
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure and redox tuning of gas adsorption properties in calixarene-supported Fe(II)-based porous cages†
Meaghan M.
Deegan
 a,
Tonia S.
Ahmed
b,
Glenn P. A.
Yap
a and
Eric D.
Bloch
*a
a,
Tonia S.
Ahmed
b,
Glenn P. A.
Yap
a and
Eric D.
Bloch
*a
aDepartment of Chemistry & Biochemistry, University of Delaware, Newark, DE 19716, USA. E-mail: edb@udel.edu
bDepartment of Chemistry and Chemical Biology, Harvard University, 12 Oxford Street, Cambridge, MA 02138, USA
First published on 4th May 2020
Abstract
We describe the synthesis of Fe(II)-based octahedral coordination cages supported by calixarene capping ligands. The most porous of these molecular cages has an argon accessible BET surface area of 898 m2 g−1 (1497 m2 g−1 Langmuir). The modular synthesis of molecular cages allows for straightforward substitution of both the bridging carboxylic acid ligands and the calixarene caps to tune material properties. In this context, the adsorption enthalpies of C2/C3 hydrocarbons ranged from −24 to −46 kJ mol−1 at low coverage, where facile structural modifications substantially influence hydrocarbon uptakes. These materials exhibit remarkable stability toward oxidation or decomposition in the presence of air and moisture, but application of a suitable chemical oxidant generates oxidized cages over a controlled range of redox states. This provides an additional handle for tuning the porosity and stability of the Fe cages.
Introduction
While the early development of metal–organic frameworks (MOFs) proceeded predominantly for non-redox active metals, recent research efforts have targeted the development of MOFs that incorporate redox-active metal sites.1 Our group is interested in expanding the chemistry of molecular metal–organic polyhedra (MOPs) or porous coordination cages (PCCs), whose development has lagged substantially behind MOFs, despite early developments in these areas proceeding over a similar time period.2–6 Relative to their extended framework counterparts, molecular cages offer comparable prospective structural diversity, with their potential solubility providing advantages for cage synthesis, purification, and processing. Like MOFs, porous cages predominantly incorporate non-redox active metal nodes.7 Recent reports of paddlewheel-based cages with redox active Cr, Mo, and Ru sites have been described, though their redox chemistry is not generally emphasized.8–13 Only octahedral and cuboctahedral Ru cages have been synthesized in multiple redox states, accessed through cage self-assembly from paddlewheel precursors in different oxidation states.14,15 In another case, the redox reactivity of Cr(II) paddlewheel sites promoted selective O2 adsorption,16,17 which, in this case, results in irreversible porosity loss and decomposition of the cuboctahedral cage structures.11Of note, porous cages that incorporate potentially redox active Fe(II)-based metal nodes remain elusive to date.7,15,18 Previous work has shown that derivatives of thiacalix[4]arene and sulfonylcalix[4]arene ligands support coordinatively saturated tetranuclear, square metal clusters.19 When these calixarene-capped clusters are generated under solvothermal conditions in the presence of a variety of multitopic, predominantly carboxylate-based ligands, the self-assembly of molecular coordination cages is accessible (Fig. 1), with structures of this type sometimes referred to as metal–organic supercontainers (MOSCs).20–23 While reported cages of this type have most frequently incorporated non-redox active Co(II), Ni(II), and Mg(II)-based cluster nodes, related structures can be accessed for Fe-based systems.24–26
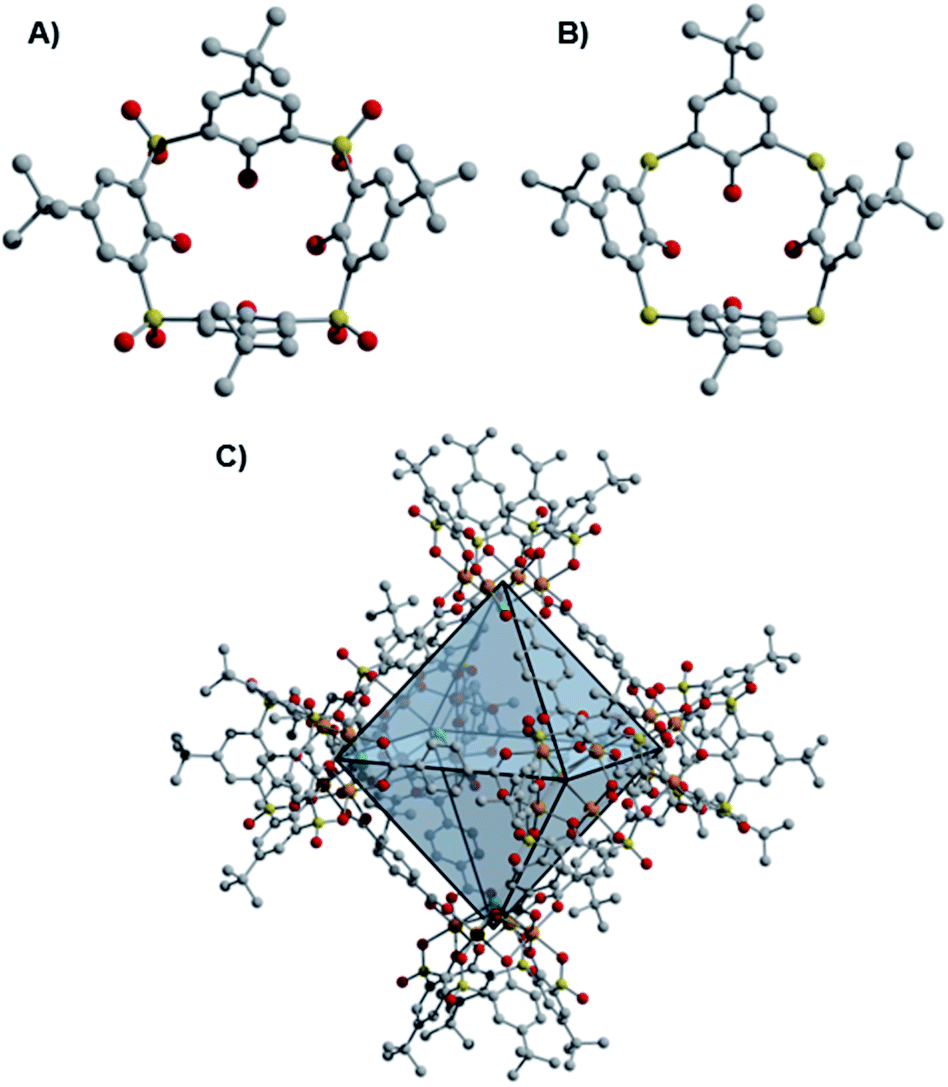 | ||
| Fig. 1 Structures of the sulfonylcalix[4]arene (sc4a, (A)) and thiacalix[4]arene (tc4a, (B)) capping ligands, as well as [(Fe24Cl6(sc4a)6(bdc)12)]6− ((sc4a)Fe(bdc); bdc2− = terephthalate, (C)) where orange, yellow, green, gray, and red spheres represent iron, sulfur, chlorine, carbon, and oxygen, respectively. Hydrogen atoms and charge-balancing cations are omitted for clarity. | ||
Herein, we describe the synthesis of octahedral, Fe(II)-based coordination cages supported by calixarene capping ligands. Activation of these cages under suitable conditions provides access to porous Fe(II)-based cages, that exhibit BET surface areas as high as 898 m2 g−1 (1497 m2 g−1 Langmuir; Ar). The modular assembly of the molecular cages allows for facile tuning of pore size and connectivity (edge vs. face-linked octahedra) without significantly perturbing their octahedral structures. The hydrocarbon adsorption properties of these materials were explored to examine the extent to which these small structural modifications can be leveraged to tune gas adsorption selectivity. Relative to previously reported Fe(II) MOFs, the Fe(II) cages described herein exhibit remarkable stability toward decomposition upon exposure to air and moisture. To access the Fe(II)/(III) redox couple in these materials, we instead apply a strong chemical oxidant (magic blue), which ultimately provides access to oxidized materials with partial or complete incorporation of Fe(III). This further provides a handle for tuning cage porosity, and factors that contribute to the observed porosity trend upon oxidation are examined.
Results and discussion
Initial access to the chemistry of Fe(II)-based cages was provided by screening conditions suitable for the assembly of t-butyl substituted sulfonylcalix[4]arene (sc4a) and thiacalix[4]arene (tc4a)-capped Fe cages. For the sc4a-capped Fe cages, heating the sc4a ligand with anhydrous FeCl2 at 100 °C in DMF, followed by the addition of bridging carboxylate ligands and EtOH (25–50%) afforded cage products as highly crystalline solids under solvothermal conditions (see ESI† for details). This approach generates a face-linked octahedral cage [(Fe24Cl6(sc4a)6(btc)8)]6− ((sc4a)Fe(btc)) with trimesic acid (H3btc) bridging ligands (Fig. 2) and an expanded structurally-related cage [(Fe24Cl6(sc4a)6(tatb)8)]6− ((sc4a)Fe(tatb)) with triazinetrisbenzoic acid (H3tatb) bridges (Fig. 3). Addition of the ditopic bridging ligand, terephtalic acid (H2bdc) generates a cage [(Fe24Cl6(sc4a)6(bdc)12)]6− ((sc4a)Fe(bdc)) with a similar overall octahedral structure, with the tetranuclear metal nodes instead connected along the edges of the octahedron (Fig. 1). Cage assembly upon substitution of the calixarene ligand with tc4a required distinct reaction conditions. In this case, prolonged heating of a mixture of tc4a, FeCl2, and H3btc at 130 °C in DMF followed by the addition of EtOH generates [(Fe24Cl6(tc4a)6(btc)8)]6− ((tc4a)Fe(btc)) as a bright yellow crystalline solid after heating at 100 °C for an additional 2 d.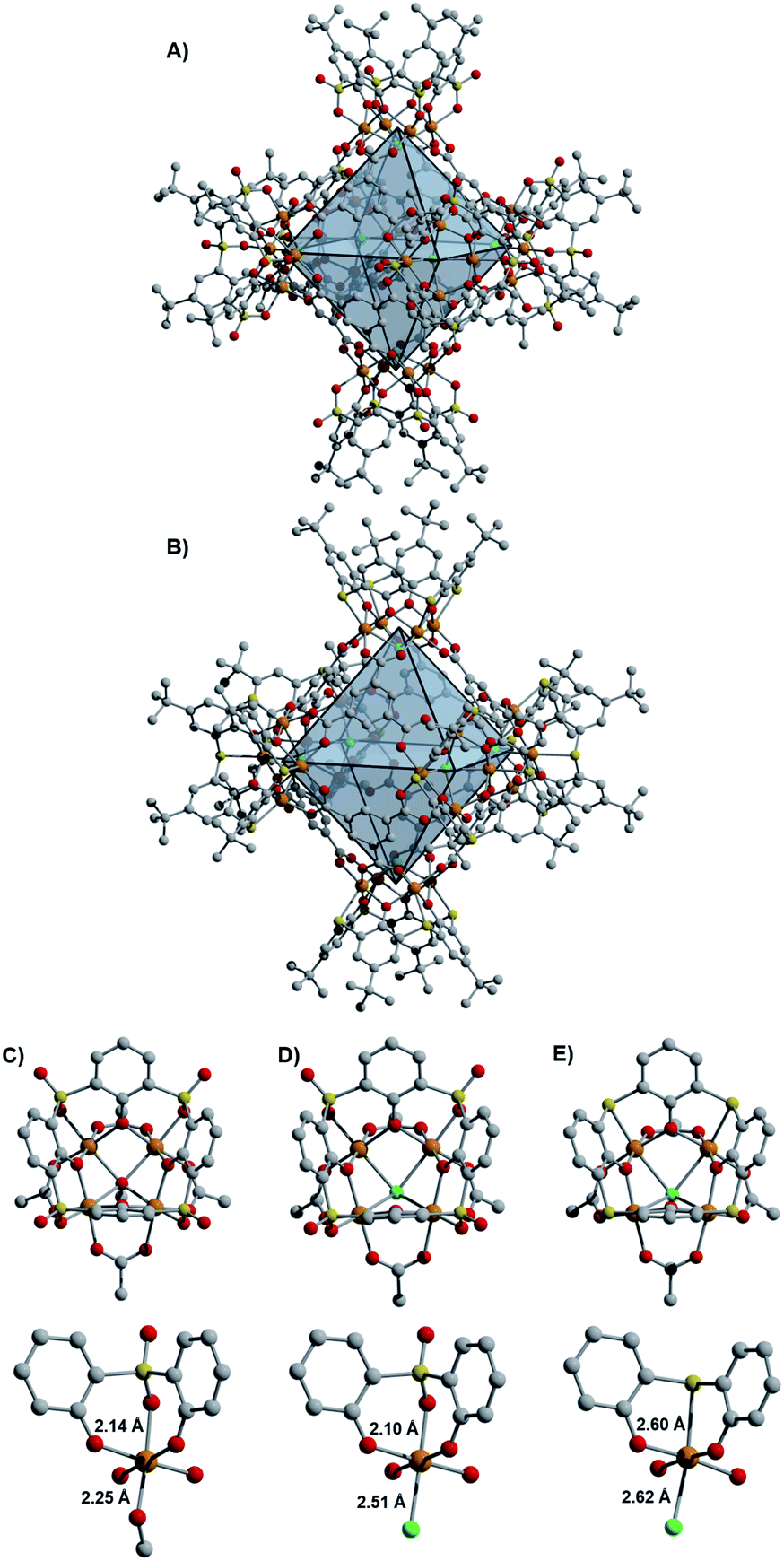 | ||
| Fig. 2 Structure of (sc4a)Fe(btc) (A) and (tc4a)Fe(btc) (B) where orange, yellow, green, gray, and red spheres represent iron, sulfur, chlorine, carbon, and oxygen, respectively. Hydrogen atoms and charge-balancing cations are omitted for clarity. Although the structures of these cages are similar, the metal building units of (sc4a)Fe(btc) (D) and (tc4a)Fe(btc) (E) are significantly distorted as compared to (sc4a)Fe(OAc) (C). | ||
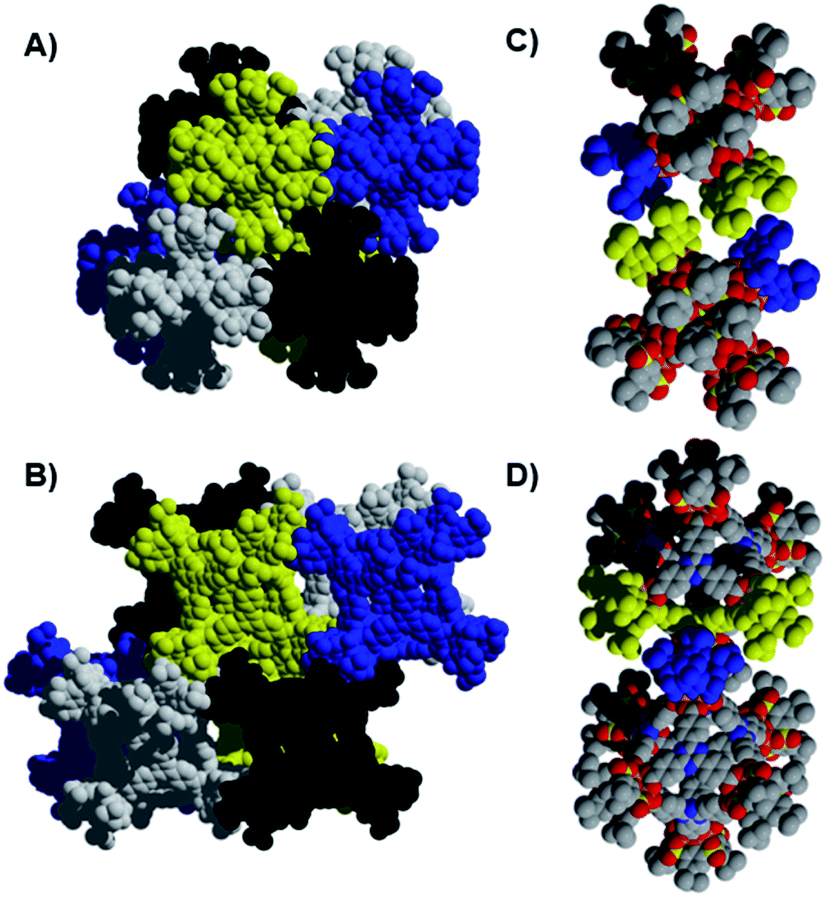 | ||
| Fig. 3 Crystal packing for (sc4a)Fe(btc) (A and C) and (sc4a)Fe(tatb) (B and D). The images on the right (C and D) show the interactions between cages where those in (sc4a)Fe(btc) (C) are purely tbutyl–tbutyl in nature while those for the expanded cage (D) involve both tbutyl–tbutyl and tbutyl–arene interactions. | ||
While the overall structures of the sc4a and tc4a-capped cages are similar, substantial differences are observed in structures of the cluster nodes (Fig. 2). This is apparent from direct comparison between the node structures and a molecular sc4a-supported acetate-capped Fe cluster (see ESI† for synthetic details).19 For the sc4a-capped systems, incorporation of a μ4-OMe, rather than a μ4-Cl ligand in the isolated cluster, gives a structure with much shorter Fe-μ4 distances (2.25 Å vs. 2.51 Å). The Fe–O bond trans to the μ4-bridging ligand is elongated in the molecular cluster (2.14 Å vs. 2.10 Å), as expected with an increased trans influence. More substantial structural perturbations are observed in the tc4a-capped cluster, which features a quite long Fe–S distance of 2.60 Å. Concurrent with the elongation of the Fe–S interaction is a substantial deviation from an idealized octahedral geometry at Fe and an expansion of the cluster relative to the sc4a-capped cage with longer Fe–Fe (3.28–3.31 Å vs. 3.16–3.18 Å) and Fe–Cl distances (2.62 Å vs. 2.51 Å). This likely contributes to a destabilization of the tc4a-capped clusters, where both the more thermally forcing reaction conditions required for cage assembly and the reported propensity for the formation of higher nuclearity Fe clusters support this supposition.27
Prior to further exploring cage properties, we set out to confirm the Fe oxidation states in the synthesized cages. For the previously reported calixarene-supported Ni(II) and Co(II) cage structures, counterions are typically too small and positionally disordered to refine reliably, with an exception where a charge-balancing metal-based counterion is observed.28 The same is true of these Fe cages where, with exclusive incorporation of Fe(II) ions and complete occupation of the μ4-Cl sites, each cage has an overall hexanionic charge, despite the absence of crystallographically observed counterions. With potential redox chemistry accessible at Fe, this could instead indicate partial cage oxidation to a mixed-valent state, where incorporation of 25% Fe(III) would generate a charge neutral material, for example [(Fe(II)18Fe(III)6Cl6(sc4a)6(btc)8]. Mössbauer spectra of the synthesized cages confirmed the exclusive incorporation of high-spin Fe(II) sites in both btc-linked cages with predominantly high-spin Fe(II) observed for both the tatb- and bdc-linked cages (see ESI† for additional details).
In order to obtain maximal surface areas for each cage, we surveyed a variety of solvent exchange and activation conditions. In contrast to solvent exchange protocols employed for many MOFs, the exact nature of the amide and/or volatile solvent used for washing is vitally important for molecular adsorbents as their potential solubility limits the utilization of many exchange solvents. For the materials reported here, it was ultimately discovered that multiple room temperature diethyl ether exchanges over the course of two to three days afforded the highest porosities (see ESI† for full synthesis, exchange, and activation procedures). Each of the synthesized Fe(II) cages was activated under optimized conditions and CO2 (195 K), N2 (77 K), and Ar (77 K) accessible surface areas were determined. Notably, for each cage, the measured surface areas are heavily gas dependent, without consistent trends observed across the materials. Among calixarene-capped cages, the btc-bridged structures prepared with Fe, Ni, Co, and Mg feature similar pore sizes, with typical cross-cage distances between μ4 atoms in these materials between 13 and 15 Å. The surface areas measured for the (sc4a)Fe(btc) cage were modest for both CO2 (368 m2 g−1 BET; 623 m2 g−1 Langmuir) and N2 (850 m2 g−1 Langmuir). A decrease in the measured surface areas was observed upon substituting the sc4a-capping ligand in the (tc4a)Fe(btc) cage. In this case, the BET (Langmuir) surface areas for CO2, N2, and Ar were determined to be 229 m2 g−1 (408 m2 g−1), 217 m2 g−1 (437 m2 g−1), and 390 m2 g−1 (802 m2 g−1), respectively. These values are comparable to the reported (sc4a)Ni(btc) cage, which has a N2 BET surface area of 230 m2 g−1. The edge-linked cage (sc4a)Fe(bdc) has a slightly expanded pore, with a cross-cage distance of approximately 16.5 Å, comparable to bdc-linked Ni and Co cages (∼17 Å). The measured surface areas (BET; Langmuir) of the (sc4a)Fe(bdc) cage toward CO2 (354 m2 g−1; 670 m2 g−1), N2 (511 m2 g−1; 787 m2 g−1), and Ar (490 m2 g−1; 989 m2 g−1) are similar to those reported for the directly analogous Ni and Co cages, which have N2 BET surface areas of 523 m2 g−1 and 423 m2 g−1 respectively.23
Incorporating the tritopic tatb ligand in place of trimesic acid results in significant expansion of the octahedral pores, with a cross-cage Cl–Cl distance of 23 Å. An increase in porosity upon cage expansion is apparent from the surface areas (BET; Langmuir) measured with CO2 (652 m2 g−1; 1082 m2 g−1), N2 (879 m2 g−1; 1248 m2 g−1), and Ar (898 m2 g−1; 1497 m2 g−1). The Ar surface area of this material is among the highest reported for calixarene-capped cages.25,29 While no directly analogous cage structures have been reported with other metals, closely related sc4a and tc4a-capped Co cages have been previously described that incorporate a benzene trisbenzoate (btb) bridging ligand, with a Cl–Cl distance of 23.5 Å for the tc4a-capped cage. Despite their similar pore sizes and overall structures, both reported cages have appreciably lower N2 BET surface areas of 250 m2 g−1 and 504 m2 g−1 (605 m2 g−1 Langmuir), respectively, for the sc4a and tc4a-capped btb-linked cages.20,21
The surface area discrepancies observed across calixarene-capped cage materials of a similar size may, in part, be attributable to differences in their solid-state packing. Both of the btc-bridged cages pack such that the external cavities of six calixarene caps are oriented toward one another, to generate additional ‘pores’ between the 0-D cage structures.20 Similar external pore structures are not observed in the packing of the tatb and bdc-linked cages, with packing of the calixarene caps offset in these structures (Fig. 3). For the (sc4a)Fe(tatb) cage, this generates parallel channels through the edges of the octahedral cage structures and through the void space between adjacent cages. Related offset cage packing is observed for a tc4a-capped bipyridyl dicarboxylate-linked octahedral Co cage, with this particular material exhibiting the highest porosity reported for cages of this type, with an N2 accessible surface area of 1211 m2 g−1 (BET).25
The structurally related porous cages reported here, namely [(Fe24Cl6(sc4a)6(btc)8)]6−, [(Fe24Cl6(sc4a)6(tatb)8)]6−, and [(Fe24Cl6(tc4a)6(btc)8)]6− offer a unique opportunity to study the gas adsorption properties of an isoreticular series of molecular adsorbents. Expanded-pore MOFs have been studied in this regard where straightforward substitution of bridging ligands can be used to achieve systematic pore size variations.30–33 Although the saturation capacities of CO2, N2, CH4, C2H6, C2H4, C3H8, and C3H6 at room temperature and one bar generally track with N2 accessible surface area for these three cages, interesting trends in the hydrocarbon adsorption are apparent. For all three cages, consistent with the relative polarizabilities of the adsorbates, the uptake capacities of the C3 hydrocarbons are higher than those of the C2 hydrocarbons across the entire pressure range (Fig. 4). Further, as these cages lack the open metal cation sites that endow MOFs with selective gas binding, the adsorption enthalpies of the saturated hydrocarbons are similar to those of their unsaturated counterparts (Fig. 5). The adsorbate–adsorbent interactions in these cages are purely physisorptive and likely ligand-based where the size and shape of the coordination cage governs the interaction strength. C2/C3 adsorption in (sc4a)Fe(btc) is illustrative of this point where the enthalpy of propane/propylene adsorption is considerably higher than that of ethane/ethylene over the entire pressure range investigated. The corresponding adsorption enthalpies in the expanded pore cage (sc4a)Fe(tatb) are more similar, as the expanded nature of the cage and its larger pore windows are unsuited for strong adsorbate–adsorbent interactions.
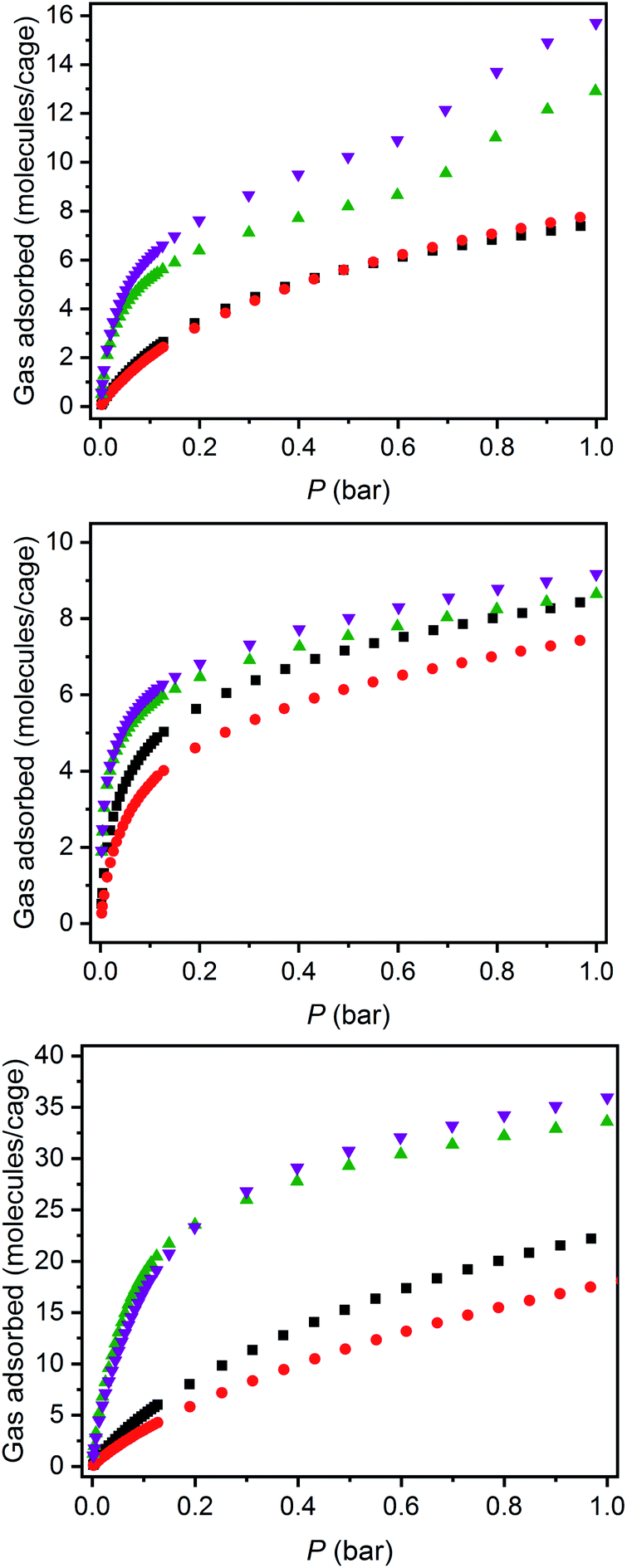 | ||
| Fig. 4 298 K hydrocarbon adsorption isotherms for (sc4a)Fe(btc) (top), (tc4a)Fe(btc) (middle), and (sc4a)Fe(tatb) (bottom) where inverted purple triangles, green triangles, black squares, and red circles represent propylene, propane, ethane, and ethylene, respectively. | ||
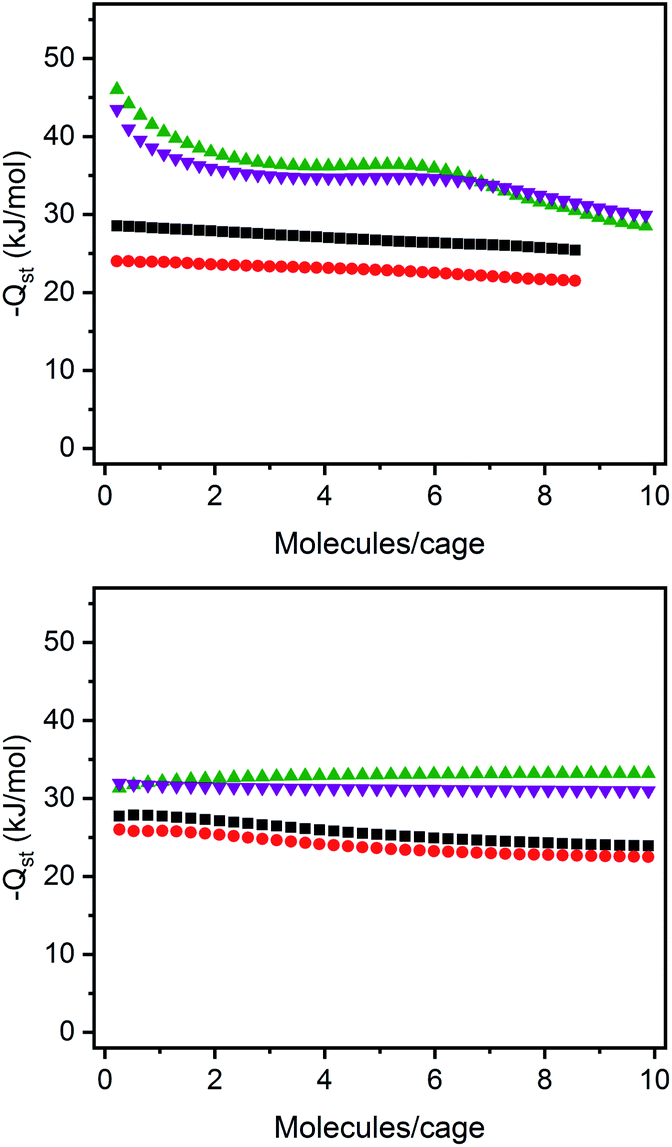 | ||
| Fig. 5 Enthalpies of propylene (purple inverted triangles), propane (green triangles), ethane (black squares), and ethylene (red circles) for (sc4a)Fe(btc) (top) and (sc4a)Fe(tatb) (bottom). | ||
While surveying the gas adsorption properties of the synthesized cages we noted that the uptake of O2 is low and reversible, with negligible loss of N2 surface area for the (sc4a)Fe(btc) and (sc4a)Fe(bdc) cages following exposure to O2. This initial observation prompted us to explore whether these materials might exhibit further stability toward air and moisture. Here, the stabilities of both the face-linked (sc4a)Fe(btc) and the edge-linked (sc4a)Fe(bdc) octahedral cages were examined. In either case, both the Et2O exchanged and activated materials exhibited minimal loss of porosity when stored under air overnight. Further, the Mössbauer spectrum of the Et2O exchanged (sc4a)Fe(btc) cage collected following storage of the material in air over two weeks is consistent with the presence of exclusively high-spin Fe(II) centres (39% δ = 1.27 mm s−1, ΔEQ = 1.52 mm s−1; 61% δ = 1.27 mm s−1, ΔEQ = 2.26 mm s−1), confirming the stability of this material toward air oxidation. To further probe the limits of cage stability, we next added large quantities of water (>1 mL per 100 mg) to both Et2O exchanged and fully activated cages overnight. Following slow removal of water in vacuo and subsequent cage reactivation at 175 °C, the materials retain most of their porosity as judged by comparison of the 77 K N2 uptakes at P/P0 = 0.8. At this relative pressure, the fully activated (sc4a)Fe(btc) cage adsorbs 7.5 mmol g−1, with the cages exposed to water before and after activation retaining 93% and 81% of this uptake upon reactivation, respectively.
The ability of these molecular Fe(II) cages to retain most of their porosity following exposure to air and, more remarkably, water contrasts sharply with reported Fe(II) MOFs. While these materials have been explored for O2/N2 separation, this has required operation under strictly anhydrous conditions.34–36 Notably, the hydrolytic stability of sc4a-supported Ni(II) and Co(II) octahedral cages has been demonstrated previously and allowed for the study of the host-guest chemistry of these materials with water soluble adsorbates, among other applications.20,23,28,37 This stability may be attributable, in part, to packing between the hydrophobic t-butyl residues on the exterior of the calixarene cages, with structural rigidity at the coordinatively saturated tetranuclear metal nodes likely contributing to cage stability.
To further tune the properties of these materials, we set out to leverage the accessibility of the Fe(II)/(III) redox couple to examine the effect of oxidation on cage stability and porosity across several charge states. With 24 Fe centres per octahedral cage, the overall charge of the cages can, in principle, range from the initial hexanionic Fe(II) cage to a cage incorporating exclusively Fe(III) centres that has an overall +18 charge. Here, we hypothesized that porosity could be maximized in a charge neutral state, where occupation of pore void space by nonporous counterions is negated. Of course, such an effect could be offset if a decrease in cage stability is observed upon partial oxidation. To explore these possibilities we targeted the oxidation of the (sc4a)Fe(btc) cage, which incorporates exclusively high-spin Fe(II) centres in its synthesized state. Given the appreciable stability of these cages toward oxidation in air, we focused on accessing partial and complete cage oxidation through the application of strong, solution-phase chemical oxidants.
In this case, treating a suspension of the all Fe(II) cage in MeCN with 6, 12, 18, or 24 equivalents of [N(4-Br(C6H4))3][SbCl6] (magic blue) provides access to materials that incorporate 25–100% Fe(III). Here, the decolorization of the solution of oxidant as the reaction proceeds to completion indicated the complete consumption of oxidizing equivalents. This is apparent in the Mössbauer spectra of the oxidation products, where the all Fe(II) cage treated with 6 equivalents of magic blue forms a material containing a mixture of high-spin Fe(II) (25% δ = 1.1 mm s−1, ΔEQ = 1.78 mm s−1; 50% δ = 1.21 mm s−1, ΔEQ = 2.33 mm s−1) and high-spin Fe(III) sites (25% δ = 0.75 mm s−1, ΔEQ = 0.72 mm s−1). Treating the starting Fe(II) cage with additional oxidizing equivalents leads to an increase in the Fe(III) component in the oxidation products, where, in the most extreme case, the addition of 24 equivalents of magic blue generates a material that incorporates exclusively high-spin Fe(III) (δ = 0.53 mm s−1, ΔEQ = 0.84 mm s−1; Fig. 6). Like their solvent-exchanged cage precursors, these materials are amorphous solids. Attempts to manipulate the oxidized materials in the solution phase invariably led to their decomposition.
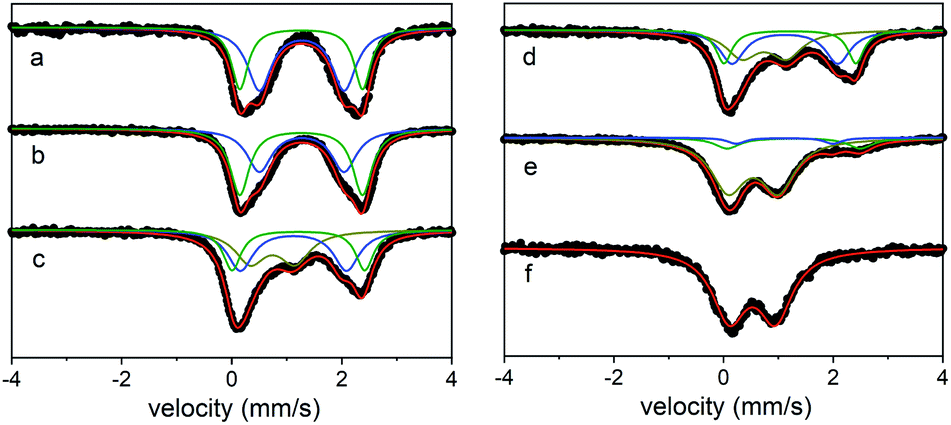 | ||
| Fig. 6 Mössbauer spectra for (sc4a)Fe(btc) as-synthesized (a), upon exposure to air (b), and after reaction with 6 (c), 12 (d), 18 (e), or 24 (f) equivalents of oxidizing agent. | ||
We next turned toward examination of the porosity of the oxidized materials, as it could provide circumstantial evidence that the materials retain their cage structures upon oxidation. To do this, we surveyed conditions for their activation and examined their porosity toward N2 at 77 K. The all Fe(II) cage has a Langmuir surface area of 850 m2 g−1 (N2), with an N2 uptake of 7.3 mmol g−1 at P/P0 = 0.90. The materials generated upon oxidation with either 6 or 12 equivalents of magic blue display comparable porosities toward N2, with BET surface areas of 397 m2 g−1 and 396 m2 g−1 and N2 uptakes of 7.8 and 6.3 mmol g−1 at P/P0 = 0.87. Direct comparison of these values is complicated by the distinct solvent exchange protocols conducted prior to their activation. Upon further oxidation to predominantly Fe(III) containing materials, an appreciable loss in porosity is observed, with N2 BET surface areas of 234 m2 g−1 (325 m2 g−1 Langmuir) and 76 m2 g−1 (165 m2 g−1 Langmuir) in the two most oxidized states. The trend in the porosity accessible for these materials is consistent with our initial hypothesis, namely that the increased occupation of structural void space with non-porous counteranions leads to a decrease in porosity. Additional evidence suggests that some loss of surface area for the two most oxidized cages can be attributed to a decrease in cage stability. Like the synthesized all Fe(II) cages, the partially oxidized materials generated upon treatment with 6 or 12 equivalents of magic blue retain most of their porosity when activated at elevated temperatures (>300 °C). The same cannot be said of the two most oxidized cages, where both materials lose substantial porosity when activated under vacuum above room temperature. This stability trend is also apparent by thermogravimetric analysis, where substantial mass loss is observed for the two most oxidized cages at lower temperatures. This observation is initially counterintuitive as, upon oxidation to Fe(III), the M–L bond strengths in the material increase. One possible rationalization for this observation is an electronic destabilization of the Fe4 cluster where, upon heating it can undergo structural rearrangement with concurrent cage collapse. Alternatively, thermal decomposition of the SbCl6 counteranion, with 12 or 18 equivalents per cage in the most oxidized materials, may contribute to their observed instability.
The octahedral pore structure in these cages is analogous to the pores observed in the PCN-9 series of frameworks that have been described by Zhou and coworkers, incorporating Co, Fe, and Mn.38,39 These frameworks feature tetranuclear metal nodes bridged by a μ4-oxo and connected by tritopic tatb ligands. As reported, the Co and Mn frameworks incorporate exclusively divalent metal centres, while predominantly trivalent metal centres are incorporated in the Fe framework, as judged by Mössbauer spectroscopy. Independent attempts to prepare PCN-9 (Fe) under related solvothermal conditions yielded similar results in our hands (see ESI† for details). Further, relative to the Co and Mn frameworks, which have BET surface areas of 1064 and 836 m2 g−1, respectively, the Fe-based MOF has an appreciably lower surface area of 682 m2 g−1. The low porosity accessible for the Fe-based material can be rationalized in light of our observations for structurally-related cages. Specifically, we hypothesize that a combination of the increased occupation of structural void space by counterions and the poorer structural integrity for the predominantly Fe(III) MOF both contribute to limiting its porosity.
Conclusions
We have described the first examples of porous Fe(II) cages, that, among all Fe(II)-based porous materials, exhibit remarkable stabilities toward air and moisture. One of these cages features an Ar BET surface area of 898 m2 g−1 (1497 m2 g−1 Langmuir), which is among the highest surface areas reported for cages of this type. The modular assembly of these cages allows for facile manipulation of hydrocarbon adsorption enthalpies, which vary depending on cage pore size, choice of capping ligand, and cage connectivity. The Fe(II)-based metal nodes provide an additional handle for tuning the gas adsorption properties of these cages through redox chemistry, where porosity and cage stability are shown to vary with Fe oxidation states. These observations motivate our ongoing interest in expanding the chemistry of molecular metal–organic materials to complement studies of MOFs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Theodore A. Betley for use of his Mössbauer spectrometer used in this work. We also thank Gerald E. Decker and Michael R. Dworzak for experimental assistance. This material is based upon work supported by the U.S. Department of Energy's Office of Energy Efficiency and Renewable Energy under the Hydrogen and Fuel Cell Technologies and Vehicle Technologies Offices under Award Number DE-EE0008813.References
- D. M. D'Alessandro, Chem. Commun., 2016, 52, 8957–8971 RSC.
- G. E. Decker, G. R. Lorzing, M. M. Deegan and E. D. Bloch, J. Mater. Chem. A, 2020, 8, 4217–4229 RSC.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 123044 CrossRef PubMed.
- M. Eddaoudi, J. Kim, J. B. Wachter, H. K. Chae, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2001, 123, 4368–4369 CrossRef CAS PubMed.
- B. Moulton, J. Lu, A. Mondal and M. J. Zaworotko, Chem. Commun., 2001, 863–864 RSC.
- S. S.-Y. Chui, S. M.-F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148–1150 CrossRef CAS PubMed.
- N. Ahmad, A. H. Chughtai, H. A. Younus and F. Verpoort, Coord. Chem. Rev., 2014, 280, 1–27 CrossRef CAS.
- J.-R. Li, A. A. Yakovenko, W. Lu, D. J. Timmons, W. Zhuang, D. Yuan and H.-C. Zhou, J. Am. Chem. Soc., 2010, 132, 17599–17610 CrossRef CAS PubMed.
- J. Park, Y.-P. Chen, Z. Perry, J.-R. Li and H.-C. Zhou, J. Am. Chem. Soc., 2014, 136, 16895–16901 CrossRef CAS PubMed.
- J. Park, Z. Perry, Y.-P. Chen, J. Bae and H.-C. Zhou, ACS Appl. Mater. Interfaces, 2017, 9, 28064–28068 CrossRef CAS PubMed.
- G. R. Lorzing, B. A. Trump, C. M. Brown and E. D. Bloch, Chem. Mater., 2017, 29, 8583–8587 CrossRef CAS.
- C. A. Rowland, G. R. Lorzing, E. J. Gosselin, B. A. Trump, G. P. A. Yap, C. M. Brown and E. D. Bloch, J. Am. Chem. Soc., 2018, 140, 11153–11157 CrossRef CAS PubMed.
- G. R. Lorzing, E. J. Gosselin, B. S. Lindner, R. Bhattacharjee, G. P. A. Yap, S. Caratzoulas and E. D. Bloch, Chem. Commun., 2019, 55, 9527–9530 RSC.
- M. D. Young, Q. Zhang and H.-C. Zhou, Inorg. Chim. Acta, 2015, 424, 216–220 CrossRef CAS.
- G. R. Lorzing, E. J. Gosselin, B. A. Trump, A. H. P. York, A. Sturluson, C. A. Rowland, G. P. A. Yap, C. M. Brown, C. M. Simon and E. D. Bloch, J. Am. Chem. Soc., 2019, 141, 12128–12138 CrossRef CAS PubMed.
- L. J. Murray, M. Dinca, J. Yano, S. Chavan, S. Bordiga, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2010, 132, 7856–7857 CrossRef CAS PubMed.
- E. D. Bloch, W. L. Queen, M. R. Hudson, J. A. Mason, D. J. Xiao, L. J. Murray, R. Flacau, C. M. Brown and J. R. Long, Angew. Chem., Int. Ed., 2016, 55, 8605–8609 CrossRef CAS PubMed.
- E. J. Gosselin, G. R. Lorzing, B. A. Trump, C. M. Brown and E. D. Bloch, Chem. Commun., 2018, 54, 6392–6395 RSC.
- T. Kajiwara, T. Kobashi, R. Shinagawa, T. Ito, S. Takaishi, M. Yamashita and N. Iki, Eur. J. Inorg. Chem., 2006, 1765–1770 CrossRef CAS.
- F.-R. Dai and Z. Wang, J. Am. Chem. Soc., 2012, 134, 8002–8005 CrossRef CAS PubMed.
- M. Liu, W. Liao, C. Hu, S. Du and H. Zhang, Angew. Chem., Int. Ed., 2012, 51, 1585–1588 CrossRef CAS PubMed.
- K. Xiong, F. Jiang, Y. Gai, D. Yuan, L. Chen, M. Wu, K. Su and M. Hong, Chem. Sci., 2012, 3, 2321–2325 RSC.
- F.-R. Dai, U. Sambasivam, A. J. Hammerstrom and Z. Wang, J. Am. Chem. Soc., 2014, 136, 7480–7491 CrossRef CAS PubMed.
- H. Tan, S. Du, Y. Bi and W. Liao, Chem. Commun., 2013, 49, 8211–8213 RSC.
- H. Tan, S. Du, Y. Bi and W. Liao, Inorg. Chem., 2014, 53, 7083–7085 CrossRef CAS PubMed.
- X. Zhu, S. Wang, H. Han and W. Liao, RSC Adv., 2018, 8, 39208–39213 RSC.
- C. Desroches, G. Pilet, P. Á Szilágyi, G. Molnár, S. A. Borshch, A. Bousseksou, S. Parola and D. Luneau, Eur. J. Inorg. Chem., 2006, 357–365 CrossRef CAS.
- F.-R. Dai, D. C. Becht and Z. Wang, Chem. Commun., 2014, 50, 5385–5387 RSC.
- W. Gong, D. Chu, H. Jiang, X. Chen, Y. Cui and Y. Liu, Nat. Commun., 2019, 10, 600 CrossRef CAS PubMed.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS PubMed.
- H. Deng, S. Grunder, K. E. Cordova, C. Valente, H. Furukawa, M. Hmadeh, F. Gándara, A. C. Whalley, Z. Liu, S. Asahina, H. Kazumori, M. O'Keeffe, O. Terasaki, J. F. Stoddart and O. M. Yaghi, Science, 2012, 336, 1018–1023 CrossRef CAS PubMed.
- Q. Yao, J. Su, O. Cheung, Q. Liu, N. Hedin and X. Zou, J. Mater. Chem., 2012, 22, 10345–10351 RSC.
- S. Yuan, L. Huang, Z. Huang, D. Sun, J.-S. Qin, L. Feng, J. Li, X. Zou, T. Cagin and H.-C. Zhou, J. Am. Chem. Soc., 2020, 142, 4732–4738 CrossRef CAS PubMed.
- E. D. Bloch, L. J. Murray, W. L. Queen, S. Chavan, S. N. Maximoff, J. P. Bigi, R. Krishna, V. K. Peterson, F. Grandjean, G. J. Long, B. Smit, S. Bordiga, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2011, 133, 14814–14822 CrossRef CAS PubMed.
- J. S. Anderson, A. T. Gallagher, J. A. Mason and T. D. Harris, J. Am. Chem. Soc., 2014, 136, 16489–16492 CrossRef CAS PubMed.
- D. A. Reed, D. J. Xiao, H. Z. H. Jiang, K. Chakarawet, J. Oktawiec and J. R. Long, Chem. Sci., 2020, 11, 1698–1702 RSC.
- N. L. Netzer, F.-R. Dai, Z. Wang and C. Jiang, Angew. Chem., Int. Ed., 2014, 53, 10965–10969 CrossRef CAS PubMed.
- S. Ma and H.-C. Zhou, J. Am. Chem. Soc., 2006, 128, 11734–11735 CrossRef CAS PubMed.
- S. Ma, D. Yuan, J.-S. Chang and H.-C. Zhou, Inorg. Chem., 2009, 48, 5398–5402 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1993711–1993715. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc01833c |
| This journal is © The Royal Society of Chemistry 2020 |