
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExpanded toolbox for directing the biosynthesis of macrocyclic peptides in bacterial cells†
Jacob A.
Iannuzzelli
 and
Rudi
Fasan
*
and
Rudi
Fasan
*
Department of Chemistry, University of Rochester, Rochester, New York 14627, USA. E-mail: rfasan@ur.rochester.edu
First published on 27th May 2020
Abstract
The macrocyclization of recombinant polypeptides by means of genetically encodable non-canonical amino acids has recently provided an attractive strategy for the screening and discovery of macrocyclic peptide inhibitors of protein–protein interactions. Here, we report the development of an expanded suite of electrophilic unnatural amino acids (eUAAs) useful for directing the biosynthesis of genetically encoded thioether-bridged macrocyclic peptides in bacterial cells (E. coli). These reagents are shown to provide efficient access to a broad range of macrocyclic peptide scaffolds spanning from 2 to 20 amino acid residues, with the different eUAAs offering complementary reactivity profiles toward mediating short- vs. long-range macrocyclizations. Swapping of the eUAA cyclization module in a cyclopeptide inhibitor of streptavidin and Keap1 led to compounds with markedly distinct binding affinity toward the respective target proteins, highlighting the effectiveness of this strategy toward tuning the structural and functional properties of bioactive macrocyclic peptides. The peptide cyclization strategies reported here expand opportunities for the combinatorial biosynthesis of natural product-like peptide macrocycles in bacterial cells or in combination with display platforms toward the discovery of selective agents capable of targeting proteins and protein-mediated interactions.
Introduction
Macrocyclic peptides have emerged as an attractive class of bioactive molecules and therapeutic agents, in particular because of their potential ability to interact with extended and shallow protein surfaces1,2 and thus provide a means to address notoriously challenging targets such as protein–protein interactions.3–7 Conformational restriction through backbone and/or side-chain cyclization8 has been shown to impart peptides with several advantageous features such as increased specificity and affinity toward the target protein,9–11 enhanced proteolytic resistance12–15 and/or improved cell permeability.16–20 Reflecting the increasing interest in this structural class, various strategies have been implemented for the construction and exploration of combinatorial libraries of these cyclic peptides. Among them, the use of genetically encoded peptide libraries has constituted an attractive strategy to generate large collections of these molecules, which have been amenable to screening through display platforms for accelerating the discovery of cyclopeptide binders of proteins and enzymes.21–26 For example, in vitro translation of cyclic peptides27–29 or chemical cyclization of mRNA- or phage-displayed peptides via chemical30–34 or enzymatic means,35,36 have been successfully applied for this purpose, resulting in the identification of cyclic peptides capable of interacting with a variety of target proteins and enzymes.This progress notwithstanding, and with the notable exception of SICLOPPS libraries,26,37,38 none of the aforementioned methodologies allows for the production of macrocyclic peptides of arbitrary sequences in living cells, thus limiting the screening of these macrocyclic peptide libraries to in vitro affinity-based selection procedures.27–34 To expand capabilities in this area, our group has introduced methodologies useful for mediating the synthesis of macrocyclic peptides (a.k.a. macrocyclic organo-peptide hybrids or MOrPHs)39 in living cells via the spontaneous, post-translational cyclization of recombinant polypeptides by means of genetically encoded non-canonical amino acids (ncAA).40–43 These methodologies have enabled the creation and screening of macrocyclic peptide libraries which can be produced in cellulo42,44 or displayed on phage particles45 for the discovery of chemical agents capable of disrupting protein–protein interactions. So far, however, only a few ncAAs40,42 were made available for these applications, thus limiting opportunities for structural diversification of the resulting macrocyclic peptides through variation of the ncAA module. Here, we report the development and characterization of an expanded suite of non-canonical amino acids suitable for directing the biosynthesis of macrocyclic peptides constrained by a non-reducible, inter-side-chain thioether linkage in bacterial cells. These cyclization strategies provide efficient access to a broader range of macrocyclic peptide scaffolds than previously possible. In addition, variation of the ncAA module is shown to furnish an effective means to modulate the functional properties of bioactive macrocyclic peptides.
Results and discussion
An overview of the MOrPH synthesis method investigated here is provided in Fig. 1a. According to this strategy, a genetically encoded non-canonical amino acid (ncAA) is introduced via (amber) stop codon suppression within a target peptide sequence containing a proximal cysteine residue. The ncAA is designed to bear an electrophilic functional group within its side chain, enabling the spontaneous formation of an inter-side-chain thioether bridge between the ncAA and nearby cysteine upon recombinant expression of the precursor polypeptide in the cell (Fig. 1a). Within this general approach,40 a key requirement concerning the choice of the electrophilic unnatural amino acid (eUAA) include its ability to enable efficient peptide cyclization via intramolecular alkylation of the nearby cysteine, while outcompeting undesirable intermolecular reactions with nucleophiles present in the crowded cellular environment (e.g. glutathione and other thiol-containing metabolites or proteins) and/or hydrolysis. In addition, such eUAAs must be amenable to genetic incorporation via an orthogonal aminoacyl-tRNA synthetase (AARS)/tRNA pair.46 Previously, we identified in O-(2-bromoethyl)-tyrosine (O2beY) a viable eUAA for this purpose, permitting the efficient cyclization of target peptide sequences encompassing one to eight intervening amino acid residues between O2beY and the cysteine residue.40,41 With the goal of expanding the range of macrocyclic peptide scaffolds accessible through this methodology, we selected four target eUAAs, namely p-(vinylsulfonamido)phenylalanine (pVsaF), p-(acrylamido)phenylalanine (pAaF), p-(2-chloroacetamido)-phenylalanine (pCaaF), and O-(4-bromobutyl)-tyrosine (O4bbY) (Fig. 1b). pVsaF and pAaF carry within their side-chains a vinylsulfonamide and acrylamide groups, respectively, which are able to undergo Michael addition reactions with thiol nucleophiles.47–49 In addition, both of these electrophilic warheads have been effectively used in the context of the development of small molecules as covalent inhibitors of proteins/enzymes,50–53 which supports their compatibility with complex environments such as the intracellular milieu. In addition to the previously described pVsaF and pAaF,54 two additional eUAAs were designed and targeted for investigation, corresponding to pCaaF and O4bbY (Fig. 1b). pCaaF carries an α-chloro-acetamido moiety which has also been exploited as an electrophilic warhead in drug discovery.55 O4bbY, on the other hand, represents a structural homologue of O2beY featuring a longer (C4vs. C2) side-chain bromoalkyl group. Altogether, this set of ncAAs was meant to serve the two-fold purpose of (i) testing a diverse range of electrophilic groups with respect to their compatibility and relative efficiency toward inducing peptide macrocyclization according to the strategy outlined in Fig. 1a, and (ii) enabling the construction of macrocyclic peptides constrained by structurally different thioether linkages as given by the different nature of the reactive group involved in the cysteine-crosslinking reaction (Fig. 1c).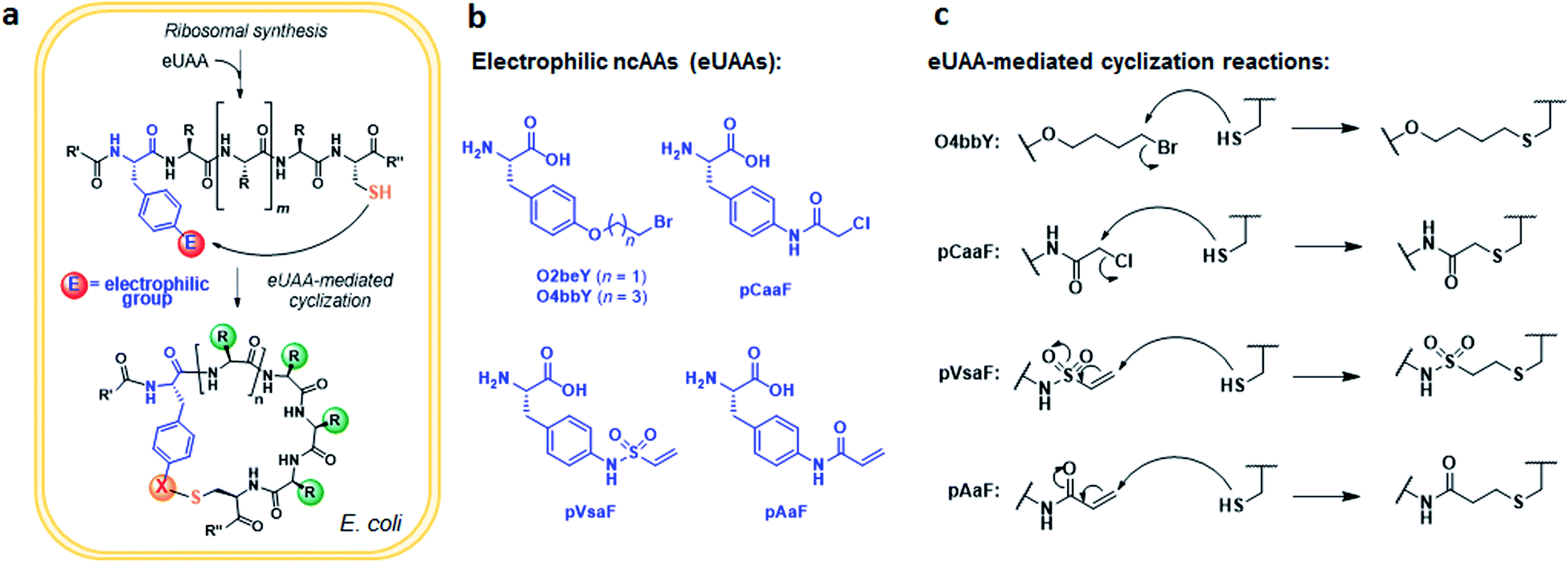 | ||
| Fig. 1 Expanded toolbox for the biosynthesis of thioether-bridged macrocyclic peptides in bacterial cells. (a) Overview of methodology for spontaneous peptide cyclization at the post-translational level by action of electrophilic unnatural amino acids (eUAA). (b and c) Chemical structures of the eUAA cyclization modules investigated in this study (b) and their corresponding cysteine crosslinking reactions (c). | ||
Conveniently, two engineered variants of Methanocaldococcus jannaschii tyrosyl-tRNA synthetase (MjTyrRS) were recently made available for the genetic incorporation of both pVsaF and pAaF for their use in the context of intermolecular protein–protein crosslinking via aza-Michael reactions.54 Accordingly, these AARS variants were prepared by site-directed mutagenesis of MjTyrRS and cloned into a pEVOL-based vector56 for expression of pVsaF-RS (Tyr32Gly, Leu65Tyr, Phe108His, Gln109Gly, Asp158Gly, Ile159Leu, Leu162Gln, Asp286Arg) or pAaF-RS (a.k.a. TyrRS42: Tyr32Val, Leu65Tyr, Phe108His, Gln109Gly, Asp158Gly, Leu162Glu, Asp286Arg) in conjunction with the cognate suppressor MjtRNATyrCUA. Both AARS/tRNA pairs were found to enable the selective incorporation of pVsaF (or pAaF) into a protein, as determined using a reporter yellow fluorescent protein containing an amber stop codon within its N-terminal region (YFP(TAG))42 (Fig. 2a). To identify a suitable AARS for genetic incorporation of pCaaF, we surmised that this ncAA may be also recognized by pVsaF-RS and/or pAaF-RS, in view of the structural similarity between pCaaF and pVsaF/pAaF and considering the polyspecificity often exhibited by engineered AARS.42,57,58 Gratifyingly, both AARSs and in particular pAaF-RS, were found to effectively charge the suppressor tRNA with pCaaF, enabling its efficient incorporation into the reporter YFP protein (Fig. 2a). In fact, both AARS showed a marked polyspecificity within this set of ncAAs, which may be facilitated by specific recognition of the amido/sulfonamido group of these substrates within the enzyme active site. To identify a AARS for the genetic encoding of O4bbY, we screened a broader in-house collection of MjTyrRS variants previously engineered for recognition of various non-canonical tyrosine and phenylalanine derivatives, including O-propargyl-tyrosine (OpgY),59 3-amino-tyrosine (3AmY),60 and 2-naphthylalanine (2NapA), among others. Using the aforementioned YFP(TAG)-based fluorescence assay, the MjTyrRS variant O2beY-RS, which we previously engineered for recognition of O2beY,40 was found to provide a viable AARS for incorporation of O4bbY in response to an amber stop codon (Fig. 2b). Interestingly, O2beY-RS differs from the OpgY-specific aminoacyl-tRNA synthetase OpgY-RS by a single mutation, Ala32Gly, which expands the active site cavity in proximity of the para position of the tyrosine substrate.40 Unlike for O2beY, no detectable incorporation of O4bbY was observed using OpgY-RS (Fig. 2b), indicating that this cavity-enlarging mutation becomes critical for accommodating the more extended para substituent in this ncAA compared to O2beY and OpgY.
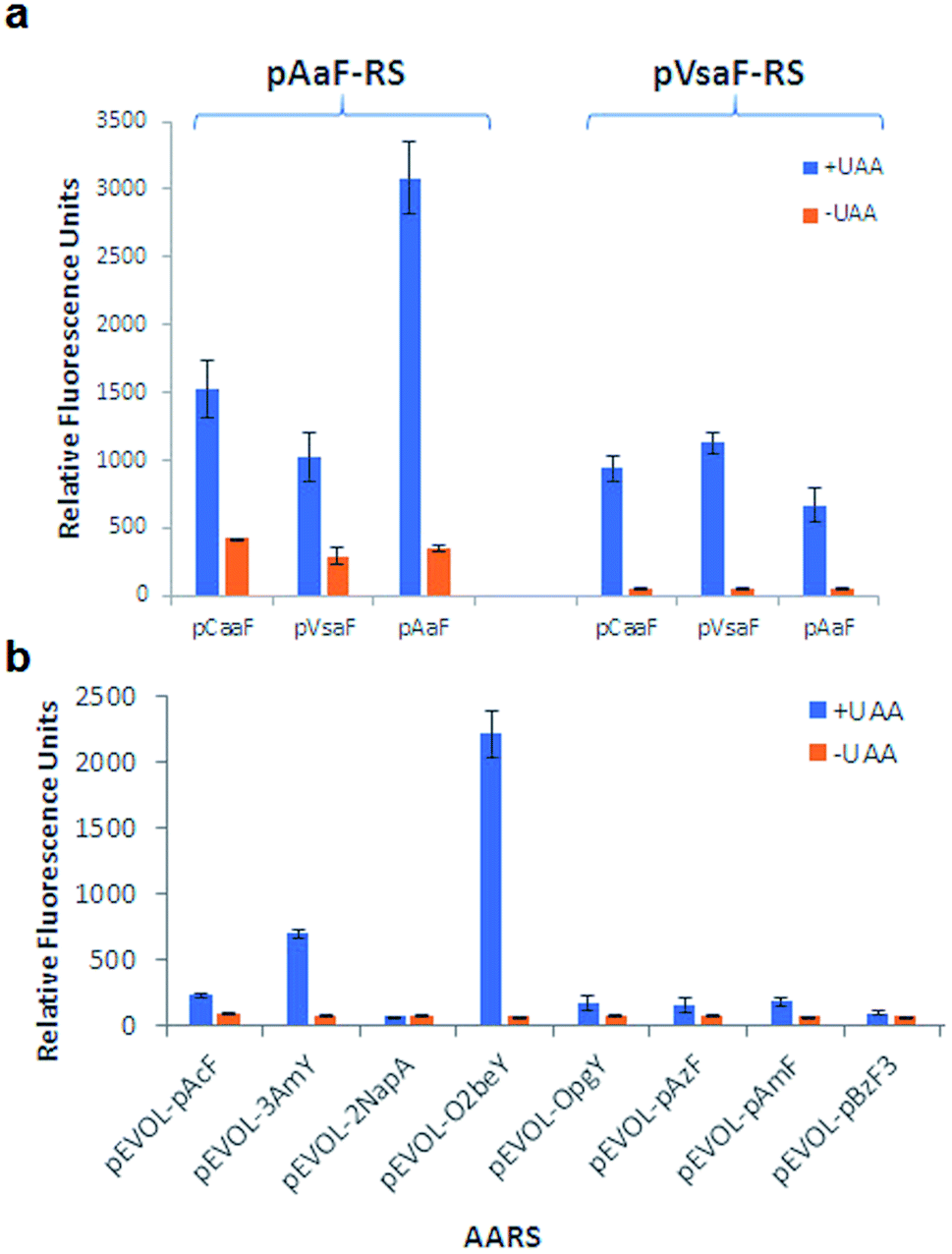 | ||
| Fig. 2 Identification of orthogonal AARS for genetic incorporation of the eUAA pVsaF, pAaF, pCaaF, and O4bbY via amber stop codon suppression using reporter yellow fluorescence protein. (a) Relative YFP yield in the presence and absence of pCaaF, pVsaF, and pAaF using AARS pVsaF-RS and pAaF-RS. (b) Relative YFP yield in the presence and absence of O4bbY using panel of engineered MjTyrRS variants. | ||
With a functional set of orthogonal AARS/tRNA pairs for the genetic encoding of the target ncAAs in hand, we set out to test the ability of these eUAAs to promote peptide cyclization according to the strategy outlined in Fig. 1a. To this end, we prepared a series of model polypeptides encompassing a macrocycle precursor sequence in which the eUAA and Cys residues are spaced from each other by an increasing number of intervening residues and leading to a i/i + 1 to i/i + 20 macrocycle, where i and n correspond to the eUAA and Cys residue, respectively (Table 1). In these constructs, the macrocycle precursor sequence was N-terminally fused to a FLAG tag for ease of detection by mass spectrometry and in the binding assays (vide infra) and C-terminally fused to a chitin-binding domain (CBD) containing a poly-histidine tag. The latter was introduced to facilitate purification of the recombinantly produced constructs and isolation of any product resulting from potential reactions of the genetically incorporated eUAA with other nucleophiles in cells. In addition, a Factor Xa cleavage site was introduced between the macrocycle precursor sequence and the CBD tag to allow for proteolytic cleavage of the tag for LC-MS analysis of cyclization efficiency.
| Entry | Name | Macrocycle precursor sequencea | Cysb |
|---|---|---|---|
| a The full protein construct contains an N-terminal FLAG tag (MDYKDDDDK-GSGSG-) and a C-terminal Factor Xa site (-GIEGR-) followed by chitin binding domain from B. circulans chitinase A1 containing a poly-His tag. b Relative position of cysteine with respect to the electrophilic unnatural amino acid (eUAA) (=position ‘i’). | |||
| 1 | Z1C |

|
i + 1 |
| 2 | Z2C |

|
i + 2 |
| 3 | Z3C |
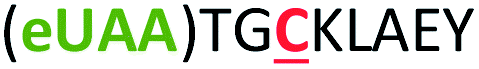
|
i + 3 |
| 4 | Z4C |
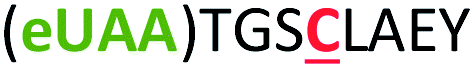
|
i + 4 |
| 5 | Z5C |
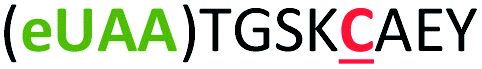
|
i + 5 |
| 6 | Z6C |
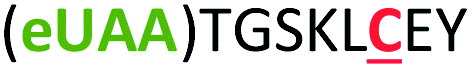
|
i + 6 |
| 7 | Z8C |
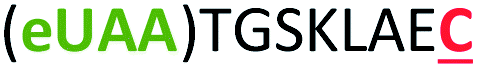
|
i + 8 |
| 8 | Z10C |

|
i + 10 |
| 9 | Z12C |

|
i + 12 |
| 10 | Z15C |

|
i + 15 |
| 11 | Z20C |

|
i + 20 |
| 12 | C6Z |

|
i − 6 |
| 13 | C8Z |

|
i − 8 |
| 14 | Strep-m3(Z) |
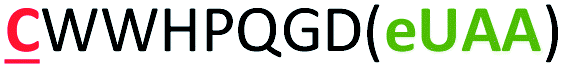
|
i − 8 |
| 15 | KDD-m1(Z) |

|
i + 7 |
The target constructs were produced in E. coli BL21(DE3) cells containing a dual plasmid system for expression of the precursor polypeptide and the orthogonal AARS/tRNA pair in the presence of the desired eUAA. The recombinant constructs were purified via Ni-affinity chromatography and analyzed by MALDI-TOF mass spectrometry. For each of the ncAAs, the corresponding proteins were obtained in high yields (e.g., pVsaF constructs (avg): ∼25 mg protein per L culture; pAaF constructs (avg): ∼65 mg L−1 culture; pCaaF constructs (avg): ∼26 mg L−1 culture; O4bbY constructs (avg): ∼72 mg L−1 culture). In addition, MALDI-TOF MS analysis of both the purified proteins and the N-terminal peptides obtained after proteolytic (Factor Xa) cleavage of the CBD tag (Fig. S2–S5†) show a m/z signal corresponding to the desired ncAA-containing construct with no detectable amounts of by-products resulting from amino acid misincorporation. These results thus confirmed the functionality and specificity, respectively, of the AARS enzymes applied for incorporation of these eUAAs. To measure the extent of eUAA-induced cyclization, the N-terminal peptides were analysed by LC-MS to quantify the amount of the desired thioether-linked macrocyclic product vs. the linear peptide (see Fig. S8–S11†). pCaaF- and O4bbY-mediated cyclization results in the loss of a HX group (X = Cl or Br), enabling direct discrimination of cyclic vs. linear peptide based on the mass difference. Since the cyclized and acyclic forms of the pVsaF- and pAaF-containing peptides are isobaric, these compounds were treated with iodoacetamide prior to LC-MS analysis. Under these conditions, the free Cys in the linear peptide is quantitatively alkylated (+57 mass units; Fig. S1†) enabling its discrimination from the cyclic form.
As shown in Fig. 3 and S2–S5,† these experiments showed that all the eUAAs are able to induce efficient macrocyclization across variable target sequence lengths, thus demonstrating their functionality and efficiency toward directing the biosynthesis of macrocyclic peptides in E. coli. Previously, O2beY-mediated cyclization was found to occur efficiently (80–100%) only across inter-residue distances ranging from i/i + 2 to i/i + 8, where i is the eUAA and the Cys residue is located at the i + n positon (Fig. 3a).40 In stark contrast, all of the new eUAAs investigated here undergo efficient cyclization from i/i + 2 up to i/i + 20 inter-residue distances, enabling the formation of peptide macrocycles spanning 3 to 21 amino acid residues in high to quantitative yields (>80–100%; Fig. 3a–d). As the only exception, modest cyclization (30%) was observed for the 16mer construct (Z15C) with pVsaF (Fig. 3c). This result, however, likely arises from a sequence-dependent effect since significantly higher macrocycle yield (85%) was observed for the longer (21mer) target sequence using this eUAA. Thus, complementing the reactivity profile of O2beY,40 the pCaaF-, O4bbY-, pVsaF-, and pAaF-based cyclization modules greatly expand the range of cyclopeptide ring sizes accessible using this methodology (Fig. 1) and provide an effective means to achieve long-range peptide macrocyclizations (i.e., i/i + 10 to i/i + 20) in addition to medium- and short-range ones (i.e., i/i + 2 to i/i + 8). To assess the impact of the orientation of the eUAA/Cys pair on cyclization efficiency, two additional constructs were examined in which the Cys/eUAA residues are installed in a i/i − 6 and i/i − 8 arrangement (Table 1, entries 12 and 13). In both cases and across all of the eUAAs, the desired macrocyclic peptide was obtained in comparably high yields as for the i/i + 6-linked and i/i + 8-linked counterparts (94–100%; Fig. 3a–d and S2–S5†), indicating that both orientations of the eUAA/Cys pair are compatible with efficient macrocyclization.
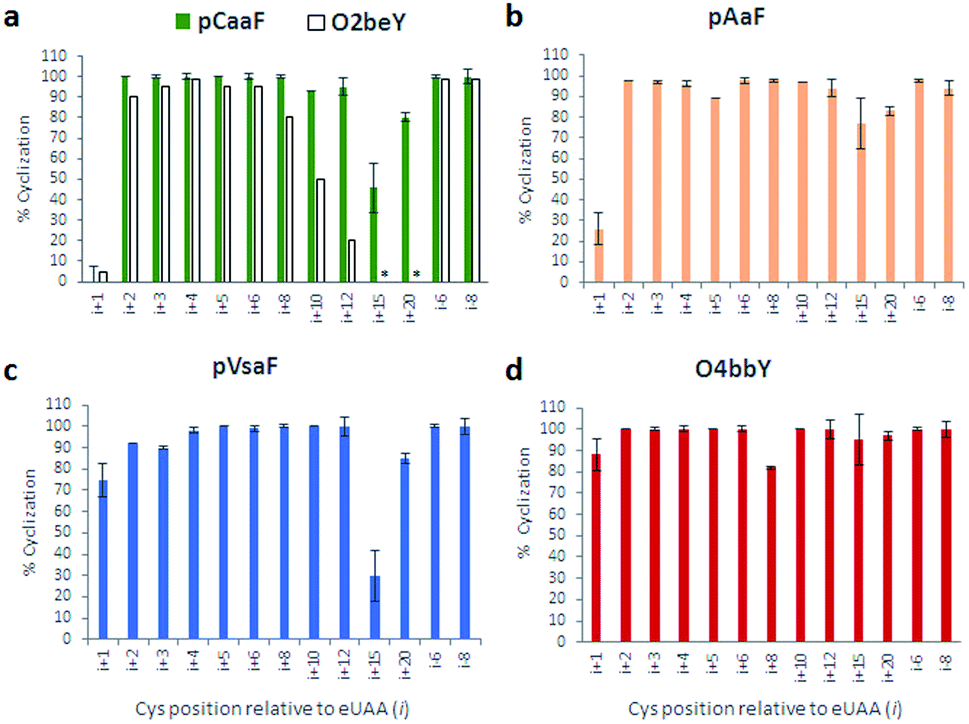 | ||
| Fig. 3 Cyclization efficiency for the different macrocycle precursor sequences with varying distances and orientations of the eUAA/Cys pair. For each graph, the eUAA cyclization module is indicated. Proteins were isolated after expression for 16–18 hours at 27 °C and the extent of cyclization was determined by LC-MS analysis after proteolytic cleavage of the CBD tag (see ESI for details†). | ||
Whereas the eUAAs performed similarly across i/i + 2 to i/i + 20 crosslinking distances, insights into their differential reactivity could be gained from analysis of the i/i + 1 constructs (Z1C(eUAA); Table 1; entry 1). Indeed, in contrast to pCaaF, pAaF, and O2beY, which undergo no to inefficient macrocyclization (0–26%) with an adjacent cysteine (Fig. 3a and b), both pVsaF and O4bbY are able to form the corresponding i/i + 1-linked macrocycles with substantially higher efficiency (75–88%; Fig. 3c and d). Notably, these reactions generate 16-membered macrocycles containing five sp2 carbon centers, whose formation is expected to be quite unfavorable due to ring strain.61 Compared to the structurally similar pAaF, the higher efficiency of pVsaF toward i/i + 1 cyclization (75% vs. 26%) can be rationalized based on the higher reactivity of vinylsulfonamides vs. acrylamides toward Michael addition reactions with thiols.62 Another noteworthy aspect is the accumulation of a certain amount of glutathione adduct or hydrolysis product for the constructs unable to undergo efficient cyclization, namely Z1C(pCaaF), Z1C(pAaF), and Z15C(pCaaF) (Fig. S2 and S3†). While this indicate that these reactions can potentially compete with intramolecular crosslinking within the cell, the high extent of cyclization observed for the vast majority of the constructs and reactions tested (i.e., 48/52 = 92%; Fig. S2–S5†) highlights the fact that eUAA-mediated cyclization effectively outcompete these side-reactions.
The different types of side-chain electrophilic groups carried by the eUAA modules result in the formation of peptide macrocycles constrained by structurally different thioether linkages (Fig. 4). Since subtle backbone modifications are known to have a large impact on the conformational (and thus functional) properties of cyclic peptides,4,63 we envisioned the different eUAAs could also provide a means to tune the functional properties of the corresponding peptide macrocycles. To investigate this aspect, we selected as testbed two bioactive cyclopeptides we have recently isolated from the screening of phage displayed libraries of O2beY-based macrocycles.45 These correspond to Strep-m3, a (i/i − 8)-linked macrocycle with nanomolar affinity for streptavidin (KD: 20 nM), and KDD-m1, a (i/i + 7)-linked macrocyclic peptide capable of targeting Kelch-like ECH-associated protein 1 (Keap1) (KD: 107 nM), which is implicated in regulating cytoprotective responses to oxidative stress in human cells.64 To examine the impact of the thioether linkage on the function of these peptides, a series of macrocyclic peptides were generated by introducing each of the new eUAAs in place of O2beY within these peptide sequences (Table 1, entries 14 and 15). Consistent with the structure–activity analyses of Fig. 3, all of the desired macrocycles containing the different eUAAs were obtained in quantitative yields as confirmed by MS analysis (Fig. S6 and S7†). After purification, the binding affinity of these macrocyclic peptides toward the respective target proteins (streptavidin or Keap1) was determined using an in vitro assay. As shown in Fig. 5, these experiments revealed a pronounced effect of the nature of the thioether linkage on the binding properties of these molecules. In the case of the Strep-m3 derived series, pAaF- and pCaaF-mediated cyclization resulted in cyclic peptides displaying a streptavidin binding affinity comparable to that of the O2beY-linked peptide (KD: 31–35 nM vs. 20 nM) (Fig. 5c), indicating that the corresponding thioether linkages (–NHC(CO)(CH2)n–, where n = 1 and 2, respectively) are isofunctional to that generated via O2beY-mediated cyclization (–O(CH2)2–) in the context of this macrocyclic scaffold (Fig. 5a). In contrast, a 17-fold lower binding affinity (KD: 343 vs. 20 nM) was measured for the pVsaF-cyclized peptide, despite the fact its thioether linkage is identical in length to that given by pAaF and differs from it by only a subtle structural feature, namely the presence of a sulfonamide (–NHSO2–) vs. amide (–NHCO–) bond (Fig. 5a). Only a modest reduction in affinity (2.5-fold) was observed instead for the O4bbY-cyclized peptide, which features an alkyl thioether linkage extended by two methylene units compared to that of O2beY. Interestingly, a completely different scenario emerged from the analysis of the Keap1-targeting peptides derived from KKD-m1 (Fig. 5b and c). Here, the best Keap1 binder among this series is the pVsaF-cyclized macrocycle, which binds Keap1 with only a slightly reduced affinity (3-fold) compared to the reference O2beY-linked macrocycle (KD: 327 nM vs. 107 nM; Fig. 5c). Whereas the O4bbY-cyclized peptide also maintained substantial affinity for Keap1 (KD: 1200 nM), both the pAaF- and pCaaF-cyclized peptide showed a dramatic drop (>100-fold) in the strength of their binding interaction with Keap1 compared to the O2beY-based peptide. Taken together, these studies illustrate the ability of the different eUAA cyclization modules and corresponding thioether linkages to tune the functional properties of a bioactive macrocyclic peptide, also displaying a differential effect in the context of different cyclopeptide scaffolds. In this regard, particularly instructive is the markedly distinct behaviour of the pVsaF- vs. pAaF-based macrocycles, which is dependent upon a small variation (–SO2– vs. –C(O)– unit) within their thioether linkages. Given that the rest of these molecules remain invariant, this effect likely arises from a subtle yet functionally important change in the conformation of the macrocycles as influenced by the nature of the intramolecular crosslink.
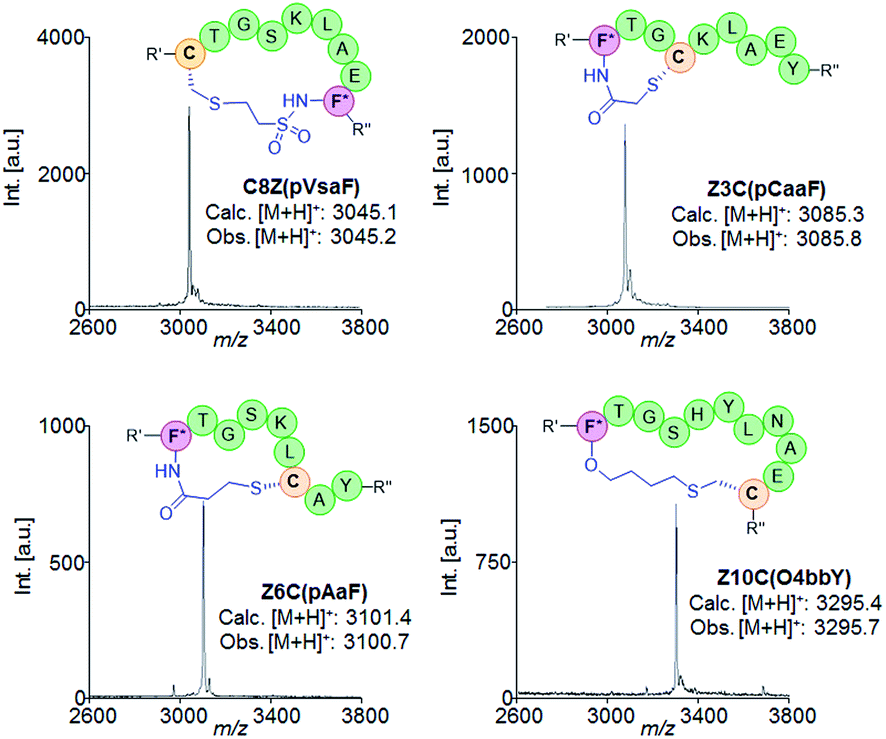 | ||
| Fig. 4 Schematic structures and MALDI-TOF MS spectra of representative macrocyclic peptides obtained using different eUAA cyclization modules and i/i ± n connectivities. F* = para-substituted Phe; R′ = flag tag; R′′ = GIEGR. | ||
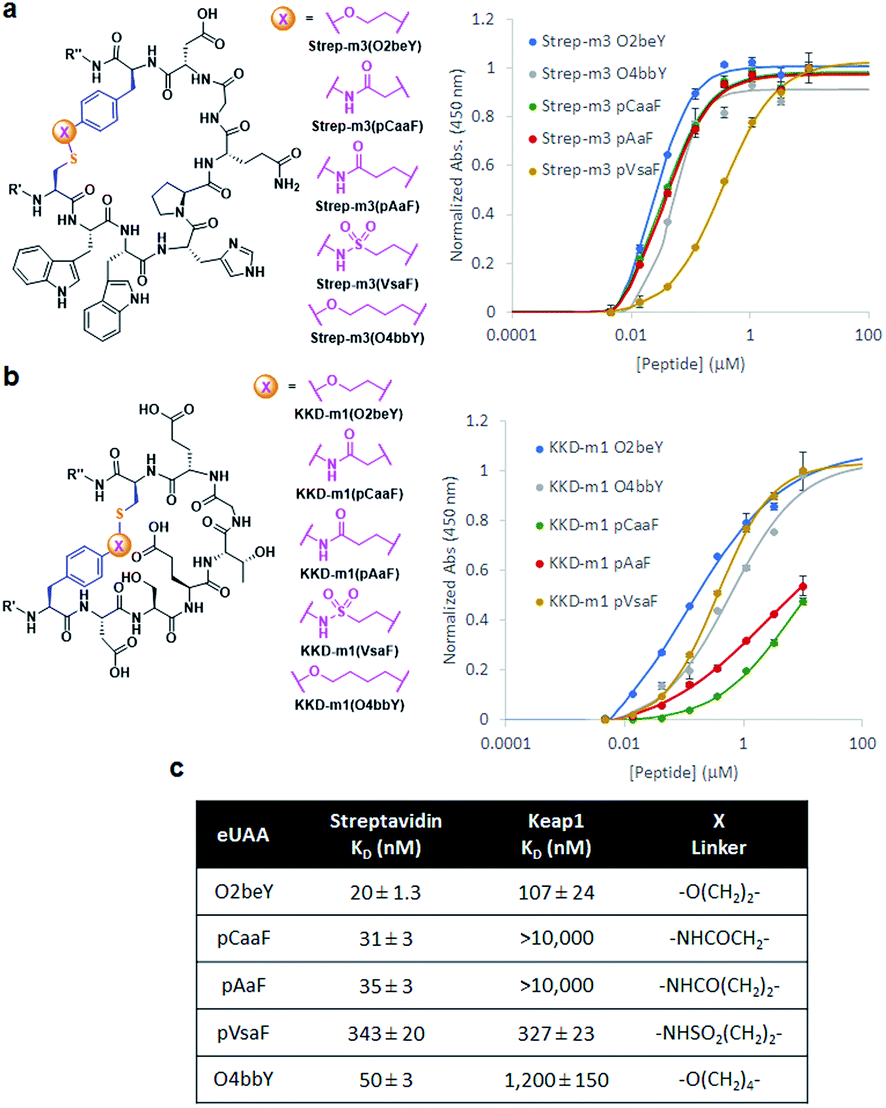 | ||
| Fig. 5 Chemical structures of (a) streptavidin-binding and (b) Keap1-binding macrocyclic peptides featuring different thioether linkages (R′ = FLAG tag; R′′ = CBD tag). The graphs report the peptide binding curves to plate-immobilized streptavidin or Keap1 Kelch domain, respectively, as determined via a colorimetric assay with HRP-conjugated anti-FLAG antibody. (c) Calculated binding dissociation constants for the different peptides. | ||
Conclusion
In summary, we have developed an expanded methodology for directing the biosynthesis of thioether-linked macrocyclic peptides via a cysteine cross-linking reaction by means of electrophilic non-canonical amino acids (Fig. 1). In particular, our studies demonstrate the functionality and high efficiency of pVsaF, pAaF, pCaaF, and O4bbY toward mediating the spontaneous formation of cyclic peptides featuring a variable inter-sidechain linkage and spanning up to 21-amino acid residues in cellulo. Notably, these eUAAs are able to mediate long-range macrocyclizations as well as medium- and short-range ones, resulting in the efficient formation of peptide macrocycles consisting of 16- to 73-membered rings. Furthermore, our results show how swapping of the eUAA cyclization module in two bioactive cyclopeptides had a profound and scaffold-dependent effect on the target binding properties, highlighting the value of this strategy toward modulating the protein recognition properties of a functional macrocyclic peptide. Overall, the peptide cyclization strategies reported here are expected to expand opportunities for the creation of structurally and functionally diverse libraries of peptide macrocycles which can be produced in bacterial cells and can be explored through functional assays42,44 or in combination with high-throughput display platforms,45 toward the discovery of chemical agents for targeting and modulating biomolecular interactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the U.S. National Institute of Health (NIH) grant R01 GM134076. MS instrumentation at the University of Rochester was supported by the National Science Foundation grants CHE-0840410 and CHE-0946653 and NIH grant S10 OD021486.References
- E. A. Villar, D. Beglov, S. Chennamadhavuni, J. A. Porco Jr, D. Kozakov, S. Vajda and A. Whitty, Nat. Chem. Biol., 2014, 10, 723–731 CrossRef CAS PubMed.
- A. K. Malde, T. A. Hill, A. Iyer and D. P. Fairlie, Chem. Rev., 2019, 119, 9861–9914 CrossRef CAS PubMed.
- E. M. Driggers, S. P. Hale, J. Lee and N. K. Terrett, Nat. Rev. Drug Discovery, 2008, 7, 608–624 CrossRef CAS PubMed.
- T. A. Hill, N. E. Shepherd, F. Diness and D. P. Fairlie, Angew. Chem., Int. Ed. Engl., 2014, 53, 13020–13041 CrossRef CAS PubMed.
- E. Valeur, S. M. Gueret, H. Adihou, R. Gopalakrishnan, M. Lemurell, H. Waldmann, T. N. Grossmann and A. T. Plowright, Angew. Chem., Int. Ed., 2017, 56, 10294–10323 CrossRef CAS PubMed.
- T. A. Cardote and A. Ciulli, ChemMedChem, 2016, 11, 787–794 CrossRef CAS PubMed.
- A. A. Vinogradov, Y. Z. Yin and H. Suga, J. Am. Chem. Soc., 2019, 141, 4167–4181 CrossRef CAS PubMed.
- C. J. White and A. K. Yudin, Nat. Chem., 2011, 3, 509–524 CrossRef CAS PubMed.
- M. A. Dechantsreiter, E. Planker, B. Matha, E. Lohof, G. Holzemann, A. Jonczyk, S. L. Goodman and H. Kessler, J. Med. Chem., 1999, 42, 3033–3040 CrossRef CAS PubMed.
- R. L. Dias, R. Fasan, K. Moehle, A. Renard, D. Obrecht and J. A. Robinson, J. Am. Chem. Soc., 2006, 128, 2726–2732 CrossRef CAS PubMed.
- R. M. Cardoso, F. M. Brunel, S. Ferguson, M. Zwick, D. R. Burton, P. E. Dawson and I. A. Wilson, J. Mol. Biol., 2007, 365, 1533–1544 CrossRef CAS PubMed.
- D. P. Fairlie, J. D. Tyndall, R. C. Reid, A. K. Wong, G. Abbenante, M. J. Scanlon, D. R. March, D. A. Bergman, C. L. Chai and B. A. Burkett, J. Med. Chem., 2000, 43, 1271–1281 CrossRef CAS PubMed.
- D. Wang, W. Liao and P. S. Arora, Angew. Chem., Int. Ed., 2005, 44, 6525–6529 CrossRef CAS PubMed.
- T. L. Aboye, H. Ha, S. Majumder, F. Christ, Z. Debyser, A. Shekhtman, N. Neamati and J. A. Camarero, J. Med. Chem., 2012, 55, 10729–10734 CrossRef CAS PubMed.
- J. M. Smith, J. R. Frost and R. Fasan, Chem. Commun., 2014, 50, 5027–5030 RSC.
- L. D. Walensky, A. L. Kung, I. Escher, T. J. Malia, S. Barbuto, R. D. Wright, G. Wagner, G. L. Verdine and S. J. Korsmeyer, Science, 2004, 305, 1466–1470 CrossRef CAS PubMed.
- D. S. Nielsen, H. N. Hoang, R. J. Lohman, T. A. Hill, A. J. Lucke, D. J. Craik, D. J. Edmonds, D. A. Griffith, C. J. Rotter, R. B. Ruggeri, D. A. Price, S. Liras and D. P. Fairlie, Angew. Chem., Int. Ed. Engl., 2014, 53, 12059–12063 CrossRef CAS PubMed.
- W. M. Hewitt, S. S. F. Leung, C. R. Pye, A. R. Ponkey, M. Bednarek, M. P. Jacobson and R. S. Lokey, J. Am. Chem. Soc., 2015, 137, 715–721 CrossRef CAS PubMed.
- H. Wu, G. Mousseau, S. Mediouni, S. T. Valente and T. Kodadek, Angew. Chem., Int. Ed. Engl., 2016, 55, 12637–12642 CrossRef CAS PubMed.
- L. Peraro, Z. J. Zou, K. M. Makwana, A. E. Cummings, H. L. Ball, H. T. Yu, Y. S. Lin, B. Levine and J. A. Kritzer, J. Am. Chem. Soc., 2017, 139, 7792–7802 CrossRef CAS PubMed.
- G. P. Smith and V. A. Petrenko, Chem. Rev., 1997, 97, 391–410 CrossRef CAS PubMed.
- K. Josephson, A. Ricardo and J. W. Szostak, Drug Discovery Today, 2014, 19, 388–399 CrossRef CAS PubMed.
- A. Angelini and C. Heinis, Curr. Opin. Chem. Biol., 2011, 15, 355–361 CrossRef CAS PubMed.
- Y. C. Huang, M. M. Wiedmann and H. Suga, Chem. Rev., 2019, 119, 10360–10391 CrossRef CAS PubMed.
- J. R. Frost, J. M. Smith and R. Fasan, Curr. Opin. Struct. Biol., 2013, 23, 571–580 CrossRef CAS PubMed.
- A. Tavassoli, Curr. Opin. Chem. Biol., 2017, 38, 30–35 CrossRef CAS PubMed.
- Y. Yamagishi, I. Shoji, S. Miyagawa, T. Kawakami, T. Katoh, Y. Goto and H. Suga, Chem. Biol., 2011, 18, 1562–1570 CrossRef CAS PubMed.
- K. Ito, K. Sakai, Y. Suzuki, N. Ozawa, T. Hatta, T. Natsume, K. Matsumoto and H. Suga, Nat. Commun., 2015, 6, 6373–6376 CrossRef CAS PubMed.
- M. Nawatha, J. M. Rogers, S. M. Bonn, I. Livneh, B. Lemma, S. M. Mali, G. B. Vamisetti, H. Sun, B. Bercovich, Y. C. Huang, A. Ciechanover, D. Fushman, H. Suga and A. Brik, Nat. Chem., 2019, 11, 644–652 CrossRef CAS PubMed.
- S. M. Howell, S. V. Fiacco, T. T. Takahashi, F. Jalali-Yazdi, S. W. Millward, B. L. Hu, P. Wang and R. W. Roberts, Sci. Rep., 2014, 4, 6008 CrossRef CAS PubMed.
- E. R. White, L. X. Sun, Z. Ma, J. M. Beckta, B. A. Danzig, D. E. Hacker, M. Huie, D. C. Williams, R. A. Edwards, K. Valerie, J. N. M. Glover and M. C. T. Hartman, ACS Chem. Biol., 2015, 10, 1198–1208 CrossRef CAS PubMed.
- C. Heinis, T. Rutherford, S. Freund and G. Winter, Nat. Chem. Biol., 2009, 5, 502–507 CrossRef CAS PubMed.
- V. Baeriswyl, S. Calzavarini, S. Y. Chen, A. Zorzi, L. Bologna, A. Angelillo-Scherrer and C. Heinis, ACS Chem. Biol., 2015, 10, 1861–1870 CrossRef CAS PubMed.
- S. Ng, E. Lin, P. I. Kitov, K. F. Tjhung, O. O. Gerlits, L. Deng, B. Kasper, A. Sood, B. M. Paschal, P. Zhang, C. C. Ling, J. S. Klassen, C. J. Noren, L. K. Mahal, R. J. Woods, L. Coates and R. Derda, J. Am. Chem. Soc., 2015, 137, 5248–5251 CrossRef CAS PubMed.
- K. J. Hetrick, M. C. Walker and W. A. van der Donk, ACS Cent. Sci., 2018, 4, 458–467 CrossRef CAS PubMed.
- J. H. Urban, M. A. Moosmeier, T. Aumuller, M. Thein, T. Bosma, R. Rink, K. Groth, M. Zulley, K. Siegers, K. Tissot, G. N. Moll and J. Prassler, Nat. Commun., 2017, 8, 1500 CrossRef PubMed.
- E. Miranda, I. K. Nordgren, A. L. Male, C. E. Lawrence, F. Hoakwie, F. Cuda, W. Court, K. R. Fox, P. A. Townsend, G. K. Packham, S. A. Eccles and A. Tavassoli, J. Am. Chem. Soc., 2013, 135, 10418–10425 CrossRef CAS PubMed.
- A. Tavassoli and S. J. Benkovic, Angew. Chem., Int. Ed. Engl., 2005, 44, 2760–2763 CrossRef CAS PubMed.
- J. M. Smith, F. Vitali, S. A. Archer and R. Fasan, Angew. Chem., Int. Ed. Engl., 2011, 50, 5075–5080 CrossRef CAS PubMed.
- N. Bionda, A. L. Cryan and R. Fasan, ACS Chem. Biol., 2014, 9, 2008–2013 CrossRef CAS PubMed.
- N. Bionda and R. Fasan, Chembiochem, 2015, 16, 2011–2016 CrossRef CAS PubMed.
- J. R. Frost, N. T. Jacob, L. J. Papa, A. E. Owens and R. Fasan, ACS Chem. Biol., 2015, 10, 1805–1816 CrossRef CAS PubMed.
- J. R. Frost, Z. J. Wu, Y. C. Lam, A. E. Owens and R. Fasan, Org. Biomol. Chem., 2016, 14, 5803–5812 RSC.
- A. E. Owens, I. de Paola, W. A. Hansen, Y. W. Liu, S. D. Khare and R. Fasan, J. Am. Chem. Soc., 2017, 139, 12559–12568 CrossRef CAS PubMed.
- A. E. Owens, J. A. Iannuzzelli, Y. Gu and R. Fasan, ACS Cent. Sci., 2020, 6, 368–381 CrossRef CAS PubMed.
- C. C. Liu and P. G. Schultz, Annu. Rev. Biochem., 2010, 79, 413–444 CrossRef CAS PubMed.
- J. C. Powers, J. L. Asgian, O. D. Ekici and K. E. James, Chem. Rev., 2002, 102, 4639–4750 CrossRef CAS PubMed.
- J. J. Reddick, J. M. Cheng and W. R. Roush, Org. Lett., 2003, 5, 1967–1970 CrossRef CAS PubMed.
- M. M. M. Santos and R. Moreira, Mini-Rev. Med. Chem., 2007, 7, 1040–1050 CrossRef CAS PubMed.
- J. Singh, R. C. Petter, T. A. Baillie and A. Whitty, Nat. Rev. Drug Discovery, 2011, 10, 307–317 CrossRef CAS PubMed.
- X. Q. Lu, S. K. Olsen, A. D. Capili, J. S. Cisar, C. D. Lima and D. S. Tan, J. Am. Chem. Soc., 2010, 132, 1748–1749 CrossRef CAS PubMed.
- A. L. Garske, U. Peters, A. T. Cortesi, J. L. Perez and K. M. Shokat, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 15046–15052 CrossRef CAS PubMed.
- L. A. Honigberg, A. M. Smith, M. Sirisawad, E. Verner, D. Loury, B. Chang, S. Li, Z. Pan, D. H. Thamm, R. A. Miller and J. J. Buggy, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13075–13080 CrossRef CAS PubMed.
- J. L. Furman, M. C. Kang, S. Choi, Y. Cao, E. D. Wold, S. B. Sun, V. V. Smider, P. G. Schultz and C. H. Kim, J. Am. Chem. Soc., 2014, 136, 8411–8417 CrossRef CAS PubMed.
- E. Resnick, A. Bradley, J. R. Gan, A. Douangamath, T. Krojer, R. Sethi, P. P. Geurink, A. Aimon, G. Amitai, D. Bellini, J. Bennett, M. Fairhead, O. Fedorov, R. Gabizon, J. Gan, J. X. Gu, A. Plotnikov, N. Reznik, G. F. Ruda, L. Diaz-Saez, V. M. Straub, T. Szommer, S. Velupillai, D. Zaidman, Y. L. Zhang, A. R. Coker, C. G. Dowson, H. M. Barr, C. Wang, K. V. M. Huber, P. E. Brennan, H. Ovaa, F. von Delft and N. London, J. Am. Chem. Soc., 2019, 141, 8951–8968 CrossRef CAS PubMed.
- T. S. Young, I. Ahmad, J. A. Yin and P. G. Schultz, J. Mol. Biol., 2010, 395, 361–374 CrossRef CAS PubMed.
- A. L. Stokes, S. J. Miyake-Stoner, J. C. Peeler, D. P. Nguyen, R. P. Hammer and R. A. Mehl, Mol. BioSyst., 2009, 5, 1032–1038 RSC.
- D. D. Young, T. S. Young, M. Jahnz, I. Ahmad, G. Spraggon and P. G. Schultz, Biochemistry, 2011, 50, 1894–1900 CrossRef CAS PubMed.
- A. Deiters and P. G. Schultz, Bioorg. Med. Chem. Lett., 2005, 15, 1521–1524 CrossRef CAS PubMed.
- M. R. Seyedsayamdost, J. Xie, C. T. Y. Chan, P. G. Schultz and J. Stubbe, J. Am. Chem. Soc., 2007, 129, 15060–15071 CrossRef CAS PubMed.
- A. R. Bogdan, S. V. Jerome, K. N. Houk and K. James, J. Am. Chem. Soc., 2012, 134, 2127–2138 CrossRef CAS PubMed.
- S. Chatani, D. P. Nair and C. N. Bowman, Polym. Chem., 2013, 4, 1048–1055 RSC.
- J. E. Bock, J. Gavenonis and J. A. Kritzer, ACS Chem. Biol., 2013, 8, 488–499 CrossRef CAS PubMed.
- K. Itoh, N. Wakabayashi, Y. Katoh, T. Ishii, K. Igarashi, J. D. Engel and M. Yamamoto, Genes Dev., 1999, 13, 76–86 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, synthetic procedures, additional MS spectra and LC-MS chromatograms, NMR spectra. See DOI: 10.1039/d0sc01699c |
| This journal is © The Royal Society of Chemistry 2020 |