
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeep-red fluorescence from isolated dimers: a highly bright excimer and imaging in vivo†
Qing
Luo‡
ac,
Lin
Li‡
b,
Huili
Ma
d,
Chunyan
Lv
a,
Xueyan
Jiang
d,
Xinggui
Gu
 *b,
Zhongfu
An
d,
Bo
Zou
*e,
Cheng
Zhang
c and
Yujian
Zhang
*a
*b,
Zhongfu
An
d,
Bo
Zou
*e,
Cheng
Zhang
c and
Yujian
Zhang
*a
aSchool of Engineering, HuZhou University, Huzhou Cent Hosp, 759 Erhuan Rd, Huzhou, Zhejiang, P. R. China. E-mail: sciencezyj@foxmail.com
bBeijing Advanced Innovation Center for Soft Matter Science and Engineering, College of Materials Science and Engineering, Beijing University of Chemical Technology, Beijing 100029, P. R. China. E-mail: guxinggui@mail.buct.edu.cn
cCollege of Chemical Engineering, Zhejiang University of Technology, Hangzhou 310014, P. R. China
dKey Laboratory of Flexible Electronics & Institute of Advanced Materials, Nanjing Tech University, 30 South Puzhu Road, Nanjing 211816, P. R. China
eState Key Laboratory of Superhard Materials, College of Physics, Jilin University, Qianjin Street 2699, Changchun, 130012, P. R. China. E-mail: zoubo@jlu.edu.cn
First published on 25th May 2020
Abstract
Restricted by the energy-gap law, the development of bright near-infrared (near-IR) fluorescent luminophors in the solid state remains a challenge. Herein, we report a new design strategy for realizing high brightness and deep-red/near-IR-emissive organic molecules based on the incorporation of a hybridized local and charge-transfer (HLCT) state and separated dimeric stacks into one aggregate. Experimental and theoretical analyses show that this combination not only contributes to high photoluminescent quantum yields (PLQYs) but also significantly lessens the energy gap. The fluorophore BTA-TPA exhibits excellent fluorescence performance, achieving a PLQY of 54.8% for the fluorescence peak at 690 nm, which is among the highest reported for near-IR fluorescent excimers. In addition, because of its bioimaging performance, the designed luminophor has potential for use as a deep-red fluorescent probe for biomedical applications. This research opens the door for developing deep-red/near-IR emissive materials with high PLQYs.
Introduction
Luminescent organic materials (LOMs) emitting in deep-red/near-IR regions have recently attracted considerable attention because of their potential applications in night vision devices, bioimaging, sensors, and lasers.1 However, the development of an efficient near-IR emitter has long been hindered by the very low photoluminescence (PL) efficiency of such a material. In principle, deep-red/near-IR fluorescence arises from a narrow band gap, which unfortunately is inconsistent with high PL quantum yields (PLQYs).2 As a consequence, it remains a great challenge to construct deep-red/near-IR LOMs with high PLQYs. To date, the universally adopted design strategy for deep-red/near-IR molecules is the construction of a polar donor–acceptor (D–A) conjugated framework with a strong intramolecular charge-transfer (CT) effect.3 However, such a CT state generally gives rise to forbidden electronic transitions, causing fluorescence quenching.4 To solve this problem, Ma’s group recently combined both local excited (LE) and CT states into a special state, namely, a hybridized local and charge-transfer (HLCT) excited state.5 This state has two complementary characteristics: a large orbital overlap from the LE state and a large dipole from the CT state. The former characteristic contributes to a high PLQY, whereas the latter guarantees the generation of a narrow band gap. Accordingly, Ma’s group reported a butterfly-shaped D–A-type fluorophore, PTZ-BZP,5a with strong near-IR fluorescence at 700 nm and a film efficiency of 16%. However, our group recently prepared a deep-red benzothiadiazole (BTA) derivative exhibiting the HLCT state.6 The luminophor presented good emissive abilities in the monomeric form, unexpectedly, which were seriously weakened in the solid state due to the formation of the H-aggregates. Clearly, even for HLCT-type LOMs, the molecular packing mode is still critical for the PL efficiency.In 1954, excimer fluorescence was first discovered by Förster in the case of pyrene and some of its derivatives.7 The excimer emission frequently presents a tremendous redshift of approximately 100 nm relative to the emission of the monomer, in principle making excimer-emitting LOMs with long-wavelength fluorescence possible. Unfortunately, the polyaromatic materials adopting continuous H-type packing give rise to feeble excimer fluorescence due to the strong π–π interactions.8 In sharp contrast, the discrete dimeric stacks of LOMs exhibit intriguing fluorescence. Recently, Yoshizawa and Yang et al., in an elegant approach, employed molecular capsules,9 cyclophane-like motifs10 and supramolecular interactions11 to generate separated dimeric stacks. However, as a consequence of the strong dipole–dipole interaction and complex polyaromatic frameworks, the assembly of intramolecular CT-type luminophors into discrete dimers has rarely been achieved.
Clearly, combining both HLCT behaviour and discrete dimeric packing into one aggregate would be an ideal strategy for generating deep-red/near-IR emitters with a high PLQY. This approach is a relay-race tactic: the large PL redshift results from the CT part of the HLCT state; then, the PL wavelength is further lengthened and falls in the deep-red/NIR region in the presence of the separated dimer. Importantly, the discrete dimers still retain high brightness, which is related to the LE part of the HLCT state. Herein, the “relay-race tactic” is achieved as follows: a monomeric HLCT-active dye (BTA-TPA), constructed from two bulky triphenylamine (TPA) groups and a planar BTA group, emits red luminescence (614 nm) with a PLQY as high as 83.2%. Then, HLCT-type molecules are assembled into a dimeric packing architecture. The bulky substituents at both ends provide a large space to separate the dimers from each other. As expected, the pairwise stacking fluorophore emits light that is further red-shifted to 690 nm with a PLQY of up to 54.8%. In addition, the in-depth relationship between the PL properties and supermolecular interactions of the discrete dimer was fully investigated by in situ high-pressure experiments. By taking advantage of this superior deep-red/near-IR emission, BTA-TPA performed well in in vitro and in vivo bioimaging, demonstrating substantial potential for use in biomedical applications.
Results and discussion
The synthetic routes to form BTA-TPA are depicted in Scheme 1. The initial step for the preparation of the luminophor involves a Suzuki coupling reaction to give the key intermediate (TTA). The subsequent Knoevenagel reaction of 2-(4-(diphenylamino)-phenyl) acetonitrile with TTA yields the desired LOMs.12 The final molecular structure was determined spectroscopically by high-resolution mass spectrometry (HRMS), NMR spectroscopy, and X-ray single-crystal analysis (Fig. S1, S2 and Table S1†). As shown in Fig. S3,† the luminophor exhibited similar absorption profiles in different solvents, with two distinct bands: one high-energy band at approximately 300 nm associated with the localized π–π* electronic transition and another band attributed to the CT transition from the donor (TPA) to the acceptor (BTA). The results presented only a slight change in the dipole of BTA-TPA in the ground state. Nevertheless, the PL spectra of BTA-TPA revealed an obvious bathochromic shift from 618 nm in n-hexane to 751 nm in high-polarity acetonitrile (Fig. S4†), implying typical CT characteristics of the excited states. We further examined the solvatochromic effect via the Lippert–Mataga model, and the dipole moments (μe values) of the S1 exciton can be estimated from the slope of the Stokes shift (νa–νf) when plotted versus the orientation polarizability (Δf). As depicted in Fig. 1a, the plot shows two sections, implying two distinct excited states in low- and high-polarity solvents. In high-polarity solvents, the excited-state dipole μe was calculated to be 23.5 D as a consequence of the CT states. In low-polarity solvents, μe was found to be 7.6 D, attributed to the LE-like state. In addition, the PLQY values of the luminophor (f < 1.6, Table S2†) were all above 82% in low-polarity solvents. Therefore, we could infer that the emissive state of BTA-TPA was an LE-like state involving a slight CT component. Interestingly, BTA-TPA exhibited a single-exponential PL decay in n-hexane and isopropyl ether solutions, as depicted in Fig. S5.† The one lifetime illustrated that the S1 state was a hybridization of LE and CT states (HLCT state) rather than two mixed states.5a,13 To further understand the HLCT state, we characterized the distribution of natural transition orbitals (NTOs) of BTA-TPA according to the single-crystal structure. For the S0 → S1 excitation, the particle and hole NTOs presented an excellent balance between the spatial separation and the orbital overlap (Fig. 1b), which implied that the coexistence of the CT and LE transition corresponded to the HLCT state. The easily distinguishable separated orbitals gave rise to CT character with a large dipole moment. However, sufficient orbital overlap was responsible for the LE character and ensured a large radiative-transition rate. Thus, the S0 → S1 transition of BTA-TPA exhibited large oscillator strength (f = 1.3418). In contrast, the particle and hole NTOs of S2 showed a clear spatial separation, leading to very small oscillator strength (f = 0.0258). In general, the “polarity environment” of the luminophor in polymethyl methacrylate (PMMA) was similar to that of the luminophore in the low-polarity solvents. Therefore, BTA-TPA should exhibit HLCT properties in low-polarity PMMA. As shown in Fig. 1c, the doped films emitted bright orange light (614 nm) with a PLQY as high as 83.2%, consistent with the HLCT character. Moreover, BTA-TPA in PMMA exhibited a Stokes shift of 108 nm. Of particular interest, the PL colour of the luminophor in the crystalline state became deep red (690 nm) upon illumination at 365 nm. The Stokes shift was further increased to 195 nm due to the formation of the excimer (discussed in detail below). The PLQY of BTA-TPA crystals was determined to be 54.8%. The results indicated that employing “relay-race tactics” to realize deep-red/near-IR fluorescence was feasible. | ||
| Scheme 1 Synthetic routes of BTA-TPA. | ||
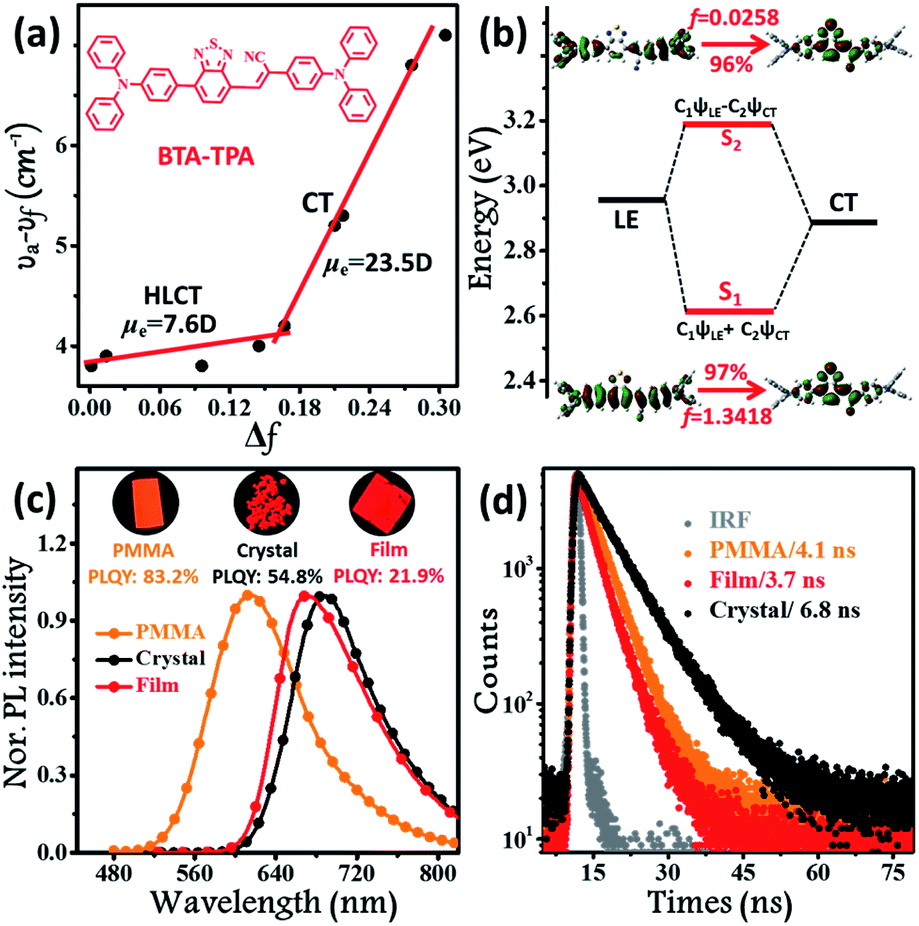 | ||
| Fig. 1 (a) Linear correlation of the orientation polarization (Δf) of solvent media with the Stokes shift (νa–νf) for BTA-TPA. See Table S2† for detailed data; the lines in the low- and high-polarity regions (HLCT and CT, respectively); (b) NTOs from S0 to S1 and S2 of BTA-TPA, where f represents the oscillator strength; steady-state PL spectra (c) and time-resolved PL spectra (d) of BTA-TPA in different states (0.1% wt/wt dye-doped film, crystals and a spin-coated film). Inset images in Fig. 2c represent the corresponding PL photographs upon irradiation with 365 nm UV light. | ||
Although the excimer species formed in the crystal decrease the emission efficiency relative to that of the monomer in doped PMMA films, the PLQY value (54.8%) was uncommonly high for the deep-red/near-IR luminophor (around 700 nm) reported so far.14 Such a result appears to be in contradiction with the conventional proposal that an excimer usually has low efficiency. To understand the high-efficiency fluorescence of the near-IR excimer, the single-crystal X-ray structure of BTA-TPA was determined, and selected crystallographic data are listed in Table S1.† Interestingly, along the b-axis of the unit cell, BTA-TPA molecules were stacked in the form of staggered discrete dimers (Fig. 2a and b). As depicted in Fig. 2c, the distance of the BTA plane between the neighbouring dimers reached as high as 4.31 Å. Importantly, the BTA plane of neighbouring dimers is displaced substantially along the short molecular axis resulting in virtually no π-overlap between the neighbouring dimers (see Fig. 2a). Clearly, these π–π dimers were spatially isolated from each other by the two-sided substituents (TPA units), substantially eliminating the π–π interaction between the dimers.15,16a For comparison, we prepared a similar luminophor, BTA-DMeO, in which the twisted TPA unit on the side of BTA-TPA was replaced by a benzene group.6 This luminophor, adopting a continuous dimeric stacking (H-aggregation; Fig. S6†), showed a PLQY as low as 9%. The results demonstrated that isolated dimeric stacking plays an important role in the high PLQY of BTA-TPA crystals. Within the discrete dimer, two BTA units were arranged in an anti-parallel fashion (Fig. 2c), revealing an interplanar distance of 3.50 Å and an overlapping area of approximately 65% (top view). This result implied that a π–π interaction existed in the dimer, which was further enhanced by the multiple C–H⋯N/π hydrogen bonds (Fig. 2d). For the spin-coated film, the luminophor, adopting a random packing mode (Fig. S7†), showed a slight blueshift in the PL spectrum with respect to that in the crystalline state (Fig. 1c). As shown in Fig. 1d, the transient PL spectra of the luminophor in the crystalline state revealed a single-exponential decay with a lifetime of 6.8 ns, which was obviously larger than that in the amorphous state (3.7 ns). The fluorescence behaviour (longer lifetime and redshift) of the crystals agreed well with the spectral characteristics of the excimer. In addition, the PLQY of this film was obviously decreased to 21.9% (see Fig. 1c). The results further proved that the presence of isolated dimers contributed to the high brightness, consistent with the previous results.9–11
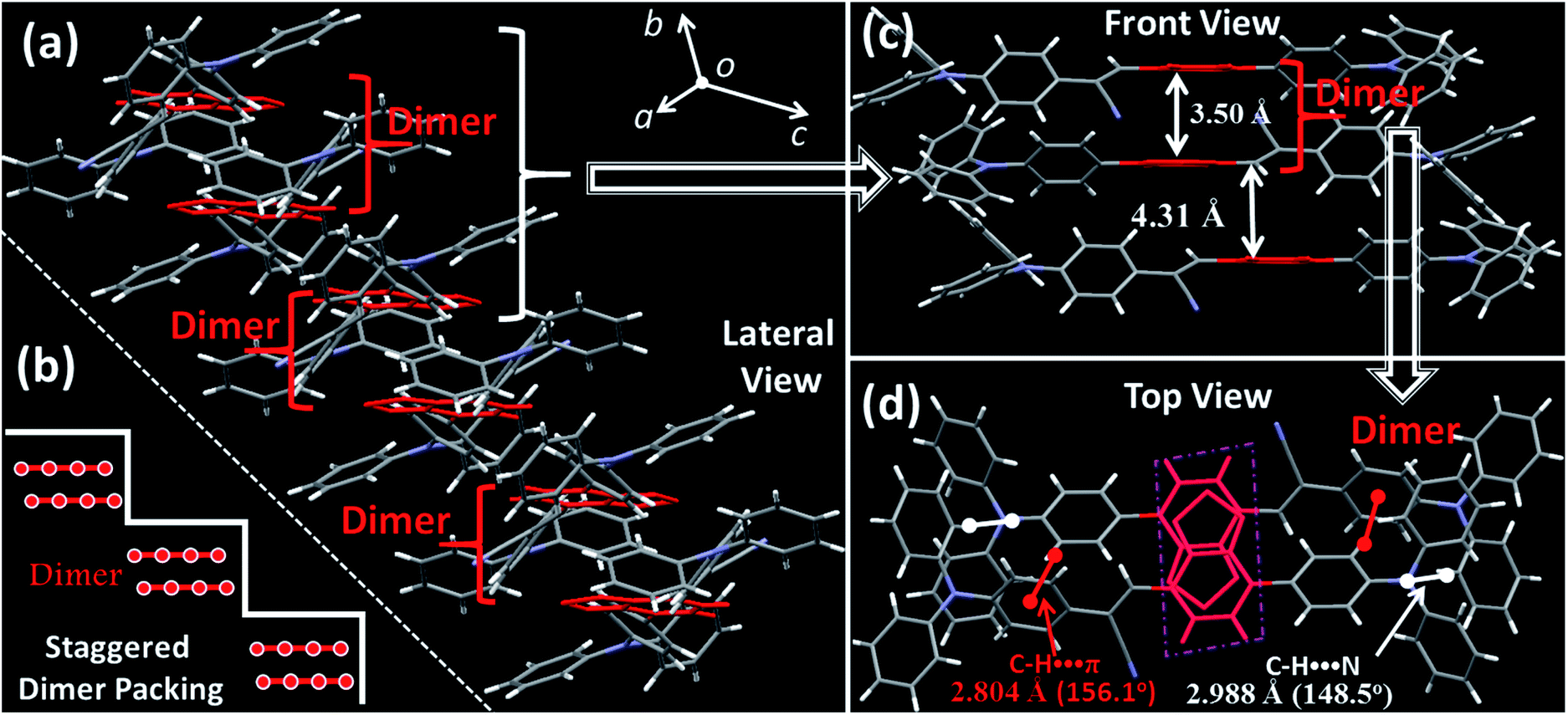 | ||
| Fig. 2 Crystal structures of BTA-TPA: (a) lateral view of the column arrangement; (b) schematic illustration of the staggered dimer packing; (c) front view of the anti-parallel arrangement along the long molecular axis; (d) top view of the BTA-TPA dimer and the illustration of the C–H⋯N/π interactions. | ||
To understand the intrinsic photophysics of discrete dimers, we investigated the NTOs (S1 → S0) of the excimer states. As depicted in Fig. S8,† the hole wave functions were mainly localized on one monomer. As a comparison, the particles were entirely delocalized over two π–π stacking BTA moieties. The results implied that a strong π–π interaction occurred within the pairwise dimer. In general, the excimer, as a low-lying energy-trapping “dark” state, is a low- or non-emissive species due to forbidden transitions (S1 → S0).4a,15a However, in the case of an isolated dimer, the S1 → S0 transition was optically allowed as a result of large oscillator strength (f = 0.6454), consistent with its high-efficiency fluorescence behavior. The potential energy curves (PECs) were further shown as a function of the interplanar distance between two BTA units for both the ground and excited states. The PECs of the ground state presented a deep potential well at an interplanar distance of Rg,PEC = 3.263 Å (Fig. S9†), indicating a typical intermolecular π–π interaction in the ground state. For the excited state, the π–π interplanar distance will be slightly decreased to Re,PEC = 3.229 Å. This fact indicates that such a dimer had a “compressed” excited state with a shortened π–π distance and increased π–π overlap, contributing to the restriction of intramolecular motions.11a,16 Considering all the experimental and theoretical data, we proposed the origin of high PL efficiency for the discrete dimer as follows: (1) upon photoexcitation, the pairwise dimer showed a “compressed” excimer, in favour of the suppression of nonradiative PL quenching; (2) the discrete dimeric π-stacking guaranteed the purity and singleness of the excimer state as a result of the exciton localization, which blocked the occurrence of a “dark” state from the nonradiative energy transfer.11a Briefly, such special dimeric stacking in favour of discrete excimer formation was highly beneficial for high-efficiency fluorescence.
To further explore the photophysical behaviour of the mono-disperse dimer, in situ UV-vis, PL and infrared spectroscopy experiments were performed under high pressure. A piece of the dimeric crystal, emitting deep-red fluorescence under atmospheric pressure, was loaded into the hole of a T301 steel gasket. During the compression process, the PL colour of this sample became dark-red at pressures below 4.6 GPa (insets of Fig. 3a and b), accompanied by continually decreasing PL brightness. At 5.4 GPa, the dark-red fluorescence was not observed by the naked eye. Interestingly, the PL spectra of the BTA-TPA crystal spanned the deep-red and near-IR regions below 8.0 GPa. As depicted in Fig. 3a and b, the PL peaks from the discrete excimer were gradually redshifted, and its intensity was constantly weakened, eventually reaching near-IR fluorescence at 802 nm when the pressure was close to 8.05 GPa.
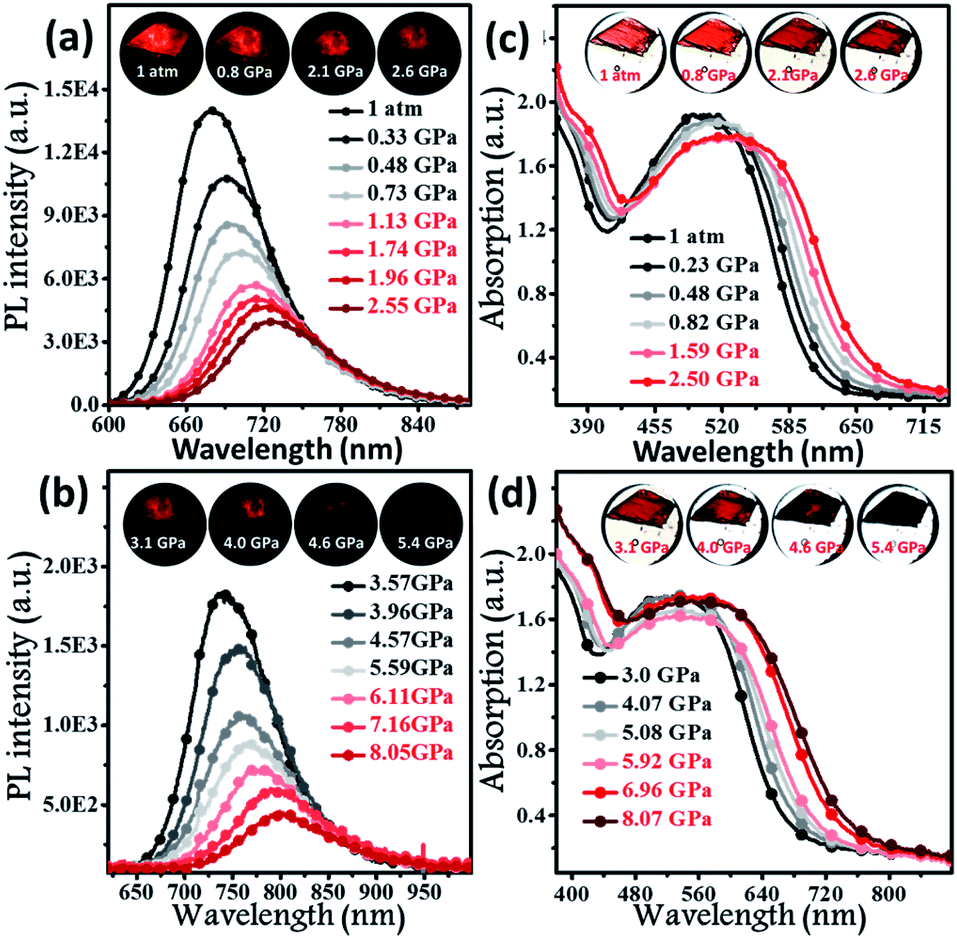 | ||
| Fig. 3 In situ PL spectra (a and b) and absorption spectra (c and d) of BTA-TPA in the crystalline state under various pressures. Inset images showing corresponding photographs of the crystal upon irradiation with 365 nm UV light (a and b) and natural light (c and d). | ||
As illustrated in Fig. 3c and d, the absorption spectra were gradually redshifted during the compression, consistent with the PL redshift. The corresponding energy gap decreased from 2.02 eV (612 nm) to 1.61 eV (706 nm), generally resulting from molecular planarization.12 In Fig. S10,† the rotation of the BTA unit to decrease the dihedral angle resulted in a decrease in the S1 excitation energy, which corresponded to a redshift in the CT bands during the compression process. The results reflected that the planarity of the molecular conformation might give rise to the redshifted fluorescence. After the pressure was fully released, the absorption spectrum reverted to the original position (Fig. S11†), demonstrating the reversibility of the change in molecular conformation. Fig. S12† indicates the change in the unit cell volume versus static pressure. The volume was constantly decreased during the compression, showing the decreased intermolecular distances. To further confirm this proposal, we performed in situ IR spectroscopy using hydrostatic pressure. As depicted in Fig. S13,† the broad IR absorption peaks located at 3030 and 1588 cm−1 could be definitively attributed to the stretching vibrations of aromatic C–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C units, respectively. Interestingly, the C–H and CC stretching frequencies were distinctly blueshifted below 7.2 GPa, indicating the decreased interatomic distances. This finding indicated that the molecular packing became closer and the intermolecular distances (such as Rg of the isolated dimer) decreased.12a,17 In comparison, the peak position of the –C
C units, respectively. Interestingly, the C–H and CC stretching frequencies were distinctly blueshifted below 7.2 GPa, indicating the decreased interatomic distances. This finding indicated that the molecular packing became closer and the intermolecular distances (such as Rg of the isolated dimer) decreased.12a,17 In comparison, the peak position of the –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N vibration was altered slightly during the compression (Fig. S13†). These results implied that the extent of electron delocalization was enhanced due to the more planar configuration of the acrylonitrile moieties, consistent with the redshift of absorption spectra. Clearly, the BTA-TPA molecules adopted a more planar conformation during compression. Moreover, the decreased interplanar distance resulted in the enhancement of intermolecular interactions. This fact, in combination with the conformational planarization, was responsible for a red-shift of the PL wavelength and a decrease in PL intensity.
N vibration was altered slightly during the compression (Fig. S13†). These results implied that the extent of electron delocalization was enhanced due to the more planar configuration of the acrylonitrile moieties, consistent with the redshift of absorption spectra. Clearly, the BTA-TPA molecules adopted a more planar conformation during compression. Moreover, the decreased interplanar distance resulted in the enhancement of intermolecular interactions. This fact, in combination with the conformational planarization, was responsible for a red-shift of the PL wavelength and a decrease in PL intensity.
In addition, BTA-TPA with excellent deep-red/near-IR emission properties with a high PL efficiency and a long PL wavelength in the aggregated state could be expected to serve as a promising optical nanoagent for biomedical applications.18 As illustrated in Fig. S14 and S15,† the pure THF solution of BTA-TPA had an intense deep-red fluorescence with a PL peak at 679 nm. Upon addition of water, the fluorescence intensity decreased in the water fraction range of 10 to 60 vol% (Fig. S15a†), which should be attributed to the twisted intramolecular charge transfer. Interestingly, the fluorescence of BTA-TPA was revitalized when a large amount of water (fw > 70%) was added into THF. As depicted in Fig. S15b,† the remarkable increase of PL intensity indicated the aggregation-induced emission (AIE) characteristic of BTA-TPA.19 The PLQYs of NPs in the THF/water mixture with a water fraction of 90 vol% reached as high as 67.8%. Moreover, their PL spectrum at 662 nm was obviously blueshifted compared to that of the crystal, which may be due to the hydrophobic microenvironment inside the aggregates. Then, as a proof of concept, the BTA-TPA nanoparticles (NPs) were prepared by nanoprecipitation.14e size of the BTA-TPA NPs was characterized by dynamic light scattering (DLS), and an average diameter of approximately 150 nm was determined (Fig. S16†). Before bioimaging of BTA-TPA NPs, cytotoxicity was evaluated by using 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyltetra-zolium bromide (MTT) at different concentrations (0, 0.4, 0.8, 1.6, 3.1, 6.3, 12.5, 25, 50, and 100 μg mL−1).20 As suggested in Fig. S17,† no significant change in the cell viability was observed even when the concentration was up to 100 μg mL−1, demonstrating the almost complete lack of cytotoxicity of BTA-TPA NPs to cells. Then, the HeLa cells were imaged with BTA-TPA NPs (Fig. 4a–c). The intense red emission of BTA-TPA was obviously observed and randomly distributed in the cytoplasm of the HeLa cells after 3 h of incubation, thus demonstrating the successful uptake of BTA-TPA NPs and underlying the enhanced permeability and retention effect (EPR). Additionally, the BTA-TPA NPs exhibit a large Stokes shift of 140 nm, which can overcome the imaging interference experienced by traditional fluorescent agents.20 To further investigate the imaging ability of BTA-TPA NPs in vivo, lymphatic mapping of BTA-TPA NPs in BALB/c mice was conducted. After the intradermal injection of BTA-TPA NPs into the right forepaw pads of mice, the fluorescence signals of the nearby lymph were continuously monitored, as shown in Fig. 4d–f. Obvious fluorescence signals emerged in the underarm, with an extremely low background that prevented autofluorescence of the body. The fluorescence intensity increased at 5 and 10 minutes, thus showing clear lymphatic mapping and imaging in vivo. Interestingly, the diffusion of BTA-TPA NPs from the injection site into the lymphatic system could be clearly observed, indicative of the potential application in sentinel lymph node (SLN) mapping based on BTA-TPA NPs. Thus, deep-red/near-IR BTA-TPA could indeed act as a superior candidate with the advantages of high brightness, deep penetration and low background for biomedical applications, and further work is underway.
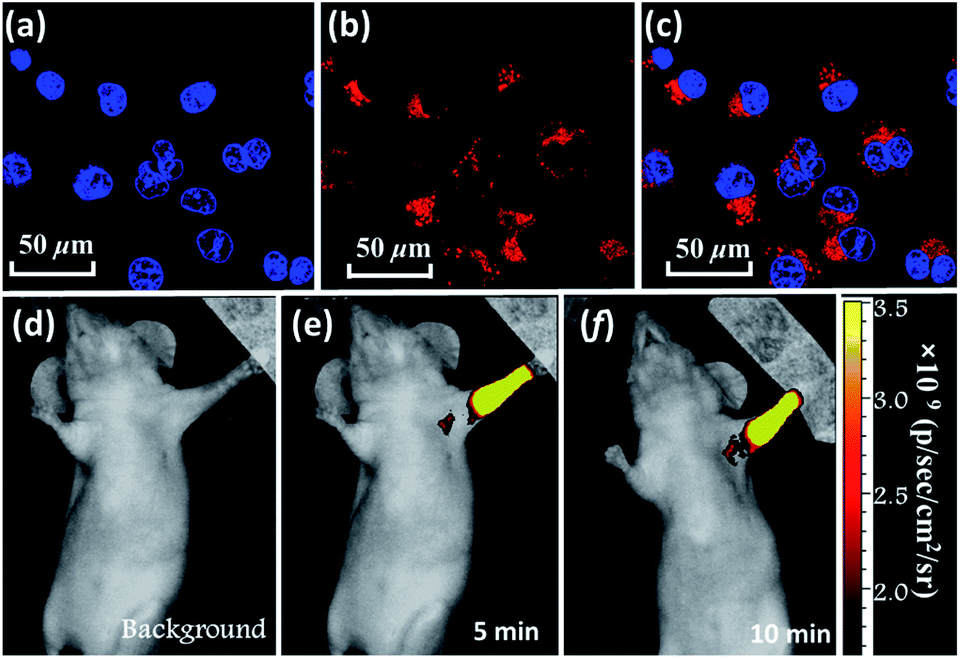 | ||
| Fig. 4 CLSM images of HeLa cells incubated with BTA-TPA NPs. (a) Hoechst 33342 channel. (b) BTA-TPA NP channel. (c) The overlay of (a and b). Hoechst 33342 channel: λex: 405 nm; BTA-TPA NP channel: λex: 514 nm; in vivo lymphatic mapping and imaging of mice injected with BTA-TPA NPs at various time points (d–f). | ||
Conclusions
In summary, an efficient strategy for highly emissive deep-red/near-IR emitters has been developed in D–π–A-type luminophores. BTA-TPA powdery crystals emit deep-red fluorescence with an excellent PLQY of 54.8%. This PLQY is the best result achieved for deep-red/near-IR fluorescence (around 700 nm) based on excimer-emitting materials. Our results demonstrate that combining the HLCT state and separated dimers can induce long-wavelength fluorescence and high PLQYs. The experimental and theoretical analyses confirm that isolated dimers exhibit a “compressed” excimer, restricting the intramolecular motions and blocking the occurrence of a “dark” state from nonradiative energy transfer, which results in very bright fluorescence. Moreover, in vitro and in vivo imaging are explored to demonstrate the potential use in biomedical applications. Therefore, we believe that our findings are valuable for guiding the development of deep-red/near-IR luminophors and materials with high PLQYs, benefiting their practical applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
All animal experiments were performed according to the Chinese Regulations for the Administration of Affairs Concerning Experimental Animals and approved by the Institutional Animal Care and Use Committee (IACUC) of the Beijing University of Chemical Technology (Beijing, China). This work was supported by the Natural Science Foundation of Zhejiang Province (LZ20E030001 and LY17F050001) and the National Natural Science Foundation of China (21725304, 21702016 and 91833302). We are grateful to the High-performance Computing Center in Nanjing Tech University for supporting the computational resources.Notes and references
- (a) D. H. Kim, A. D’Aléo, X. K. Chen, A. D. S. Sandanayaka, D. Yao, L. Zhao, T. Komino, E. Zaborova, G. Canard, Y. Tsuchiya, E. Choi, J. W. Wu, F. Fages, J. L. Brédas, J. C. Ribierre and C. Adachi, Nat. Photonics, 2018, 12, 98–104 CrossRef CAS; (b) T. Seki, N. Tokodai, S. Omagari, T. Nakanishi, Y. Hasegawa, T. Iwasa, T. Taketsugu and H. Ito, J. Am. Chem. Soc., 2017, 139, 6514–6517 CrossRef CAS PubMed; (c) S. Wang, X. Yan, Z. Cheng, H. Zhang, Y. Liu and Y. Wang, Angew. Chem., Int. Ed., 2015, 54, 13068–13072 CrossRef CAS PubMed; (d) S. Fateminia, Z. Mao, S. Xu, Z. Yang, Z. Chi and B. Liu, Angew. Chem., Int. Ed., 2017, 56, 12160–12164 CrossRef CAS PubMed; (e) H. Luo, S. Chen, Z. Liu, C. Zhang, Z. Cai, X. Chen, G. Zhang, Y. Zhao, S. Decurtins, S. Liu and D. Zhang, Adv. Funct. Mater., 2014, 24, 4250–4258 CrossRef CAS.
- (a) J. V. Caspar, E. M. Kober, B. P. Sullivan and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 630–632 CrossRef CAS; (b) S. Cummings and R. Eisenberg, J. Am. Chem. Soc., 1996, 118, 1949–1960 CrossRef CAS; (c) J. S. Wilson, N. Chawdhury, M. Al-Mandhary, M. Younus, M. Khan, P. Raithby, A. Köhler and R. H. Friend, J. Am. Chem. Soc., 2001, 123, 9412–9417 CrossRef CAS PubMed.
- (a) Q. Wei, N. Fei, A. Islam, T. Lei, L. Hong, R. Peng, X. Fan, L. Chen, P. Gao and Z. Ge, Adv. Opt. Mater., 2018, 6, 1800512 CrossRef; (b) Q. Zhao and J. Z. Sun, J. Mater. Chem. C, 2016, 4, 10588–10609 RSC.
- (a) M. Shimizu and T. Hiyama, Chem.–Asian J., 2010, 5, 1516–1531 CrossRef CAS PubMed; (b) M. V. d. Auweraer, Z. R. Grabowski and W. Rettig, J. Phys. Chem., 1991, 95, 2083–2092 CrossRef.
- (a) L. Yao, S. Zhang, R. Wang, W. Li, F. Shen, B. Yang and Y. Ma, Angew. Chem., Int. Ed., 2014, 53, 2119–2123 CrossRef CAS PubMed; (b) S. Zhang, L. Yao, Q. Peng, W. Li, Y. Pan, R. Xiao, Y. Gao, C. Gu, Z. Wang, P. Lu, F. Li, S. Su, B. Yang and Y. Ma, Adv. Funct. Mater., 2015, 25, 1755–1762 CrossRef CAS.
- Z. Sun, Q. Zang, Q. Luo, C. Lv, Q. Song, R. Zhao, Y. Zhang and W. Wong, Chem. Commun., 2019, 55, 4735–4738 RSC.
- T. Förster, Angew. Chem., Int. Ed., 1969, 8, 333–343 CrossRef.
- (a) T. Hinoue, M. Miyata, I. Hisaki and N. Tohnai, Angew. Chem., Int. Ed., 2012, 51, 155–158 CrossRef CAS PubMed; (b) J. Zhang, B. Xu, J. Chen, S. Ma, Y. Dong, L. Wang, B. Li, L. Ye and W. Tian, Adv. Mater., 2014, 26, 739–745 CrossRef PubMed; (c) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed; (d) G. Han, D. Kim, Y. Park, J. Bouffard and Y. Kim, Angew. Chem., Int. Ed., 2015, 54, 3912–3916 CrossRef CAS PubMed.
- M. Inouye, K. Hayashi, Y. Yonenaga, T. Itou, K. Fujimoto, T. Uchida, M. Iwamura and K. Nozaki, Angew. Chem., Int. Ed., 2014, 53, 14392–14396 CrossRef CAS PubMed.
- H. Osaki, C. Chou, M. Taki, K. Welke, D. Yokogawa, S. Irle, Y. Sato, T. Higashiyama, S. Saito, A. Fukazawa and S. Yamaguchi, Angew. Chem., Int. Ed., 2016, 55, 7131–7135 CrossRef CAS PubMed.
- (a) H. Liu, L. Yao, B. Li, X. Chen, Y. Gao, S. Zhang, W. Li, P. Lu, B. Yang and Y. Ma, Chem. Commun., 2016, 52, 7356–7359 RSC; (b) S. Sekiguchi, K. Kondo, Y. Sei, M. Akita and M. Yoshizawa, Angew. Chem., Int. Ed., 2016, 55, 6906–6910 CrossRef CAS PubMed.
- (a) Y. Zhang, J. Zhang, J. Shen, J. Sun, K. Wang, Z. Xie, H. Gao and B. Zou, Adv. Opt. Mater., 2018, 6, 1800956 CrossRef; (b) Y. Sagara, K. Kubo, T. Nakamura, N. Tamaoki and C. Weder, Chem. Mater., 2017, 29, 1273–1278 CrossRef CAS.
- W. Li, Y. Pan, R. Xiao, Q. Peng, S. Zhang, D. Ma, F. Li, F. Shen, Y. Wang, B. Yang and Y. Ma, Adv. Funct. Mater., 2014, 24, 1609–1614 CrossRef CAS.
- (a) X. Han, Q. Bai, L. Yao, H. Liu, Y. Gao, J. Li, L. Liu, Y. Liu, X. Li, P. Lu and B. Yang, Adv. Funct. Mater., 2015, 25, 7521–7529 CrossRef; (b) C. Li, R. Duan, B. Liang, G. Han, S. Wang, K. Ye, Y. Liu, Y. Yi and Y. Wang, Angew. Chem., Int. Ed., 2017, 56, 11525–11529 CrossRef CAS PubMed; (c) T. Liu, L. Zhu, C. Zhong, G. Xie, S. Gong, J. Fang, D. Ma and C. Yang, Adv. Funct. Mater., 2017, 27, 1606384 CrossRef; (d) J. Kim, J. Yun and J. Lee, Adv. Opt. Mater., 2018, 6, 1800255 CrossRef; (e) H. Lu, Y. Zheng, X. Zhao, L. Wang, S. Ma, X. Han, B. Xu, W. Tian and H. Gao, Angew. Chem., Int. Ed., 2016, 55, 155–159 CrossRef CAS PubMed.
- (a) S. Varghese and S. Das, J. Phys. Chem. Lett., 2011, 2, 863–873 CrossRef CAS PubMed; (b) Q. Luo, C. Lv, H. Sheng, F. Cao, J. Sun, C. Zhang, M. Ouyang, B. Zou and Y. Zhang, Adv. Opt. Mater., 2020, 8, 1901836 CrossRef CAS; (c) J. Cornil, D. Beljonne, J. Calbert and J. Brédas, Adv. Mater., 2001, 13, 1053–1067 CrossRef CAS.
- (a) Y. Gao, H. Liu, S. Zhang, Q. Gu, Y. Shen, Y. Ge and B. Yang, Phys. Chem. Chem. Phys., 2018, 20, 12129–12137 RSC; (b) Z. An, C. Zheng, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng, X. Liu and W. Huang, Nat. Mater., 2015, 14, 685–690 CrossRef CAS PubMed; (c) J. Yang, X. Zhen, B. Wang, X. Gao, Z. Ren, J. Wang, Y. Xie, J. Li, Q. Peng, K. Pu and Z. Li, Nat. Commun., 2018, 9, 840 CrossRef PubMed; (d) Y. Shen, H. Liu, S. Zhang, Y. Gao, B. Li, Y. Yan, Y. Hu, L. Zhao and B. Yang, J. Mater. Chem. C, 2017, 5, 10061–10067 RSC.
- H. Liu, Y. Dai, Y. Gao, H. Gao, L. Yao, S. Zhang, Z. Xie, K. Wang, B. Zou, B. Yang and Y. Ma, Adv. Opt. Mater., 2018, 6, 1800085 CrossRef.
- (a) C. Wang, L. Xu, C. Liang, J. Xiang, R. Peng and Z. Liu, Adv. Mater., 2014, 26, 8154–8162 CrossRef CAS PubMed; (b) X. Gu, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Biomaterials, 2017, 146, 115–135 CrossRef CAS PubMed; (c) D. Ding, K. Li, B. Liu and B. Z. Tang, Acc. Chem. Res., 2013, 46, 2441–2453 CrossRef CAS PubMed; (d) R. Tian, H. Ma, Q. Yang, H. Wan, S. Zhu, S. Chandra, H. Sun, D. Kiesewetter, G. Niu, Y. Liang and X. Chen, Chem. Sci., 2019, 10, 326–332 RSC.
- (a) W. Qin, D. Ding, J. Liu, W. Yuan, Y. Hu, B. Liu and B. Tang, Adv. Funct. Mater., 2012, 22, 771–779 CrossRef CAS; (b) S. Wang, J. Liu, C. Goh, L. Ng and B. Liu, Adv. Mater., 2019, 31, 1904447 CrossRef CAS PubMed; (c) L. Zong, H. Zhang, Y. Li, Y. Gong, D. Li, J. Wang, Z. Wang, Y. Xie, M. Han, Q. Peng, X. Li, J. Dong, J. Qian, Q. Li and Z. Li, ACS Nano, 2018, 12, 9532–9540 CrossRef CAS PubMed; (d) W. Tsai, C. Wang, C. Liao, C. Yao, T. Kuo, M. Liu, C. Hsu, S. Lin, C. Wu, J. Pyle, J. Chen and Y. Chan, Chem. Sci., 2019, 10, 198–207 RSC.
- (a) C. W. T. Leung, Y. Hong, S. Chen, E. Zhao, J. W. Y. Lam and B. Z. Tang, J. Am. Chem. Soc., 2013, 135, 62–65 CrossRef CAS PubMed; (b) X. Gu, E. Zhao, T. Zhao, M. Kang, C. Gui, J. W. Y. Lam, S. Du, M. M. T. Loy and B. Z. Tang, Adv. Mater., 2016, 28, 5064–5071 CrossRef CAS PubMed; (c) Z. Zheng, H. Liu, S. Zhai, H. Zhang, G. Shan, R. Kwok, C. Ma, H. Sung, I. Williams, J. Lam, K. Wong, X. Hu and B. Tang, Chem. Sci., 2020, 11, 2494–2503 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1888552. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc01873b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |