
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNickel catalysis enables convergent paired electrolysis for direct arylation of benzylic C–H bonds†
Lei
Zhang
and
Xile
Hu
 *
*
Laboratory of Inorganic Synthesis and Catalysis, Institute of Chemical Sciences and Engineering, Ecole Polytechnique Fédérale de Lausanne (EPFL), ISCI-LSCI BCH 3305, 1015 Lausanne, Switzerland. E-mail: xile.hu@epfl.ch; Web: http://lsci.epfl.ch
First published on 27th April 2020
Abstract
Convergent paired electrosynthesis is an energy-efficient approach in organic synthesis; however, it is limited by the difficulty to match the innate redox properties of reaction partners. Here we use nickel catalysis to cross-couple the two intermediates generated at the two opposite electrodes of an electrochemical cell, achieving direct arylation of benzylic C–H bonds. This method yields a diverse set of diarylmethanes, which are important structural motifs in medicinal and materials chemistry. Preliminary mechanistic study suggests oxidation of a benzylic C–H bond, Ni-catalyzed C–C coupling, and reduction of a Ni intermediate as key elements of the catalytic cycle.
Electrochemical organic synthesis has drawn much attention in recent years.1 Compared to processes using stoichiometric redox agents, electrosynthesis can potentially be more selective and safe, generate less waste, and operate under milder conditions.1b In the majority of examples, the reaction of interest occurs at one electrode (anode for oxidation or cathode for reduction), while a sacrificial reaction occurs at the counter electrode to fulfil electron neutrality.1a,2 Paired electrolysis uses both anodic and cathodic reactions for the target synthesis, thereby maximizing energy efficiency.1a,3 However, there are comparatively few examples of paired electrolysis for organic synthesis.1a,3,4
Paired electrolysis might be classified into three types: parallel, sequential, and convergent (Fig. 1).1a,3a In parallel paired electrolysis (Fig. 1a), the two half reactions are simultaneous but non-interfering. In sequential paired electrolysis (Fig. 1b), a substrate is oxidized and reduced (or vice versa) sequentially. In convergent paired electrolysis (Fig. 1c), intermediates generated by the anodic and cathodic processes react with one another to yield the product.1a,3a,4b,c,5 The activation mode of all three types of paired electrolysis is based on the innate redox reactivity of substrates. As a result, the types of reactions that could be conducted by paired electrolysis remain limited. We proposed a catalytic version of convergent paired electrolysis, where a catalyst is used to cross-couple the two intermediates generated at the two separated electrodes (Fig. 1d). Although mediators have been used in paired electrosynthesis,3a,4c,6 catalytic coupling of anodic and cathodic intermediates remains largely undeveloped. This mode of action will leverage the power of cross-coupling to electrosynthesis, opening up a wide substrate and product space. Here we report the development of such a process, where cooperative nickel catalysis and paired electrolysis enable direct arylation of benzylic C–H bonds (Fig. 1e).
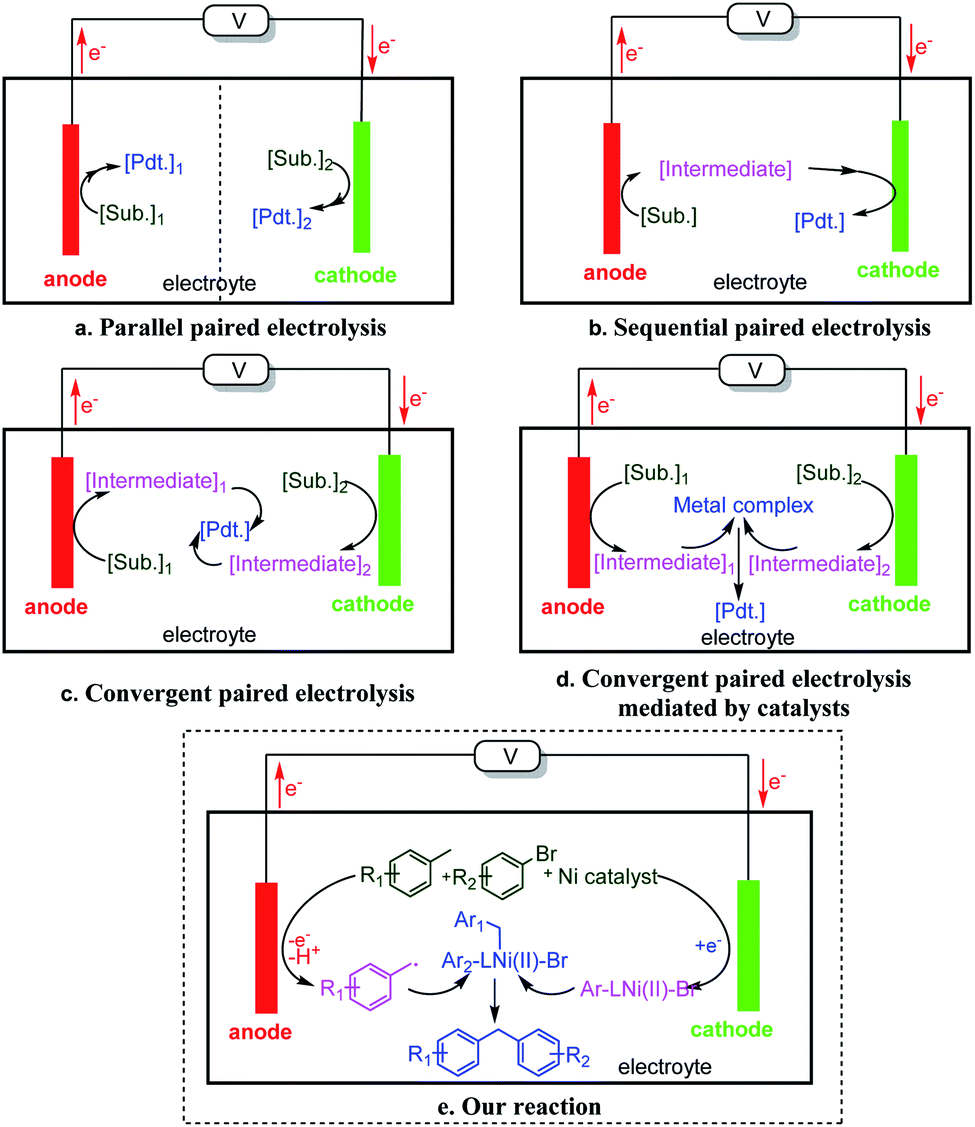 | ||
| Fig. 1 Different types of paired electrolysis: (a) parallel paired electrolysis, (b) sequential paired electrolysis, (c) convergent paired electrolysis, (d) catalytic convergent paired electrolysis and (e) this work. | ||
Our method can be used to synthesize diarylmethanes, which are important structural motifs in bioactive compounds,7 natural products8 and materials.9 Direct arylation of benzylic C–H bonds has been recognized as an efficient strategy to synthesize diarylmethanes, and methods using metal catalysis10 and in particular combined photoredox and transition-metal catalysis have been reported.11 Electrosynthesis provides a complementary approach to these methods, with the potential advantages outlined above. The groups of Yoshida12 and Waldvogel13 previously developed synthesis of diarylmethanes via a Friedel–Crafts-type reaction of a benzylic cation and a nucleophile. The benzylic cations were generated by anodic oxidation of benzylic C–H bonds.14 To avoid the overoxidation of products and to stabilize the very reactive benzylic cations, the reactions had to be conducted in two steps, where the benzylic cations generated in the anodic oxidation step had to be trapped by a reagent. We thought a Ni catalyst could be used to trap the benzyl radical to form an organonickel intermediate, which is then prone to a Ni-catalyzed C–C cross-coupling reaction. Although such a coupling scheme was unprecedented, Ni-catalyzed electrochemical reductive coupling of aryl halides was well established.15 We were also encouraged by a few recent reports of combined Ni catalysis and electrosynthesis for C–N,16 C–S,17 and C–P18 coupling reactions.
We started our investigations using the reaction between 4-methylanisole 1a and 4-bromoacetophenone 2a as a test reaction (Table 1). Direct arylation of benzylic C–H bonds was challenging and was typically conducted using toluene derivatives in large excess, e.g., as a solvent.11a,b,11d–f To improve the reaction efficiency, we decided to use only 3 equivalents of 4-methylanisole 1a relative to 2a. After some initial trials, we decided to conduct the reaction in an undivided cell using a constant current of 3 mA. These conditions are straightforward from a practical point of view. After screening various reaction parameters, we found that a combination of 4,4′-dimethoxy-2-2′-bipyridine (L1) and (DME)NiBr2 as a catalyst, THF/CH3CN (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as a solvent, fluorine-doped tin oxide (FTO) coated glass as an anode and carbon fibre as a cathode gave a 50% GC yield of 1-(4-(4-methoxybenzyl)phenyl)ethanone 3a after 18 h (Table 1, entry 1). Extending the reaction time to 36 h improved the yield to 76% (isolated yield) (entry 2). The target products were formed in a diminished yield with other bipyridine type ligands (entries 3–5). Solvents commonly used in Ni-catalyzed cross-coupling reactions, such as DMA and DMF, were less effective (entries 7–8). Replacing carbon fibre by nickel foam or platinum foil as the cathode was detrimental to the coupling, but substantial yields were still obtained (entries 9–10). On the other hand, FTO could not be replaced as the anode. Using carbon fibre as the anode shut down the reaction (entry 11). Likewise, using Pt foil as the anode gave only a 7% GC yield (entry 12). The sensitivity of the reaction outcomes to the electrodes originates from the electrode-dependent redox properties of reaction components (see below). Additional data showing the influence of other reaction parameters such as nickel sources, current, concentration, and electrolytes are provided in the ESI (Table S1, ESI†).
:1) as a solvent, fluorine-doped tin oxide (FTO) coated glass as an anode and carbon fibre as a cathode gave a 50% GC yield of 1-(4-(4-methoxybenzyl)phenyl)ethanone 3a after 18 h (Table 1, entry 1). Extending the reaction time to 36 h improved the yield to 76% (isolated yield) (entry 2). The target products were formed in a diminished yield with other bipyridine type ligands (entries 3–5). Solvents commonly used in Ni-catalyzed cross-coupling reactions, such as DMA and DMF, were less effective (entries 7–8). Replacing carbon fibre by nickel foam or platinum foil as the cathode was detrimental to the coupling, but substantial yields were still obtained (entries 9–10). On the other hand, FTO could not be replaced as the anode. Using carbon fibre as the anode shut down the reaction (entry 11). Likewise, using Pt foil as the anode gave only a 7% GC yield (entry 12). The sensitivity of the reaction outcomes to the electrodes originates from the electrode-dependent redox properties of reaction components (see below). Additional data showing the influence of other reaction parameters such as nickel sources, current, concentration, and electrolytes are provided in the ESI (Table S1, ESI†).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | Anode | Cathode | Solvent | Yield (%) |
| a Reaction conditions: 1a (0.6 mmol), 2a (0.2 mmol), (DME)NiBr2 (6 mol%), ligand (7.2 mol mol%), LutHClO4 (0.1 M), and lutidine (0.8 mmol) in solvent (2 mL) at 40 °C. GC yield. b Reaction time: 36 h. Isolated yield. | |||||
| 1 | L1 | FTO | Carbon fibre | THF/CH3CN = 4:1 |
56 |
| 2 | L1 | FTO | Carbon fibre | THF/CH3CN = 4:1 |
76b |
| 3 | L2 | FTO | Carbon fibre | THF/CH3CN = 4:1 |
43 |
| 4 | L3 | FTO | Carbon fibre | THF/CH3CN = 4:1 |
46 |
| 5 | L4 | FTO | Carbon fibre | THF/CH3CN = 4:1 |
21 |
| 6 | L1 | FTO | Carbon fibre | CH3CN | 4 |
| 7 | L1 | FTO | Carbon fibre | DMA | 15 |
| 8 | L1 | FTO | Carbon fibre | DMF | 6 |
| 9 | L1 | FTO | Ni foam | THF/CH3CN = 4:1 |
45 |
| 10 | L1 | FTO | Pt foil | THF/CH3CN = 4:1 |
28 |
| 11 | L1 | Carbon fibre (1 cm2) | Carbon fibre | THF/CH3CN = 4:1 |
0 |
| 12 | L1 | Pt foil (cm2) | Carbon fibre | THF/CH3CN = 4:1 |
7 |
|
|||||
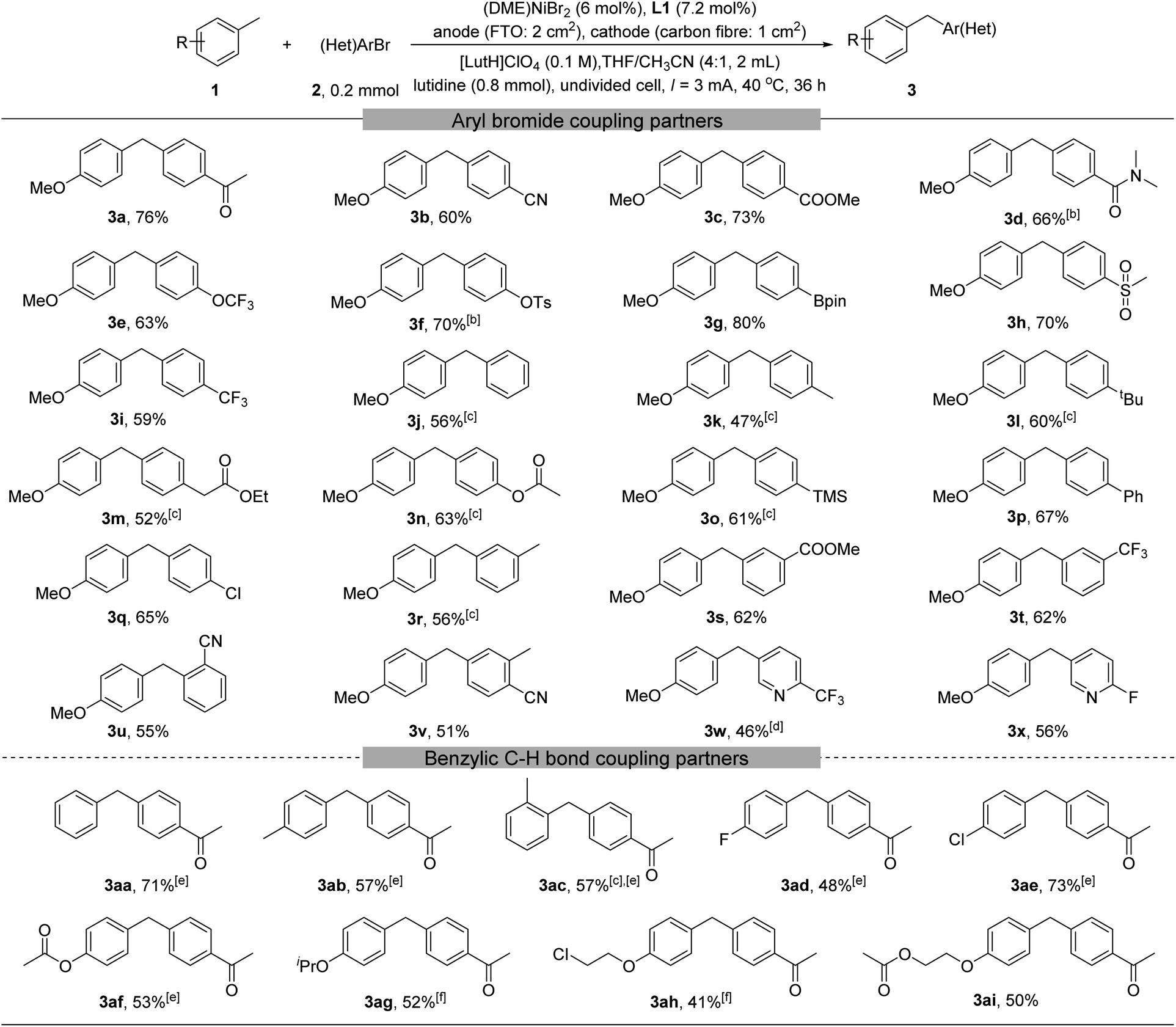
With the optimized reaction conditions in hand, we explored the substrate scope (Table 2). A large number of aryl and heteroaryl bromides could be coupled (3a–3x). These substrates may contain electron-rich, neutral, or poor groups. For aryl bromide bearing electron-donating groups, replacing (DME)NiBr2 by Ni(acac)2 gave higher yields (3k–3o). The method tolerates numerous functional groups in the (hetero)aryl bromides, including for example ketone (3a), nitrile (3b, 3u, and 3v), ester (3c, 3m, 3n, and 3s), amide (3d), aryl-Cl (3q), CF3(3i, 3t, and 3w), OCF3(3e), aryl-F(3x), pyridine (3w and 3x), and arylboronic ester (3g). We then probed the scope of benzylic substrates using 4-bromoacetophenone 2a as the coupling partner (3aa–3ai). Toluene and electron-rich toluene derivatives were readily arylated (3aa–3ac). Toluene derivatives containing an electron-withdrawing group such as fluoride (3ad) and chloride (3ae) could also be arylated, although a higher excess of them (10 equiv.) was necessary. More elaborated toluene derivatives containing an additional ester (3af, 3ai) or ether (3ag, 3ah, and 3ai) were also viable.
|
a Reaction conditions: 1 (0.6 mmol), 2 (0.2 mmol), (DME)NiBr2 (6 mol%), L1 (7.2 mol mol%), LutHClO4 (0.1 M), and lutidine (0.8 mmol) in THF/CH3CN (4:1, 2 mL) at 40 °C. Isolated yield.
b (DME)NiBr2 (5 mol%) and L1 (6 mol%) were used as the catalysts.
c Ni(acac)2 was used instead of (DME)NiBr2.
d Solvent: THF/CH3CN (3:1, 2 mL).
e 2 mmol toluene or its derivative was used as the substrate.
f Reaction time: 60 h.
|
|---|
|
Linear sweep voltammetry (LSV) was applied to probe the possible processes at both the anode and cathode. The measurements were made in THF/CN3CN (4:1, 2 mL) using [LutH]ClO4 (0.1 M) as the electrolyte and lutidine (0.4 M) as an additional base to mimic the coupling conditions. The LSV curves of individual reaction components indicate that only 4-methylanisole 1a and the Ni catalyst may be oxidized at the anode (Fig. 2a). The current at 3 mA appears to be the sum of the oxidation currents of 1a and the Ni catalyst. Meanwhile, LSV curves indicate that only the Ni catalyst might be reduced at the cathode (Fig. 2b).
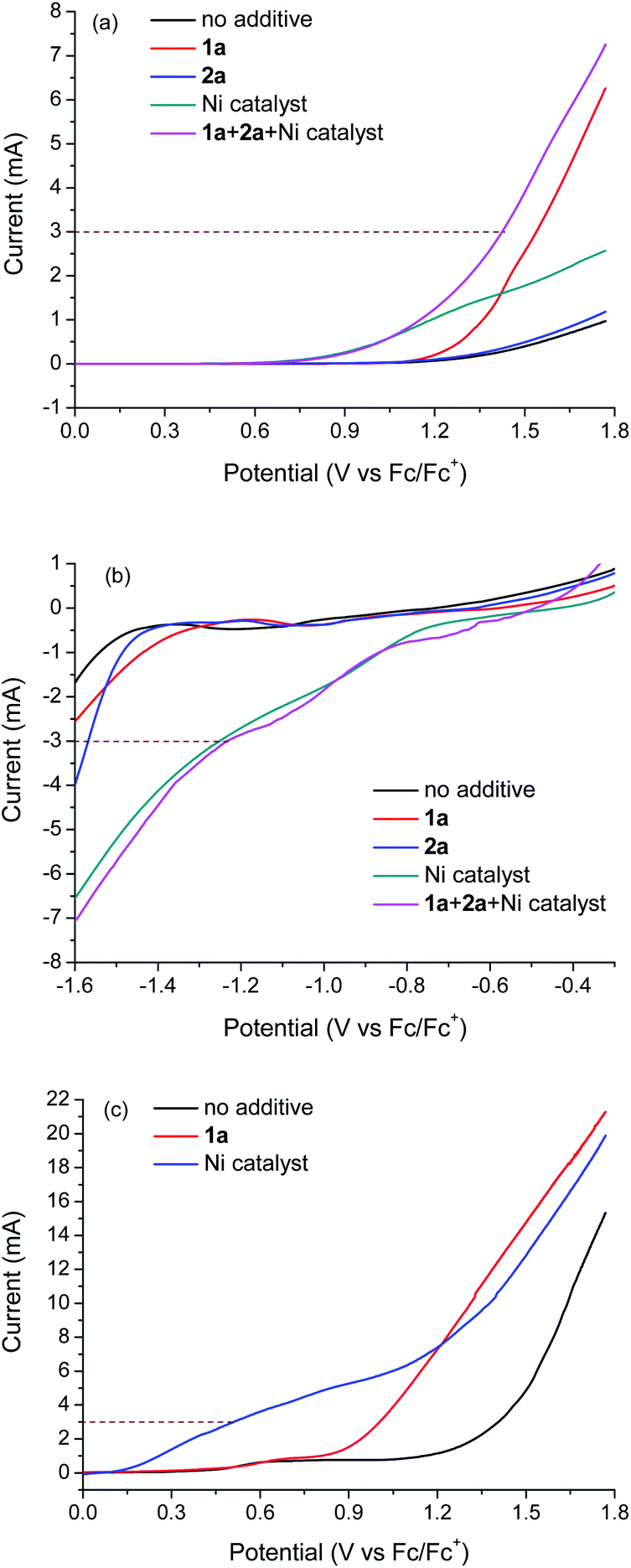 | ||
| Fig. 2 The LSV curves of different reaction components at the anode or cathode. The components were dissolved in THF/CN3CN (4:1, 2 mL); the solution also contained [LutH]ClO4 (0.1 M) and lutidine (0.4 M). Scan rate: 50 mV s−1. (a) The LSV curves of different reaction components at the FTO anode; (b) the LSV curves of different reaction components at the carbon fibre cathode; (c) the LSV curves of different reaction components at the carbon fibre anode. | ||
It was observed that FTO was an essential anode for the reactions. If FTO was replaced by a carbon fibre anode, no coupling product was obtained. LSV was performed to probe the oxidation of 1a and the Ni catalyst on a carbon fibre anode (Fig. 2c). The oxidation of the Ni catalyst was much easier on carbon fibre than on FTO. At 3 mA, the oxidation is exclusively due to the Ni catalyst. This result suggests that the absence of coupling on the carbon fibre anode is due to no oxidation of 1a. The different redox properties of 1a and the Ni catalyst observed on different electrodes might be attributed to the different nature of surface species which influence the electron transfer. Although FTO is rarely used in electrosynthesis, it is widely used in electrocatalysis and photoelectrocatalysis for energy conversion.19 FTO is stable, commercially available and inexpensive. In our reactions, the FTO anode could be reused at least three times.
The LSV curves in Fig. 2 revealed the issue of “short-circuit” of catalyzed/mediated paired electrolysis in an undivided cell, as the catalyst or mediator can be reduced and oxidized at both the cathode and anode. When carbon fibre or graphite was used as the anode, the short-circuit problem was very severe so that nearly no current was used for electrosynthesis. However, by using an appropriate anode such as FTO, the short-circuit problem was alleviated and around half of the current was used to oxidize the substrate (1a) while the other half was used to oxidize the nickel complex. The remaining short-circuit is one of the reasons why the current efficiencies of the reactions are low (<10%). Another factor contributing to the low current efficiency is the instability of the benzyl radical, which can abstract hydrogen from the solvent to regenerate the substrate. Nevertheless, useful products could be obtained in synthetically useful yields under conditions advantageous to previous methods.
For the test reaction (Table 1), a small amount of homo-coupling product bis(4-methoxyphenyl)methane (<2%) was detected by GC-MS under the optimized conditions. In the absence of ligand L1, the yield of the homo-coupling products increased (∼8%). In the presence of a radical acceptor, the electron-withdrawing alkene vinyl benzoate, the product originating from the addition of a benzyl radical to the olefin was obtained in about 12% GC yield (ESI, Scheme S1†). These data support the formation of a benzyl radical intermediate. As bromide existed in our reaction system, it is possible to be oxidized to form a bromine radical. Previous studies showed that a bromine radical can react with a toluene derivative to give a benzyl radical.11b,g,20 To probe the involvement of the Br radical, we conducted a coupling of 4-methylanisole 1a with 4′-Iodoacetophenone, using Ni(acac)2 instead of (DME)NiBr2 as the Ni source. We obtained a GC yield of 24% for the coupling after 18 h (Scheme S2†). This result suggests that a Br-free path exists for the coupling, although a non-decisive involvement of Br−/Br˙ cannot be ruled out.
Based on the data described above, we propose a mechanism for the coupling (Scheme 1). The oxidation of a toluene derivative at the anode gives a benzyl radical. This radical is trapped by a LNi(II)(Ar)(Br) species (B) in the solution to give a LNi(III)(Ar)(benzyl)(Br) intermediate (C). The latter undergoes reductive elimination to give a diarylmethane and a LNi(I)(Br) species (A). There are at least two ways A can be convert to B to complete the catalytic cycle: either by oxidative addition of ArBr followed by a 1-e reduction at the cathode or by first 1-e reduction to form a Ni(0) species followed by oxidation addition of ArBr. In addition to a toluene derivative, a Ni species is oxidized at the anode. We propose that this oxidation is an off cycle event, which reduces the faradaic and catalytic efficiency but does not shut down the productive coupling.
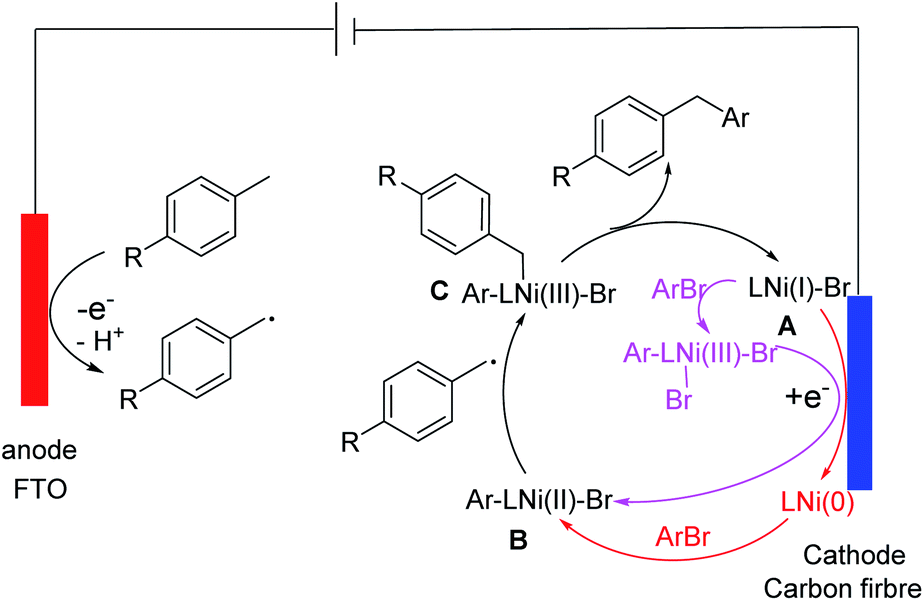 | ||
| Scheme 1 Proposed mechanism of the direct arylation of benzylic C–H bonds. | ||
Conclusions
In summary, by integrating Ni-catalyzed benzyl-aryl coupling into paired electrolysis, we achieved direct arylation of benzylic C–H bonds under simple electrochemical conditions. The direct arylation method has a broad scope and high functional group tolerance, giving access to a diverse range of diarylmethanes. Our work demonstrates the utility of metal catalysis in convergent paired electrolysis, which has much untapped potential in organic electrosynthesis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the European Research Council (no. 681292).Notes and references
- (a) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230 CrossRef CAS PubMed; (b) A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594 CrossRef CAS PubMed; (c) S. Möhle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 6018 CrossRef PubMed; (d) N. Sauermann, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Catal., 2018, 8, 7086 CrossRef CAS; (e) G. S. Sauer and S. Lin, ACS Catal., 2018, 8, 5175 CrossRef CAS; (f) R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492 RSC.
- (a) H. Wang, X. Gao, Z. Lv, T. Abdelilah and A. Lei, Chem. Rev., 2019, 119, 6769 CrossRef CAS PubMed; (b) Y. Yuan and A. Lei, Acc. Chem. Res., 2019, 52, 3309 CrossRef CAS PubMed; (c) S. Tang, L. Zeng and A. Lei, J. Am. Chem. Soc., 2018, 140, 13128 CrossRef CAS PubMed; (d) S. Tang, Y. Liu and A. Lei, Chem, 2018, 4, 27 CrossRef CAS; (e) C. Ma, P. Fang and T.-S. Mei, ACS Catal., 2018, 8, 7179 CrossRef CAS.
- (a) C. A. Paddon, M. Atobe, T. Fuchigami, P. He, P. Watts, S. J. Haswell, G. J. Pritchard, S. D. Bull and F. Marken, J. Appl. Electrochem., 2006, 36, 617 CrossRef CAS; (b) T. Wu, B. H. Nguyen, M. C. Daugherty and K. D. Moeller, Angew. Chem., Int. Ed., 2019, 58, 3562 CrossRef CAS PubMed.
- (a) K. Park, P. N. Pintauro, M. M. Baizer and K. Nobe, J. Electrochem. Soc., 1985, 132, 1850 CrossRef CAS; (b) G. Hilt, Angew. Chem., Int. Ed., 2003, 42, 1720 CrossRef CAS PubMed; (c) Y. Ma, X. Yao, L. Zhang, P. Ni, R. Cheng and J. Ye, Angew. Chem., Int. Ed., 2019, 58, 16548 CrossRef CAS PubMed; (d) H. Pütter and H. Hannebaum, Production of phthalides by cathodic reduction of phthalic acid or its derivatives, 1997, BASF patent, DE 19618854(13-11-1997); Chem. Abstr. 127, P338559y.
- (a) S. Liang, C.-C. Zeng, X.-G. Luo, F.-z. Ren, H.-Y. Tian, B.-G. Sun and R. D. Little, Green Chem., 2016, 18, 2222 RSC; (b) W. Jud, C. O. Kappe and D. Cantillo, Chem.–Eur. J., 2018, 24, 17234 CrossRef CAS PubMed.
- (a) S. Ito, R. Katayama, A. Kunai and K. Sasaki, Tetrahedron Lett., 1989, 30, 205 CrossRef CAS; (b) C. Belmont and H. H. Giault, Electrochim. Acta, 1995, 40, 2505 CrossRef CAS.
- Y.-Q. Long, X.-H. Jiang, R. Dayam, T. Sanchez, R. Shoemaker, S. Sei and N. Neamati, J. Med. Chem., 2004, 47, 2561 CrossRef CAS PubMed.
- K. L. McPhail, D. E. A. Rivett, D. E. Lack and M. T. Davies-Coleman, Tetrahedron, 2000, 56, 9391 CrossRef CAS.
- S. Wang, C. Zhang, Y. Shu, S. Jiang, Q. Xia, L. Chen, S. Jin, I. Hussain, A. I. Cooper and B. Tan, Sci. Adv., 2017, 3, e1602610 CrossRef PubMed.
- (a) A. Vasilopoulos, S. L. Zultanski and S. S. Stahl, J. Am. Chem. Soc., 2017, 139, 7705 CrossRef CAS PubMed; (b) W. Zhang, P. Chen and G. Liu, J. Am. Chem. Soc., 2017, 139, 7709 CrossRef CAS PubMed; (c) S.-C. Sha, S. Tcyrulnikov, M. Li, B. Hu, Y. Fu, M. C. Kozlowski and P. J. Walsh, J. Am. Chem. Soc., 2018, 140, 12415 CrossRef CAS PubMed; (d) H. Jiang, S.-C. Sha, S. A. Jeong, B. C. Manor and P. J. Walsh, Org. Lett., 2019, 21, 1735 CrossRef CAS PubMed.
- (a) B. J. Shields and A. G. Doyle, J. Am. Chem. Soc., 2016, 138, 12719 CrossRef CAS PubMed; (b) D. R. Heitz, J. C. Tellis and G. A. Molander, J. Am. Chem. Soc., 2016, 138, 12715 CrossRef CAS PubMed; (c) I. B. Perry, T. F. Brewer, P. J. Sarver, D. M. Schultz, D. A. DiRocco and D. W. C. MacMillan, Nature, 2018, 560, 70 CrossRef CAS PubMed; (d) Y. Shen, Y. Gu and R. Martin, J. Am. Chem. Soc., 2018, 140, 12200 CrossRef CAS PubMed; (e) N. Ishida, Y. Masuda, N. Ishikawa and M. Murakami, Asian J. Org. Chem., 2017, 6, 669 CrossRef CAS; (f) A. Dewanji, P. E. Krach and M. Rueping, Angew. Chem., Int. Ed., 2019, 58, 3566 CrossRef CAS PubMed; (g) X. Cheng, H. Lu and Z. Lu, Nat. Commun., 2019, 10, 3549 CrossRef PubMed.
- R. Hayashi, A. Shimizu and J.-i. Yoshida, J. Am. Chem. Soc., 2016, 138, 8400 CrossRef CAS PubMed.
- Y. Imada, J. L. Röckl, A. Wiebe, T. Gieshoff, D. Schollmeyer, K. Chiba, R. Franke and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 12136 CrossRef CAS PubMed.
- J.-i. Yoshida, A. Shimizu and R. Hayashi, Chem. Rev., 2018, 118, 4702 CrossRef CAS PubMed.
- (a) C. Amatore and A. Jutand, Organometallics, 1988, 7, 2203–2214 CrossRef CAS; (b) C. Amatore and A. Jutand, J. Am. Chem. Soc., 1991, 113, 2819 CrossRef CAS; (c) C. Amatore, A. Jutand and L. Mottier, J. Electroanal. Chem., 1991, 306, 125 CrossRef CAS; (d) C. Amatore and A. Jutand, J. Electroanal. Chem., 1991, 306, 141 CrossRef CAS; (e) M. Durandetti, J.-Y. Nédélec and J. Périchon, J. Org. Chem., 1996, 61, 1748 CrossRef CAS PubMed; (f) H. Li, C. P. Breen, H. Seo, T. F. Jamison, Y.-Q. Fang and M. M. Bio, Org. Lett., 2018, 20, 1338 CrossRef CAS PubMed; (g) R. J. Perkins, D. J. Pedro and E. C. Hansen, Org. Lett., 2017, 19, 3755 CrossRef CAS PubMed; (h) T. Koyanagi, A. Herath, A. Chong, M. Ratnikov, A. Valiere, J. Chang, V. Molteni and J. Loren, Org. Lett., 2019, 21, 816 CrossRef CAS PubMed; (i) T. J. DeLano and S. E. Reisman, ACS Catal., 2019, 9, 6751 CrossRef CAS PubMed; (j) K.-J. Jiao; D. Liu; H.-X. Ma; H. Qiu; P. Fang; T.-S. Mei, Angew. Chem., Int. Ed., 2020, 59, 6520 Search PubMed.
- (a) C. Li, Y. Kawamata, H. Nakamura, J. C. Vantourout, Z. Liu, Q. Hou, D. Bao, J. T. Starr, J. Chen, M. Yan and P. S. Baran, Angew. Chem., Int. Ed., 2017, 56, 13088 CrossRef CAS PubMed; (b) Y. Kawamata, J. C. Vantourout, D. P. Hickey, P. Bai, L. Chen, Q. Hou, W. Qiao, K. Barman, M. A. Edwards, A. F. Garrido-Castro, J. N. deGruyter, H. Nakamura, K. Knouse, C. Qin, K. J. Clay, D. Bao, C. Li, J. T. Starr, C. Garcia-Irizarry, N. Sach, H. S. White, M. Neurock, S. D. Minteer and P. S. Baran, J. Am. Chem. Soc., 2019, 141, 6392 CrossRef CAS PubMed.
- D. Liu, H.-X. Ma, P. Fang and T.-S. Mei, Angew. Chem., Int. Ed., 2019, 58, 5033 CrossRef CAS PubMed.
- Y. Bai, N. Liu, S. Wang, S. Wang, S. Ning, L. Shi, L. Cui, Z. Zhang and J. Xiang, Org. Lett., 2019, 21, 6835 CrossRef CAS PubMed.
- (a) A. E. Rakhshani, Y. Makdisi and H. A. Ramazaniyan, J. Appl. Phys., 1998, 83, 1049 CrossRef CAS; (b) C. G. Morales-Guio, L. Liardet and X. Hu, J. Am. Chem. Soc., 2016, 138, 8946 CrossRef CAS PubMed; (c) C. G. Morales-Guio, M. T. Mayer, A. Yella, S. D. Tilley, M. Grätzel and X. Hu, J. Am. Chem. Soc., 2015, 137, 9927 CrossRef CAS PubMed.
- J. K. Matsui, S. B. Lang, D. R. Heitz and G. A. Molander, ACS Catal., 2017, 7, 2563 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc01445a |
| This journal is © The Royal Society of Chemistry 2020 |