
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering plasticization resistant gas separation membranes using metal–organic nanocapsules†
Hongliang
Wang
a,
Kexin
Zhang
a,
Jerry Pui
Ho Li
 ad,
Jingyu
Huang
b,
Biao
Yuan
a,
Chen
Zhang
c,
Yi
Yu
a,
Yong
Yang
a,
Yongjin
Lee
a and
Tao
Li
*a
ad,
Jingyu
Huang
b,
Biao
Yuan
a,
Chen
Zhang
c,
Yi
Yu
a,
Yong
Yang
a,
Yongjin
Lee
a and
Tao
Li
*a
aSchool of Physical Science and Technology, ShanghaiTech University, Shanghai 201210, China. E-mail: litao1@shanghaitech.edu.cn
bDepartment of Materials Science, University of California, Berkeley, California 94720, USA
cDepartment of Chemical and Biomolecular Engineering, North Carolina State University, Raleigh, North Carolina 27695, USA
dSchool of Chemistry and Chemical Engineering, Queen's University Belfast, Belfast, BT9 5AG, UK
First published on 15th April 2020
Abstract
Membrane technologies hold great potential for industrial gas separation. Nevertheless, plasticization, a common phenomenon that is responsible for the loss of gas pair selectivity and the decrease of membrane lifespan, is one of the top challenges withholding the deployment of advanced membrane materials in realistic applications. Here, we report a highly generalizable approach, that utilizes PgC5Cu, a copper metal–organic nanocapsule (MONC) containing 24 open metal sites (OMSs) as a multi-dentate node to coordinatively crosslink polymers. By adding merely 1–3 wt% of PgC5Cu, a wide range of carbonyl group-containing polymers can be effectively crosslinked. Through rigorous dissolution tests, molecular dynamic simulations, and in situ FT-IR spectroscopy, we qualitatively and quantitatively unveiled the coordinative binding nature at the polymer–MONC interface. As a result, we produced a series of composite membranes showing near complete plasticization resistance to CO2, C2H4, and C2H6 under high pressure with no loss of mechanical and gas transport properties.
Introduction
Membrane separation has been widely considered as a promising alternative to current separation technologies due to its low energy consumption, small footprint, mechanical simplicity and low cost.1,2 To date, many high-performance polymeric materials such as polysulfone (PSF), polycarbonate (PC), cellulose acetate (CA), polyimide (PI) and polymer of intrinsic microporosity (PIM) have been extensively developed to meet the industrial demands from natural gas sweetening and air separation to hydrogen recovery, etc.3–5 Nevertheless, compared to the overall gas separation market, membrane-based separation only accounts for a small fraction. Taking CO2/natural gas separation as an example, only about 10% of the separation was accomplished using membrane technology.6,7 One of the primary reasons is because the gas streams in refineries and petrochemical plants often contain high concentrations of plasticizing vapors including C2 to C6 hydrocarbons and BTEX aromatics (benzene, toluene, ethylbenzene and xylenes).6,8 These vapors tend to condense and dissolve in polymeric matrices under high pressure resulting in considerable swelling of the polymers. The swelling results in increasing polymer chain mobility and free volume. Consequently, the membrane loses its initial packing structure along with its gas pair selectivity and membrane lifespan. This phenomenon is commonly referred to as plasticization.9–11 A membrane separation material market research report recently published by MarketsandMarkets™ specifically pointed out that “stringent regulations on the plasticization of polymeric membranes is a major restraint for market growth”.12 Therefore, developing plasticization resistant membranes is of paramount importance for the expended deployment of new polymeric membrane materials in industry.To address this challenge, the most common practice is to crosslink the polymer network using covalent bonds. Specific methods include thermal cross-linking,13,14 UV cross-linking,15,16 esterification,17 azide-based cross-linking18 decarboxylation-induced cross-linking19,20etc. These methods often require specific chemical modification on polymers using crosslinkable functional moieties. However, the introduction of small molecular fragments will reduce the free volume of the matrix polymers (e.g. PIMs and PIs) that comprise rigid backbones, thereby negatively impacting membrane permeability.18,21–24 The thermally rearranged (TR) polymer is a special case that increases polymer chain rigidity through intramolecular ring closure reactions.25 The resultant membranes exhibit a strong anti-plasticization capability. But, mechanical stability, especially ductility, is often sacrificed.5,25–28 Incorporating inorganic fillers into polymeric matrices to form hybrid membranes is a promising alternative to enhance membrane plasticization resistance.7,21,22 For instance, Long et al. reported a series of mixed matrix membranes (MMMs) containing ultra-small MOF nanocrystals with open metal sites (OMSs) that exhibited strong solvent/plasticization resistant properties and record breaking performance for C2 separation.29 The high density OMSs on the surface of MOF-74 nanocrystallites provide strong interaction with the polymer matrix through coordinative crosslinking thereby effectively suppressing plasticization in CO2, C2H4 and C2H6. However, due to the limited external surface area of these filler materials (zeolites, MOFs, silica etc.), high loading (>20%) is typically required to achieve satisfactory plasticization resistant capability.29–34 Consequently, not only does filler dispersibility become a major concern during membrane processing but the mechanical properties of the membranes are also often severely compromised.35–38 Moreover, from a fundamental point of view, the introduction of a porous filler such as MOFs brings numerous new variables (particle size distribution, defects, grain boundaries, dispersibility, pore orientation, batch-to-batch variation etc.) into consideration further complicating the rational design of advanced composite membranes.
Metal–organic nanocapsules (MONCs), also known as metal–organic polyhedra (MOPs), are a class of discrete yet precise supramolecular assemblies of metal ions and organic ligands.39–44 They feature ultra-small particle size (typically <5 nm) and often exhibit multiple OMSs for coordination. They are soluble and dispersible in a wide range of solvents and matrices. Meanwhile, their molecularly discrete nature eliminates complications originating from particle size distribution, defects, and batch-to-batch variation. These features render them ideal additives for coordinative crosslinking of polymers.
Here, we demonstrate a highly generalizable interfacial coordinative crosslinking (ICC) approach that utilizes a copper MONC, PgC5Cu (PgC5, C-pentylpyrogallol[4]arene) containing multiple OMSs as a multi-dentate crosslinking node to achieve ICC of polymers thereby enhancing the plasticization resistance of their respective membranes. PgC5Cu can be easily dispersed in a wide range of polymeric matrices without agglomeration. The ultra-high external surface area of PgC5Cu allows a coordinative crosslinking network (CCN) to be formed with many polymers including PC, polylactic acid (PLA), CA, polymethyl methacrylate (PMMA), PSF, PIs, and even PIM-1 etc. The CCN can be established with mild heating even with a MONC loading as low as 1 wt%. The crosslinked composite membranes show solvent resistant properties and reduced polymer chain mobility. Most importantly, at such low additive loading, the mechanical properties of the matrix polymer are fully preserved after crosslinking. As a result of reduced chain mobility, the composite membranes show near complete plasticization resistance for C2H6, C2H4 and CO2 under high pressure making high pressure C2 separation possible.
Results and discussion
PgCnCu (PgCn, C-alkylpyrogallol[4]arenes; n represents the length of the associated alkyl tail), an octahedron-shaped MONC assembled from 6 polyphenol ligand molecules, PgCn, with 24 Cu2+, was first developed by Atwood's group (Scheme 1).45–48 Interestingly, all 24 Cu sites are coordinated by four planar phenol groups leaving 24 exposed Cu OMSs available for coordinative binding. Such geometry maximizes the utilization of OMSs for crosslinking and the possibility of polymer–MONC interaction at their interface. The dangling alkyl chains, on the other hand, play a key role in governing the solubility of PgCnCu in various media. Following a reported method,47 PgCnCu (n = 3, 5, 7 and 9) was synthesized as single crystals. Matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) spectra were collected to confirm the formation of these assemblies (Fig. S2†). Simple drop-casting of PgC5Cu onto a TEM grid from a diluted dichloromethane (DCM) solution affords an even distribution of capsules with an estimate size of 2 nm as shown in the scanning transmission electron microscopy (STEM) image (Fig. 1a). Despite the presence of an ∼0.4 nm cavity within each capsule, it is, however, inaccessible by any gas molecules because the opening toward that cavity is only 1.9 Å in diameter (Fig. S3†). This is confirmed by an N2 adsorption experiment as PgC5Cu exhibits a typical type II isotherm suggesting its non-porous nature (Fig. S4†). Thermogravimetric analyses (TGA) show that PgC5Cu is thermally stable up to ∼250 °C (Fig. S5†).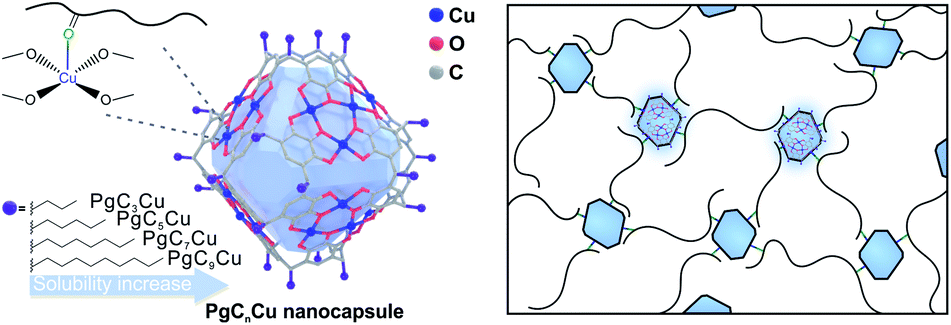 | ||
| Scheme 1 Structure of PgCnCu nanocapsules. | ||
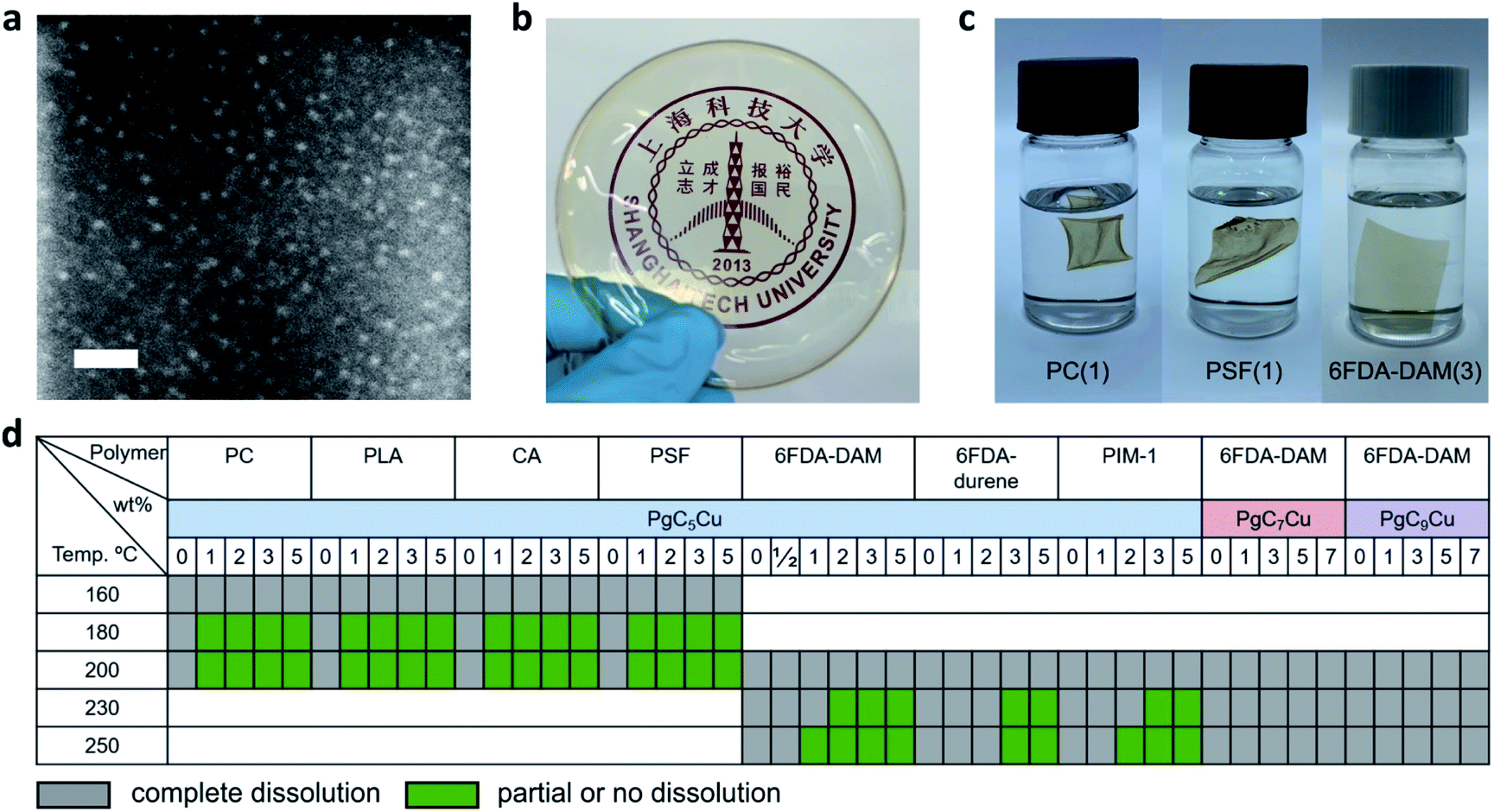 | ||
| Fig. 1 Qualitative dissolution analyses for various PgCnCu–polymer composite membranes. (a) STEM image of PgC5Cu, scale bar 10 nm. (b) A photograph of a 6FDA-DAM(2) membrane. (c) Photographs of PC(1), PSF(1) and 6FDA-DAM(3) immersed in DCM. (d) Dissolution table of various neat polymers and their composite membranes with PgCnCu. | ||
After mixing MONCs into polymer solutions to form clear mixture solutions, a solution casting technique was employed to prepare dense composite membranes containing PgCnCu (n = 5, 7, 9) and various polymers. PgC3Cu was not employed for membrane making due to its limited solubility in various organic solvents (Table S1†). For ease of discussion, composite membranes containing PgC5Cu were denoted as polymer(X) where X indicates the weight percentage of PgC5Cu within the matrices. Fig. 1b shows a highly transparent polyimide membrane, 6FDA-DAM(2), suggesting that the agglomeration of PgC5Cu is negligible. To unveil the dispersibility of PgC5Cu at a microscopic level, the 6FDA-DAM(2) membrane was sliced into 20 nm sections using an ultramicrotome. The energy dispersion X-ray spectroscopy (EDS) mapping displays a homogeneous distribution of Cu throughout the 6FDA-DAM matrix (Fig. S6†). Meanwhile, after surveying multiple locations using STEM, we only found occasional agglomerates with sizes < 3.5 nm (Fig. S6†) which firmly proves that nearly all PgC5Cu capsules were individually dispersed within the polymer matrix. To obtain crosslinked membranes, the coordination bond between the MONC and polymer needs to be established at the interface. A mild thermal treatment (180–250 °C) under vacuum is sufficient to remove the coordinated solvent molecules (e.g. H2O) on the OMSs, therefore triggering ICC.
Membrane dissolution tests were carried out to qualitatively evaluate the effect of crosslinking. It is apparent that after introducing 1–3 wt% PgC5Cu, PC(1) (polycarbonate), PSF(1) (polysulfone) and 6FDA-DAM(3) were no longer able to completely dissolve in DCM (Fig. 1c). To gain a deeper understanding of the ICC phenomenon, we systematically investigated the effect of activation temperature, MONC loading and alkyl chain length on the solubility of various polymers. Fig. 1d shows that the addition of 1–5 wt% of PgC5Cu can effectively prevent a range of polymers from dissolving in a good solvent which demonstrates the generalizability of this approach. Among the listed polymers, PC, PLA, CA and PSF require a relatively low activation temperature (180–200 °C) to achieve crosslinking whereas 6FDA-DAM, 6FDA-durene and PIM-1 require a slightly higher activation temperature (210–250 °C). This is likely because polyimides and PIM-1 have a much higher glass transition temperature (Tg) therefore requiring a higher temperature to achieve sufficient polymer chain mobility for coordinative binding. Interestingly, after replacing PgC5Cu with longer alkyl chain analogues, PgC7Cu and PgC9Cu, the CCN can no longer form below 250 °C. This phenomenon can be attributed to the steric hindrance of longer alkyl groups that blocks the polymer chain from accessing OMSs. More in depth analysis regarding this phenomenon will be discussed later in the simulation section (vide infra).
Next, we sought to quantitatively evaluate the swelling of PSF–PgC5Cu composite membranes in chloroform vapor. The time dependent swelling ratio profiles show that with increasing MONC loading from 0 to 2 wt%, the swelling ratio continuously decreased (Fig. 2a). For PSF(2), the swelling ratio peaked after ∼600 s of exposure to chloroform vapor and plateaued afterwards at around 200% suggesting that maximum swelling had reached. We further compared the swelling ratio of neat PMMA, 6FDA-durene, and 6FDA-DAM versus their corresponding composite membranes with PgC5Cu after ∼900–1200 s of exposure to chloroform vapor. In all cases, the films containing PgC5Cu showed a lower swelling ratio compared to their neat polymer counterparts (Fig. 2b and S11†). To compare the swelling behavior of the polymer–PgC5Cu composite before and after thermal activation, tetrahydrofuran (THF) vapor adsorption experiments were performed on a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 6FDA-DAM and PgC5Cu (Fig. 2c). The non-activated sample manifests a continuous uptake curve throughout the whole pressure range. Interestingly, the activated sample exhibits a distinct two-step uptake behavior. In a low partial pressure region (<0.4 P/Po), the THF uptake was considerably lower than that of the non-activated sample. This is because thermal activation induced crosslinking suppresses polymer swelling. However, after a steep rise in THF uptake at around 0.4 P/Po, the isotherm followed the same pathway as that of the non-activated sample. This is likely due to THF competitively binding with the OMSs, out competing the polymer–OMS interaction under higher vapor pressure.
:1 mixture of 6FDA-DAM and PgC5Cu (Fig. 2c). The non-activated sample manifests a continuous uptake curve throughout the whole pressure range. Interestingly, the activated sample exhibits a distinct two-step uptake behavior. In a low partial pressure region (<0.4 P/Po), the THF uptake was considerably lower than that of the non-activated sample. This is because thermal activation induced crosslinking suppresses polymer swelling. However, after a steep rise in THF uptake at around 0.4 P/Po, the isotherm followed the same pathway as that of the non-activated sample. This is likely due to THF competitively binding with the OMSs, out competing the polymer–OMS interaction under higher vapor pressure.
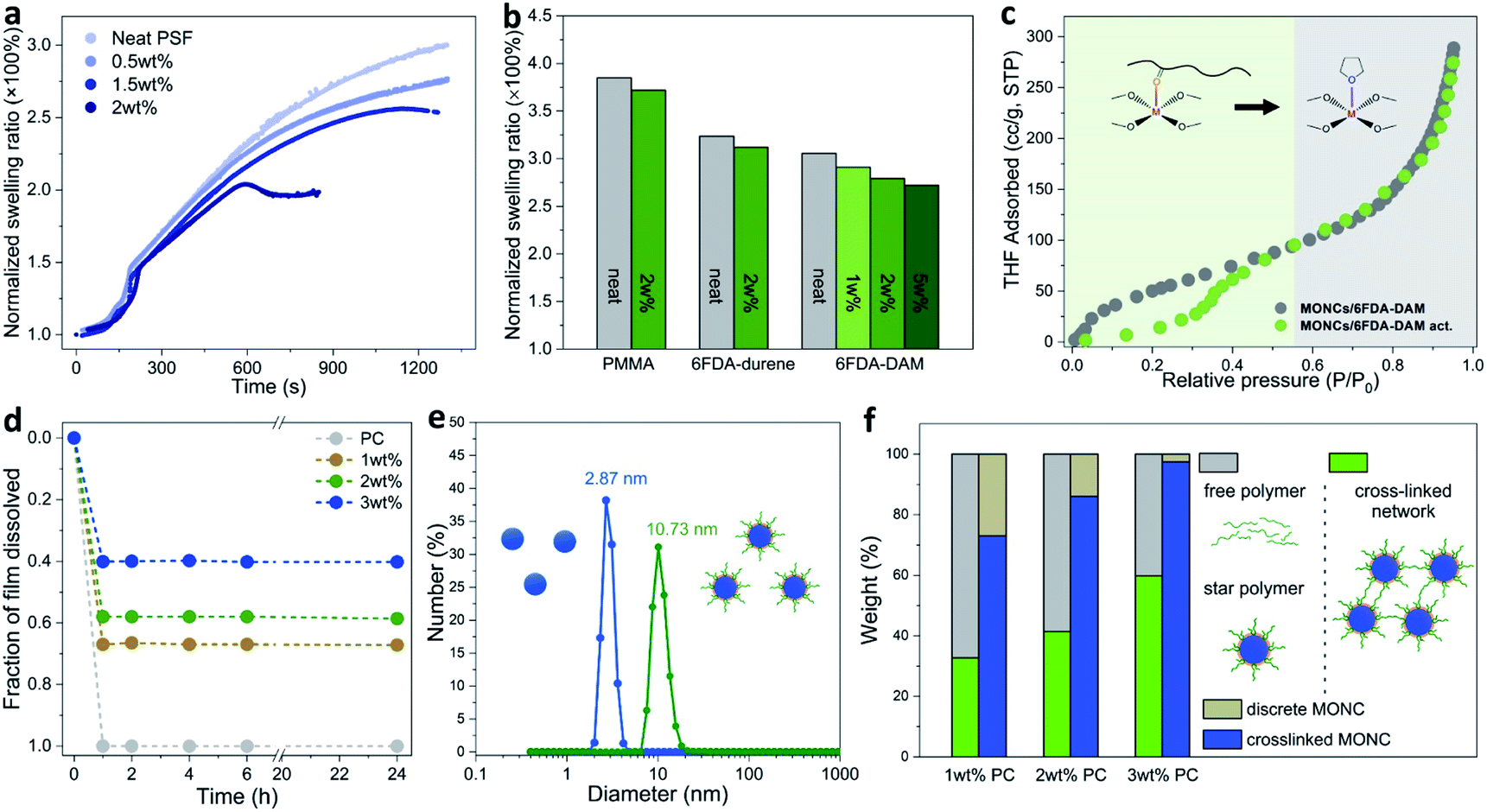 | ||
| Fig. 2 Quantitative analyses of swelling kinetics, THF vapor adsorption and dissolution experiments for PgCnCu–polymer composite membranes. (a) Time dependent swelling ratio profiles of PgC5Cu–PSF composite membranes in chloroform vapor. (b) Swelling ratio comparisons between neat polymers and their composite membranes. The data were collected after 935, 1170 and 1170 s of exposure to chloroform vapor for PMMA, 6FDA-durene and 6FDA-DAM, respectively. (c) THF vapor adsorption isotherms for a 2:1 mixture of 6FDA-DAM and PgC5Cu before (grey) and after (green) thermal activation (230 °C for 12 hours under vacuum). (d) Time-dependent dissolution profiles for neat PC and PgC5Cu–PC composite membranes. (e) hydrodynamic particle size distribution of a DCM solution of neat PgC5Cu (blue) and a DCM solution of dissolved PC(0.5) films (green). (f) Solubility of PC(x) membranes in DCM and quantitative analysis of a crosslinked MONC versus a discrete MONC. Green and grey columns represent the percentage of the insoluble and soluble part of the membranes, respectively. Blue and yellow columns represent the percentage of crosslinked versus discrete PgC5Cu, respectively. | ||
Quantitative membrane dissolution tests were performed to dissect different forms of PgC5Cu present in PC composite membranes. Remarkably, the time dependent dissolution profiles show that as neat PC quickly dissolved in DCM, the dissolution process of PC(1), PC(2), and PC(3) ceased after 1 h as no further weight loss was observed until 24 hours. The weight of the residual membranes was 33, 41 and 60% for PC(1), PC(2), and PC(3), respectively (Fig. 2d). It is thus reasonable to speculate that the insoluble part of the membrane is a CCN in which each polymer chain was bridged by multiple MONCs. On the other hand, the soluble part was composed of a free polymer and a PC star polymer (PC chains coordinatively grafted on a discrete MONC). To verify the presence of a star polymer, the amount of PgC5Cu in the supernatant and residual membrane was independently quantified by UV-vis spectroscopy (detailed quantification procedures can be found in the characterization section in the ESI†). As Fig. 2f shows, at 1 and 2 wt% loading, 73 and 86% PgC5Cu formed a crosslinked network whereas the rest remained discrete. When the loading increased to 3 wt%, nearly all PgC5Cu (97%) participated in crosslinking as a result of closer inter-capsule distance in the matrix. Furthermore, dynamic light scattering (DLS) analysis of a dissolved solution of PC(0.5) in DCM gives a unimodal hydrodynamic particle size of 10.73 nm, significantly larger than the size of neat PgC5Cu (2.87 nm) (Fig. 2e). As a control experiment, a physical mixture of PgC5Cu and PC in DCM shows an average particle size of ∼3.3 nm (Fig. S12†), suggesting that no polymer–MONC coordination bond was established in solution. Moreover, when PC(1) was dissolved in dimethylformamide (DMF) at an elevated temperature, the competitive coordination of DMF with the OMSs led to the disassembly of the star polymers. Consequently, the particle size measured by DLS dropped back down to 2.3 nm (Fig. S13†). This evidence once again confirmed the existence of coordinative PC star polymers in composite membranes.
Molecular dynamic (MD) simulations were performed to investigate the interaction at the PgCnCu–polymer interface. Here, PSF was selected as a model polymer because of its structural simplicity. Fig. 3b shows the number of atomic pairs between the Cu OMSs from PgCnCu (n = 3, 5, 7, and 9) and the polymer within a 4 Å distance per capsule at an equilibrium state. It is apparent that the most significant interaction comes from the sulfonyl oxygen (O1) as PgC3Cu and PgC5Cu contributed 3.6 and 3.2 Cu–O1 interaction sites per capsule, respectively. When the alkyl chains extended to C7 and C9, the number of Cu–O1 interaction sites drastically decreased to 0.4 and 0.2 per capsule due to steric hindrance. This result perfectly explained the inability of PgC7Cu and PgC9Cu to crosslink 6FDA-DAM. Apart from the sulfonyl oxygen, C1 and H1 on the adjacent phenyl ring can also be found within close proximity to the Cu OMSs. However, they are not likely to profoundly contribute to the interfacial interaction due to the lack of free electron pairs. We also investigated the time dependent evolution of Cu–O1 coordination bond formation per PgCnCu capsule (Fig. 3c). As expected, PgC3Cu and PgC5Cu formed 5 and 4 coordination bonds with O1 after 2.2 and 1.9 ns, respectively, and they reached an equilibrium state thereafter. In contrast, only occasional Cu–O1 bond formation was observed on PgC7Cu and PgC9Cu over the course of 10 ns of simulation time. This is again consistent with the membrane solubility results.
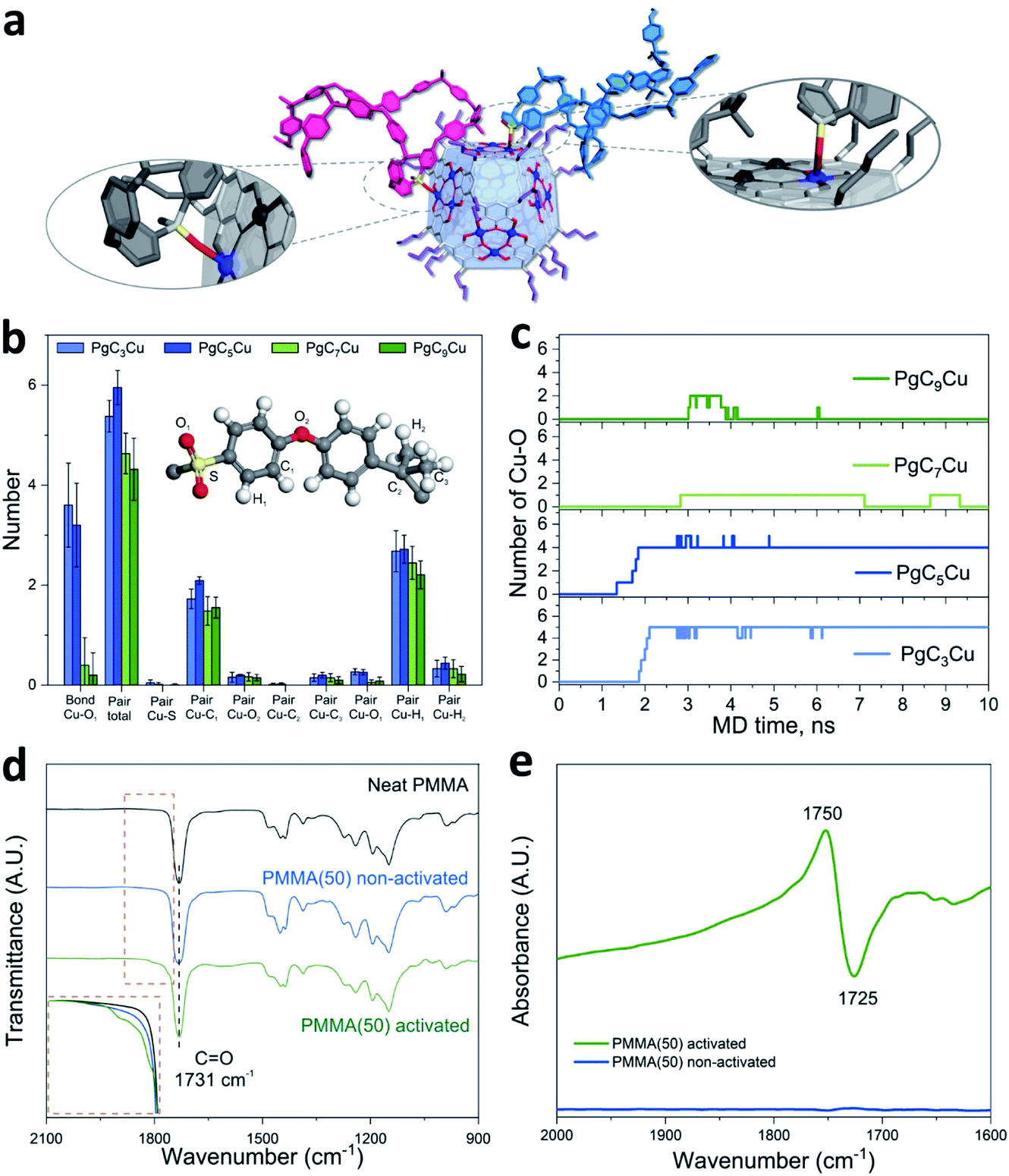 | ||
Fig. 3 Molecular level characterization experiments of the interaction at the PgCnCu/polymer interface. (a–c) MD simulation results. (a) A snapshot of the structure of a PgC5Cu–PSF composite system at an equilibrium state. The Cu–O coordination bonds are highlighted. (b) Number of atomic pairs between Cu OMSs on PgCnCu (n = 3, 5, 7, and 9) and various atoms on PSF within a 4 Å distance per capsule at an equilibrium state. (c) Time dependent evolution of the number of Cu–O1 coordination bonds (bond length < 2.5 Å) formed per capsule over the course of 10 ns. (d) Transmition FTIR spectra for neat PMMA (black), PMMA(50) non-activated (blue) and PMMA(50) activated (green). The inset shows the zoom-in comparison of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching region. (e) Stacked plot of the FTIR spectra of PMMA(50) before (blue) and after (green) activation. O stretching region. (e) Stacked plot of the FTIR spectra of PMMA(50) before (blue) and after (green) activation. | ||
To experimentally monitor the Cu–O bonding at the interface, Fourier transform infrared (FTIR) spectroscopy was performed. A 1:1 mixture of PgC5Cu–PMMA composite was chosen for this study because (1) PMMA has a relatively simple IR spectrum and (2) PMMA has a soft backbone that will maximize its interaction with the MONC at the interface. The carbonyl group in PMMA shows a signature CO stretching peak at 1731 cm−1. After mixing with PgC5Cu as a composite, no carbonyl peak shift was observed. Interestingly, after thermal activation, several shoulder peaks appeared in the range of 1755 to 1821 cm−1 suggesting that the chemical environment of part of the carbonyl groups has changed (Fig. 3d). To better understand the origin of these new peaks, the PgC5Cu–PMMA composite was activated in an in situ FTIR sample cell. By taking the spectra of the non-activated samples as background, we observed a negative peak at 1725 cm−1 and a symmetrical positive peak at 1750 cm−1 (Fig. 3e). By comparison, the position of the positive peak matches well with the newly observed shoulder peaks in the activated sample while the negative peak matches with the native carbonyl stretching frequency. This observation strongly indicates that the 1760 cm−1 shoulder peak is a result of Cu–O coordination.
Encouraged by previous results, we were keen to investigate the influence of ICC on the mechanical properties of the membranes. This is a critical aspect that will profoundly impact the membrane processability which is of paramount importance to industrial application. An ideal crosslinking method should not sacrifice the mechanical properties of the membrane. In our case, the elongation at break values of 6FDA-DAM(1), 6FDA-DAM(2), and 6FDA-DAM(3) are 4.23 ± 1.21, 3.92 ± 0.45, and 3.87 ± 1.08%. These values are identical to that of a neat 6FDA-DAM membrane (4.37 ± 1.12%). Similarly, the break strength values of 6FDA-DAM(1), 6FDA-DAM(2), and 6FDA-DAM(3) are 58.54 ± 3.62, 58.13 ± 1.77 and 56.86 ± 1.91 MPa respectively again very close to their neat polymer counterparts (56.36 ± 3.13 MPa). These results suggest that at low MONC loading, the ICC process has no negative impact on the mechanical properties of the matrix polymer (Fig. 4a and b and S16†).
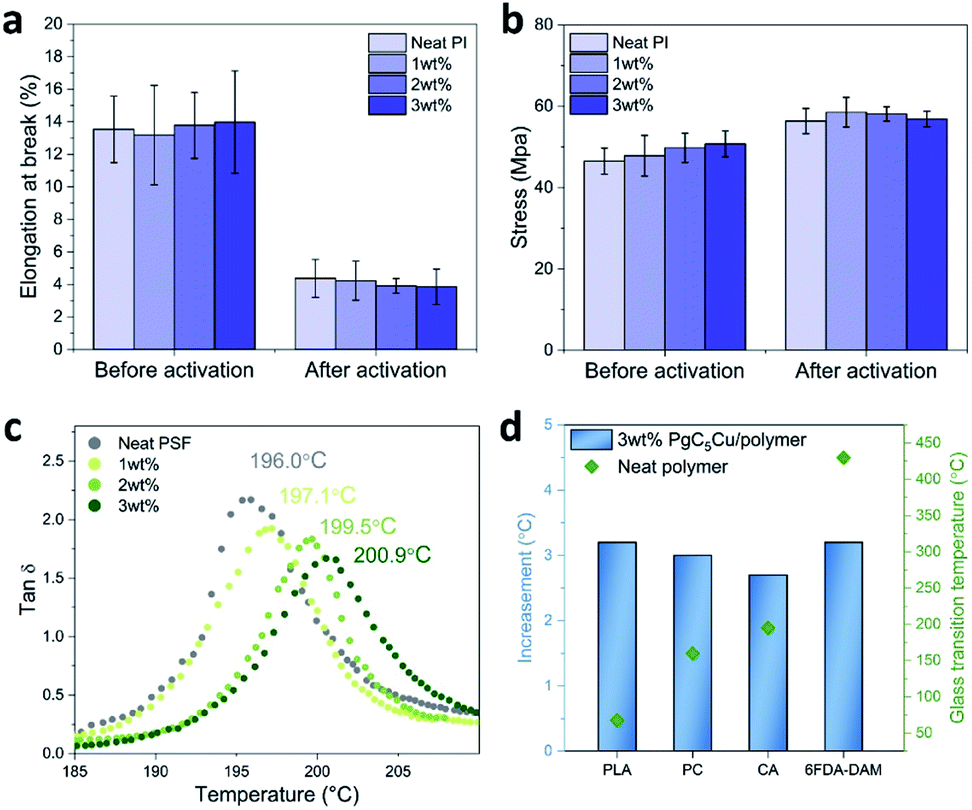 | ||
| Fig. 4 Mechanical properties and glass transition temperature (Tg). (a and b) Elongation at break (a) and break strength (b) of neat 6FDA-DAM and its composite membranes before and after activation. The error bars were obtained from three consecutive measurements of three individual samples. (c) DMA plots showing a tanδ response to changing temperature for neat PSF, PSF(1), PSF(2) and PSF(3) with Tg values labeled aside. (d) Tg of various polymers and their composites with 3 wt% PgC5Cu. | ||
Another aspect that ICC led to is reduced polymer chain mobility. To gain more insight, dynamic mechanical analysis (DMA) was performed to acquire the Tg of various PgC5Cu–polymer composites. Fig. 4c shows the tanδ response to the changing temperature for neat PSF, PSF(1), PSF(2) and PSF(3). With increasing MONC loading, the Tg also increased almost linearly from 196.0 °C for neat PSF to 197.1, 199.5 and 200.9 °C for PSF(1), PSF(2) and PSF(3), respectively (Fig. S17†). Similarly, PLA, PC, CA, and 6FDA-DAM all exhibited a 2.7–3.2 °C increase of Tg with 3 wt% loading of PgC5Cu (Fig. 4d and S18†). These results firmly established that ICC using PgC5Cu is a general way to reduce the chain mobility of polymers. To this end, we are confident to proceed to investigate the plasticization suppression effect of PgC5Cu–polymer composites.
High-pressure single gas permeation experiments were carried out to evaluate the plasticization resistance of 6FDA-DAM(X) composite membranes using C2H4, C2H6 and CO2. As Fig. 5a–c show, the neat 6FDA-DAM membrane exhibits a plasticization pressure of 15 bar for C2H6, 25 bar for C2H4, and 15 bar for CO2. By incorporating merely 0.5 wt% PgC5Cu, the plasticization pressure was significantly elevated to 25, 35 and 30 bar for C2H6, C2H4 and CO2, respectively. Recalling previous results, 0.5 wt% PgC5Cu loading was not enough to form a CCN within the 6FDA-DAM matrix as the composite membrane can fully dissolve in DCM. Therefore, such a dramatic enhancement of plasticization resistance can be ascribed to the physical crosslinking between the star polymer branches and the matrix polymer. A similar phenomenon was also observed in an MOF-based MMM system where polyimide brushes were covalently grafted on the MOF surface.49 However, compared to MOF particles, MONC particles are considerably smaller thereby allowing plasticization resistance to be achieved at extremely low loading. After increasing PgC5Cu loading to 1 wt%, 2 wt% and 3 wt%, plasticization was almost completely suppressed for C2H6, C2H4 and CO2 as no apparent increase of permeability was observed even at 35, 54, and 50 bar, respectively. Besides, given that the real gases deviate significantly from ideal gas behavior at high pressure, CO2, C2H6 and C2H4 fugacity-based permeability isotherms of neat PI and their composite membranes were plotted (Fig. S21 and S24†). As expected, the increase of fugacity-based permeability at high pressure for 6FDA-DAM(1–3) is inconsequential compared to that of the neat 6FDA-DAM membrane. Such strong plasticization resistance capability can be attributed to the synergistic effect of the presence of the ICC network in the composite membranes and the star polymer induced physical crosslinking. To demonstrate the generalizability of this method, we further extended this strategy to 6FDA-durene, CA and PC. Without surprise, 6FDA-durene(3), CA(3) and PC(3) all showed complete plasticization resistance to CO2 all the way to 50 bar with the addition of only 3 wt% PgC5Cu (Fig. 5d).
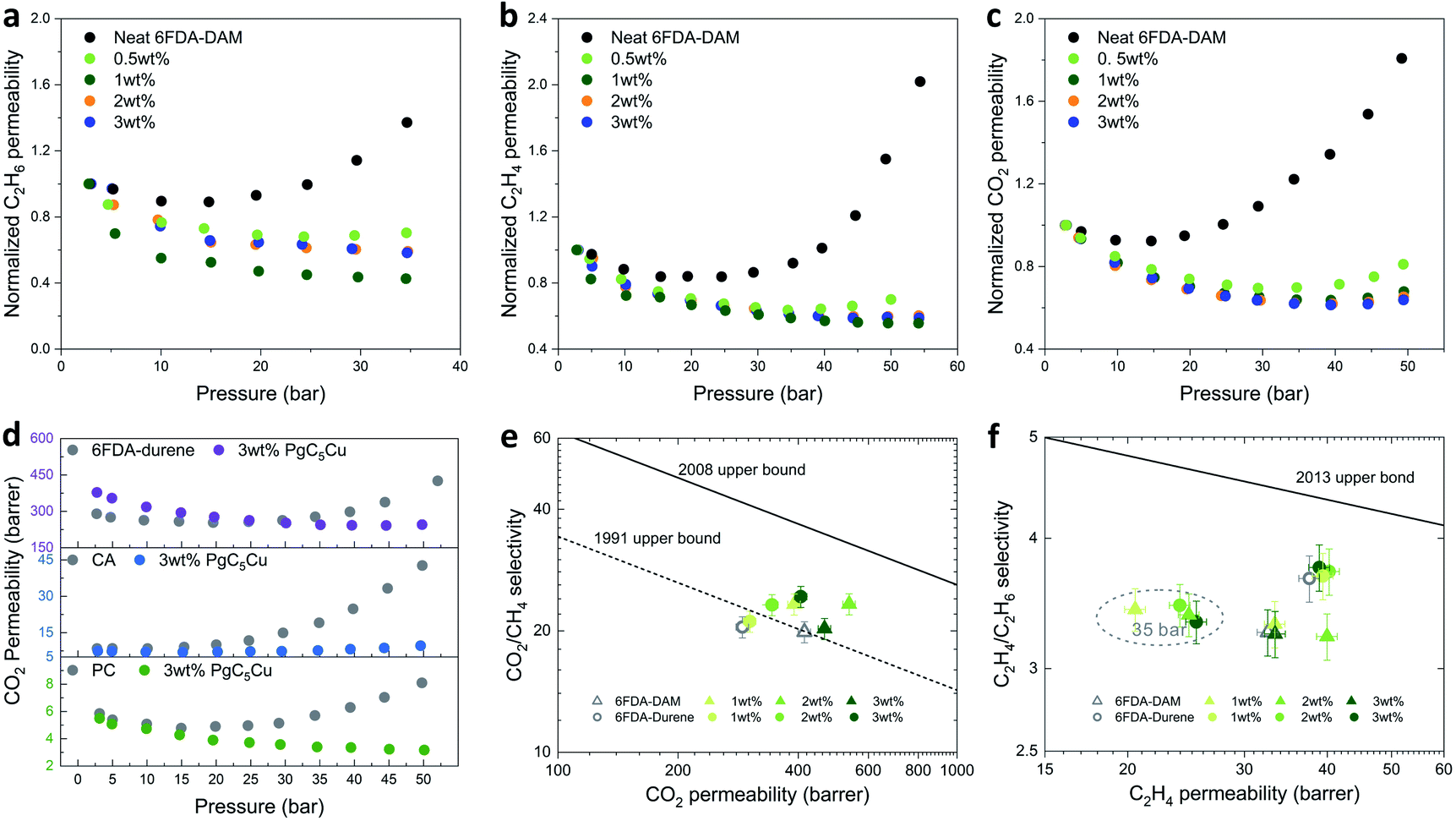 | ||
| Fig. 5 Pure gas transport properties. (a–c) Single gas permeation at 35 °C for neat 6FDA-DAM and their composite membranes. The pressure dependent permeability profiles of C2H6 (a), C2H4 (b) and CO2 (c) were normalized to the permeability values at 2.8 bar. (d) Pressure dependent CO2 permeability of neat 6FDA-durene, CA, PC membranes (grey) and their composite membranes containing 3 wt% PgC5Cu. (e) CO2 permeability versus CO2/CH4 ideal selectivity for 6FDA-DAM (triangle) and 6FDA-durene (cycle) and their composite membranes. (f) C2H4 permeability versus C2H4/C2H6 ideal selectivity for 6FDA-DAM (triangle) and 6FDA-durene (cycle) and their composite membranes. Error bars correspond to propagation of uncertainty from the measured membrane thickness, area and upstream pressure. All the unspecified data points in (e–f) were collected at 2.8 bar and 35 °C. | ||
For pure gas transport experiments, two high performance polyimides, 6FDA-DAM and 6FDA-durene, and their composite membranes with 1–3 wt% PgC5Cu were studied. The permeability and ideal selectivity of two gas pairs, CO2/CH4 and C2H4/C2H6, were investigated (Fig. 5e and f). Specifically, under 2.8 bar upstream pressure, the CO2 permeability and CO2/CH4 selectivity of 6FDA-DAM(1), 6FDA-DAM(2), and 6FDA-DAM(3) were 391, 536, 465 barrer and 23.3, 23.3, 20.2, respectively. These values are comparable with the transport properties of the neat 6FDA-DAM membrane (PCO2 = 415, SCH4/CO2 = 20.0). For 6FDA-durene, CO2 permeability increased from 290 barrer for the neat polyimide to 302, 344, and 406 barrer for 6FDA-durene(1), 6FDA-durene(2), and 6FDA-durene(3), respectively. Meanwhile, CO2/CH4 ideal selectivity also slightly increased from 20.4 for the neat polyimide to 21.1, 23.3, and 24.3 for 6FDA-durene composite membranes with increasing PgC5Cu loading. When plotted on the Robeson upper bound, it is clear that the introduction of PgC5Cu led to an appreciable increase of both permeability and selectivity, positioning some of the data points above the 1991 upper bound. Note that the PgC5Cu inner cavity does not contribute to the increase of membrane free volume; the increase of CO2 permeability can be attributed to the change of polymer packing as a result of ICC. For C2H4/C2H6 separation, neat 6FDA-DAM and 6FDA-durene membranes exhibit C2H4 permeability of 33 and 38 barrer, and a C2 selectivity of 3.3 and 3.7, respectively. After introducing PgC5Cu, both permeability and selectivity slightly increased, similar to that of CO2/CH4 separation. These results strongly suggest that ICC using low loading MONC will, to say the least, not affect the free volume of the matrix polymer thereby making the gas transport properties after crosslinking highly predictable. Because of the strong plasticization resistant properties of the PgC5Cu–polyimide composite membranes, we were able to collect C2 separation data at 35 bar and 35 °C. As shown in Fig. 5f, four composite membranes exhibit a C2H4/C2H6 ideal selectivity of ∼3.3 and C2H4 permeability in the range of 20–25 barrer, comparable to their transport behaviors at 2.8 bar.
Lastly, the physical aging as well as the long term storage stability of the MONC crosslinked membrane was investigated. Physical aging in glassy polymers is a spontaneous and continuous evolution towards the equilibrium state of polymer chain packing driven by a dissipation of nonequilibrium free volume.50 Here, CO2 permeability and CO2/CH4 selectivity of methanol treated 6FDA-DAM and 6FDA-DAM(2) were monitored. It is known that methanol treatment of the membranes increases the likelihood of physical aging due to increased free volume.51 Over the course of 42 days, the neat 6FDA-DAM membrane exhibited continuous decrease in CO2 permeability from 461 to 257 barrer indicating the occurrence of aging. In contrast, 6FDA-DAM(2) only showed a slight decrease in CO2 permeability (from 453 to 411 barrer) (Fig. S25 and Table S5†). This proves that ICC using MONC is an effective approach to mitigate physical aging in polymeric membranes. In addition, to evaluate the storage stability of the membrane, a piece of the 6FDA-DAM(1) membrane was stored in a humid environment (R.H. ∼ 73%) for 266 days. A dissolution test suggests that 6FDA-DAM(1) retained its solvent resistance properties against DCM even after long term storage. Analysing the supernatant using DLS revealed that the discrete PgC5Cu capsules were still covered with PI branches suggesting that the coordination bonds were largely intact (Fig. S26†). This implies that MONC crosslinked membranes can be potentially used in applications involving humid gas streams.
Conclusions
In summary, we demonstrate the use of PgC5Cu nanocapsules to coordinatively crosslink a wide range of polymers through Cu–O binding therefore endowing the polymeric membrane with strong plasticization resistant capability toward C2H6, C2H4 and CO2. Due to the high external surface area and high OMS density of PgC5Cu, only 1–3 wt% loading was required. As a result, both the mechanical properties and transport properties of the polymeric membranes are fully retained. This approach may serve as a universal way to improve the plasticization resistance of polymeric membranes. Meanwhile, the molecular insights revealed at the coordination interface may shed light on the rational design of anti-plasticization membranes to meet the demand for realistic industry applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21701110) and the start-up funding from ShanghaiTech University. The authors acknowledge the support from the Analytical Instrumentation Center (# SPST-AIC10112914), SPST, ShanghaiTech University. This research used resources of the Centre for High-resolution Electron Microscopy (CħEM) of SPST at ShanghaiTech University under Grant No. EM02161943. The computing resources were made available through the high-performance computing platform of ShanghaiTech University and the Shanghai Supercomputer Center.Notes and references
- R. W. Baker and B. T. Low, Macromolecules, 2014, 47, 6999–7013 CrossRef CAS.
- H. B. Park, J. Kamcev, L. M. Robeson, M. Elimelech and B. D. Freeman, Science, 2017, 356, eaab0530 CrossRef PubMed.
- R. W. Baker and K. Lokhandwala, Ind. Eng. Chem. Res., 2008, 47, 2109–2121 CrossRef CAS.
- N. W. Ockwig and T. M. Nenoff, Chem. Rev., 2007, 107, 4078–4110 CrossRef CAS PubMed.
- D. F. Sanders, Z. P. Smith, R. Guo, L. M. Robeson, J. E. McGrath, D. R. Paul and B. D. Freeman, Polymer, 2013, 54, 4729–4761 CrossRef CAS.
- M. Galizia, W. S. Chi, Z. P. Smith, T. C. Merkel, R. W. Baker and B. D. Freeman, Macromolecules, 2017, 50, 7809–7843 CrossRef CAS.
- M. Zhang, L. Deng, D. Xiang, B. Cao, S. Hosseini and P. Li, Processes, 2019, 7, 51 CrossRef CAS.
- R. W. Baker, Ind. Eng. Chem. Res., 2002, 41, 1393–1411 CrossRef CAS.
- M. Wessling, S. Schoeman, T. van der Boomgaard and C. A. Smolders, Gas Sep. Purif., 1991, 5, 222–228 CrossRef CAS.
- M. Wessling, I. Huisman, T. v. d. Boomgaard and C. A. Smolders, J. Polym. Sci., Part B: Polym. Phys., 1995, 33, 1371–1384 CrossRef CAS.
- R. Swaidan, B. Ghanem, E. Litwiller and I. Pinnau, Macromolecules, 2015, 48, 6553–6561 CrossRef CAS.
- MarketsandMarkets, Gas Separation Membranes Market by Material Type (Polyimide & Polyaramide, Polysulfone, and Cellulose Acetate), Application (Nitrogen Generation & Oxygen Enrichment, Carbon Dioxide Removal, and Hydrogen Recovery), and Region - Global Forecast to 2024, 2019, report code CH 5549 Search PubMed.
- F. Y. Li, Y. Xiao, T.-S. Chung and S. Kawi, Macromolecules, 2012, 45, 1427–1437 CrossRef CAS.
- Q. Song, S. Cao, R. H. Pritchard, B. Ghalei, S. A. Al-Muhtaseb, E. M. Terentjev, A. K. Cheetham and E. Sivaniah, Nat. Commun., 2014, 5, 4813 CrossRef PubMed.
- H. Kita, T. Inada, K. Tanaka and K.-i. Okamoto, J. Membr. Sci., 1994, 87, 139–147 CrossRef CAS.
- H. Lin, E. Van Wagner, B. D. Freeman, L. G. Toy and R. P. Gupta, Science, 2006, 311, 639–642 CrossRef CAS PubMed.
- G. Liu, N. Li, S. J. Miller, D. Kim, S. Yi, Y. Labreche and W. J. Koros, Angew. Chem., Int. Ed., 2016, 55, 13754–13758 CrossRef CAS PubMed.
- N. Du, M. M. Cin, I. Pinnau, A. Nicalek, G. P. Robertson and M. D. Guiver, Macromol. Rapid Commun., 2011, 32, 631–636 CrossRef CAS PubMed.
- W. Qiu, C.-C. Chen, L. Xu, L. Cui, D. R. Paul and W. J. Koros, Macromolecules, 2011, 44, 6046–6056 CrossRef CAS.
- N. Du, M. M. Dal-Cin, G. P. Robertson and M. D. Guiver, Macromolecules, 2012, 45, 5134–5139 CrossRef CAS.
- Z. X. Low, P. M. Budd, N. B. McKeown and D. A. Patterson, Chem. Rev., 2018, 118, 5871–5911 CrossRef CAS PubMed.
- C. Z. Liang, T. S. Chung and J. Y. Lai, Prog. Polym. Sci., 2019, 97, 101141 CrossRef CAS.
- S. D. Kelman, S. Matteucci, C. W. Bielawski and B. D. Freeman, Polymer, 2007, 48, 6881–6892 CrossRef CAS.
- W. F. Yong, F. Y. Li, T. S. Chung and Y. W. Tong, J. Mater. Chem. A, 2013, 1, 13914–13925 RSC.
- H. B. Park, C. H. Jung, Y. M. Lee, A. J. Hill, S. J. Pas, S. T. Mudie, E. Van Wagner, B. D. Freeman and D. J. Cookson, Science, 2007, 318, 254–258 CrossRef CAS PubMed.
- D. F. Sanders, Z. P. Smith, C. P. Ribeiro, R. Guo, J. E. McGrath, D. R. Paul and B. D. Freeman, J. Membr. Sci., 2012, 409–410, 232–241 CrossRef CAS.
- Q. Liu, D. R. Paul and B. D. Freeman, Polymer, 2016, 82, 378–391 CrossRef CAS.
- L. Ye, L. Wang, X. Jie, C. Yu, G. Kang and Y. Cao, J. Membr. Sci., 2019, 573, 21–35 CrossRef CAS.
- J. E. Bachman, Z. P. Smith, T. Li, T. Xu and J. R. Long, Nat. Mater., 2016, 15, 845–849 CrossRef CAS PubMed.
- R. T. Adams, J. S. Lee, T.-H. Bae, J. K. Ward, J. R. Johnson, C. W. Jones, S. Nair and W. J. Koros, J. Membr. Sci., 2011, 367, 197–203 CrossRef CAS.
- S. Shahid and K. Nijmeijer, J. Membr. Sci., 2014, 470, 166–177 CrossRef CAS.
- J. E. Bachman and J. R. Long, Energy Environ. Sci., 2016, 9, 2031–2036 RSC.
- W. S. Chi, B. J. Sundell, K. Zhang, D. J. Harrigan, S. C. Hayden and Z. P. Smith, ChemSusChem, 2019, 12, 2355–2360 CAS.
- T. C. Merkel, Z. He, I. Pinnau, B. D. Freeman, P. Meakin and A. J. Hill, Macromolecules, 2003, 36, 6844–6855 CrossRef CAS.
- M. J. C. Ordoñez, K. J. Balkus, J. P. Ferraris and I. H. Musselman, J. Membr. Sci., 2010, 361, 28–37 CrossRef.
- E. M. Mahdi and J.-C. Tan, J. Membr. Sci., 2016, 498, 276–290 CrossRef CAS.
- S. J. D. Smith, C. H. Lau, J. I. Mardel, M. Kitchin, K. Konstas, B. P. Ladewig and M. R. Hill, J. Mater. Chem. A, 2016, 4, 10627–10634 RSC.
- Z. P. Smith, J. E. Bachman, T. Li, B. Gludovatz, V. A. Kusuma, T. Xu, D. P. Hopkinson, R. O. Ritchie and J. R. Long, Chem. Mater., 2018, 30, 1484–1495 CrossRef CAS.
- L. J. Barbour, G. W. Orr and J. L. Atwood, Nature, 1998, 393, 671–673 CrossRef CAS.
- B. Olenyuk, J. A. Whiteford, A. Fechtenkotter and P. J. Stang, Nature, 1999, 398, 796–799 CrossRef CAS PubMed.
- N. Takeda, K. Umemoto, K. Yamaguchi and M. Fujita, Nature, 1999, 398, 794–796 CrossRef CAS.
- M. Eddaoudi, J. Kim, J. B. Wachter, H. K. Chae, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2001, 123, 4368–4369 CrossRef CAS PubMed.
- D. J. Tranchemontagne, Z. Ni, M. O'Keeffe and O. M. Yaghi, Angew. Chem., Int. Ed., 2008, 47, 5136–5147 CrossRef CAS PubMed.
- J. R. Li and H. C. Zhou, Nat. Chem., 2010, 2, 893–898 CrossRef CAS PubMed.
- R. M. McKinlay, G. W. Cave and J. L. Atwood, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 5944–5948 CrossRef CAS PubMed.
- S. J. Dalgarno, N. P. Power and J. L. Atwood, Coord. Chem. Rev., 2008, 252, 825–841 CrossRef CAS.
- S. J. Dalgarno, N. P. Power, J. E. Warren and J. L. Atwood, Chem. Commun., 2008, 1539–1541 RSC.
- H. Kumari, S. R. Kline, N. J. Schuster and J. L. Atwood, Chem. Commun., 2011, 47, 12298–12300 RSC.
- H. Wang, S. He, X. Qin, C. Li and T. Li, J. Am. Chem. Soc., 2018, 140, 17203–17210 CrossRef CAS PubMed.
- R. Swaidan, B. Ghanem, E. Litwiller and I. Pinnau, Macromolecules, 2015, 48, 6553–6561 CrossRef CAS.
- M. Carta, P. Bernardo, G. Clarizia, J. C. Jansen and N. B. McKeown, Macromolecules, 2014, 47, 8320–8327 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc01498b |
| This journal is © The Royal Society of Chemistry 2020 |