig. 21D). The NFP/C-3 electrocatalyst also exhibited good stability over the full pH range. The carbon support is important to structural integrity and catalytic performance. NFP-3 without carbon showed degradation of electrocatalytic activity and stability. XPS analysis and DFT calculations demonstrated that Ni doping could weaken the strong hydrogen adsorption on pure FeP to favour the HER.
Highly active and robust bimetallic MOF-derived multifunctional electrocatalysts for the ORR, OER and/or HER can be applied for efficient rechargeable metal–air batteries185,219,224,315 and overall water splitting.230,233,258,328–330 Very recently, Xu and co-workers synthesized a capsular carbon embedded with iron-nickel phosphide NPs (FeNiP/NCH) through pyrolysis of capsular-MOF followed by phosphidation with melamine.94 FeNiP/NCH was employed as a trifunctional electrocatalyst for the ORR/OER/HER. In 1.0 M KOH solution, FeNiP/NCH obtained a current density of 10 mA cm−2 at overpotentials of 250 and 216 mV for the OER and HER, respectively. For ORR electrocatalysis in 0.1 M KOH solution, FeNiP/NCH exhibited an E1/2 of 0.75 V. When FeNiP/NCH was applied as both cathode and anode electrocatalysts for overall water splitting, a small cell voltage of 1.59 V was achieved at 10 mA cm−2 (Fig. 22). When FeNiP/NCH was applied as the air electrode electrocatalyst in a Zn–air battery, a high power density (250 mW cm−2) could be achieved. In a galvanostatic discharge–charge cycling test at 5 mA cm−2, the charge/discharge potentials were 1.89 and 1.23 V with a small voltage gap of 0.66 V. Moreover, no obvious voltage gap change was observed for 500 h, indicating the high stability of FeNiP/NCH.
 | ||
| Fig. 22 (A) HER polarization of FeNiP/NCH and control samples in 1 M KOH at a scan rate of 10 mV s−1. Inset: corresponding Tafel curves. (B) Polarization curve of the FeNiP/NCH‖FeNiP/NCH electrode in a two-electrode configuration for water splitting. Inset: representation of the water-splitting device. (D) Catalytic stability at different current densities for around 40 h. Inset: the enlarged region of the curves for 100 mA cm−2. Notably, in each increment of current density, the voltage could increase accordingly and then stabilize rapidly, whereafter, the cell voltage was also completely restored when the current density finally decreased to 10 mA cm−2. (D) ORR polarization of FeNiP/NCH and control samples in O2-saturated 0.1 M KOH solution with a RDE rotation rate of 1600 rpm. (E) Representation of a Zn–air battery (GDF: gas diffusion film). (F) Battery voltage and power density of Zn–air batteries. (G) Galvanostatic discharge–charge cycling curves of rechargeable Zn–air batteries with FeNiP/NCH and Pt/C catalysts on Ni foam, respectively. Reproduced with permission from ref. 94. Copyright 2019, American Chemical Society. | ||
Electrochemical CO2 reduction is a potentially effective approach to produce synthetic fuels utilizing waste CO2.331 Bimetallic MOF-derived nanomaterials demonstrated impressive FE for CO2 reduction.238,240,332,333 Pan, Liu, Jiang and co-workers employed single-atom Ni implanted N doped carbon catalysts (NiSA–Nx–C) with a controlled N coordination number (x = 2, 3, 4) derived from a MgNi-MOF-74 confined polypyrrole (PPy) composite for the electrochemical reduction of CO2 to CO.240 The CO FE of NiSA–Nx–C followed a trend of NiSA–N2–C > NiSA–N3–C > NiSA–N4–C. The TOF values for CO production of NiSA–N2–C, NiSA–N3–C and NiSA–N4–C are 1622, 1120, and 225 h−1 at −0.8 V, respectively. NiSA–N2–C possessed the highest CO2 reduction activity among the NiSA–Nx–C catalysts, attributed to the favorable formation of the COOH* intermediate on low coordinated Ni–N2 sites. Sun, Han and co-workers applied MOF-derived In-Cu bimetallic oxide catalysts (InCuO-x, where x is the Cu/In molar ratio) for CO2 electroreduction.332 InCuO-0.92 showed a highest FE of 92.1% at a potential of −0.8 V, which was 1.1, 1.3, 1.7, and 3.0 times higher than that of InCuO-0.72, InCuO-0.55, InCuO-0.37, and InCuO-0.15, respectively. The increase of x led to enhanced CO2 reduction activity, indicating the synergistic effect between In oxides and Cu oxides to lead to stronger CO2 adsorption, a higher electrochemical surface area and lower charge transfer resistance.
Electrochemical N2 reduction (NNR) under ambient conditions is a promising sustainable route for ammonia synthesis.334 The related study on bimetallic MOF derivatives for NNR is still in its infancy. Recently, Qin, Cho and co-workers synthesized MoFe embedded in phosphorus-doped carbon microspheres (MoFe-PC) derived from bimetallic MoFe-MOFs for electrocatalytic NNR.335 The MoFe-PC catalyst gave a NH3 formation rate of 34.23 μg h−1 mgcat.−1 with a high FE of 16.83% at −0.5 V, surpassing those of Mo–C (12.52 μg h−1 mgcat.−1, FE: 9.67%), Fe–C (17.83 μg h−1 mgcat.−1, FE: 7.69%), and MoFe–C (24.73 μg h−1 mgcat.−1, FE: 12.47%) prepared from pyrolysis of Mo-MOF, Fe-MOF, and MoFe-MOF without phosphorization, respectively. The superior performance of MoFe-PC for NNR can be attributed to the synergistic effect of Mo and Fe oxides and P-doped carbon.
5.2 Energy storage and conversion
Bimetallic MOF-derived nanomaterials have attracted significant attention for energy storage applications such as supercapacitors and rechargeable batteries.336–339Superior to monometallic compounds, bimetallic compounds can enable richer redox reactions and higher electrical conductivity owing to charge transfer between different ions. Bimetallic compounds with tuneable compositions reveal facile and reversible faradaic behaviors for excellent electrochemical capacitors. Bimetal oxides derived from bimetallic MOFs have high specific capacities but may suffer from unsatisfactory conductivity. Converting bimetallic MOFs into phosphides and sulphides can improve the electrical conductivity. For example, Xia and co-workers applied cobalt/nickel boride/sulphide (Co–Ni–B–S) derived from bimetallic Co–Ni MOFs for supercapacitors.255 A Co–Ni MOF was first treated with sodium borohydride to lower the valence state of Co/Ni species (Co–Ni–B), followed by a sulfurization process to yield Co–Ni–B–S. When tested for supercapacitors, the Co–Ni–B–S electrode exhibited a high specific capacitance of 1281 F g−1 at 1 A g−1, with a high retention of 92.1% after 10000 cycles (Fig. 23A). The energy density of the Co–Ni–B–S electrode could reach as high as 50.0 Wh kg−1 at a power density of 857.7 W kg−1, with a capacity retention of 87.7% at a 12 A g−1 after 5000 cycles.
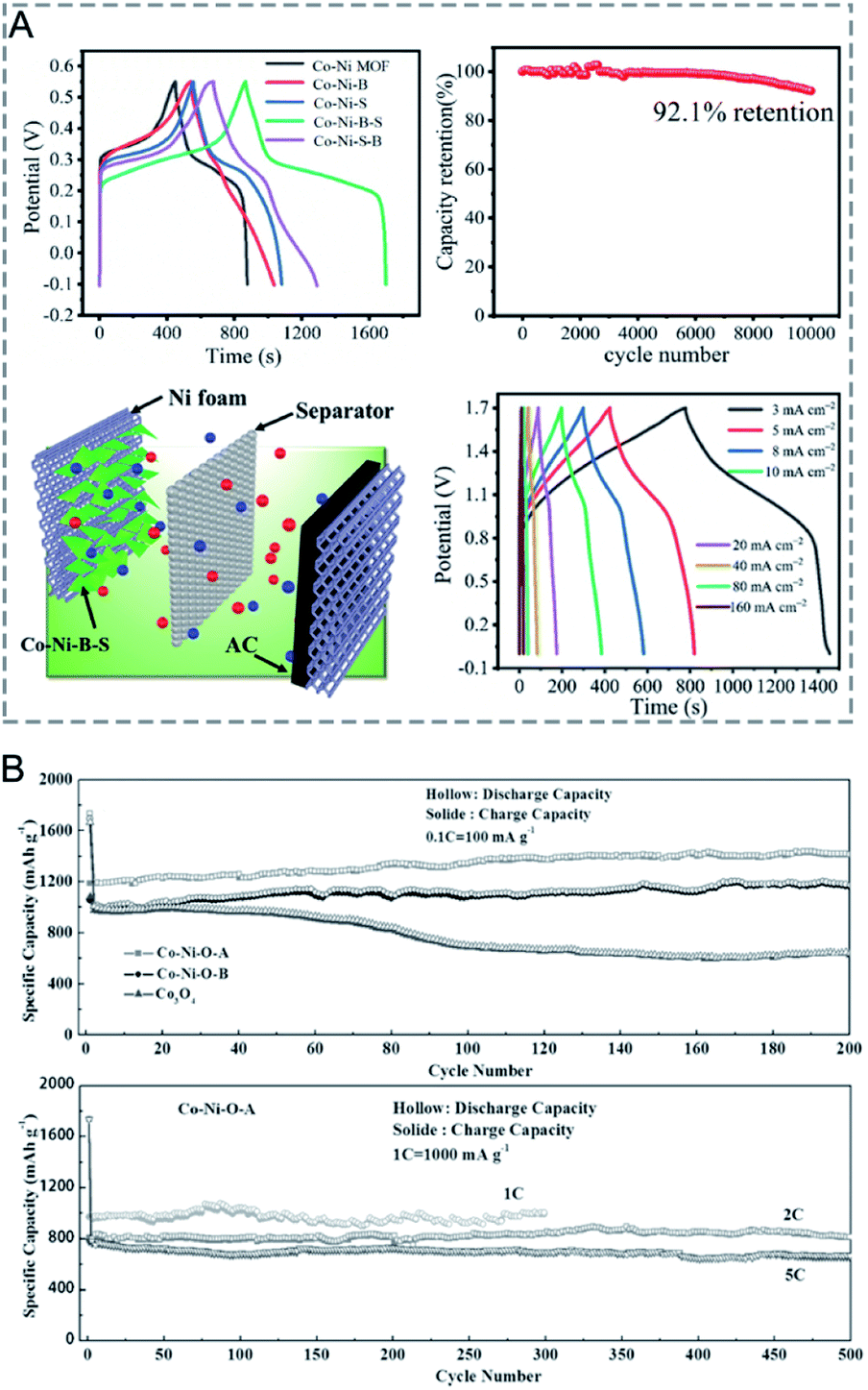 | ||
| Fig. 23 (A) GCD plots of the prepared samples and cycling life of Co–Ni–B–S at 15 A g−1 (top). Schematic illustration of the asymmetric Co–Ni–B–S/NF//AC/NF device and GCD profiles at various current densities in the voltage range of 0 to 1.7 V (bottom). Reproduced with permission from ref. 255. Copyright 2019, Wiley-VCH. (B) Cycling performances of various products of Co–Ni–O–A, Co–Ni–O–B, and Co3O4 at 100 mA g−1 (0.1C) and high-rate cycling performances of Co–Ni–O–A. Reproduced with permission from ref. 243. Copyright 2015, Wiley-VCH. | ||
Metal compounds with complex structures can induce abundant hetero-interfaces and tune the electronic structure to facilitate ion/electron transfer and improve the specific capacity.245,252,348 Hu, Lou and co-workers fabricated CoO/Co–Cu–S hierarchical tubular heterostructures (HTHSs) for hybrid supercapacitors.348 CoO/Co–Cu–S was obtained by calcination of polyacrylonitrile (PAN)@MOF-74(Co/Cu) in air followed by annealing with S powder in a N2 atmosphere. CoO/Co–Cu–S-2 (2 indicates the molar ratio of Co2+/Cu2+ in the precursors) delivered a specific capacity of 320 mA h g−1 at a current density of 2.0 A g−1, higher than that of CoO/Co–Cu–S-1 (192 mA h g−1), CoO/Co–Cu–S-0.5 (167 mA h g−1), CoO/Co–Cu–S-2 nanoneedles without a tubular structure (135 mA h g−1), monometallic CoO/CoSx nanofibers (110 mA h g−1) and Cu1.81S nanofibers (97 mA h g−1). A hybrid supercapacitor constructed with CoO/Co–Cu–S-2 and activated carbon electrodes showed a high and stable energy density of 90.7 Wh kg−1 at a power density of 800 W kg−1.
Constructing metal compound and carbon composites can improve the electrical conductivity, accommodate the volumetric change, and prevent the aggregation and dissolution of the metal compounds, thereby improving the rate capability and cycling stability. Cai, Zhang and co-workers synthesized bimetallic nickel cobalt sulphide embedded nitrogen-doped carbon composites (Ni–Co–S-n/NC, n represents the Ni/Co molar ratio) with a hollow spherical structure for supercapacitors.344 The Ni–Co–S-0.5/NC composite showed high specific capacities of 543.9 and 366.0C g−1 at 1 and 20 A g−1, respectively. An asymmetric supercapacitor based on the hollow Ni–Co–S-0.5/NC composite as the cathode exhibited good electrochemical performance with a high energy density of 39.6 W h kg−1 at a power density of 808 W kg−1.
For LIBs, bimetallic oxides with complex chemical compositions have been designed to provide enhanced electrical conductivity and rich redox-active sites.243,352–354 Sun, Wang and co-workers used NixCo3−xO4 nanorods derived from Co/Ni-MOF-74 for lithium storage.243 Co/Ni-MOF-74 A (Co:Ni = 9:1) and Co/Ni-MOF-74 B (Co:Ni = 2:1) afforded Ni0.3Co2.7O4 and NiCo2O4 after calcination at 450 °C in air, respectively. Ni0.3Co2.7O4 nanorods exhibited a large reversible Li-storage capacity of 1410 mA h g−1 after 200 cycles at 100 mA g−1, which was higher than that of NiCo2O4 (1157 mA h g−1) and Co3O4 (625 mA h g−1). Ni0.3Co2.7O4 also showed large reversible capacities of 812 and 656 mA h g−1 after 500 cycles at large current densities of 2 and 5 A g−1, respectively (Fig. 23B). The excellent performances of Ni0.3Co2.7O4 could be attributed to the mesoporous nanorod structure and the synergistic effect of two active metal oxides. Hybrid metal oxides with complex structures, such as ZnO/ZnFe2O4 sub-microcubes,355 Fe2O3@NiCo2O4 nanocages,246 and CuO@NiO hollow spheres,247 have been designed to accommodate large volume variation and shorten the lithium-ion diffusion length.
Embedding bimetal oxides in porous carbon can accommodate volume change and enhance electron/ion transport to greatly enhance the lithium storage performance. In this regard, bimetallic MOF-derived bimetallic oxide/carbon composites, such as CuCo2O4/C,356 ZnFe2O4/C@N-doped carbon nanotubes,357 and ZnO/ZnFe2O4/C,358 have been designed as anode materials for LIBs. In addition, other bimetal alloys and compounds (sulphides and phosphides) supported on carbon composites234,235,344,345,359,360 derived from bimetallic MOFs were also reported as anode materials for LIBs.
MOF-derived bimetallic compounds and their carbon composites can also be applied as anodes for SIBs. Bimetallic MOF-derived metal compounds, such as CoFe2O4 (ref. 361) and Co3O4/ZnO,362 could deliver high capacities but may suffer from poor rate capability and cycling stability due to a low electrical conductivity and large volume variation during charge/discharge processes. Bimetallic MOF-derived metal compound/carbon composites such as (Co0.5Ni0.5)9S8/N–C231 and Ti-doped-CoO@C,363 have been demonstrated as high-performance anode materials for SIBs. The porous carbon components can facilitate electron/ion transport and improve the structural integrity, and the metal compound components can deliver high capacities for sodium storage.
Bimetallic MOF-derived materials have also been applied as cathode materials for LOBs. Bimetallic MOF-derived metal compounds and their carbon composites, such as Co–Mn–O nanocubes,352 Co3O4@graphitic porous carbon,364 and ZnO/ZnFe2O4/C nanocages,365 have showed high performance in LOBs. In metal compound/carbon composites, the carbon supports with a large pore volume are beneficial for mass transportation and accommodation of discharge products (e.g., LiO2), and the well distributed catalysts accelerate oxygen-related redox reactions.
6. Conclusions and prospects
Bimetallic MOFs increase the complexity of MOF materials. Bimetallic MOFs can be divided into two categories according to the metal distribution, namely, solid solution and core–shell bimetallic MOFs. In solid solution bimetallic MOFs, two different metal ions have delocalized or even homogeneous distributions throughout the whole MOF crystals. Direct synthesis, post-synthetic modifications, and template synthesis have been applied for the preparation of solid solution bimetallic MOFs. A variety of techniques should be combined to well characterize the composition, location, and arrangement of metals in the synthesized bimetallic MOFs. Many studies lack detailed characterization to identify the metal arrangements within the frameworks, either mixed in the same SBUs or separated in different SBUs. The identification of the metal arrangements in bimetallic MOFs is of vital importance to correlate the metal mixing patterns with properties.Two MOFs with different metal centres can be assembled into core–shell bimetallic MOFs. Seed-induced growth, post-synthetic exchange, and one-pot synthesis have been developed for the synthesis of core–shell bimetallic MOFs. The assembly of core–shell structures usually leads to distorted interfaces with defects and modified crystal structures and thus induces new mechanical, electronic, and catalytic properties. In future work, detailed characterization of the interfaces is expected, which will improve the fundamental understanding of the assembly process and establish structure–property relationships.
Bimetallic MOFs with complex compositions and structures frequently show superior properties to their monometallic counterparts. Metal substitution in the SBUs can tune the stability, flexibility, pore structure, and electronic structure of bimetallic MOFs. The possibility to tune the physical and chemical properties provides bimetallic MOFs with great promise in many applications, including gas adsorption, catalysis, energy storage and conversion, and luminescence sensing. Moreover, bimetallic MOFs with well-defined crystal structures are suitable for atomically precise structural characterization and computational modelling to achieve fundamental understanding of the structure–performance relationship. Despite these advantages, the industrial applications of bimetallic MOFs are restricted due to their low stability, microporous structure, blockage of active sites, and poor conductivity.
Especially, the stability issue of bimetallic MOFs should be addressed if they are to be applied in industrial applications. Much effort has been devoted to improving the stability of MOFs, including direct synthesis of high-valent metal-carboxylate or low-valent metal-azolate MOFs and post-synthetic modification of organic linkers or coating of a protective layer. General and facile strategies are to be developed to be applicable to different bimetallic MOF systems. It is necessary to systematically investigate the chemical, thermal, and mechanical stability of bimetallic MOFs to guide the synthesis of very stable bimetallic MOFs for targeted applications.
Moreover, bimetallic MOFs can serve as excellent precursors/templates for the synthesis of a variety of nanostructured materials, including carbon composites, metal compounds, and MOF composites. The achieved success in controlling the compositions and distributions of metal ions in bimetallic MOFs can allow the tuning of compositions and structures of the obtained metal atoms/NPs/compounds. The homogeneous doping of heteroatoms (N, P, S, etc.) in the organic linkers can be inherited to the derived carbon materials to stabilize the metal species and facilitate the activity. The different coordination abilities of metal ions with the organic linkers in bimetallic MOFs offer the possibility to selectively transform part of the framework to metal NPs/compounds while preserving the rest of the framework to retain the porosity to a certain extent.
Bimetallic MOF derivatives show exposed active sites, good stability and conductivity, enabling them to extend their applications in the catalysis of more challenging reactions under harsh conditions and electrochemical energy storage and conversion. Despite the achieved success in the transformation of bimetallic MOFs to functional materials, several common drawbacks persist during the transformation process: (1) the poor control over the pore structure of bimetallic MOF-derived materials and (2) the inevitable aggregation of metal NPs/compounds. To address these issues, template-assisted synthesis and controlled transformation to “quasi-MOFs” are currently emerging to boost the development of bimetallic MOF-derived nanomaterials. Template-assisted synthesis utilizing SiO2, etc., can provide robust skeletons to prevent the architecture from collapsing and serious aggregation of metal NPs/compounds during calcination. Besides, controlled transformation into quasi-MOFs can not only allow the exposure of active sites, but also preserve the porous structures to a certain extent. The unique features of quasi-MOFs remain to be explored, which will pave the way to the development of the next-generation of functional materials.
Although many challenges still exist, the rapid development of bimetallic MOFs and their derivatives in recent years will lead to a promising future. Continued research and development in this exciting area can be expected to enable the practical applications of bimetallic MOF-based materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the editor for the kind invitation. We would like to thank the AIST and National Natural Science Foundation of China (NSFC-21875207) for financial support.Notes and references
- S. Kitagawa, R. Kitaura and S.-i. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed
.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 974–986 CrossRef CAS PubMed
- H. Li, L. Li, R.-B. Lin, W. Zhou, Z. Zhang, S. Xiang and B. Chen, EnergyChem, 2019, 1, 100006 CrossRef
- M. Woellner, S. Hausdorf, N. Klein, P. Mueller, M. W. Smith and S. Kaskel, Adv. Mater., 2018, 30, 1704679 CrossRef PubMed
- X. Zhao, Y. Wang, D.-S. Li, X. Bu and P. Feng, Adv. Mater., 2018, 30, 1705189 CrossRef PubMed
- L. Chen and Q. Xu, Matter, 2019, 1, 57–89 CrossRef
- D. Li, H.-Q. Xu, L. Jiao and H.-L. Jiang, EnergyChem, 2019, 1, 100005 CrossRef
- C. Xu, R. Fang, R. Luque, L. Chen and Y. Li, Coord. Chem. Rev., 2019, 388, 268–292 CrossRef CAS
- W. Liu, W. P. Lustig and J. Li, EnergyChem, 2019, 1, 100008 CrossRef
- K. Lu, T. Aung, N. Guo, R. Weichselbaum and W. Lin, Adv. Mater., 2018, 30, 1707634 CrossRef PubMed
- T. Simon-Yarza, A. Mielcarek, P. Couvreur and C. Serre, Adv. Mater., 2018, 30, 1707365 CrossRef PubMed
- H. Wang, Q.-L. Zhu, R. Zou and Q. Xu, Chem, 2017, 2, 52–80 CAS
- Z. Liang, R. Zhao, T. Qiu, R. Zou and Q. Xu, EnergyChem, 2019, 1, 100001 CrossRef
- X. Li, X. Yang, H. Xue, H. Pang and Q. Xu, EnergyChem, 2020, 2, 100027 CrossRef
- X. Yang and Q. Xu, Cryst. Growth Des., 2017, 17, 1450–1455 CrossRef CAS
- A. D. Burrows, CrystEngComm, 2011, 13, 3623–3642 RSC
- M. Y. Masoomi, A. Morsali, A. Dhakshinamoorthy and H. Garcia, Angew. Chem., Int. Ed., 2019, 58, 15188–15205 CrossRef CAS PubMed
- L. Feng, K.-Y. Wang, G. S. Day and H.-C. Zhou, Chem. Soc. Rev., 2019, 48, 4823–4853 RSC
- M. L. Foo, R. Matsuda and S. Kitagawa, Chem. Mater., 2014, 26, 310–322 CrossRef CAS
- J. E. Mondloch, W. Bury, D. Fairen-Jimenez, S. Kwon, E. J. DeMarco, M. H. Weston, A. A. Sarjeant, S. T. Nguyen, P. C. Stair, R. Q. Snurr, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2013, 135, 10294–10297 CrossRef CAS PubMed
- P. Ji, Y. Song, T. Drake, S. S. Veroneau, Z. Lin, X. Pan and W. Lin, J. Am. Chem. Soc., 2018, 140, 433–440 CrossRef CAS PubMed
- L. Chen, Z. Gao and Y. Li, Catal. Today, 2015, 245, 122–128 CrossRef CAS
- L. Chen, S. Rangan, J. Li, H. Jiang and Y. Li, Green Chem., 2014, 16, 3978–3985 RSC
- S. Wu, L. Chen, B. Yin and Y. Li, Chem. Commun., 2015, 51, 9884–9887 RSC
- A. Aijaz, A. Karkamkar, Y. J. Choi, N. Tsumori, E. Rönnebro, T. Autrey, H. Shioyama and Q. Xu, J. Am. Chem. Soc., 2012, 134, 13926–13929 CrossRef CAS PubMed
- L. Chen, H. Chen, R. Luque and Y. Li, Chem. Sci., 2014, 5, 3708–3714 RSC
- L. Chen, X. Chen, H. Liu and Y. Li, Small, 2015, 11, 2642–2648 CrossRef CAS PubMed
- L. Chen, R. Luque and Y. Li, Chem. Soc. Rev., 2017, 46, 4614–4630 RSC
- L. Chen, R. Luque and Y. Li, Dalton Trans., 2018, 47, 3663–3668 RSC
- A. M. Rice, G. A. Leith, O. A. Ejegbavwo, E. A. Dolgopolova and N. B. Shustova, ACS Energy Lett., 2019, 4, 1938–1946 CrossRef CAS
- A. Dhakshinamoorthy, A. M. Asiri and H. Garcia, Catal. Sci. Technol., 2016, 6, 5238–5261 RSC
- S. Abednatanzi, P. Gohari Derakhshandeh, H. Depauw, F.-X. Coudert, H. Vrielinck, P. Van Der Voort and K. Leus, Chem. Soc. Rev., 2019, 48, 2535–2565 RSC
- S. Dang, Q.-L. Zhu and Q. Xu, Nat. Rev. Mater., 2017, 3, 17075 CrossRef
- J.-K. Sun and Q. Xu, Energy Environ. Sci., 2014, 7, 2071–2100 RSC
- A. F. Sapnik, H. S. Geddes, E. M. Reynolds, H. H. M. Yeung and A. L. Goodwin, Chem. Commun., 2018, 54, 9651–9654 RSC
- M. Lammert, C. Glißmann and N. Stock, Dalton Trans., 2017, 46, 2425–2429 RSC
- M. Kim, J. F. Cahill, H. Fei, K. A. Prather and S. M. Cohen, J. Am. Chem. Soc., 2012, 134, 18082–18088 CrossRef CAS PubMed
- H. Fei, J. F. Cahill, K. A. Prather and S. M. Cohen, Inorg. Chem., 2013, 52, 4011–4016 CrossRef CAS PubMed
- M. A. Gotthardt, R. Schoch, S. Wolf, M. Bauer and W. Kleist, Dalton Trans., 2015, 44, 2052–2056 RSC
- Q. Liu, H. Cong and H. Deng, J. Am. Chem. Soc., 2016, 138, 13822–13825 CrossRef CAS PubMed
- K. A. Lomachenko, J. Jacobsen, A. L. Bugaev, C. Atzori, F. Bonino, S. Bordiga, N. Stock and C. Lamberti, J. Am. Chem. Soc., 2018, 140, 17379–17383 CrossRef CAS PubMed
- C. Castillo-Blas, V. A. de la Peña-O’Shea, I. Puente-Orench, J. R. de Paz, R. Sáez-Puche, E. Gutiérrez-Puebla, F. Gándara and Á. Monge, Sci. Adv., 2017, 3, e1700773 CrossRef PubMed
- M. A. Syzgantseva, C. P. Ireland, F. M. Ebrahim, B. Smit and O. A. Syzgantseva, J. Am. Chem. Soc., 2019, 141, 6271–6278 CrossRef CAS PubMed
- F. Trousselet, A. Archereau, A. Boutin and F.-X. Coudert, J. Phys. Chem. C, 2016, 120, 24885–24894 CrossRef CAS
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS PubMed
- M.-H. Zeng, B. Wang, X.-Y. Wang, W.-X. Zhang, X.-M. Chen and S. Gao, Inorg. Chem., 2006, 45, 7069–7076 CrossRef CAS PubMed
- Y. Wang, B. Bredenkötter, B. Rieger and D. Volkmer, Dalton Trans., 2007, 689–696 RSC
- A. Nowacka, P. Briantais, C. Prestipino and F. X. Llabrés i Xamena, Cryst. Growth Des., 2019, 19, 4981–4989 CrossRef CAS
- J. A. Botas, G. Calleja, M. Sánchez-Sánchez and M. G. Orcajo, Langmuir, 2010, 26, 5300–5303 CrossRef CAS PubMed
- T. Wang, X. Li, W. Dai, Y. Fang and H. Huang, J. Mater. Chem. A, 2015, 3, 21044–21050 RSC
- J. A. Botas, G. Calleja, M. Sánchez-Sánchez and M. G. Orcajo, Int. J. Hydrogen Energy, 2011, 36, 10834–10844 CrossRef CAS
- H. Yang, X.-W. He, F. Wang, Y. Kang and J. Zhang, J. Mater. Chem., 2012, 22, 21849–21851 RSC
- A. Schejn, A. Aboulaich, L. Balan, V. Falk, J. Lalevée, G. Medjahdi, L. Aranda, K. Mozet and R. Schneider, Catal. Sci. Technol., 2015, 5, 1829–1839 RSC
- Z. Zhou, L. Mei, C. Ma, F. Xu, J. Xiao, Q. Xia and Z. Li, Chem. Eng. Sci., 2016, 147, 109–117 CrossRef CAS
- F. Nouar, T. Devic, H. Chevreau, N. Guillou, E. Gibson, G. Clet, M. Daturi, A. Vimont, J. M. Grenèche, M. I. Breeze, R. I. Walton, P. L. Llewellyn and C. Serre, Chem. Commun., 2012, 48, 10237–10239 RSC
- F. Nouar, M. I. Breeze, B. C. Campo, A. Vimont, G. Clet, M. Daturi, T. Devic, R. I. Walton and C. Serre, Chem. Commun., 2015, 51, 14458–14461 RSC
- L. J. Wang, H. Deng, H. Furukawa, F. Gándara, K. E. Cordova, D. Peri and O. M. Yaghi, Inorg. Chem., 2014, 53, 5881–5883 CrossRef CAS PubMed
- S.-T. Zheng, T. Wu, C. Chou, A. Fuhr, P. Feng and X. Bu, J. Am. Chem. Soc., 2012, 134, 4517–4520 CrossRef CAS PubMed
- D. Feng, K. Wang, Z. Wei, Y.-P. Chen, C. M. Simon, R. K. Arvapally, R. L. Martin, M. Bosch, T.-F. Liu, S. Fordham, D. Yuan, M. A. Omary, M. Haranczyk, B. Smit and H.-C. Zhou, Nat. Commun., 2014, 5, 5723 CrossRef CAS PubMed
- S. Wongsakulphasatch, F. Nouar, J. Rodriguez, L. Scott, C. Le Guillouzer, T. Devic, P. Horcajada, J. M. Grenèche, P. L. Llewellyn, A. Vimont, G. Clet, M. Daturi and C. Serre, Chem. Commun., 2015, 51, 10194–10197 RSC
- D. Kim, K. S. Song, O. Buyukcakir, T. Yildirim and A. Coskun, Chem. Commun., 2018, 54, 12218–12221 RSC
- M. Dincǎ and J. R. Long, J. Am. Chem. Soc., 2007, 129, 11172–11176 CrossRef PubMed
- T. K. Prasad, D. H. Hong and M. P. Suh, Chem. – Eur. J., 2010, 16, 14043–14050 CrossRef CAS PubMed
- Q. Yao, J. Sun, K. Li, J. Su, M. V. Peskov and X. Zou, Dalton Trans., 2012, 41, 3953–3955 RSC
- S. R. Caskey and A. J. Matzger, Inorg. Chem., 2008, 47, 7942–7944 CrossRef CAS PubMed
- M. Lalonde, W. Bury, O. Karagiaridi, Z. Brown, J. T. Hupp and O. K. Farha, J. Mater. Chem. A, 2013, 1, 5453–5468 RSC
- C. K. Brozek and M. Dincă, Chem. Soc. Rev., 2014, 43, 5456–5467 RSC
- P. Deria, J. E. Mondloch, O. Karagiaridi, W. Bury, J. T. Hupp and O. K. Farha, Chem. Soc. Rev., 2014, 43, 5896–5912 RSC
- C. K. Brozek and M. Dincă, J. Am. Chem. Soc., 2013, 135, 12886–12891 CrossRef CAS PubMed
- J.-H. Liao, W.-T. Chen, C.-S. Tsai and C.-C. Wang, CrystEngComm, 2013, 15, 3377–3384 RSC
- Z. Wei, W. Lu, H.-L. Jiang and H.-C. Zhou, Inorg. Chem., 2013, 52, 1164–1166 CrossRef CAS PubMed
- C. K. Brozek, V. K. Michaelis, T.-C. Ong, L. Bellarosa, N. López, R. G. Griffin and M. Dincă, ACS Cent. Sci., 2015, 1, 252–260 CrossRef CAS PubMed
- L. Mi, H. Hou, Z. Song, H. Han, H. Xu, Y. Fan and S.-W. Ng, Cryst. Growth Des., 2007, 7, 2553–2561 CrossRef CAS
- C. K. Brozek, A. F. Cozzolino, S. J. Teat, Y.-S. Chen and M. Dincă, Chem. Mater., 2013, 25, 2998–3002 CrossRef CAS
- S. Das, H. Kim and K. Kim, J. Am. Chem. Soc., 2009, 131, 3814–3815 CrossRef CAS PubMed
- Z. Zhang, L. Zhang, L. Wojtas, P. Nugent, M. Eddaoudi and M. J. Zaworotko, J. Am. Chem. Soc., 2012, 134, 924–927 CrossRef CAS PubMed
- L. Mi, H. Hou, Z. Song, H. Han and Y. Fan, Chem. – Eur. J., 2008, 14, 1814–1821 CrossRef CAS PubMed
- X. Song, T. K. Kim, H. Kim, D. Kim, S. Jeong, H. R. Moon and M. S. Lah, Chem. Mater., 2012, 24, 3065–3073 CrossRef CAS
- X.-S. Wang, M. Chrzanowski, L. Wojtas, Y.-S. Chen and S. Ma, Chem. – Eur. J., 2013, 19, 3297–3301 CrossRef CAS PubMed
- C. Hon Lau, R. Babarao and M. R. Hill, Chem. Commun., 2013, 49, 3634–3636 RSC
- P. Á. Szilágyi, P. Serra-Crespo, I. Dugulan, J. Gascon, H. Geerlings and B. Dam, CrystEngComm, 2013, 15, 10175–10178 RSC
- J. G. Santaclara, A. I. Olivos-Suarez, A. Gonzalez-Nelson, D. Osadchii, M. A. Nasalevich, M. A. van der Veen, F. Kapteijn, A. M. Sheveleva, S. L. Veber, M. V. Fedin, A. T. Murray, C. H. Hendon, A. Walsh and J. Gascon, Chem. Mater., 2017, 29, 8963–8967 CrossRef CAS
- M. S. Denny, L. R. Parent, J. P. Patterson, S. K. Meena, H. Pham, P. Abellan, Q. M. Ramasse, F. Paesani, N. C. Gianneschi and S. M. Cohen, J. Am. Chem. Soc., 2018, 140, 1348–1357 CrossRef CAS PubMed
- R. Li, X. Ren, H. Ma, X. Feng, Z. Lin, X. Li, C. Hu and B. Wang, J. Mater. Chem. A, 2014, 2, 5724–5729 RSC
- T. Panda, S. Horike, K. Hagi, N. Ogiwara, K. Kadota, T. Itakura, M. Tsujimoto and S. Kitagawa, Angew. Chem., Int. Ed., 2017, 56, 2413–2417 CrossRef CAS PubMed
- J.-S. M. Lee, Y.-i. Fujiwara, S. Kitagawa and S. Horike, Chem. Mater., 2019, 31, 4205–4212 CrossRef CAS
- D. Kim and A. Coskun, Angew. Chem., Int. Ed., 2017, 56, 5071–5076 CrossRef CAS PubMed
- D. Liu, J. Wan, G. Pang and Z. Tang, Adv. Mater., 2019, 31, 1803291 CrossRef PubMed
- H. J. Lee, W. Cho and M. Oh, Chem. Commun., 2012, 48, 221–223 RSC
- K. Shen, L. Zhang, X. Chen, L. Liu, D. Zhang, Y. Han, J. Chen, J. Long, R. Luque, Y. Li and B. Chen, Science, 2018, 359, 206–210 CrossRef CAS PubMed
- M. Qiao, Y. Wang, Q. Wang, G. Hu, X. Mamat, S. Zhang and S. Wang, Angew. Chem., Int. Ed., 2020, 59, 2688–2694 CrossRef CAS PubMed
- J. Teng, M. Chen, Y. Xie, D. Wang, J.-J. Jiang, G. Li, H.-P. Wang, Y. Fan, Z.-W. Wei and C.-Y. Su, Chem. Mater., 2018, 30, 6458–6468 CrossRef CAS
- Z. Zhang, Y. Chen, S. He, J. Zhang, X. Xu, Y. Yang, F. Nosheen, F. Saleem, W. He and X. Wang, Angew. Chem., Int. Ed., 2014, 53, 12517–12521 CAS
- Y.-S. Wei, M. Zhang, M. Kitta, Z. Liu, S. Horike and Q. Xu, J. Am. Chem. Soc., 2019, 141, 7906–7916 CrossRef CAS PubMed
- Z. Zhang, Y. Chen, X. Xu, J. Zhang, G. Xiang, W. He and X. Wang, Angew. Chem., Int. Ed., 2014, 53, 429–433 CrossRef CAS PubMed
- L. Chen, B. Huang, X. Qiu, X. Wang, R. Luque and Y. Li, Chem. Sci., 2016, 7, 228–233 RSC
- L. Chen, W. Huang, X. Wang, Z. Chen, X. Yang, R. Luque and Y. Li, Chem. Commun., 2017, 53, 1184–1187 RSC
- S. Furukawa, K. Hirai, K. Nakagawa, Y. Takashima, R. Matsuda, T. Tsuruoka, M. Kondo, R. Haruki, D. Tanaka, H. Sakamoto, S. Shimomura, O. Sakata and S. Kitagawa, Angew. Chem., Int. Ed., 2009, 48, 1766–1770 CrossRef CAS PubMed
- J. Tang, R. R. Salunkhe, J. Liu, N. L. Torad, M. Imura, S. Furukawa and Y. Yamauchi, J. Am. Chem. Soc., 2015, 137, 1572–1580 CrossRef CAS PubMed
- M. Zhao, K. Yuan, Y. Wang, G. Li, J. Guo, L. Gu, W. Hu, H. Zhao and Z. Tang, Nature, 2016, 539, 76–80 CrossRef CAS PubMed
- T.-Y. Luo, C. Liu, X. Y. Gan, P. F. Muldoon, N. A. Diemler, J. E. Millstone and N. L. Rosi, J. Am. Chem. Soc., 2019, 141, 2161–2168 CrossRef CAS PubMed
- H. J. Lee, Y. J. Cho, W. Cho and M. Oh, ACS Nano, 2013, 7, 491–499 CrossRef PubMed
- J. Zhuang, L.-Y. Chou, B. T. Sneed, Y. Cao, P. Hu, L. Feng and C.-K. Tsung, Small, 2015, 11, 5551–5555 CrossRef CAS PubMed
- F. Wang, S. He, H. Wang, S. Zhang, C. Wu, H. Huang, Y. Pang, C.-K. Tsung and T. Li, Chem. Sci., 2019, 10, 7755–7761 RSC
- F. Wang, H. Wang and T. Li, Nanoscale, 2019, 11, 2121–2125 RSC
- Y. Gu, Y.-n. Wu, L. Li, W. Chen, F. Li and S. Kitagawa, Angew. Chem., Int. Ed., 2017, 56, 15658–15662 CrossRef CAS PubMed
- L. Feng, S. Yuan, J.-L. Li, K.-Y. Wang, G. S. Day, P. Zhang, Y. Wang and H.-C. Zhou, ACS Cent. Sci., 2018, 4, 1719–1726 CrossRef CAS PubMed
- X. Song, S. Jeong, D. Kim and M. S. Lah, CrystEngComm, 2012, 14, 5753–5756 RSC
- L. Chen, H. Chen and Y. Li, Chem. Commun., 2014, 50, 14752–14755 RSC
- L. Chen, X. Chen, H. Liu, C. Bai and Y. Li, J. Mater. Chem. A, 2015, 3, 15259–15264 RSC
- H. Liu, L. Chang, C. Bai, L. Chen, R. Luque and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 5019–5023 CrossRef CAS PubMed
- W. Guo, W. Xia, K. Cai, Y. Wu, B. Qiu, Z. Liang, C. Qu and R. Zou, Small, 2017, 13, 1702049 CrossRef PubMed
- H. Depauw, I. Nevjestić, J. De Winne, G. Wang, K. Haustraete, K. Leus, A. Verberckmoes, C. Detavernier, F. Callens, E. De Canck, H. Vrielinck and P. Van Der Voort, Chem. Commun., 2017, 53, 8478–8481 RSC
- T. Fukushima, S. Horike, H. Kobayashi, M. Tsujimoto, S. Isoda, M. L. Foo, Y. Kubota, M. Takata and S. Kitagawa, J. Am. Chem. Soc., 2012, 134, 13341–13347 CrossRef CAS PubMed
- H. Li, W. Shi, K. Zhao, H. Li, Y. Bing and P. Cheng, Inorg. Chem., 2012, 51, 9200–9207 CrossRef CAS PubMed
- X. Song, M. Oh and M. S. Lah, Inorg. Chem., 2013, 52, 10869–10876 CrossRef CAS PubMed
- J. Yang, X. Wang, F. Dai, L. Zhang, R. Wang and D. Sun, Inorg. Chem., 2014, 53, 10649–10653 CrossRef CAS PubMed
- Y. Jiao, C. R. Morelock, N. C. Burtch, W. P. Mounfield, J. T. Hungerford and K. S. Walton, Ind. Eng. Chem. Res., 2015, 54, 12408–12414 CrossRef CAS
- J.-P. Zhang, H.-L. Zhou, D.-D. Zhou, P.-Q. Liao and X.-M. Chen, Natl. Sci. Rev., 2017, 5, 907–919 CrossRef
- J. H. Choi, Y. J. Choi, J. W. Lee, W. H. Shin and J. K. Kang, Phys. Chem. Chem. Phys., 2009, 11, 628–631 RSC
- L. Sun, M. G. Campbell and M. Dincă, Angew. Chem., Int. Ed., 2016, 55, 3566–3579 CrossRef CAS PubMed
- E. A. Dolgopolova, A. J. Brandt, O. A. Ejegbavwo, A. S. Duke, T. D. Maddumapatabandi, R. P. Galhenage, B. W. Larson, O. G. Reid, S. C. Ammal, A. Heyden, M. Chandrashekhar, V. Stavila, D. A. Chen and N. B. Shustova, J. Am. Chem. Soc., 2017, 139, 5201–5209 CrossRef CAS PubMed
- G. Kaur, R. K. Rai, D. Tyagi, X. Yao, P.-Z. Li, X.-C. Yang, Y. Zhao, Q. Xu and S. K. Singh, J. Mater. Chem. A, 2016, 4, 14932–14938 RSC
- D.-D. Zhou, X.-W. Zhang, Z.-W. Mo, Y.-Z. Xu, X.-Y. Tian, Y. Li, X.-M. Chen and J.-P. Zhang, EnergyChem, 2019, 1, 100016 CrossRef
- P.-P. Cui, X.-D. Zhang, P. Wang, Y. Zhao, M. Azam, S. I. Al-Resayes and W.-Y. Sun, Inorg. Chem., 2017, 56, 14157–14163 CrossRef CAS PubMed
- Q.-G. Zhai, X. Bu, C. Mao, X. Zhao and P. Feng, J. Am. Chem. Soc., 2016, 138, 2524–2527 CrossRef CAS PubMed
- R.-Q. Zou, H. Sakurai, S. Han, R.-Q. Zhong and Q. Xu, J. Am. Chem. Soc., 2007, 129, 8402–8403 CrossRef CAS PubMed
- V. Bernales, M. A. Ortuño, D. G. Truhlar, C. J. Cramer and L. Gagliardi, ACS Cent. Sci., 2018, 4, 5–19 CrossRef CAS PubMed
- D. J. Xiao, E. D. Bloch, J. A. Mason, W. L. Queen, M. R. Hudson, N. Planas, J. Borycz, A. L. Dzubak, P. Verma, K. Lee, F. Bonino, V. Crocellà, J. Yano, S. Bordiga, D. G. Truhlar, L. Gagliardi, C. M. Brown and J. R. Long, Nat. Chem., 2014, 6, 590–595 CrossRef CAS PubMed
- P. Verma, K. D. Vogiatzis, N. Planas, J. Borycz, D. J. Xiao, J. R. Long, L. Gagliardi and D. G. Truhlar, J. Am. Chem. Soc., 2015, 137, 5770–5781 CrossRef CAS PubMed
- D. Sun, F. Sun, X. Deng and Z. Li, Inorg. Chem., 2015, 54, 8639–8643 CrossRef CAS PubMed
- A. Nowacka, P. Briantais, C. Prestipino and F. X. Llabrés i Xamena, ACS Sustainable Chem. Eng., 2019, 7, 7708–7715 CrossRef CAS
- A. W. Stubbs, L. Braglia, E. Borfecchia, R. J. Meyer, Y. Román- Leshkov, C. Lamberti and M. Dincă, ACS Catal., 2018, 8, 596–601 CrossRef CAS
- Á. Szécsényi, G. Li, J. Gascon and E. A. Pidko, Chem. Sci., 2018, 9, 6765–6773 RSC
- D. Y. Osadchii, A. I. Olivos-Suarez, Á. Szécsényi, G. Li, M. A. Nasalevich, I. A. Dugulan, P. S. Crespo, E. J. M. Hensen, S. L. Veber, M. V. Fedin, G. Sankar, E. A. Pidko and J. Gascon, ACS Catal., 2018, 8, 5542–5548 CrossRef CAS
- D. M. Shakya, O. A. Ejegbavwo, T. Rajeshkumar, S. D. Senanayake, A. J. Brandt, S. Farzandh, N. Acharya, A. M. Ebrahim, A. I. Frenkel, N. Rui, G. L. Tate, J. R. Monnier, K. D. Vogiatzis, N. B. Shustova and D. A. Chen, Angew. Chem., Int. Ed., 2019, 58, 16533–16537 CrossRef CAS PubMed
- H. G. T. Nguyen, L. Mao, A. W. Peters, C. O. Audu, Z. J. Brown, O. K. Farha, J. T. Hupp and S. T. Nguyen, Catal. Sci. Technol., 2015, 5, 4444–4451 RSC
- C. Huang, R. Liu, W. Yang, Y. Li, J. Huang and H. Zhu, Inorg. Chem. Front., 2018, 5, 1923–1932 RSC
- Y. Fu, L. Xu, H. Shen, H. Yang, F. Zhang, W. Zhu and M. Fan, Chem. Eng. J., 2016, 299, 135–141 CrossRef CAS
- L. Mitchell, P. Williamson, B. Ehrlichová, A. E. Anderson, V. R. Seymour, S. E. Ashbrook, N. Acerbi, L. M. Daniels, R. I. Walton, M. L. Clarke and P. A. Wright, Chem. – Eur. J., 2014, 20, 17185–17197 CrossRef CAS PubMed
- J. M. Moreno, A. Velty, U. Díaz and A. Corma, Chem. Sci., 2019, 10, 2053–2066 RSC
- B. Iqbal, M. Saleem, S. N. Arshad, J. Rashid, N. Hussain and M. Zaheer, Chem. – Eur. J., 2019, 25, 10490–10498 CrossRef CAS PubMed
- S. Smolders, J. Jacobsen, N. Stock and D. De Vos, Catal. Sci. Technol., 2020, 10, 337–341 RSC
- M. Giménez-Marqués, A. Santiago-Portillo, S. Navalón, M. Álvaro, V. Briois, F. Nouar, H. Garcia and C. Serre, J. Mater. Chem. A, 2019, 7, 20285–20292 RSC
- M. N. Timofeeva, V. N. Panchenko, N. A. Khan, Z. Hasan, I. P. Prosvirin, S. V. Tsybulya and S. H. Jhung, Appl. Catal., A, 2017, 529, 167–174 CrossRef CAS
- F.-N. Shi, A. R. Silva, T.-H. Yang and J. Rocha, CrystEngComm, 2013, 15, 3776–3779 RSC
- R. R. Kuruppathparambil, R. Babu, H. M. Jeong, G.-Y. Hwang, G. S. Jeong, M.-I. Kim, D.-W. Kim and D.-W. Park, Green Chem., 2016, 18, 6349–6356 RSC
- C. P. Krap, R. Newby, A. Dhakshinamoorthy, H. García, I. Cebula, T. L. Easun, M. Savage, J. E. Eyley, S. Gao, A. J. Blake, W. Lewis, P. H. Beton, M. R. Warren, D. R. Allan, M. D. Frogley, C. C. Tang, G. Cinque, S. Yang and M. Schröder, Inorg. Chem., 2016, 55, 1076–1088 CrossRef CAS PubMed
- L. M. Aguirre-Díaz, F. Gándara, M. Iglesias, N. Snejko, E. Gutiérrez-Puebla and M. Á. Monge, J. Am. Chem. Soc., 2015, 137, 6132–6135 CrossRef PubMed
- D. Saha, D. K. Hazra, T. Maity and S. Koner, Inorg. Chem., 2016, 55, 5729–5731 CrossRef CAS PubMed
- A. M. Rasero-Almansa, M. Iglesias and F. Sánchez, RSC Adv., 2016, 6, 106790–106797 RSC
- Y. Hu, J. Zhang, H. Huo, Z. Wang, X. Xu, Y. Yang, K. Lin and R. Fan, Catal. Sci. Technol., 2020, 10, 315–322 RSC
- J. Castells-Gil, N. M. Padial, N. Almora-Barrios, J. Albero, A. R. Ruiz-Salvador, J. González-Platas, H. García and C. Martí-Gastaldo, Angew. Chem., Int. Ed., 2018, 57, 8453–8457 CrossRef CAS PubMed
- J. Tu, X. Zeng, F. Xu, X. Wu, Y. Tian, X. Hou and Z. Long, Chem. Commun., 2017, 53, 3361–3364 RSC
- R. Navarro Amador, M. Carboni and D. Meyer, RSC Adv., 2017, 7, 195–200 RSC
- Y. Lee, S. Kim, J. K. Kang and S. M. Cohen, Chem. Commun., 2015, 51, 5735–5738 RSC
- D. Sun, W. Liu, M. Qiu, Y. Zhang and Z. Li, Chem. Commun., 2015, 51, 2056–2059 RSC
- L. Zou, D. Feng, T.-F. Liu, Y.-P. Chen, S. Yuan, K. Wang, X. Wang, S. Fordham and H.-C. Zhou, Chem. Sci., 2016, 7, 1063–1069 RSC
- C. Guo, J. Guo, Y. Zhang, D. Wang, L. Zhang, Y. Guo, W. Ma and J. Wang, CrystEngComm, 2018, 20, 7659–7665 RSC
- H. Yoon, S. Lee, S. Oh, H. Park, S. Choi and M. Oh, Small, 2019, 15, 1805232 CrossRef PubMed
- F. Zheng, D. Xiang, P. Li, Z. Zhang, C. Du, Z. Zhuang, X. Li and W. Chen, ACS Sustainable Chem. Eng., 2019, 7, 9743–9749 CrossRef CAS
- Q. Wang, C. Wei, D. Li, W. Guo, D. Zhong and Q. Zhao, Microporous Mesoporous Mater., 2019, 286, 92–97 CrossRef CAS
- X.-L. Wang, L.-Z. Dong, M. Qiao, Y.-J. Tang, J. Liu, Y. Li, S.-L. Li, J.-X. Su and Y.-Q. Lan, Angew. Chem., Int. Ed., 2018, 57, 9660–9664 CrossRef CAS PubMed
- J. Li, W. Huang, M. Wang, S. Xi, J. Meng, K. Zhao, J. Jin, W. Xu, Z. Wang, X. Liu, Q. Chen, L. Xu, X. Liao, Y. Jiang, K. A. Owusu, B. Jiang, C. Chen, D. Fan, L. Zhou and L. Mai, ACS Energy Lett., 2019, 4, 285–292 CrossRef CAS
- W. Zhou, D.-D. Huang, Y.-P. Wu, J. Zhao, T. Wu, J. Zhang, D.-S. Li, C. Sun, P. Feng and X. Bu, Angew. Chem., Int. Ed., 2019, 58, 4227–4231 CrossRef CAS PubMed
- F.-L. Li, Q. Shao, X. Huang and J.-P. Lang, Angew. Chem., Int. Ed., 2018, 57, 1888–1892 CrossRef CAS PubMed
- J. Duan, S. Chen and C. Zhao, Nat. Commun., 2017, 8, 15341 CrossRef CAS PubMed
- H.-W. Lin, D. Senthil Raja, X.-F. Chuah, C.-T. Hsieh, Y.-A. Chen and S.-Y. Lu, Appl. Catal., B, 2019, 258, 118023 CrossRef CAS
- J. Albo, M. Perfecto-Irigaray, G. Beobide and A. Irabien, J. CO2 Util., 2019, 33, 157–165 CrossRef CAS
- R. Wang, F. Kapteijn and J. Gascon, Chem.–Asian J., 2019, 14, 3452–3461 CrossRef CAS PubMed
- A. Mahmood, W. Guo, H. Tabassum and R. Zou, Adv. Energy Mater., 2016, 6, 1600423 CrossRef
- Z. Zhao, J. Ding, R. Zhu and H. Pang, J. Mater. Chem. A, 2019, 7, 15519–15540 RSC
- K. Rui, G. Zhao, Y. Chen, Y. Lin, Q. Zhou, J. Chen, J. Zhu, W. Sun, W. Huang and S. X. Dou, Adv. Funct. Mater., 2018, 28, 1801554 CrossRef
- S. Zhao, Y. Wang, J. Dong, C.-T. He, H. Yin, P. An, K. Zhao, X. Zhang, C. Gao, L. Zhang, J. Lv, J. Wang, J. Zhang, A. M. Khattak, N. A. Khan, Z. Wei, J. Zhang, S. Liu, H. Zhao and Z. Tang, Nat. Energy, 2016, 1, 16184 CrossRef CAS
- M. Zhao, Y. Wang, Q. Ma, Y. Huang, X. Zhang, J. Ping, Z. Zhang, Q. Lu, Y. Yu, H. Xu, Y. Zhao and H. Zhang, Adv. Mater., 2015, 27, 7372–7378 CrossRef CAS PubMed
- L. Wang, Y. Wu, R. Cao, L. Ren, M. Chen, X. Feng, J. Zhou and B. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 16736–16743 CrossRef CAS PubMed
- G. Hai, X. Jia, K. Zhang, X. Liu, Z. Wu and G. Wang, Nano Energy, 2018, 44, 345–352 CrossRef CAS
- Q.-L. Zhu and Q. Xu, Chem. Soc. Rev., 2014, 43, 5468–5512 RSC
- X. Li, D.-D. Ma, C. Cao, R. Zou, Q. Xu, X.-T. Wu and Q.-L. Zhu, Small, 2019, 15, 1902218 CrossRef PubMed
- F. Sun, G. Wang, Y. Ding, C. Wang, B. Yuan and Y. Lin, Adv. Energy Mater., 2018, 8, 1800584 CrossRef
- D. Senthil Raja, X.-F. Chuah and S.-Y. Lu, Adv. Energy Mater., 2018, 8, 1801065 CrossRef
- R. Zhao, Z. Liang, R. Zou and Q. Xu, Joule, 2018, 2, 2235–2259 CrossRef CAS
- Y. Zhang, J. Liu, S.-L. Li, Z.-M. Su and Y.-Q. Lan, EnergyChem, 2019, 1, 100021 CrossRef
- K.-B. Wang, Q. Xun and Q. Zhang, EnergyChem, 2020, 2, 100025 CrossRef
- B. Zhu, Z. Liang, D. Xia and R. Zou, Energy Storage Mater., 2019, 23, 757–771 CrossRef
- G. Xu, P. Nie, H. Dou, B. Ding, L. Li and X. Zhang, Mater. Today, 2017, 20, 191–209 CrossRef CAS
- R. Díaz, M. G. Orcajo, J. A. Botas, G. Calleja and J. Palma, Mater. Lett., 2012, 68, 126–128 CrossRef
- J. Yang, C. Zheng, P. Xiong, Y. Li and M. Wei, J. Mater. Chem. A, 2014, 2, 19005–19010 RSC
- H. Gholipour-Ranjbar, M. Soleimani and H. R. Naderi, New J. Chem., 2016, 40, 9187–9193 RSC
- Y. Jiao, J. Pei, D. Chen, C. Yan, Y. Hu, Q. Zhang and G. Chen, J. Mater. Chem. A, 2017, 5, 1094–1102 RSC
- L. Z. Shaofei Zhao, G. Cheng, L. Yu and H. Zeng, Chin. Chem. Lett., 2019, 30, 605–609 CrossRef
- D. Tian, N. Song, M. Zhong, X. Lu and C. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 1280–1291 CrossRef CAS PubMed
- C. Chen, M.-K. Wu, K. Tao, J.-J. Zhou, Y.-L. Li, X. Han and L. Han, Dalton Trans., 2018, 47, 5639–5645 RSC
- P. Nie, L. Shen, H. Luo, B. Ding, G. Xu, J. Wang and X. Zhang, J. Mater. Chem. A, 2014, 2, 5852–5857 RSC
- K. Saravanan, M. Nagarathinam, P. Balaya and J. J. Vittal, J. Mater. Chem., 2010, 20, 8329–8335 RSC
- P. Xiong, G. Zeng, L. Zeng and M. Wei, Dalton Trans., 2015, 44, 16746–16751 RSC
- Y. s. Nanba, D. Asakura, M. Okubo, Y. Mizuno, T. Kudo, H. Zhou, K. Amemiya, J. Guo and K. Okada, J. Phys. Chem. C, 2012, 116, 24896–24901 CrossRef CAS
- S. H. Kim, Y. J. Lee, D. H. Kim and Y. J. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 660–667 CrossRef CAS PubMed
- Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 112, 1126–1162 CrossRef CAS PubMed
- H.-Q. Yin and X.-B. Yin, Acc. Chem. Res., 2020, 53, 485–495 CrossRef CAS
- L. K. Cadman, M. F. Mahon and A. D. Burrows, Dalton Trans., 2018, 47, 2360–2367 RSC
- S. Wu, H. Min, W. Shi and P. Cheng, Adv. Mater., 2020, 32, 1805871 CrossRef CAS
- W. P. Lustig, S. Mukherjee, N. D. Rudd, A. V. Desai, J. Li and S. K. Ghosh, Chem. Soc. Rev., 2017, 46, 3242–3285 RSC
- B. Yan, Acc. Chem. Res., 2017, 50, 2789–2798 CrossRef CAS PubMed
- Y. Cui, F. Zhu, B. Chen and G. Qian, Chem. Commun., 2015, 51, 7420–7431 RSC
- Y. Cui, H. Xu, Y. Yue, Z. Guo, J. Yu, Z. Chen, J. Gao, Y. Yang, G. Qian and B. Chen, J. Am. Chem. Soc., 2012, 134, 3979–3982 CrossRef CAS PubMed
- S.-Y. Zhang, W. Shi, P. Cheng and M. J. Zaworotko, J. Am. Chem. Soc., 2015, 137, 12203–12206 CrossRef CAS PubMed
- S. G. Dunning, A. J. Nuñez, M. D. Moore, A. Steiner, V. M. Lynch, J. L. Sessler, B. J. Holliday and S. M. Humphrey, Chem, 2017, 2, 579–589 CAS
- B. Liu, H. Shioyama, T. Akita and Q. Xu, J. Am. Chem. Soc., 2008, 130, 5390–5391 CrossRef CAS PubMed
- B. Liu, X. Zhang, H. Shioyama, T. Mukai, T. Sakai and Q. Xu, J. Power Sources, 2010, 195, 857–861 CrossRef CAS
- Y. V. Kaneti, J. Tang, R. R. Salunkhe, X. Jiang, A. Yu, K. C. W. Wu and Y. Yamauchi, Adv. Mater., 2016, 1604898 Search PubMed
- L. Chen, L. Zhang, Z. Chen, H. Liu, R. Luque and Y. Li, Chem. Sci., 2016, 7, 6015–6020 RSC
- H.-L. Jiang, B. Liu, Y.-Q. Lan, K. Kuratani, T. Akita, H. Shioyama, F. Zong and Q. Xu, J. Am. Chem. Soc., 2011, 133, 11854–11857 CrossRef CAS PubMed
- A. Aijaz, N. Fujiwara and Q. Xu, J. Am. Chem. Soc., 2014, 136, 6790–6793 CrossRef CAS PubMed
- W. Zhong, H. Liu, C. Bai, S. Liao and Y. Li, ACS Catal., 2015, 5, 1850–1856 CrossRef CAS
- X. Wang, J. Zhou, H. Fu, W. Li, X. Fan, G. Xin, J. Zheng and X. Li, J. Mater. Chem. A, 2014, 2, 14064–14070 RSC
- N. L. Torad, M. Hu, S. Ishihara, H. Sukegawa, A. A. Belik, M. Imura, K. Ariga, Y. Sakka and Y. Yamauchi, Small, 2014, 10, 2096–2107 CrossRef CAS PubMed
- Y. Z. Chen, C. Wang, Z. Y. Wu, Y. Xiong, Q. Xu, S. H. Yu and H. L. Jiang, Adv. Mater., 2015, 27, 5010–5016 CrossRef CAS
- C.-C. Hou, L. Zou and Q. Xu, Adv. Mater., 2019, 31, 1904689 CrossRef CAS PubMed
- Y. Yang, Z. Lun, G. Xia, F. Zheng, M. He and Q. Chen, Energy Environ. Sci., 2015, 8, 3563–3571 RSC
- J. Long, K. Shen, L. Chen and Y. Li, J. Mater. Chem. A, 2016, 4, 10254–10262 RSC
- D. Sun, L. Ye, F. Sun, H. García and Z. Li, Inorg. Chem., 2017, 56, 5203–5209 CrossRef CAS PubMed
- R. Das, P. Pachfule, R. Banerjee and P. Poddar, Nanoscale, 2012, 4, 591–599 RSC
- X. F. Lu, Y. Chen, S. Wang, S. Gao and X. W. Lou, Adv. Mater., 2019, 31, 1902339 CrossRef PubMed
- J. Qian, T.-T. Li, Y. Hu and S. Huang, Chem. Commun., 2017, 53, 13027–13030 RSC
- X. Li, X. Wang, J. Zhou, L. Han, C. Sun, Q. Wang and Z. Su, J. Mater. Chem. A, 2018, 6, 5789–5796 RSC
- Y. Wang, W. Wu, Y. Rao, Z. Li, N. Tsubaki and M. Wu, J. Mater. Chem. A, 2017, 5, 6170–6177 RSC
- X. F. Lu, L. Yu and X. W. Lou, Sci. Adv., 2019, 5, eaav6009 CrossRef CAS PubMed
- T. Zhang, J. Du, P. Xi and C. Xu, ACS Appl. Mater. Interfaces, 2017, 9, 362–370 CrossRef CAS PubMed
- Y. Lian, H. Sun, X. Wang, P. Qi, Q. Mu, Y. Chen, J. Ye, X. Zhao, Z. Deng and Y. Peng, Chem. Sci., 2019, 10, 464–474 RSC
- D. Cao, W. Kang, S. Wang, Y. Wang, K. Sun, L. Yang, X. Zhou, D. Sun and Y. Cao, J. Mater. Chem. A, 2019, 7, 8268–8276 RSC
- W. Hong, M. Kitta and Q. Xu, Small Methods, 2018, 2, 1800214 CrossRef
- J. Song, C. Zhu, B. Z. Xu, S. Fu, M. H. Engelhard, R. Ye, D. Du, S. P. Beckman and Y. Lin, Adv. Energy Mater., 2017, 7, 1601555 CrossRef
- J.-c. Zheng, Y.-y. Yao, G.-q. Mao, H.-z. Chen, H. Li, L. Cao, X. Ou, W.-j. Yu, Z.-y. Ding and H. Tong, J. Mater. Chem. A, 2019, 7, 16479–16487 RSC
- H. Li, Y. Su, W. Sun and Y. Wang, Adv. Funct. Mater., 2016, 26, 8345–8353 CrossRef CAS
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong, Z. Deng, G. Zhou, S. Wei and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 10800–10805 CrossRef CAS PubMed
- H. Zhang, S. Hwang, M. Wang, Z. Feng, S. Karakalos, L. Luo, Z. Qiao, X. Xie, C. Wang, D. Su, Y. Shao and G. Wu, J. Am. Chem. Soc., 2017, 139, 14143–14149 CrossRef CAS PubMed
- C. Yan, H. Li, Y. Ye, H. Wu, F. Cai, R. Si, J. Xiao, S. Miao, S. Xie, F. Yang, Y. Li, G. Wang and X. Bao, Energy Environ. Sci., 2018, 11, 1204–1210 RSC
- J. Li, M. Chen, D. A. Cullen, S. Hwang, M. Wang, B. Li, K. Liu, S. Karakalos, M. Lucero, H. Zhang, C. Lei, H. Xu, G. E. Sterbinsky, Z. Feng, D. Su, K. L. More, G. Wang, Z. Wang and G. Wu, Nat. Catal., 2018, 1, 935–945 CrossRef CAS
- Y.-N. Gong, L. Jiao, Y. Qian, C.-Y. Pan, L. Zheng, X. Cai, B. Liu, S.-H. Yu and H.-L. Jiang, Angew. Chem., Int. Ed., 2020, 59, 2705–2709 CrossRef CAS
- J. Wang, Z. Huang, W. Liu, C. Chang, H. Tang, Z. Li, W. Chen, C. Jia, T. Yao, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 17281–17284 CrossRef CAS PubMed
- R. Wu, X. Qian, K. Zhou, J. Wei, J. Lou and P. M. Ajayan, ACS Nano, 2014, 8, 6297–6303 CrossRef CAS PubMed
- H. Li, M. Liang, W. Sun and Y. Wang, Adv. Funct. Mater., 2016, 26, 1098–1103 CrossRef CAS
- C. Castillo-Blas, N. López-Salas, M. C. Gutiérrez, I. Puente-Orench, E. Gutiérrez-Puebla, M. L. Ferrer, M. Á. Monge and F. Gándara, J. Am. Chem. Soc., 2019, 141, 1766–1774 CrossRef CAS PubMed
- S. Chen, M. Xue, Y. Li, Y. Pan, L. Zhu and S. Qiu, J. Mater. Chem. A, 2015, 3, 20145–20152 RSC
- G. Huang, L. Zhang, F. Zhang and L. Wang, Nanoscale, 2014, 6, 5509–5515 RSC
- W. Guo, W. Sun and Y. Wang, ACS Nano, 2015, 9, 11462–11471 CrossRef CAS PubMed
- X. Wang, L. Yu, B. Y. Guan, S. Song and X. W. Lou, Adv. Mater., 2018, 30, 1801211 CrossRef PubMed
- W. Liu, R. Yin, W. Shi, X. Xu, X. Shen, Q. Yin, L. Xu and X. Cao, ACS Appl. Energy Mater., 2019, 2, 579–586 CrossRef CAS
- C. Qu, B. Zhao, Y. Jiao, D. Chen, S. Dai, B. M. deglee, Y. Chen, K. S. Walton, R. Zou and M. Liu, ACS Energy Lett., 2017, 2, 1263–1269 CrossRef CAS
- S. He, Z. Li, J. Wang, P. Wen, J. Gao, L. Ma, Z. Yang and S. Yang, RSC Adv., 2016, 6, 49478–49486 RSC
- H. Mei, Y. Mei, S. Zhang, Z. Xiao, B. Xu, H. Zhang, L. Fan, Z. Huang, W. Kang and D. Sun, Inorg. Chem., 2018, 57, 10953–10960 CrossRef CAS PubMed
- X. Feng, H. Wang, X. Bo and L. Guo, ACS Appl. Mater. Interfaces, 2019, 11, 8018–8024 CrossRef CAS PubMed
- Z.-F. Huang, J. Song, K. Li, M. Tahir, Y.-T. Wang, L. Pan, L. Wang, X. Zhang and J.-J. Zou, J. Am. Chem. Soc., 2016, 138, 1359–1365 CrossRef CAS PubMed
- Q. Wang, Y. Luo, R. Hou, S. Zaman, K. Qi, H. Liu, H. S. Park and B. Y. Xia, Adv. Mater., 2019, 31, 1905744 CrossRef CAS PubMed
- Q. Mao, J. Chen, H. Chen, Z. Chen, J. Chen and Y. Li, J. Mater. Chem. A, 2019, 7, 8472–8484 RSC
- J. Hao, W. Yang, Z. Zhang and J. Tang, Nanoscale, 2015, 7, 11055–11062 RSC
- L. Yan, L. Cao, P. Dai, X. Gu, D. Liu, L. Li, Y. Wang and X. Zhao, Adv. Funct. Mater., 2017, 27, 1703455 CrossRef
- Y. Du, M. Zhang, Z. Wang, Y. Liu, Y. Liu, Y. Geng and L. Wang, J. Mater. Chem. A, 2019, 7, 8602–8608 RSC
- Y. Du, Z. Li, Y. Liu, Y. Yang and L. Wang, Appl. Surf. Sci., 2018, 457, 1081–1086 CrossRef CAS
- X. Xiao, C.-T. He, S. Zhao, J. Li, W. Lin, Z. Yuan, Q. Zhang, S. Wang, L. Dai and D. Yu, Energy Environ. Sci., 2017, 10, 893–899 RSC
- M. Lan, R.-M. Guo, Y. Dou, J. Zhou, A. Zhou and J.-R. Li, Nano Energy, 2017, 33, 238–246 CrossRef CAS
- N. Tsumori, L. Chen, Q. Wang, Q.-L. Zhu, M. Kitta and Q. Xu, Chem, 2018, 4, 845–856 CAS
- Y. Shen, L.-W. Bao, F.-Z. Sun and T.-L. Hu, Mater. Chem. Front., 2019, 3, 2363–2373 RSC
- X. Wang, H. Xiao, A. Li, Z. Li, S. Liu, Q. Zhang, Y. Gong, L. Zheng, Y. Zhu, C. Chen, D. Wang, Q. Peng, L. Gu, X. Han, J. Li and Y. Li, J. Am. Chem. Soc., 2018, 140, 15336–15341 CrossRef CAS PubMed
- X. Yang, J. Chen, J. Hu, S. Zhao, J. Zhao and X. Luo, Catal. Sci. Technol., 2018, 8, 573–579 RSC
- H. Huang, J.-R. Li, K. Wang, T. Han, M. Tong, L. Li, Y. Xie, Q. Yang, D. Liu and C. Zhong, Nat. Commun., 2015, 6, 8847 CrossRef CAS PubMed
- J. Yang, F. Zhang, H. Lu, X. Hong, H. Jiang, Y. Wu and Y. Li, Angew. Chem., Int. Ed., 2015, 54, 10889–10893 CrossRef CAS PubMed
- C. Rösler, A. Aijaz, S. Turner, M. Filippousi, A. Shahabi, W. Xia, G. Van Tendeloo, M. Muhler and R. A. Fischer, Chem. – Eur. J., 2016, 22, 3304–3311 CrossRef PubMed
- A. Li, K. Shen, J. Chen, Z. Li and Y. Li, Chem. Eng. Sci., 2017, 166, 66–76 CrossRef CAS
- B. N. Bhadra and S. H. Jhung, Nanoscale, 2018, 10, 15035–15047 RSC
- L. Hu, R. Zhang, L. Wei, F. Zhang and Q. Chen, Nanoscale, 2015, 7, 450–454 RSC
- Y.-H. Qin, L. Huang, J.-X. Zheng and Q. Ren, Inorg. Chem. Commun., 2016, 72, 78–82 CrossRef CAS
- L. Zhang, L. Shi, L. Huang, J. Zhang, R. Gao and D. Zhang, ACS Catal., 2014, 4, 1753–1763 CrossRef CAS
- J. Zhang, B. An, Y. Hong, Y. Meng, X. Hu, C. Wang, J. Lin, W. Lin and Y. Wang, Mater. Chem. Front., 2017, 1, 2405–2409 RSC
- H. Chen, K. Shen, Q. Mao, J. Chen and Y. Li, ACS Catal., 2018, 8, 1417–1426 CrossRef CAS
- L.-P. Yang, J.-L. Mi, H.-J. Wang, J.-H. Liang, X.-J. Yang, Y.-H. Feng, P. Zhang and L. Liu, ChemCatChem, 2020, 12, 584–592 CrossRef CAS
- Q. Wang, Z. Li, M. A. Bañares, L.-T. Weng, Q. Gu, J. Price, W. Han and K. L. Yeung, Small, 2019, 15, 1903525 CrossRef CAS PubMed
- H. Jiang, C. Zhu, Y. Yuan, C. Yue, C. Ling, F. Liu and A. Li, Chem. Eng. J., 2020, 384, 123302 CrossRef CAS
- S. Li, Y. Hou, Q. Chen, X. Zhang, H. Cao and Y. Huang, ACS Appl. Mater. Interfaces, 2020, 12, 2581–2590 CrossRef CAS PubMed
- M. Yadav and Q. Xu, Energy Environ. Sci., 2012, 5, 9698–9725 RSC
- Q.-L. Zhu and Q. Xu, Energy Environ. Sci., 2015, 8, 478–512 RSC
- Z. Li and Q. Xu, Acc. Chem. Res., 2017, 50, 1449–1458 CrossRef CAS PubMed
- Q. Wang, L. Chen, Z. Liu, N. Tsumori, M. Kitta and Q. Xu, Adv. Funct. Mater., 2019, 29, 1903341 CrossRef
- M.-J. Chen, D.-X. Zhang, D. Li, S.-C. Ke, X.-C. Ma, G.-G. Chang, J. Chen and X.-Y. Yang, New J. Chem., 2020, 44, 3021–3027 RSC
- C.-C. Hou, Q.-Q. Chen, K. Li, C.-J. Wang, C.-Y. Peng, R. Shi and Y. Chen, J. Mater. Chem. A, 2019, 7, 8277–8283 RSC
- X.-L. Zhang, D.-X. Zhang, G.-G. Chang, X.-C. Ma, J. Wu, Y. Wang, H.-Z. Yu, G. Tian, J. Chen and X.-Y. Yang, Ind. Eng. Chem. Res., 2019, 58, 7209–7216 CrossRef CAS
- X. Meng, C. Wan, Y. Wang and X. Ju, J. Alloys Compd., 2018, 735, 1637–1647 CrossRef CAS
- P. Tan, G. Li, R. Fang, L. Chen, R. Luque and Y. Li, ACS Catal., 2017, 7, 2948–2955 CrossRef CAS
- R. Fang, R. Luque and Y. Li, Green Chem., 2016, 18, 3152–3157 RSC
- S. Zhang, H. Ma, Y. Sun, Y. Luo, X. Liu, M. Zhang, J. Gao and J. Xu, Green Chem., 2019, 21, 1702–1709 RSC
- K. W. Golub, T. P. Sulmonetti, L. A. Darunte, M. S. Shealy and C. W. Jones, ACS Appl. Nano Mater., 2019, 2, 6040–6056 CrossRef CAS
- H. Wang, X. Li, X. Lan and T. Wang, ACS Catal., 2018, 8, 2121–2128 CrossRef CAS
- J. Zhu, F. Chen, Z. Zhang, M. Li, Q. Yang, Y. Yang, Z. Bao and Q. Ren, ACS Sustainable Chem. Eng., 2019, 7, 12955–12963 CrossRef CAS
- H. Chen, K. Shen, Y. Tan and Y. Li, ACS Nano, 2019, 13, 7800–7810 CrossRef CAS PubMed
- P. Mahata, T. Aarthi, G. Madras and S. Natarajan, J. Phys. Chem. C, 2007, 111, 1665–1674 CrossRef CAS
- H. Chen, K. Shen, J. Chen, X. Chen and Y. Li, J. Mater. Chem. A, 2017, 5, 9937–9945 RSC
- B. Hu, J.-Y. Yuan, J.-Y. Tian, M. Wang, X. Wang, L. He, Z. Zhang, Z.-W. Wang and C.-S. Liu, J. Colloid Interface Sci., 2018, 531, 148–159 CrossRef CAS PubMed
- Y. Xu, S. Wu, X. Li, Y. Huang, Z. Wang, Y. Han, J. Wu, H. Meng and X. Zhang, New J. Chem., 2017, 41, 15433–15438 RSC
- G. Liu, Y. Sheng, J. W. Ager, M. Kraft and R. Xu, EnergyChem, 2019, 1, 100014 CrossRef
- S. Zhang, H. Ye, J. Hua and H. Tian, EnergyChem, 2019, 1, 100015 CrossRef
- S. Li, J. Tan, Z. Jiang, J. Wang and Z. Li, Chem. Eng. J., 2020, 384, 123354 CrossRef CAS
- J. Lu, J. Zhang, Q. Chen and H. Liu, J. Photochem. Photobiol., A, 2019, 380, 111853 CrossRef CAS
- X. Tang, J.-H. Zhao, Y.-H. Li, Z.-J. Zhou, K. Li, F.-T. Liu and Y.-Q. Lan, Dalton Trans., 2017, 46, 10553–10557 RSC
- Y. Su, D. Ao, H. Liu and Y. Wang, J. Mater. Chem. A, 2017, 5, 8680–8689 RSC
- X. Zhao, J. Feng, J. Liu, W. Shi, G. Yang, G.-C. Wang and P. Cheng, Angew. Chem., Int. Ed., 2018, 57, 9790–9794 CrossRef CAS PubMed
- B. Ma, T.-T. Chen, Q.-Y. Li, H.-N. Qin, X.-Y. Dong and S.-Q. Zang, ACS Appl. Energy Mater., 2019, 2, 1134–1148 CrossRef CAS
- W. Chen, J. Fang, Y. Zhang, G. Chen, S. Zhao, C. Zhang, R. Xu, J. Bao, Y. Zhou and X. Xiang, Nanoscale, 2018, 10, 4463–4474 RSC
- T. Wang, L. Shi, J. Tang, V. Malgras, S. Asahina, G. Liu, H. Zhang, X. Meng, K. Chang, J. He, O. Terasaki, Y. Yamauchi and J. Ye, Nanoscale, 2016, 8, 6712–6720 RSC
- Q. Mu, W. Zhu, G. Yan, Y. Lian, Y. Yao, Q. Li, Y. Tian, P. Zhang, Z. Deng and Y. Peng, J. Mater. Chem. A, 2018, 6, 21110–21119 RSC
- H. Jiang, S. Gong, S. Xu, P. Shi, J. Fan, V. Cecen, Q. Xu and Y. Min, Dalton Trans., 2020, 49, 5074–5086 RSC
- S. Chen, J. Yu and J. Zhang, J. CO2 Util., 2018, 24, 548–554 CrossRef CAS
- H.-F. Wang, L. Chen, H. Pang, S. Kaskel and Q. Xu, Chem. Soc. Rev., 2020, 49, 1414–1448 RSC
- S. Zhu, J. Ge, C. Liu and W. Xing, EnergyChem, 2019, 1, 100018 CrossRef
- H.-F. Wang and Q. Xu, Matter, 2019, 1, 565–595 CrossRef
- Q. Ren, H. Wang, X.-F. Lu, Y.-X. Tong and G.-R. Li, Adv. Sci., 2018, 5, 1700515 CrossRef PubMed
- H. Zhang, H. Osgood, X. Xie, Y. Shao and G. Wu, Nano Energy, 2017, 31, 331–350 CrossRef CAS
- X. Han, X. Ling, Y. Wang, T. Ma, C. Zhong, W. Hu and Y. Deng, Angew. Chem., Int. Ed., 2019, 58, 5359–5364 CrossRef CAS PubMed
- L. Du, L. Luo, Z. Feng, M. Engelhard, X. Xie, B. Han, J. Sun, J. Zhang, G. Yin, C. Wang, Y. Wang and Y. Shao, Nano Energy, 2017, 39, 245–252 CrossRef CAS
- J. Zhou, Y. Dou, A. Zhou, L. Shu, Y. Chen and J.-R. Li, ACS Energy Lett., 2018, 3, 1655–1661 CrossRef CAS
- H. Xu, Z.-X. Shi, Y.-X. Tong and G.-R. Li, Adv. Mater., 2018, 30, 1705442 CrossRef PubMed
- X. Wang, L. Chai, J. Ding, L. Zhong, Y. Du, T.-T. Li, Y. Hu, J. Qian and S. Huang, Nano Energy, 2019, 62, 745–753 CrossRef CAS
- Q. Shi, S. Fu, C. Zhu, J. Song, D. Du and Y. Lin, Mater. Horiz., 2019, 6, 684–702 RSC
- Y. Qian, I. A. Khan and D. Zhao, Small, 2017, 13, 1701143 CrossRef PubMed
- X. Li, L. Yang, T. Su, X. Wang, C. Sun and Z. Su, J. Mater. Chem. A, 2017, 5, 5000–5006 RSC
- X.-Y. Yu, Y. Feng, Y. Jeon, B. Guan, X. W. Lou and U. Paik, Adv. Mater., 2016, 28, 9006–9011 CrossRef CAS PubMed
- B. Zhu, R. Zou and Q. Xu, Adv. Energy Mater., 2018, 8, 1801193 CrossRef
- W. Ahn, M. G. Park, D. U. Lee, M. H. Seo, G. Jiang, Z. P. Cano, F. M. Hassan and Z. Chen, Adv. Funct. Mater., 2018, 28, 1802129 CrossRef
- Y. Pan, K. Sun, S. Liu, X. Cao, K. Wu, W.-C. Cheong, Z. Chen, Y. Wang, Y. Li, Y. Liu, D. Wang, Q. Peng, C. Chen and Y. Li, J. Am. Chem. Soc., 2018, 140, 2610–2618 CrossRef CAS PubMed
- B. Zhu, D. Xia and R. Zou, Coord. Chem. Rev., 2018, 376, 430–448 CrossRef CAS
- L. Li, Y. Huang and Y. Li, EnergyChem, 2020, 2, 100024 CrossRef
- W. Guo, X. Sun, C. Chen, D. Yang, L. Lu, Y. Yang and B. Han, Green Chem., 2019, 21, 503–508 RSC
- X. Wang, Z. Chen, X. Zhao, T. Yao, W. Chen, R. You, C. Zhao, G. Wu, J. Wang, W. Huang, J. Yang, X. Hong, S. Wei, Y. Wu and Y. Li, Angew. Chem., Int. Ed., 2018, 57, 1944–1948 CrossRef CAS PubMed
- R. Zhao, H. Xie, L. Chang, X. Zhang, X. Zhu, X. Tong, T. Wang, Y. Luo, P. Wei, Z. Wang and X. Sun, EnergyChem, 2019, 1, 100011 CrossRef
- S. Chen, H. Jang, J. Wang, Q. Qin, X. Liu and J. Cho, J. Mater. Chem. A, 2020, 8, 2099–2104 RSC
- A. Indra, T. Song and U. Paik, Adv. Mater., 2018, 30, 1705146 CrossRef PubMed
- H. B. Wu and X. W. Lou, Sci. Adv., 2017, 3, eaap9252 CrossRef PubMed
- Y. Li, Y. Xu, W. Yang, W. Shen, H. Xue and H. Pang, Small, 2018, 14, 1704435 CrossRef PubMed
- X. Cao, C. Tan, M. Sindoro and H. Zhang, Chem. Soc. Rev., 2017, 46, 2660–2677 RSC
- R. R. Salunkhe, Y. V. Kaneti and Y. Yamauchi, ACS Nano, 2017, 11, 5293–5308 CrossRef CAS PubMed
- S. Chen, M. Xue, Y. Li, Y. Pan, L. Zhu, D. Zhang, Q. Fang and S. Qiu, Inorg. Chem. Front., 2015, 2, 177–183 RSC
- R. R. Salunkhe, Y. V. Kaneti, J. Kim, J. H. Kim and Y. Yamauchi, Acc. Chem. Res., 2016, 49, 2796–2806 CrossRef CAS PubMed
- M. S. Javed, H. Lei, J. Li, Z. Wang and W. Mai, J. Mater. Chem. A, 2019, 7, 17435–17445 RSC
- M. Yi, C. Zhang, C. Cao, C. Xu, B. Sa, D. Cai and H. Zhan, Inorg. Chem., 2019, 58, 3916–3924 CrossRef CAS PubMed
- M. K. Aslam, S. S. Ahmad Shah, M. S. Javed, S. Li, S. Hussain, B. Hu, N. A. Khan and C. Chen, J. Electroanal. Chem., 2019, 855, 113615 CrossRef CAS
- C. Young, J. Kim, Y. V. Kaneti and Y. Yamauchi, ACS Appl. Energy Mater., 2018, 1, 2007–2015 CrossRef CAS
- T. Huang, X.-Z. Song, X. Chen, X.-L. Chen, F.-F. Sun, Q.-F. Su, L.-D. Li and Z. Tan, New J. Chem., 2018, 42, 5128–5134 RSC
- W. Lu, J. Shen, P. Zhang, Y. Zhong, Y. Hu and X. W. Lou, Angew. Chem., Int. Ed., 2019, 58, 15441–15447 CrossRef CAS PubMed
- D. P. Dubal, K. Jayaramulu, J. Sunil, Š. Kment, P. Gomez-Romero, C. Narayana, R. Zbořil and R. A. Fischer, Adv. Funct. Mater., 2019, 29, 1900532 CrossRef
- G. Zou, H. Hou, P. Ge, Z. Huang, G. Zhao, D. Yin and X. Ji, Small, 2018, 14, 1702648 CrossRef PubMed
- X. Zhang, P. Dong and M.-K. Song, Batteries Supercaps, 2019, 2, 591–626 CrossRef CAS
- J. Zhang, L. Wang, L. Xu, X. Ge, X. Zhao, M. Lai, Z. Liu and W. Chen, Nanoscale, 2015, 7, 720–726 RSC
- H. Guo, T. Li, W. Chen, L. Liu, X. Yang, Y. Wang and Y. Guo, Nanoscale, 2014, 6, 15168–15174 RSC
- Y. Xia, B. Wang, G. Wang, X. Liu and H. Wang, ChemElectroChem, 2016, 3, 299–308 CrossRef CAS
- L. Hou, L. Lian, L. Zhang, G. Pang, C. Yuan and X. Zhang, Adv. Funct. Mater., 2015, 25, 238–246 CrossRef CAS
- J. Ma, H. Wang, X. Yang, Y. Chai and R. Yuan, J. Mater. Chem. A, 2015, 3, 12038–12043 RSC
- J. Wu, Y. Song, R. Zhou, S. Chen, L. Zuo, H. Hou and L. Wang, J. Mater. Chem. A, 2015, 3, 7793–7798 RSC
- F. Zou, X. Hu, Z. Li, L. Qie, C. Hu, R. Zeng, Y. Jiang and Y. Huang, Adv. Mater., 2014, 26, 6622–6628 CrossRef CAS PubMed
- M. K. Aslam, S. S. A. Shah, S. Li and C. Chen, J. Mater. Chem. A, 2018, 6, 14083–14090 RSC
- W. C. Haoying Yang, Y. Han and B. Wang, Chin. Chem. Lett., 2018, 29, 842–844 CrossRef
- X. Zhang, D. Li, G. Zhu, T. Lu and L. Pan, J. Colloid Interface Sci., 2017, 499, 145–150 CrossRef CAS PubMed
- G. Fang, J. Zhou, Y. Cai, S. Liu, X. Tan, A. Pan and S. Liang, J. Mater. Chem. A, 2017, 5, 13983–13993 RSC
- J. Li, D. Wang, J. Zhou, L. Hou and F. Gao, ChemElectroChem, 2019, 6, 917–927 CrossRef CAS
- J. Tang, S. Wu, T. Wang, H. Gong, H. Zhang, S. M. Alshehri, T. Ahamad, H. Zhou and Y. Yamauchi, ACS Appl. Mater. Interfaces, 2016, 8, 2796–2804 CrossRef CAS PubMed
- W. Yin, Y. Shen, F. Zou, X. Hu, B. Chi and Y. Huang, ACS Appl. Mater. Interfaces, 2015, 7, 4947–4954 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2020 |