
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Concerted aryl-sulfur reductive elimination from PNP pincer-supported Co(III) and subsequent Co(I)/Co(III) comproportionation†
Bryan J.
Foley
 a,
Chandra Mouli
Palit
a,
Nattamai
Bhuvanesh
a,
Jia
Zhou
*b and
Oleg V.
Ozerov
*a
a,
Chandra Mouli
Palit
a,
Nattamai
Bhuvanesh
a,
Jia
Zhou
*b and
Oleg V.
Ozerov
*a
aDepartment of Chemistry, Texas A&M University, 3255 TAMU, College Station, Texas 77842, USA. E-mail: ozerov@chem.tamu.edu
bSchool of Science, Harbin Institute of Technology, Shenzhen 518055, China. E-mail: jiazhou@hit.edu.cn
First published on 19th May 2020
Abstract
This report discloses a combined experimental and computational study aimed at understanding C–S reductive elimination from Co(III) supported by a diarylamido/bis(phosphine) PNP pincer ligand. Divalent (PNP)Co-aryl complexes could be easily oxidized to five-coordinate Co(III) derivatives, and anion metathesis provided five-coordinate (PNP)Co(Ar)(SAr′) complexes of Co(III). In contrast to their previously described (POCOP)Co(Ar)(SAr′) analogs, but similarly to the (PNP)Rh(Ar)(SAr′) and (POCOP)Rh(Ar)(SAr′) analogs, (PNP)Co(Ar)(SAr′) undergo C–S reductive elimination with the formation of the desired diarylsulfide product ArSAr′. DFT studies and experimental observations are consistent with a concerted process. However, in contrast to the Rh analogs, the immediate product of such reductive elimination, the unobserved Co(I) complex (PNP)Co, un-dergoes rapid comproportionation with the (PNP)Co(Ar)(SAr′) starting material to give Co(II) compounds (PNP)Co–Ar and (PNP)Co-SAr′.
Introduction
Carbon-heteroatom cross coupling has become an immensely powerful synthetic tool in recent years.1–4 The existing art on cross-coupling reactions is historically dominated by palladium,5 with additional prominence by another group 10 metal Ni,6,7 as well as a group 11 metal Cu.8,9 There has recently been a renewed push to find alternatives to homogeneous precious metal catalysts from among cheaper, more Earth-abundant metals.10–12Cross-coupling reactions of aryl (pseudo)halides with nucleophiles typically rely on the oxidative addition (OA) – transmetallation (TM) – reductive elimination (RE) cycles such as depicted in Fig. 1. The OA and RE steps are two-electron processes that are well established for Pd. Our group has been interested in the potential of the analogous OA–TM–RE cycle to enable cross-coupling catalysis by group 9 metals. Pd (and Ni) go through the Pd0/PdII oxidation states corresponding to the d10/d8 configurations. For group 9 metals, we have targeted the MI/MIII oxidation states (d8/d6). In particular, we were able to establish that a T-shaped RhI center supported by an anionic pincer ligand possesses a rather striking similarity in its reactivity to the LPd0 fragment.13–18 This approach with Rh proved especially fruitful in catalytic C–S coupling.19 Catalytic C–S coupling with Pd has received a considerable amount of attention.20,21
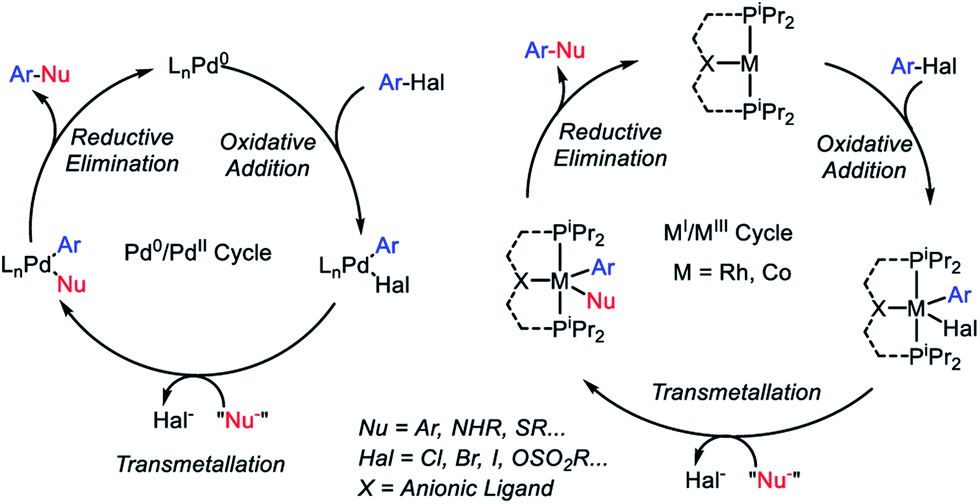 | ||
| Fig. 1 Cross-coupling mechanistic cycles for Pd0/PdII (left) and for RhI/RhIII or CoI/CoIII (right). | ||
It is easy to envisage the steps of the analogous pincer-supported CoI/III cycle (Fig. 1). However, in the chemistry of 3d metals, competition from one-electron pathways to the desired two-electron steps is something that must be closely considered.22 In principle, there is a substantial body of literature describing Co-based cross-coupling catalysis,23 and CoI/CoIII cycles are often proposed.24–28 However, firm mechanistic information remains rather limited. Fout and coworkers analyzed a Co-catalyzed aryl halide amination system in 2014 (ref. 29) where the CoI/CoIII cycle was strongly implicated but the individual steps of OA and RE were not observed. Chirik et al. reported on the C–C coupling of aryl triflates in 2016, where it appears that the CoI/CoIII cycle should operate but the details were not uncovered.30 Bernskoetter's group reported a well-defined example of C–C RE from CoIII in 2011,31 but this involved coupling of two CH3 groups, only indirectly related to aryl halide reactions. However, outside of aryl halide coupling reactions, there have in recent years appeared examples of homogeneous catalysis by pincer-supported Co complexes where two-electron OA/RE steps are either well understood or strongly suggested.32–42
In 2018, we reported on the reactivity of (POCOP)Co(Ph)(SPh).43 In contrast to (PNP)Rh(Ph)(SPh)18 or (POCOP)Rh(Ph)(SPh),19 it did not undergo C–S RE but instead a RE of the phenyl with the pincer aryl (Scheme 1). Because of this, the POCOP system did not allow for the investigation of the C–S RE. We surmised that the analogous RE with the amido of a PNP pincer should be less likely and set off to examine the reactivity of (PNP)Co(Ar)(SAr) complexes. The present report details our efforts in the synthesis of five-coordinate CoIII-aryl/thiolate complexes supported by the PNP ligand, their propensity to undergo concerted C–S RE, and the subsequent comproportionation reactivity that again diverges from the Rh system.
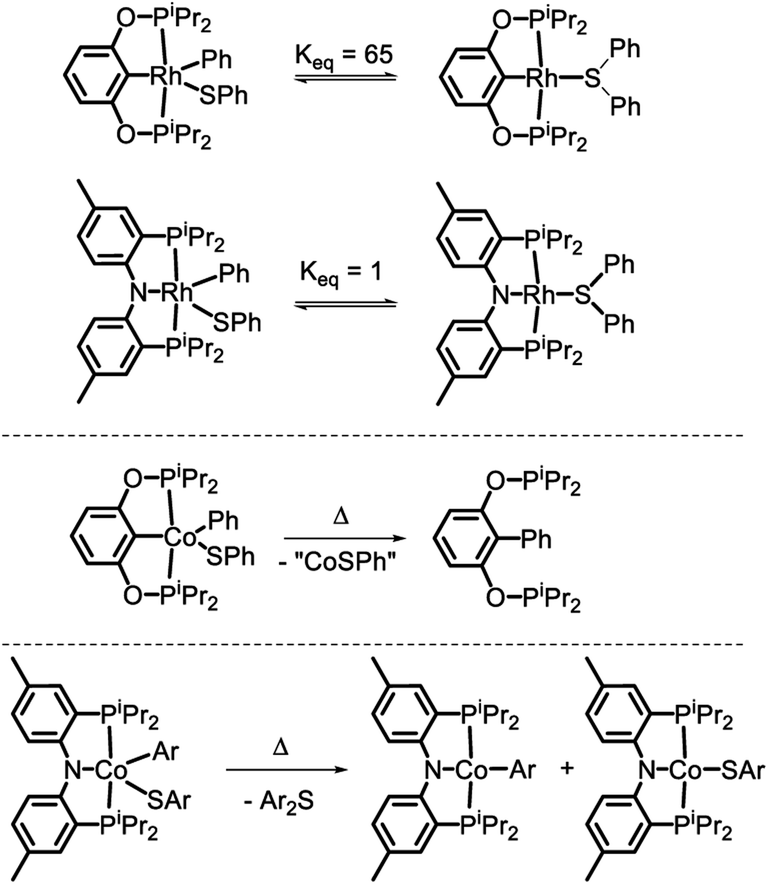 | ||
| Scheme 1 C–S reductive coupling observed for the (POCOP)Rh(Ph)(SPh) and (PNP)Rh(Ph)(SPh) complexes (ref. 18 and 19, top), C–C coupling with the pincer carbon in (POCOP)Co(Ph)(SPh) (ref. 43, middle), and the subject of this work (bottom). | ||
Results and discussion
Synthesis and characterization of (PNP)CoII complexes
Treatment of the previously reported square planar, low spin, S = 1/2 (PNP)CoCl44 (1) with selected aryl nucleophiles resulted in the formation of the corresponding CoII aryl complexes 2a–c (Scheme 2). Clean transmetallation of 1 was also accomplished using sodium thiophenolate reagents to give Co(II) thiolate complexes 3a–c (Scheme 2). Complexes 2a–c and 3a–c were green to dark teal in color. They exhibited paramagnetically shifted 1H NMR resonances contained in the +40 to −30 ppm range, except for the resonances at around −90 ppm in complexes 2a–2c which we tentatively assign as ortho-hydrogens of the Co-bound aryl rings. No 31P NMR resonances were detected for these complexes.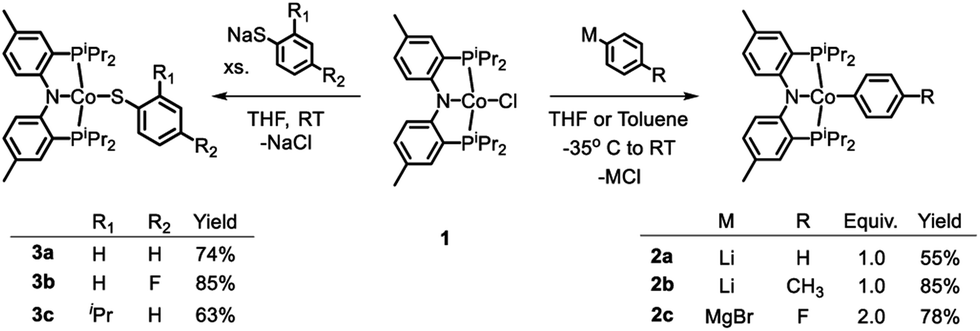 | ||
| Scheme 2 Transmetallation reactions among (PNP)CoIIX. | ||
Examination of the literature shows that four-coordinate Co(II) complexes of anionic pincer ligands are known with both a low-spin S = 1/2 configuration (square-planar geometry) and a high-spin S = 3/2 configuration (pseudotetrahedral geometry).43–52 Low-spin, square-planar Co(II) complexes give rise to paramagnetically shifted 1H NMR resonances that are broad compared to diamagnetic compounds, but are typically interpretable in terms of their relative integration and chemical (in)equivalence. High-spin Co(II) compounds tend to produce 1H NMR spectra that are broadened beyond useful interpretation. Complexes 2a–c and 3a–c in the present work and the (POCOP)CoX complexes (Fig. 2) recently reported by us and Heinekey et al.45 are all low-spin compounds. The same is true for the (PNP1)CoX complexes of the pyrrolyl-based PNP ligand by Tonzetich et al.46 and Nishibayashi et al.,47 as well as the (PNP2)CoX complexes reported by Arnold et al. and Hazari et al.48,49 In contrast, Co(II) halide complexes of the Fryzuk-type PNP3 and Gade's carbazole-based PNP4 ligands are high-spin.50–52 On the other hand, (PNP3)Co(CH2Ph) and (PNP4)CoH are low spin.50,52 It appears that the presence of even one very strong-field ligand such as hydride or aryl/alkyl is sufficient for the low-spin preference. It is interesting that in their absence, the various CoII complexes in Fig. 2 contain low and high-spin complexes with essentially the same set of donors (e.g., high-spin (PNP3)CoCl vs. low-spin (PNP)CoCl (1) or (PNP1)CoBr or (PNP2)CoCl). All the complexes in Fig. 2 possess trans-disposed phosphines or phosphinites in the pincer, and it does not appear that their presence alone is sufficient to ensure low-spin configurations. We surmise that the geometric constraint of the ligand plays a role in enforcing the corresponding geometry and thus spin state, with the less flexible PNP and PNP1 favoring square planar, low-spin CoII.53 The PNP4 ligand contains the most π-donating (dialkylamido) central N donor of the selection in Fig. 2, which may help stabilize a low-spin configuration. Amido/bis(phosphine) PNP pincer ligands can be oxidized at the ligand and thus are potentially redox non-innocent,54 but we see no evidence of it in this work.
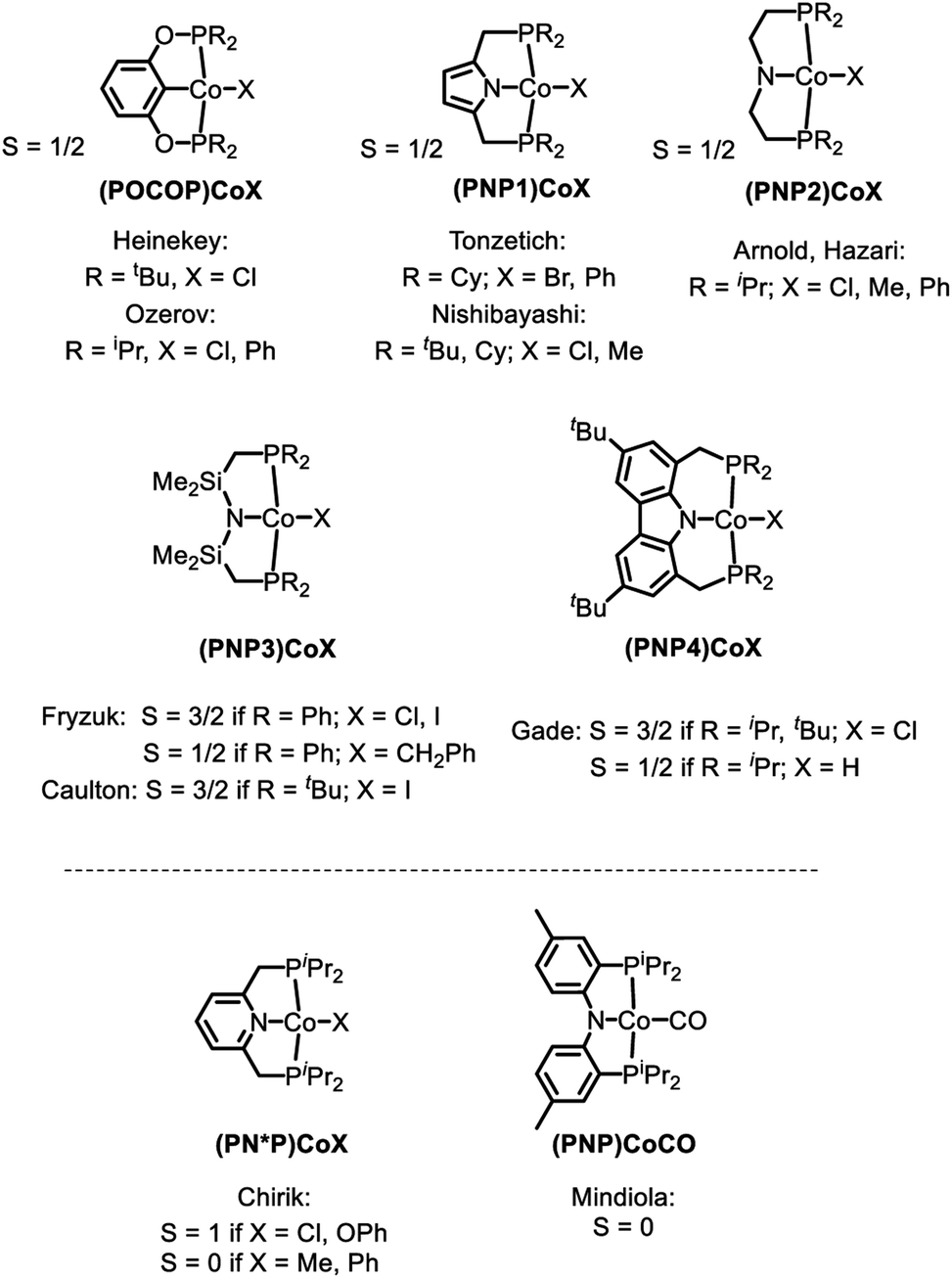 | ||
| Fig. 2 High and low spin dichotomy in POCOP and PNP pincer-supported four-coordinate complexes of CoII (top) and CoI (bottom). | ||
Synthesis and characterization of (PNP)CoIII(Ar)(X) complexes
As is the case in our (POCOP)Co system,43 reacting (PNP)Co(Aryl) complexes 2a–b with 0.5 eq. of PhI(OAc)2 led to the clean formation of (PNP)Co(Aryl)(OAc) (4a–b), isolated in good yield as tan solids after workup (Scheme 3). Treatment of 4a with Me3SiI furnished dark blue-green (PNP)Co(Ph)(I) (5a). 5a can also be prepared via reaction of 2a with 0.5 equiv. of I2. Transmetallation from 5a using sodium thiophenolate proceeded smoothly providing (PNP)Co(Ph)(SPh) complex 6a as a dark blue solid in good yields after recrystallization (Scheme 3).55 The analogous synthesis of (PNP)Co(Ar)(SAr′) complexes 6b–c from 4b–c can be achieved without the isolation of the intermediate 5b–c.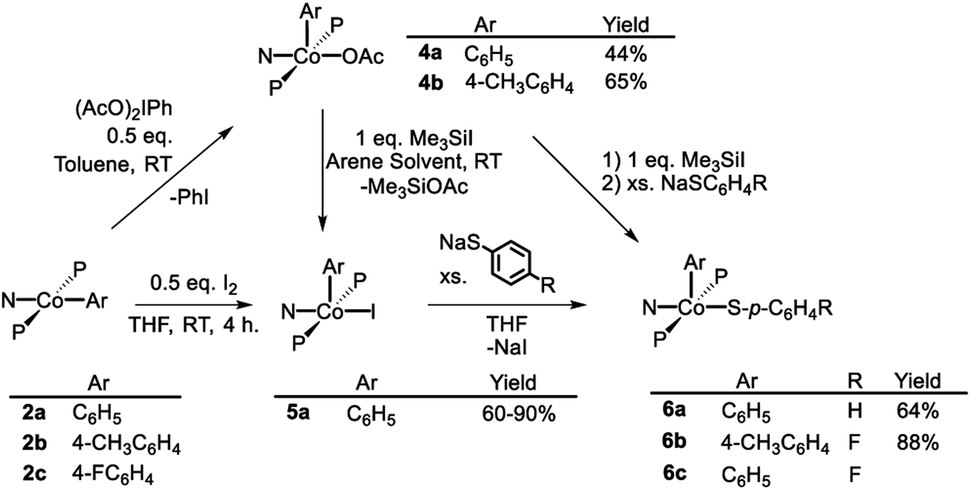 | ||
| Scheme 3 Oxidation of (PNP)CoII and synthesis of target (PNP)Co(Ar)(S–Ar). | ||
The (PNP)Co(Ar)(X) compounds (4a–b, 5a, and 6a–c) gave rise to 1H, 31P, and 13C NMR spectra expected for these diamagnetic complexes. The resonances arising from the Co-bound aryl group exhibited inequivalence between the two ortho- and between the two meta-hydrogens, characteristic of restricted rotation of the metal-bound aryl oriented cis to the central donor of a pincer ligand with two side –PiPr2 arms.14,43,56 In the cases of the aryl/-thiolate complexes 6a–6c, these aromatic resonances were broad humps, whereas in the aryl/-halide 5a and aryl/-acetato complexes 4a–4b, sharp resonances with well-resolved fine structure were observed.
Synthesis and characterization of (PNP)CoI complexes
The dimeric compound [(PNP)Co]2 (7) (Scheme 4) was previously reported by Mindiola et al.44 We were also able to observe a CoI complex (PNP)Co(PPh3) (8) by treatment of (Ph3P)2CoN(SiMe3)2 (9)29 with (PNP)H (10). This reaction liberated triphenyl phosphine and HN(SiMe3)2 (Scheme 4). A wide 1H NMR spectral window revealed a new set of paramagnetically shifted 1H NMR resonances which we have assigned to 8. Compound 8 was not isolated as it appears to be in equilibrium with diamagnetic 7 on the timescale of experimental handling. For example, freshly made 8 was observed to produce 7 when left overnight in a −35 °C freezer, while addition of 12 equiv. of PPh3 to 7 led to the observation of 8 (Scheme 4). Addition of 12 eq. of tris(4-methoxyphenyl)phosphine to this mixture gave rise to a second set of distinct but very similar paramagnetically shifted 1H NMR resonances we interpret as belonging to 11 (Scheme 4, Fig. S12†). This observation supports the notion that 8 is a PPh3-bound Co complex.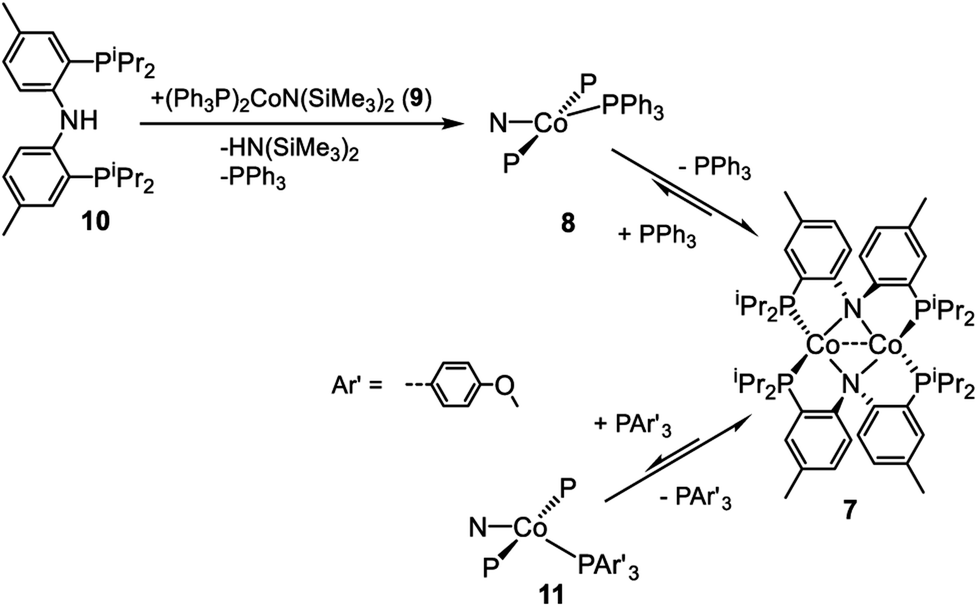 | ||
| Scheme 4 Formation of proposed (PNP)Co(PPh3) and equilibrium between (PNP)2Co2 and (PNP)Co(P(Ar)3). | ||
Based on the paramagnetically shifted 1H NMR spectra, we assume that both 8 and 11 possess an S = 1 ground state. As with four-coordinate Co(II), there are examples of both high-spin and low-spin pincer complexes of Co(I) (S = 1 or 0) (Fig. 2, bottom). With the pyridine-centered PN*P ligand, complexes substituted with stronger field Me or Ph are low-spin, while Cl or OAr as the fourth donor are high-spin.30,57 With the PNP ligand, CO in place of PPh3 in 8 was reported to give a low-spin carbonyl complex (PNP)Co(CO). Thus, it appears that the presence of at least one strong-field ligand (CO, or hydride/alkyl/aryl) is needed to ensure an S = 0 ground state.
X-ray structural studies
Single crystals of 2b, 4a, and 6a suitable for X-ray diffraction were grown from hydrocarbon solvents at −35 °C (Fig. 3). The geometry about the cobalt center in the solid-state structure of 2b is slightly distorted square planar. The Co-bound tolyl ring in 2b is approximately perpendicular to the Co/P/N/P plane.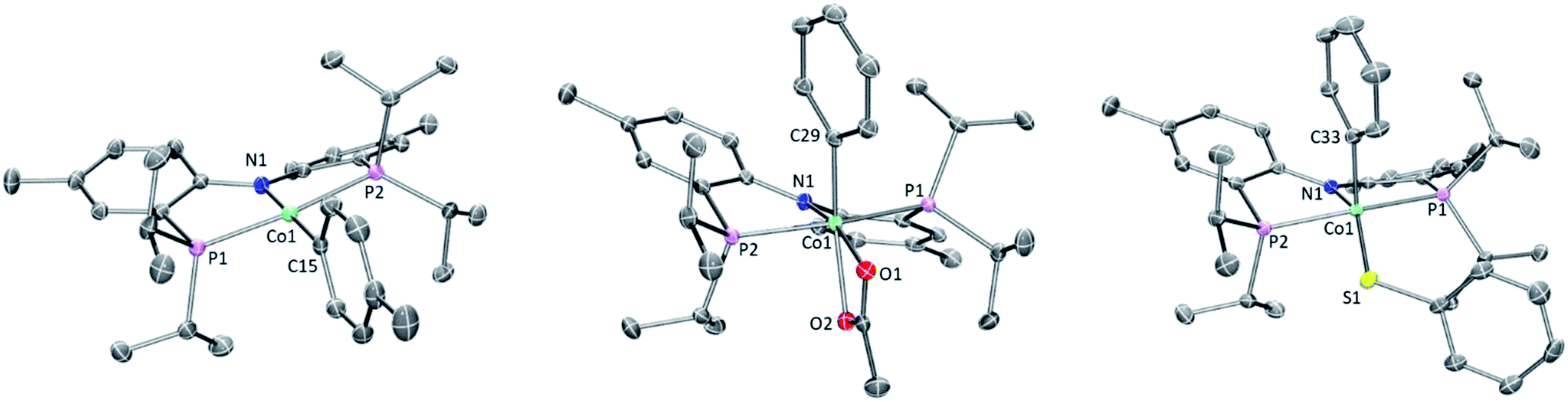 | ||
| Fig. 3 POV-ray renditions of ORTEP drawings (50% probability ellipsoids) of (PNP)Co(Tol), (PNP)Co(Ph)(OAc), and (PNP)Co(Ph)(SPh). All hydrogen atoms omitted. Selected bond distances (Å) and angles (degrees) for (PNP)Co(Tol) (2b, left): N1–Co1, 1.9262(17); P1–Co1, 2.1756(7); P2–Co1, 2.1849(7); C15–Co1, 1.939(2); N1–Co1–C15, 178.99(9); P1–Co1–P2, 172.63(3). Selected bond distances (Å) and angles (degrees) for (PNP)Co(Ph)(OAc) (4a, middle): N1–Co1, 1.9333(12); O1–Co1, 1.9896(12); O2–Co1, 2.1166(11); P1–Co1, 2.2619(7); P2–Co1, 2.2353(6); C29–Co1, 1.9403(15); C29–Co1–N1, 97.59(6); N1–Co1–O1, 165.17(5). Selected bond distances (Å) and angles (degrees) for (PNP)Co(Ph)(SPh) (6a, right): N1–Co1, 1.9497(18); P1–Co1, 2.2597(7); P2–Co1, 2.2313(7); C33–Co1, 1.933(2); S1–Co1, 2.2069(6); N1–Co1–C33, 98.54(8); N1–Co1–S1, 149.60(6). | ||
The structure of 4a is pseudo-octahedral about the metal center, with a κ2 acetate coordination. The two oxygens of the acetate are bound trans to two donors of markedly different trans-influence (amido N vs. phenyl C), which is reflected in the large difference between the two Co–O bond distances (ca. 0.13 Å). The geometry about Co in 6a is intermediate between square-pyramidal with the phenyl trans to the empty site and Y-shaped (with the thiolate at the base of the Y). The preference of low-spin five-coordinate d6 complexes for square-pyramidal and Y-shaped geometries have been discussed elsewhere.58 The angles, bond lengths, and orientation of the thioaryl ligand about the cobalt center for 6a are similar to those that we reported for (POCOP)Co(Ph)(SPh).43
Thermolysis of (PNP)CoIII(Ar)(SAr) complexes
Thermolysis of 6a in benzene led to the formation of 2a, 3a, and A in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 ratio (Scheme 5, top). Further investigation showed that this process is first order in 6a (Fig. S1†). Thermolysis of this complex in the presence of 1 eq. of 2,6-di-tert-butyl-4-methylphenol (BHT) resulted in the same distribution of products in the same time period, providing evidence against generation of free aryl radicals. Similarly, thermolysis of 6b in benzene resulted in the formation of 2b, 3b, and C in a 1:1:1 ratio (Scheme 5, bottom; Fig. S3 and S4†).
:1:1 ratio (Scheme 5, top). Further investigation showed that this process is first order in 6a (Fig. S1†). Thermolysis of this complex in the presence of 1 eq. of 2,6-di-tert-butyl-4-methylphenol (BHT) resulted in the same distribution of products in the same time period, providing evidence against generation of free aryl radicals. Similarly, thermolysis of 6b in benzene resulted in the formation of 2b, 3b, and C in a 1:1:1 ratio (Scheme 5, bottom; Fig. S3 and S4†).
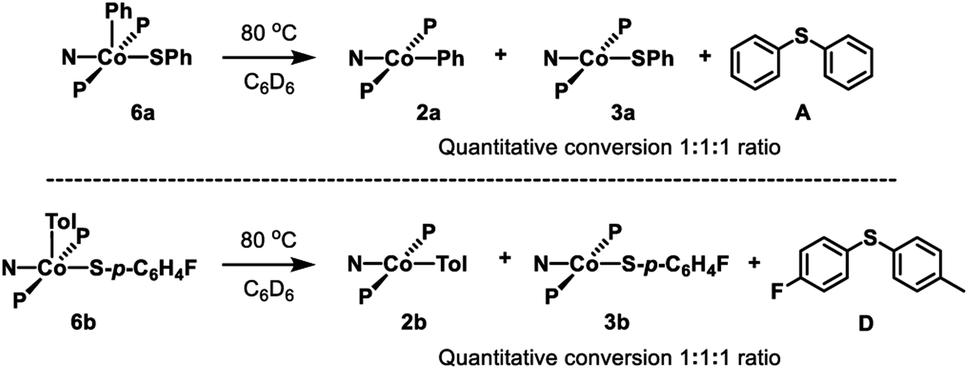 | ||
| Scheme 5 Top: Thermolysis of 6a and observed products. Bottom: Thermolysis of 6b and observed products. | ||
Attempting to determine whether the C–S bond formation step happened at a single Co center, thermolysis of 6a in a 1:1 ratio with 6b was carried out. In principle, strict unimolecular C–S reductive elimination should lead to only two diarylsulfide products as a result of this thermolysis. In the event, formation of four diarylsulfides was instead observed, along with the four expected CoII products: 2a, 2b, 3a, and 3b (Scheme 6). 19F NMR analysis during the course of the reaction at 80 °C revealed the formation of the Co(III) crossover product 6c (Fig. S5†). This suggested that during the thermolysis, thiolate ligands can exchange between the Co(III) centers prior to RE. This exchange would then lead to the formation of crossover diarylsulfides, even if RE happens unimolecularly, and thus prevent us from firmly excluding crossover via other pathways. Performing this reaction at double the initial concentration of CoIII complexes still showed 6c during thermolysis and a very similar distribution of CoII products and diaryl sulfides after the reaction had completed.
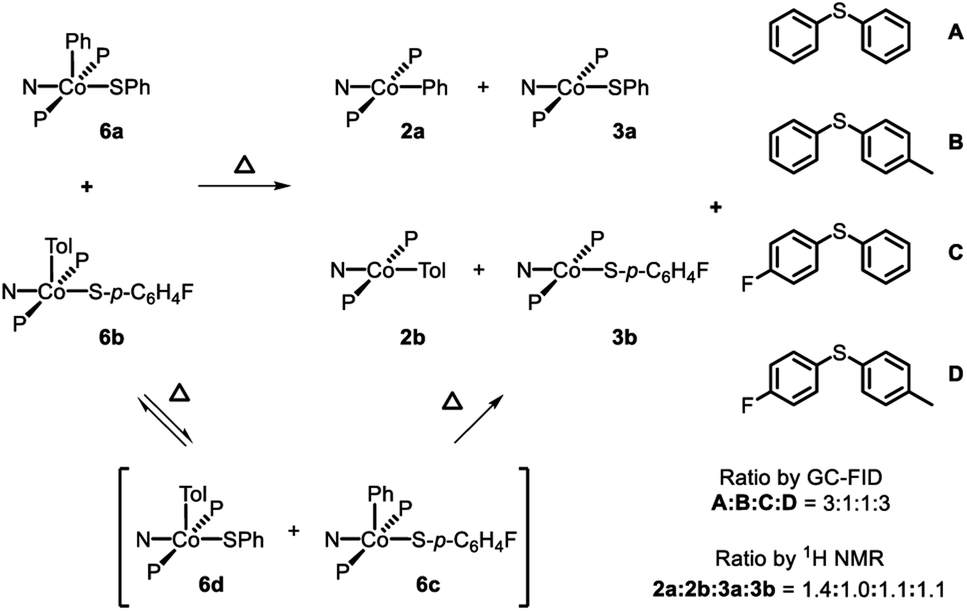 | ||
| Scheme 6 Thermolysis of a mixture of 6a and 6b. | ||
The thiolate exchange between Co(III) complexes could occur in at least59 two non-exclusive ways: (1) direct thiolate exchange between two Co(III) complexes, (2) via exchange between Co(II) and Co(III) thiolate complexes. To probe the ability of Co(III) to exchange thiolate ligands with Co(II), 6a was thermolyzed in the presence of 1 eq. 3b. In situ19F NMR observation at 80 °C revealed the formation 6c during the reaction (Scheme 7, top). After the thermolysis was complete, 6% of the total starting fluorinated thiolate was found as C demonstrating that CoII and CoIII can swap thiolates. The conditions required for the observation of thiolate swapping between two Co(III) complexes inevitably led to at least some Co(II) thiolate, which prevented us from establishing whether Co(III) thiolate complexes can exchange thiolates without the involvement of Co(II).
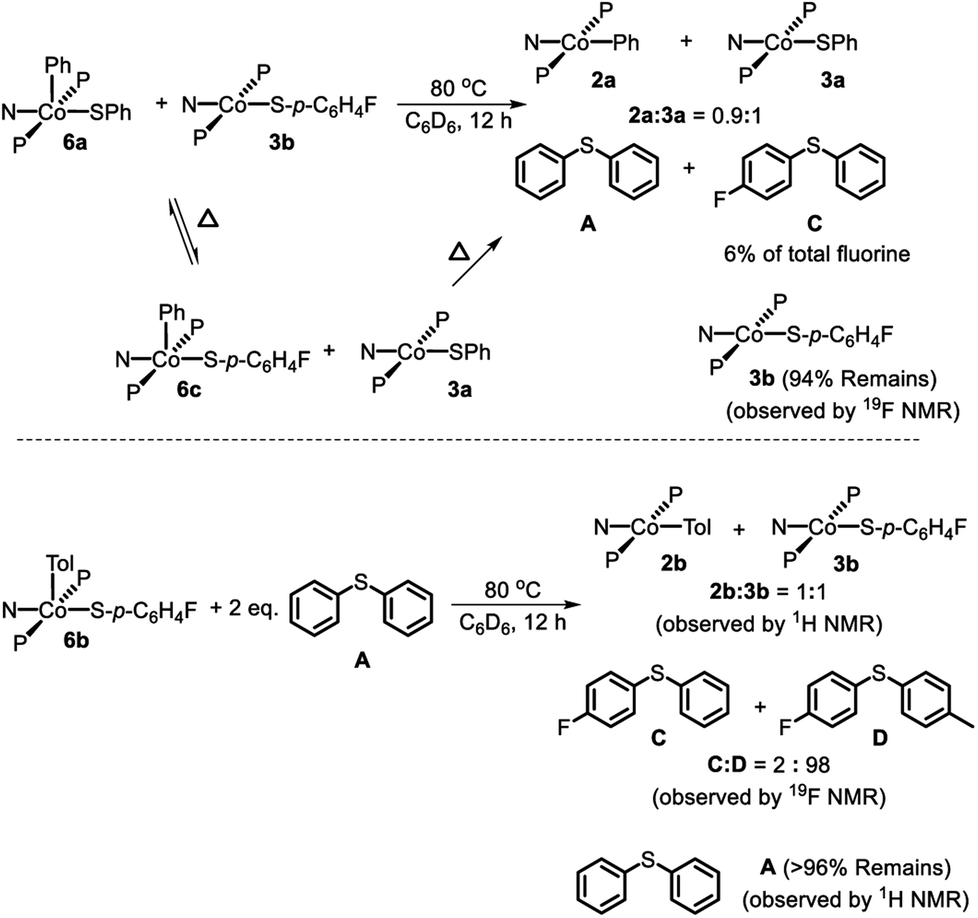 | ||
| Scheme 7 Top: Thermolysis of (PNP)Co(Ph)(SPh) (6a) in the presence of (PNP)Co(S-p-C6H4F) (3b) and observed products. Bottom: Thermolysis of 6b in the presence of A and observed products. | ||
Comproportionation hypothesis and reactions with CoI compounds
By analogy with our work on pincer rhodium complexes,13,15,19 we envisioned that after the concerted C–S reductive elimination from 6a, an unsaturated (PNP)Co fragment (12) would be generated. We further hypothesized that this unsaturated (PNP)Co species 12 undergoes rapid comproprotionation with the remaining 6a to generate the observed Co(II) products.60–62 To test this hypothesis, 7 was combined with 6a in benzene at ambient temperature. An immediate color change to green was observed upon mixing, indicating the formation of (PNP)CoII complexes (Scheme 8). 1H NMR spectroscopic observation confirmed the formation of 2a and 3a in a 1:1 ratio. Similarly, mixing freshly made 8 with 6a resulted in an immediate comproportionation producing 2a and 3a in a 1:1 ratio by 1H NMR spectroscopy (Scheme 8). In this case, free triphenylphosphine was also observed by 1H and 31P{1H} NMR spectroscopy.
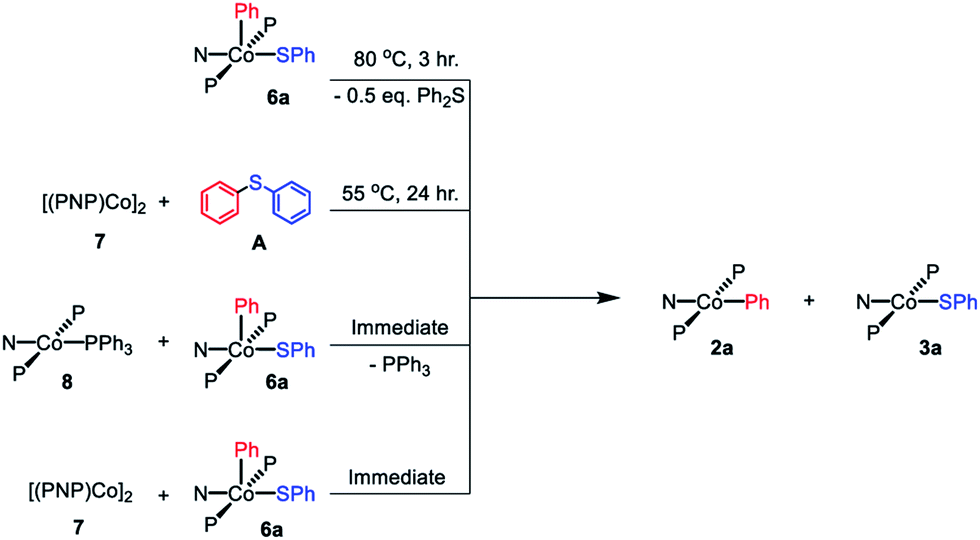 | ||
| Scheme 8 Reaction pathways shown to generate 1:1 mixture of 2a and 3a. | ||
Reversibility of C–S RE?
Treatment of 7 with diphenyl sulfide (A) and heating overnight63 in a 55 °C oil bath resulted in the formation of 2a and 3a in a 1:1 ratio (Scheme 8). This experiment shows that Co(I) here can cleave a C–S bond in a diarylsulfide, suggesting that C–S RE might be reversible. A related observation is that thermolysis of 6b in the presence of A resulted in the formation of a small amount of C in addition to D (Scheme 7, bottom); which can be interpreted as occasional trapping of a Co(I) species formed in the RE of D by Ph2S (A) as opposed to by the Co(III) starting material 6b. By way of a control experiment, thermolysis of 2b and 3b with A at 80 °C for 7 d resulted in no detectable change, establishing that CoII compounds do not react with a diarylsulfide.
In order to gain some insight into whether this C–S cleavage by Co(I) occurs via concerted OA, we subjected 7 to thermolysis with 2-isopropylphenyl-4′-fluorophenyl sulfide (E). Based on what we learned of the preferences of the (pincer)Rh systems in OA with aryl halides,13,14,19 it seemed reasonable to assume that the concerted OA mechanism with (PNP)Co should favor the C–S bond unencumbered by the ortho-isopropyl substituent (CF–S, Scheme 9). DFT calculations (see ESI†) predicted that homolytic cleavage of the CF–S bond is 1.6 kcal mol−1 less thermodynamically favorable than the cleavage of the C–S bond connecting to the 2-isopropylphenyl substituent (CR–S, Scheme 9). Thus, a radical abstraction mechanism for the C–S cleavage might be expected to favor the cleavage of CR–S.
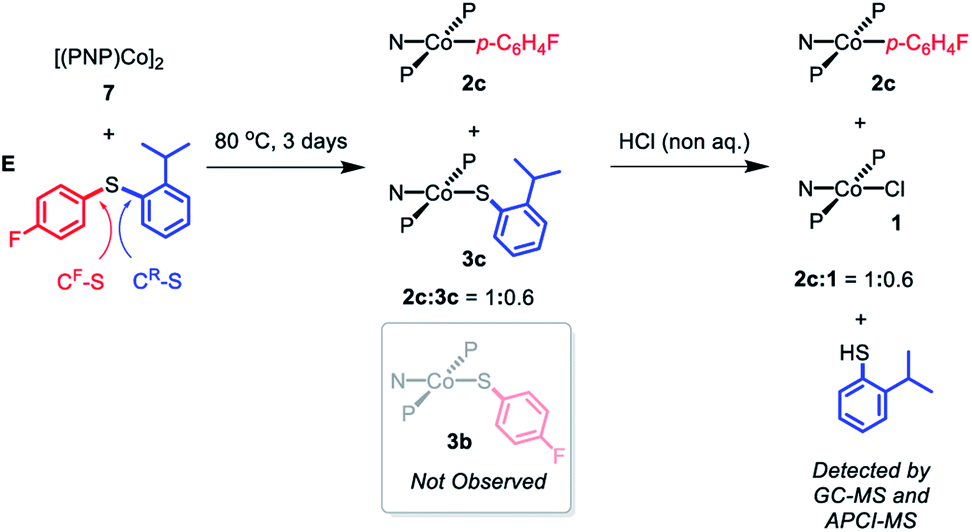 | ||
| Scheme 9 Thermolysis of [(PNP)Co]2 (7) with 2-isoropylphenyl-4′-fluorophenyl sulfide (E) and the following treatment with non-aqueous HCl. | ||
Heating the mixture of 7 with E in at 80 °C for three days resulted in the complete consumption of 7 with the formation of a 1:1 mixture of 2c and 3c (Scheme 9). Compound 3b was not observed by either 1H or 19F NMR spectroscopy. Treatment of this solution with anhydrous HCl resulted in the formation of 1 along with 2c with presumed liberation of 2-isopropylthiophenol. GC-MS and APCI-MS analysis of this solution revealed the formation of 2-isopropylthiophenol and E as the only two volatile components; p-FC6H4SH was not detected. We did not detect any biaryl or bisulfide products by GC-MS. These observations show that only the CF–S bond was cleaved, consistent with a concerted OA C–S activation pathway.64
DFT calculations
In order to gain better understanding of the system, DFT calculations were carried out with the Gaussian 09 (ref. 65) program to address a few salient points (Scheme 10). The geometries were optimized using [B3LYP/LANL2DZ/6-31G(d)] level of theory66 in the gas phase and the energies for these geometries were then determined with the [M06/SDD/6-311+G(d,p)] method67 incorporating the benzene solvent effect via the SMD model.68 Further details are given in the ESI.† We first evaluated the thermodynamics of the overall observed reaction. Conversion of 1 equiv. of 6a into a 0.5:0.5:0.5 mixture of 2a:3a:A was calculated to be favorable by −22.1 kcal mol−1 in free energy. The geometries of 2a and 3a in the doublet ground state were approximately square-planar geometry and consistent with the X-ray structure of 2b. The calculated structure of 6a reproduced the overall geometry determined in the XRD study, as well. Fig. 4 shows the calculated SOMO's and spin density profiles fo 2a and 3a. Interestingly, the nature of the SOMO's in these two compounds differs. In 2a, it is essentially a pure dz2, whereas in 3a it is primarily dxz (x axis along Co–S) with small contributions from the amido and thiolate ligands. This disparity reflects the fact that a stronger σ-donor Ph elevates the energy of dz2 in 2a relative to dxz. In either case, the SOMO is firmly metal-based.
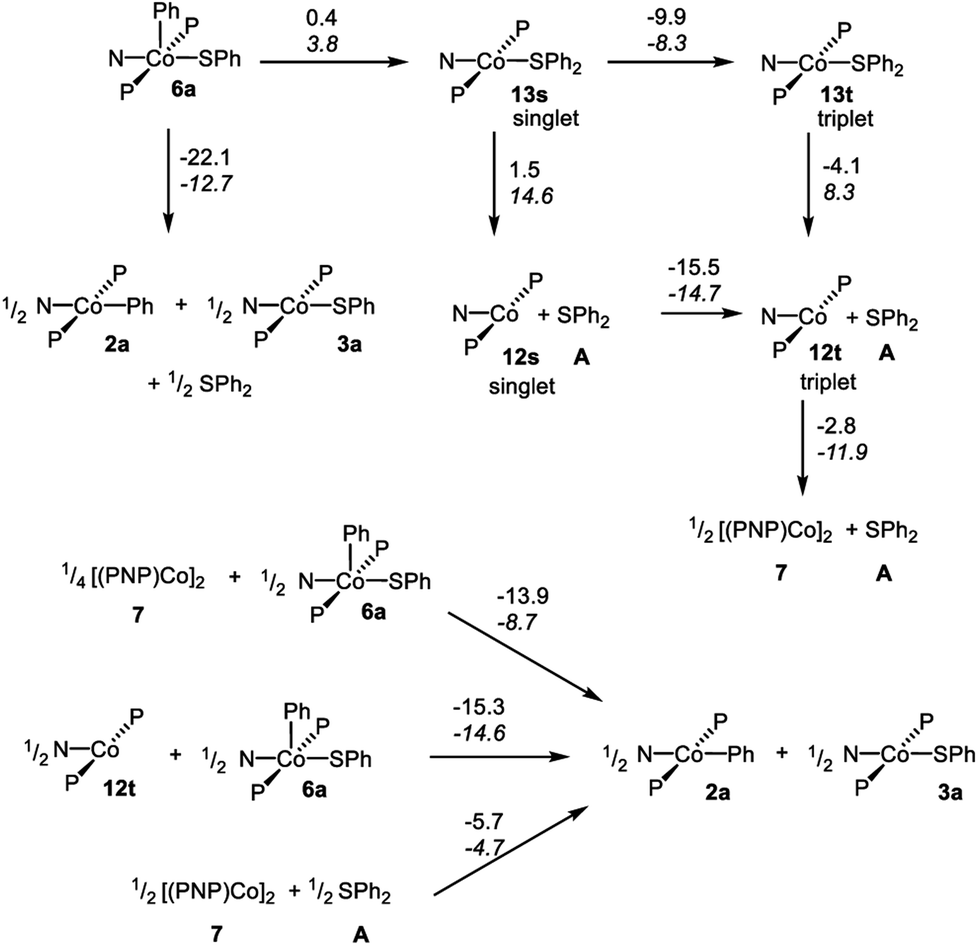 | ||
| Scheme 10 DFT calculated energies for the various transformations. Reaction free energies (at 298 K) are given over the arrows on top; reaction enthalpies in italic below. All the energies are in kcal mol−1, normalized per one mole of Co. | ||
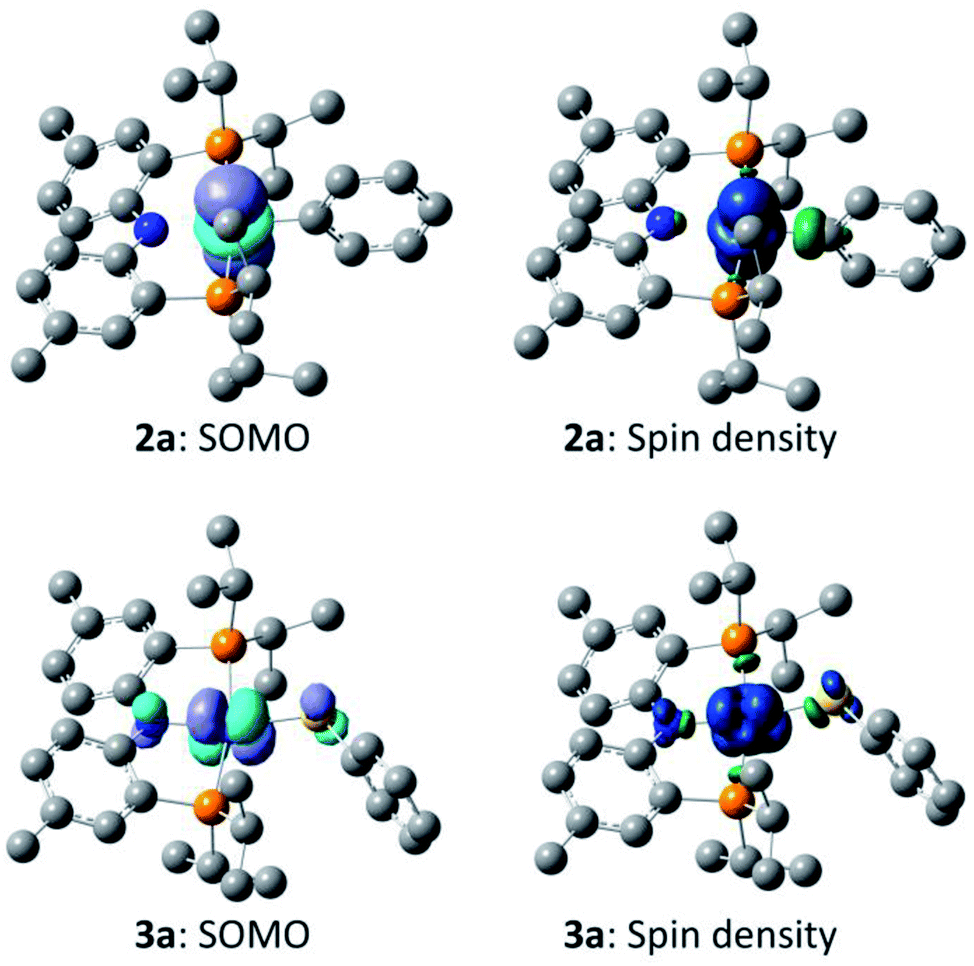 | ||
| Fig. 4 Depictions of the calculated singly occupied molecular orbital (SOMO) for 2a and 3a (left, isovalue 0.04), and of the calculated spin-density plots for 2a and 3a (right, isovalue 0.004). Hydrogen atoms are omitted for clarity. | ||
We then considered the monomeric Co(I) intermediates in the reaction. Both the “naked” (PNP)Co fragment 12 and its SPh2 adduct 13 were calculated to favor a triplet ground state (by 15.5 kcal mol−1 and 9.9 kcal mol−1 in free energy, respectively). The geometry of the triplet (PNP)Co(SPh2) (13t) about Co is decidedly not planar, and can be described as attempting to approach tetrahedral within the constraint of the pincer. The array of donor atoms in 13t is the same as in the low-spin doublet 3a, but all the calculated bond distances to Co are considerably longer, especially that for the C–S bond (2.538 Å in 13tvs. 2.264 Å in 3a).
The reductive C–S coupling in 6a to give singlet (PNP)Co(SPh2) (13s) is nearly ergoneutral, but the conversion of 6a to 13t is favorable on the enthalpy (by 4.5 kcal mol−1) and on the free energy (by 9.5 kcal mol−1) surfaces. The dissociation of SPh2 from 13t to give triplet 12t and free SPh2 is endothermic, but is of course favored entropically, resulting in a favorable free energy of dissociation. Thus the complete RE from 6a to give 12t and A is exoergic by 13.6 kcal mol−1, but that is less favorable than the formation of a mixture of 2a:3a:A.
The dimerization of 12t to form 7 was calculated to be enthalpically favorable, but disfavored entropically and overall slightly exoergic (by 2.8 kcal mol−1 per Co). This is consistent with the experimental observation of the dimer 7 as the ground state.
The thermodynamics of the standalone Co(I)/Co(III) comproprotionation reactions were calculated to be consistent with our hypothesis outlined above. The reaction of 12t with 6a to give 2a and 3a was found to be exothermic and exoergic (by −14.6 and −15.3 kcal mol−1, respectively). A similar comproportionation starting from 7 instead of 12t was also found to be favorable (by −13.9 kcal mol−1 per mole of Co).
The substantial (ca. 15 kcal mol−1) calculated preference for the triplet state of (PNP)Co (12t) is at odds with the recent report by Lee and coworkers, which presented three-coordinate (PNP5)Co as a singlet species (Fig. 5).69 This interpretation by Lee et al. is also at odds with the unambiguously established triplet ground states for the (PNP3)Co and (PNP4)Co by the Caulton70 and Gade groups,52 respectively (Fig. 5). Both (PNP3)Co70 and (PNP4)Co52 were isolated and fully characterized, including by X-ray crystallography, magnetic moment measurement, as well as by elemental analysis for (PNP4)Co. (PNP5)Co was purported to be isolated, but no structural determination or magnetic moment was reported, and satisfactory elemental analysis was not obtained. (PNP5)Co was analyzed by DFT calculations as a singlet, but the calculations examining the viability of the triplet state were not carried out. Given these facts and the very close similarity of the PNP and PNP4 ligands, it would be very surprising indeed if they led to different spin state preferences in 12vs. (PNP5)Co. Although we do not observe free 12, it is also worth pointing out that even its adduct with PPh3 (8) does not present as a low-spin complex based on the appearance of its NMR spectra. It is possible that the (PNP5)Co system needs to be reexamined more closely.
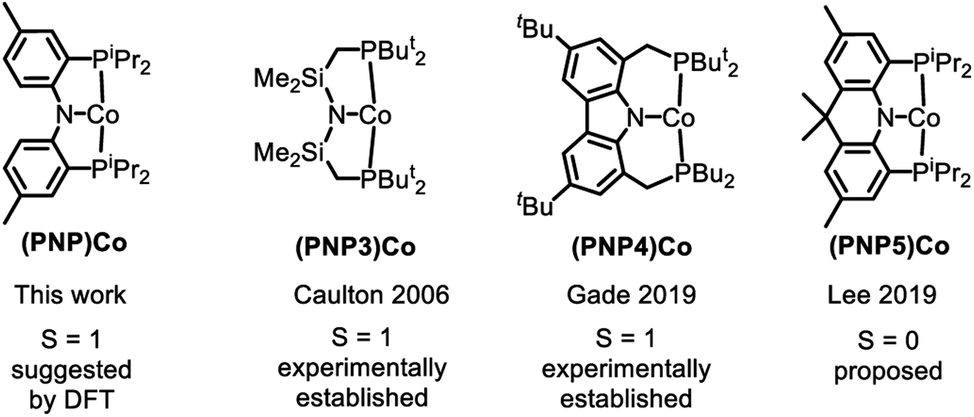 | ||
| Fig. 5 Examples of three-coordinate Co complexes supported by various anionic PNP ligands. | ||
For the C–S reductive coupling en route to 13 from 6a, a transition state was found, lying 24.8 kcal mol−1 above 6a in free energy (TS, Fig. 6). This transformation requires spin crossover in the process, which we propose happens after the singlet TS on the reaction coordinate. A singlet state of the reductive coupling product (PNP)Co(SPh2) (13s) is only 0.4 kcal mol−1 endergonic relative to 6a. However, 13t is lower in energy still. The activation barrier magnitude calculated by DFT agrees reasonably well with the experimental observations. The observed half-life of 0.6 h at 80 °C for 6a corresponds to ca. 26 kcal mol−1 in free energy barrier 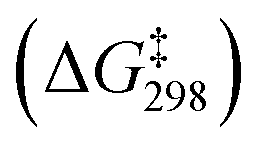 .
.
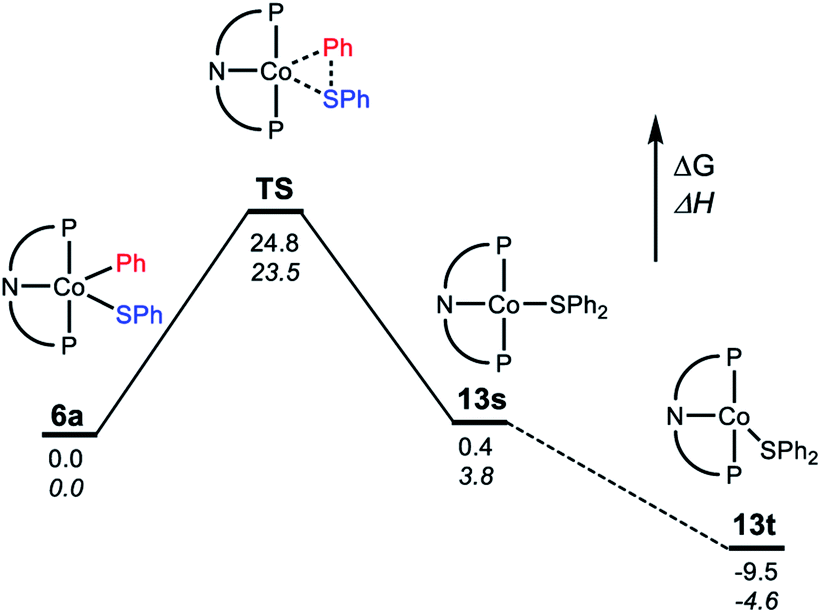 | ||
| Fig. 6 Representation of the reaction coordinate for the RE of SPh2 (A) from (PNP)Co(Ph)(SPh) (6a). Energy values are given below the bars: free energy on top, enthalpy on the bottom in italic. | ||
Examination of the geometry of TS (Fig. 7) shows that it can be thought of as reflecting the migration of the Co-bound Ph group onto the S atom, which in turn is brought more closely into the plane defined by P/N/P/Co. The Co–S distance in TS is actually slightly shorter than in 6a, and much shorter than calculated in 13t. The Co–CPh distance elongates by ca. 0.13 Å in TS (2.055 Å) vs.6a (1.929 Å), while the newly forming C–S distance (2.085 Å) is about 0.29 Å longer than the expected C–S distances of ca. 1.80 Å in Ph2S or its complexes. The other geometric feature of TS that needs to be emphasized is the necessary rotation of the Co-bound phenyl ring from edge-on relative to S in 6a to side-on in TS. The hindrance of this rotation by the iPr groups is a major contributor to the magnitude of the activation barrier. This is a rather general observation for the reductive elimination of R–X from five-coordinate d6 complexes (pincer)M(R)(X) where R = aryl or alkenyl, first articulated by Goldman and Krogh-Jespersen for the (PCP)Ir system.71 We previously discussed this issue for the closely related RE reactions from (pincer)Rh(Ar)(X) complexes.15,16,19
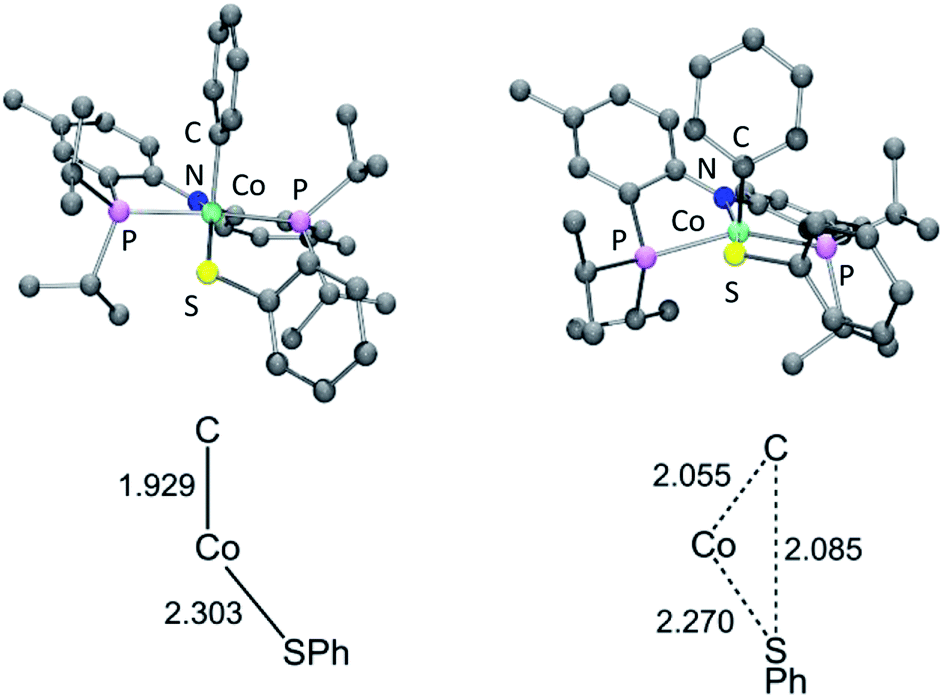 | ||
| Fig. 7 Top: DFT calculated structures of 6a (left) and TS (right). Bottom: Key distances (in Å) in 6a and TS; bottom structures not to scale. | ||
Lastly, we considered the experimental observations of the apparent reversibility of C–S RE in reactions of A with Co(I) complexes. Thermodynamically, the experimentally observed reaction of [(PNP)Co]2 with Ph2S to give 2a and 3a was indeed calculated to be favorable (Scheme 10). At first glance, the microscopic reverse C–S OA might appear kinetically feasible as the energy of 13s is similar to that of 6a. However, given that (1) 13t, (2) 12t + free SPh2, and (3) 7 + free SPh2 are all considerably lower in energy than 13s, the barriers for the microscopic reverse C–S OA starting from these states are prohibitively high.
In rationalization, two possibilities might be considered. First, it is possible that our DFT calculations do not accurately describe the relative energies of compounds in different spin states. The second option is that the reaction of A with 7 proceeds as an C–S OA within the dimer, without the formation of monomeric intermediates. The putative single C–S OA dicobalt product may then comproportionate intramolecularly to give 2a and 3a without the intermediacy of free 6a. This reaction pathway would thus not be a microscopic reverse of the monomolecular C–S RE. The complexity of the many potential pathways that would need to be considered to properly analyze the reaction of [(PNP)Co]2 with A has deterred us from pursuing this problem computationally within the scope of this report.
Conclusion
In summary, (PNP)Co complexes in the +1, +2, and +3 oxidations states relevant to potential cross-coupling reactions were prepared and fully characterized. A switch to a PNP ligand prevented intramolecular reductive elimination of the Co–Ar unit with the central donor of the pincer and permitted observation of concerted C–S reductive elimination. However, it appears that the PNP supporting ligand does not have a strong enough ligand field strength to prevent promotion of an electron from the (PNP)Co fragment to a triplet ground state. This fundamental realization is probably related to the swift Co(I)/Co(III) comproportionation investigated in this work, which removes potentially catalytically competent odd-oxidation state cobalt complexes from the reaction.Interestingly, the Co(I)/Co(III) comproportionation observed here directly mimics the Ni(0)/Ni(II) comproportionation observed and studied by the Hazari group.72 The similarity further underscores the close parallels in reactivity that exist between group 9 metals in the d8/d6 manifold and the group 10 metals in the d10/d8 manifold, as well as the contrast between the 3d metals (Co or Ni comproportionate) and the 4d metals (Rh or Pd do not comproprotionate) within the same group.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Welch Foundation (grant A-1717 to O. V. O.) for support of this research. This work was also supported by the startup of Harbin Institute of Technology (Shenzhen) through the Talent Development Starting Fund from Shenzhen Government. Computer time made available by the National Supercomputing Center of China in Shenzhen (Shenzhen Cloud Computing Center) is gratefully acknowledged. We thank Samuel R. Lee for assistance with the composition of the manuscript.Notes and references
- A. N. Desnoyer and J. A. Love, Chem. Soc. Rev., 2017, 46, 197–238 RSC.
- V. Ritleng, M. Henrion and M. J. Chetcuti, ACS Catal., 2016, 6, 890–906 CrossRef CAS.
- Q. Zeng, L. Zhang and Y. Zhou, Chem. Rec., 2018, 18, 1278–1291 CrossRef CAS PubMed.
- J. F. Hartwig, Nature, 2008, 455, 314–322 CrossRef CAS PubMed.
- Handbook of organopalladium chemistry for organic synthesis, ed. E. Negishi and A. de Meijere, Wiley-Interscience, New York, 2002 Search PubMed.
- S. Z. Tasker, E. A. Standley and T. F. Jamison, Recent advances in homogeneous nickel catalysis, Nature, 2014, 509, 299–309 CrossRef CAS PubMed.
- N. Hazari, P. R. Melvin and M. Mohadjer Beromi, Nat. Rev. Chem., 2017, 1, 1–16 CrossRef.
- Copper-Mediated Cross-Coupling Reactions, ed. G. Evano and N. Blanchard, John Wiley & Sons, Inc., 2014 Search PubMed.
- S. Thapa, B. Shrestha, S. K. Gurunga and R. Giri, Org. Biomol. Chem., 2015, 13, 4816–4827 RSC.
- J. D. Hayler, D. K. Leahy and E. M. Simmons, Organometallics, 2019, 38, 36–46 CrossRef CAS.
- Catalysis without Precious Metals, ed. R. M. Bullock, Wiley-VCH Verlag GmbH & Co. KGaA, 2010 Search PubMed.
- P. Chirik and R. Morris, Acc. Chem. Res., 2015, 48, 2495 CrossRef CAS PubMed , and the other articles in this special issue.
- S. Gatard, R. Çelenligil-Çetin, C. Guo, B. M. Foxman and O. V. Ozerov, J. Am. Chem. Soc., 2006, 128, 2808–2809 CrossRef CAS PubMed.
- M. Puri, S. Gatard, D. A. Smith and O. V. Ozerov, Organometallics, 2011, 30, 2472–2482 CrossRef CAS.
- S. D. Timpa, C. J. Pell, J. Zhou and O. V. Ozerov, Organometallics, 2014, 33, 5254–5262 CrossRef CAS.
- S. D. Timpa, J. Zhou, N. Bhuvanesh and O. V. Ozerov, Organometallics, 2014, 33, 6210–6217 CrossRef CAS.
- S. D. Timpa, C. M. Fafard, D. E. Herbert and O. V. Ozerov, Dalton Trans., 2011, 40, 5426–5429 RSC.
- S. Gatard, C. Guo, B. M. Foxman and O. V. Ozerov, Organometallics, 2007, 26, 6066–6075 CrossRef CAS.
- S. D. Timpa, C. J. Pell and O. V. Ozerov, J. Am. Chem. Soc., 2014, 136, 14772–14779 CrossRef CAS PubMed.
- M. Sayah and M. G. Organ, Chem.–Eur. J., 2011, 17, 11719–11722 CrossRef CAS PubMed.
- J. F. Hartwig, Acc. Chem. Res., 2008, 41, 1534–1544 CrossRef CAS PubMed.
- P. J. Chirik, Angew. Chem., Int. Ed., 2017, 56, 5170–5181 CrossRef CAS PubMed.
- G. Cahiez and A. Moyeux, Chem. Rev., 2010, 110, 1435–1462 CrossRef CAS PubMed.
- Y.-C. Wong, T. T. Jayanth and C.-H. Cheng, Org. Lett., 2006, 8, 5613–5616 CrossRef CAS PubMed.
- M. Amatore and C. Gosmini, Angew. Chem., Int. Ed., 2008, 47, 2089–2092 CrossRef CAS PubMed.
- H. A. Duong, Z.-H. Yeow, Y.-L. Tiong, N. H. B. M. Kamal and W. Wu, J. Org. Chem., 2019, 84, 12686–12691 CrossRef CAS PubMed.
- F. H. Lutter, L. Grokenberger, P. Spieß, J. M. Hammann, K. Karaghiosoff and P. Knochel, Angew. Chem., Int. Ed., 2020, 59, 5546–5550 CrossRef CAS PubMed.
- S. Liu, W. Huang, D. Wang, P. Wei and Q. Shen, Org. Chem. Front., 2019, 6, 2630–2634 RSC.
- M. R. Brennan, D. Kim and A. R. Fout, Chem. Sci., 2014, 5, 4831–4839 RSC.
- J. M. Neely, M. J. Bezdek and P. J. Chirik, ACS Cent. Sci., 2016, 2, 935–942 CrossRef CAS PubMed.
- H. Xu and W. H. Bernskoetter, J. Am. Chem. Soc., 2011, 133, 14956–14959 CrossRef CAS PubMed.
- K. Tokmic, C. R. Markus, L. Zhu and A. R. Fout, J. Am. Chem. Soc., 2016, 138, 11907–11913 CrossRef CAS PubMed.
- A. D. Ibrahim, S. W. Entsminger and A. R. Fout, ACS Catal., 2016, 6, 3589–3593 CrossRef CAS.
- K. Tokmic and A. R. Fout, J. Am. Chem. Soc., 2016, 138, 13700–13705 CrossRef CAS PubMed.
- A. D. Ibrahim, S. W. Entsminger and A. R. Fout, ACS Catal., 2017, 7, 3730–3734 CrossRef CAS.
- S. R. Muhammed, J. W. Nugent, K. Tokmik, L. Zhu, J. Mahmoud and A. R. Fout, Organometallics, 2019, 38, 3132–3138 CrossRef.
- N. G. Léonard, W. N. Palmer, M. R. Friedfeld, M. J. Bezdek and P. J. Chirik, ACS Catal., 2019, 9, 9034–9044 CrossRef.
- J. V. Obligacion, S. P. Semproni and P. J. Chirik, J. Am. Chem. Soc., 2016, 138, 10645–10653 CrossRef CAS PubMed.
- J. V. Obligacion and P. J. Chirik, ACS Catal., 2017, 7, 4366–4371 CrossRef CAS PubMed.
- J. V. Obligacion, S. P. Semproni and P. J. Chirik, J. Am. Chem. Soc., 2014, 136, 4133–4136 CrossRef CAS PubMed.
- R. Arevalo and P. J. Chirik, J. Am. Chem. Soc., 2019, 141, 9106–9123 CrossRef PubMed.
- T. P. Pabst, J. V. Obligacion, É. Rochette, I. Pappas and P. J. Chirik, J. Am. Chem. Soc., 2019, 141, 15378–15389 CrossRef CAS PubMed.
- B. J. Foley, C. M. Palit, S. D. Timpa and O. V. Ozerov, Organometallics, 2018, 37, 3803–3812 CrossRef CAS.
- A. R. Fout, F. Basuli, H. Fan, J. Tomaszewski, J. C. Huffman, M. -H. Baik and D. J. Mindiola, Angew. Chem., Int. Ed., 2006, 45, 3291–3295 CrossRef CAS PubMed.
- L. M. Guard, T. J. Hebden, D. E. Linn and D. M. Heinekey, Organometallics, 2017, 36, 3104–3109 CrossRef CAS.
- V. M. Krishnan, H. D. Arman and Z. J. Tonzetich, Dalton Trans., 2018, 47, 1435–1441 RSC.
- S. Kuriyama, K. Arashiba, H. Tanaka, Y. Matsuo, K. Nakajima, K. Yoshizawa and Y. Nishibayashi, Angew. Chem., Int. Ed., 2016, 55, 14291–14295 CrossRef CAS PubMed.
- S. S. Rozenel, R. Padilla and J. Arnold, Inorg. Chem., 2013, 52, 11544–11550 CrossRef CAS PubMed.
- T. M. Townsend, W. H. Bernskoetter, G. W. Brudvig, N. Hazari, H. M. C. Lant and B. Q. Mercado, Polyhedron, 2020, 177, 114308 CrossRef CAS.
- M. D. Fryzuk, D. B. Leznoff, R. C. Thompson and S. J. Rettig, J. Am. Chem. Soc., 1998, 120, 10126–10135 CrossRef CAS.
- M. J. Ingleson, M. Pink and K. G. Caulton, J. Am. Chem. Soc., 2006, 128, 4248–4249 CrossRef CAS PubMed.
- L. S. Merz, C. K. Blasius, H. Wadepohl and L. H. Gade, Inorg. Chem., 2019, 58, 6102–6113 CrossRef CAS PubMed.
- Steric bulk of the side donors in the pincer ligand may also be a factor that influences the spin state of the Co complex, see: S. P. Semproni, C. Milsmann and P. J. Chirik, J. Am. Chem. Soc., 2014, 136, 9211–9224 CrossRef CAS PubMed.
- J. J. Davidson, J. C. DeMott, C. Douvris, C. M. Fafard, N. Bhuvanesh, C.-H. Chen, D. E. Herbert, C.-I. Lee, B. J. McCulloch, B. M. Foxman and O. V. Ozerov, Inorg. Chem., 2015, 54, 2916–2935 CrossRef CAS PubMed.
- Compounds 5b and 6c were not isolated.
- Y. Zhu, D. A. Smith, D. E. Herbert, S. Gatard and O. V. Ozerov, Chem. Commun., 2012, 48, 218–220 RSC.
- S. P. Semproni, C. C. H. Atienza and P. J. Chirik, Chem. Sci., 2014, 5, 1956–1960 RSC.
- (a) W. H. Lam, S. Shimada, A. S. Batsanov, Z. Lin, T. B. Marder, J. A. Cowan, J. A. K. Howard, S. A. Mason and G. J. McIntyre, Organometallics, 2003, 22, 4557–4568 CrossRef CAS; (b) I. E.-I. Rachidi, O. Eisenstein and Y. A. Jean, New J. Chem., 1990, 14, 671–677 CAS; (c) J.-F. Riehl, Y. Jean, O. Eisenstein and M. Pelissier, Organometallics, 1992, 11, 729–737 CrossRef CAS; (d) M. Olivan, O. Eisenstein and K. G. Caulton, Organometallics, 1997, 16, 2227–2229 CrossRef CAS.
- It also cannot be excluded that undetectable traces of NaSAr or HSAr may be catalyzing the thiolate exchange.
- For an example of CoI/CoIII comproportionation, see: S. M. Rummelt, H. Zhong, N. G. Léonard, S. P. Semproni and P. J. Chirik, Organometallics, 2019, 38, 1081–1090 CrossRef CAS PubMed.
- For an example of Co0/CoII comproportionation, see: J. V. Obligacion, H. Zhong and P. J. Chirik, Isr. J. Chem., 2017, 57, 1032–1036 CrossRef CAS PubMed.
- For a study of CoII disproportionation, see: S. M. Socol and J. G. Verkade, Inorg. Chem., 1986, 25, 2658–2663 CrossRef CAS.
- No observable change was noted within 10 min after mixing the reagents at RT.
- For a recent study of non-concerted OA of benzyl halides to CoI, see: C. Sandford, L. R. Fries, T. E. Ball, S. D. Minteer and M. S. Sigman, J. Am. Chem. Soc., 2019, 141, 18877–18889 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- (a) A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS PubMed; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- Y. Zhao and D. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6796 CrossRef CAS PubMed.
- J. Choi and Y. A. Lee, Angew. Chem., Int. Ed., 2019, 58, 6938–6942 CrossRef CAS PubMed.
- M. Ingleson, H. Fan, M. Pink, J. Tomaszewski and K. G. Caulton, J. Am. Chem. Soc., 2006, 128, 1804–1805 CrossRef CAS PubMed.
- R. Ghosh, X. Zhang, P. Achord, T. J. Emge, K. Krogh-Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2007, 129, 853–866 CrossRef CAS PubMed.
- M. Mohadjer Beromi, A. Nova, D. Balcells, A. M. Brasacchio, G. W. Brudvig, L. M. Guard, N. Hazari and D. J. Vinyard, J. Am. Chem. Soc., 2017, 139, 922–936 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and pictorial NMR spectra, details of the computational studies and the coordinate files. CCDC 1868266–1868268. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc01813a |
| This journal is © The Royal Society of Chemistry 2020 |