
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarboxylate breaks the arene C–H bond via a hydrogen-atom-transfer mechanism in electrochemical cobalt catalysis†
Xin-Ran
Chen
a,
Shuo-Qing
Zhang
 a,
Tjark H.
Meyer
b,
Chun-Hui
Yang
d,
Qin-Hao
Zhang
a,
Ji-Ren
Liu
a,
Hua-Jian
Xu
*d,
Fa-He
Cao
*c,
Lutz
Ackermann
*b and
Xin
Hong
*a
a,
Tjark H.
Meyer
b,
Chun-Hui
Yang
d,
Qin-Hao
Zhang
a,
Ji-Ren
Liu
a,
Hua-Jian
Xu
*d,
Fa-He
Cao
*c,
Lutz
Ackermann
*b and
Xin
Hong
*a
aDepartment of Chemistry, Zhejiang University, Hangzhou, 310027, China. E-mail: hxchem@zju.edu.cn
bInstitut für Organische und Biomolekulare Chemie, Georg-August-Universität Göttingen, Tammannstraße 2, 37077 Göttingen, Germany. E-mail: Lutz.Ackermann@chemie.uni-goettingen.de
cSchool of Materials, Sun Yat-sen University, Guangzhou, 510006, China. E-mail: caofh5@mail.sysu.edu.cn
dSchool of Food and Biological Engineering, Hefei University of Technology, Hefei, 230009, China. E-mail: hjxu@hfut.edu.cn
First published on 19th May 2020
Abstract
Combined computational and experimental studies elucidated the distinctive mechanistic features of electrochemical cobalt-catalyzed C–H oxygenation. A sequential electrochemical–chemical (EC) process was identified for the formation of an amidylcobalt(III) intermediate. The synthesis, characterization, cyclic voltammetry studies, and stoichiometric reactions of the related amidylcobalt(III) intermediate suggested that a second on-cycle electro-oxidation occurs on the amidylcobalt(III) species, which leads to a formal Co(IV) intermediate. This amidylcobalt(IV) intermediate is essentially a cobalt(III) complex with one additional single electron distributed on the coordinating heteroatoms. The radical nature of the coordinating pivalate allows the formal Co(IV) intermediate to undergo a novel carboxylate-assisted HAT mechanism to cleave the arene C–H bond, and a CMD mechanism could be excluded for a Co(III/I) catalytic scenario. The mechanistic understanding of electrochemical cobalt-catalyzed C–H bond activation highlights the multi-tasking electro-oxidation and the underexplored reaction channels in electrochemical transition metal catalysis.
Introduction
Due to the remarkable availability and sustainability of electricity, electrochemical C–H bond activation and functionalization has recently emerged as a powerful approach of green synthesis.1 The merging of electrochemical C–H bond activation and 3d transition metal catalysis has successfully realized a series of electrochemical cobalt-catalyzed C–H bond transformations,2 including C–H oxygenation3 (Scheme 1a) and C–H amination4 (Scheme 1b), as well as C–H/N–H annulation5 (Scheme 1c) and carbonylation6 (Scheme 1d). These transformations avoid stoichiometric oxidants, presenting an appealing synthetic strategy with exceptional resource-economy.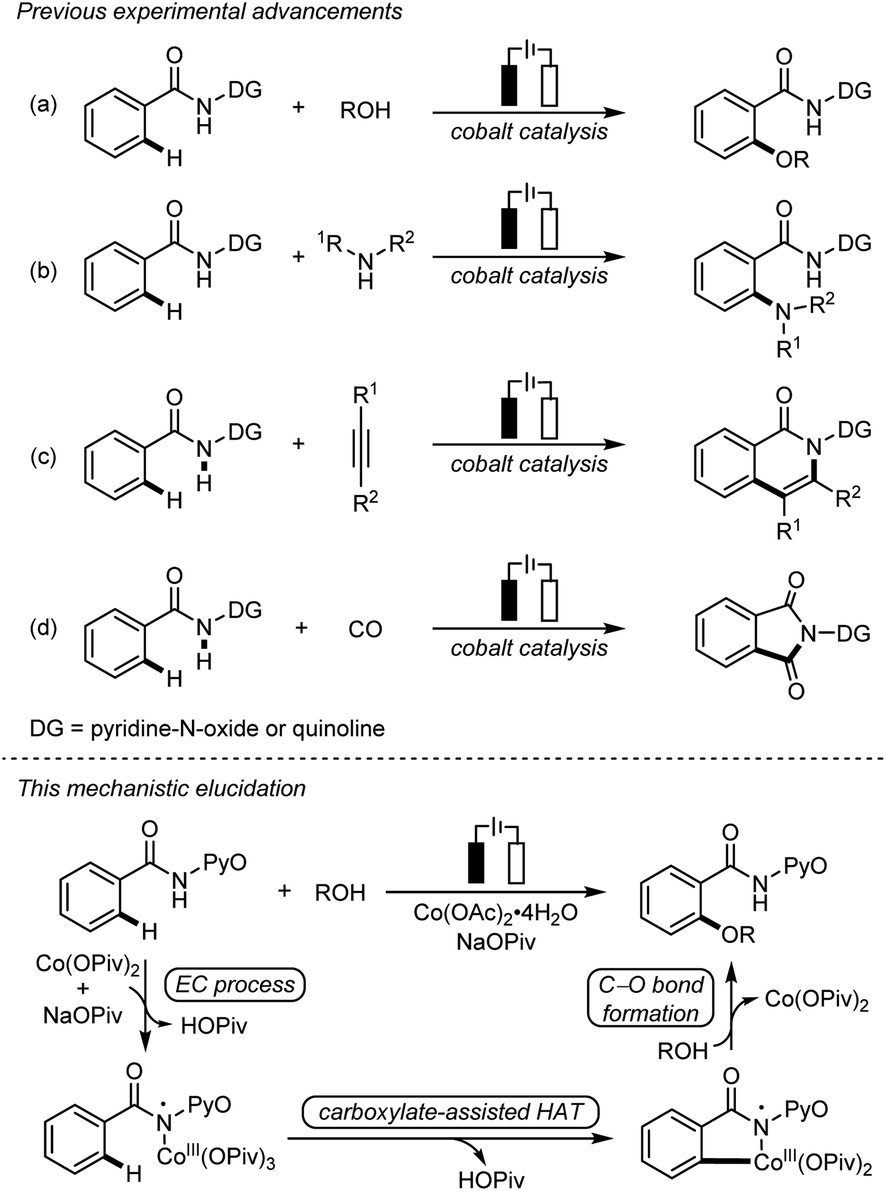 | ||
| Scheme 1 Electrochemical cobalt-catalyzed C–H oxygenation. | ||
Despite the rich history and fruitful advances in the mechanistic studies on transition metal-catalyzed C–H bond activation,7 the mechanistic understanding of electrochemical C–H bond activation still remains primitive.8 Currently, little molecular-level understanding and controlling factors of electrochemical transition metal-catalyzed C–H bond activations are available, which presents a significant challenge for rational reaction design in this field. Herein, we report a mechanistic elucidation on electrochemical cobalt-catalyzed C–H oxygenation with combined computational and experimental studies (Scheme 1). A key electrochemical–chemical (EC) process was identified for the generation of amidylcobalt(III) species, based on density functional theory (DFT) calculations, cyclic voltammetry studies, characterization and stoichiometric transformations of the proposed amidylcobalt(III) species which allows a novel carboxylate-assisted HAT mechanism to cleave the arene C–H bond. These mechanistic features highlight the distinctive reaction channels in electrochemical transition metal catalysis, providing the molecular basis for rational reaction design in this field.
Results and discussion
Two mechanisms were proposed for electrochemical cobalt-catalyzed C–H functionalization (Scheme 2).3–6,9 Mechanism A is a Co(III)–Co(I) catalytic cycle. It involves initial anodic oxidation of Co(II)pivalate to the corresponding Co(III) species. Subsequent N–H cleavage leads to the amidylcobalt(III) intermediate. This intermediate undergoes C–H bond activation, and subsequent C–O bond formation via the arylcobalt(III) species leads to the amidylcobalt(I) intermediate. Further proto-demetalation produces the oxygenated product and regenerates the cobalt catalyst. Mechanism B is a Co(IV)–Co(II) catalytic cycle. From the Co(III)pivalate, an anodic oxidation and N–H cleavage lead to the cationic amidylcobalt(IV) intermediate. This intermediate undergoes similar C–H bond activation, C–O bond formation and proto-demetalation to produce the oxgenated product.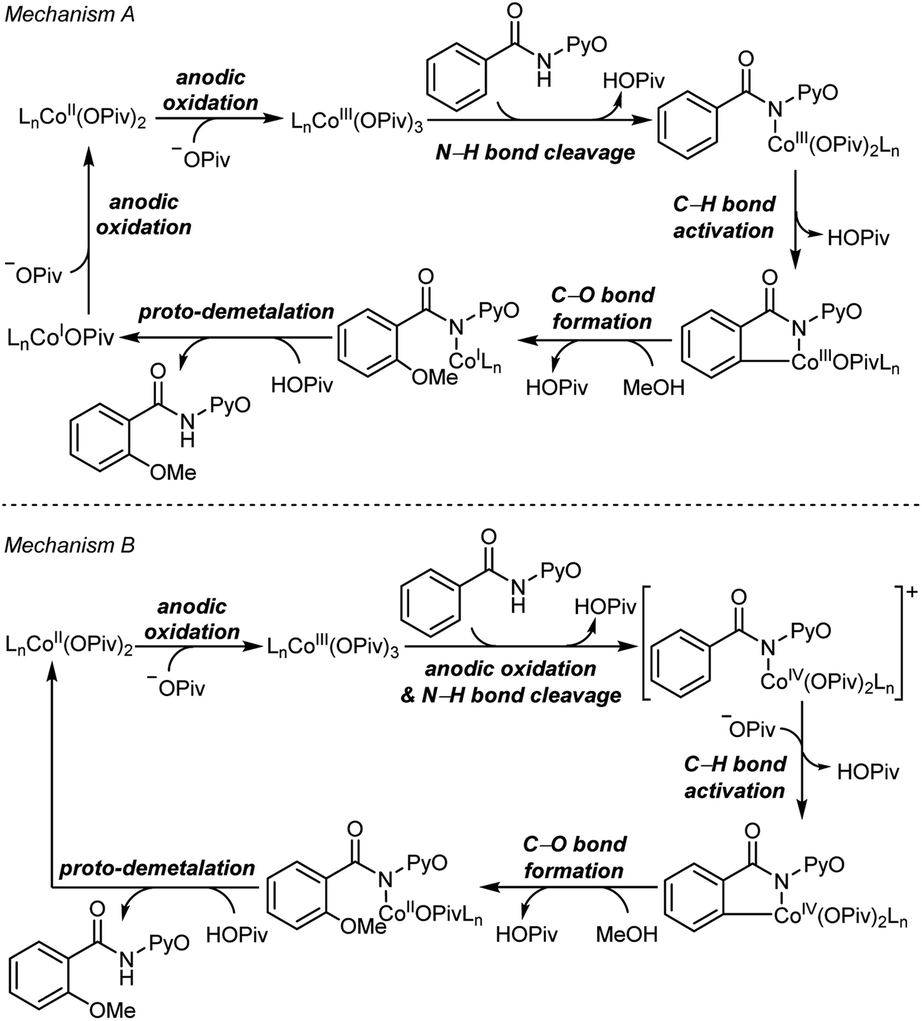 | ||
| Scheme 2 Proposed catalytic cycles of electrochemical cobalt-catalyzed C–H oxygenation. | ||
The DFT-computed free energy changes of mechanism A and the oxidation potential of the involved intermediates are shown in Fig. 1.10 The exergonic complexation of the quintet model complex Co(OPiv)3 with amide substrate 1 leads to the quintet int2. This intermediate undergoes a facile carboxylate-assisted N–H cleavage viaTS3, generating the amidylcobalt(III) intermediate int4. A subsequent carboxylate-assisted metalation process cleaves the arene C–H bond through TS5 and generates the arylcobalt(III) intermediate int6. int6 further complexes with methanol through hydrogen-bonding to form int7.
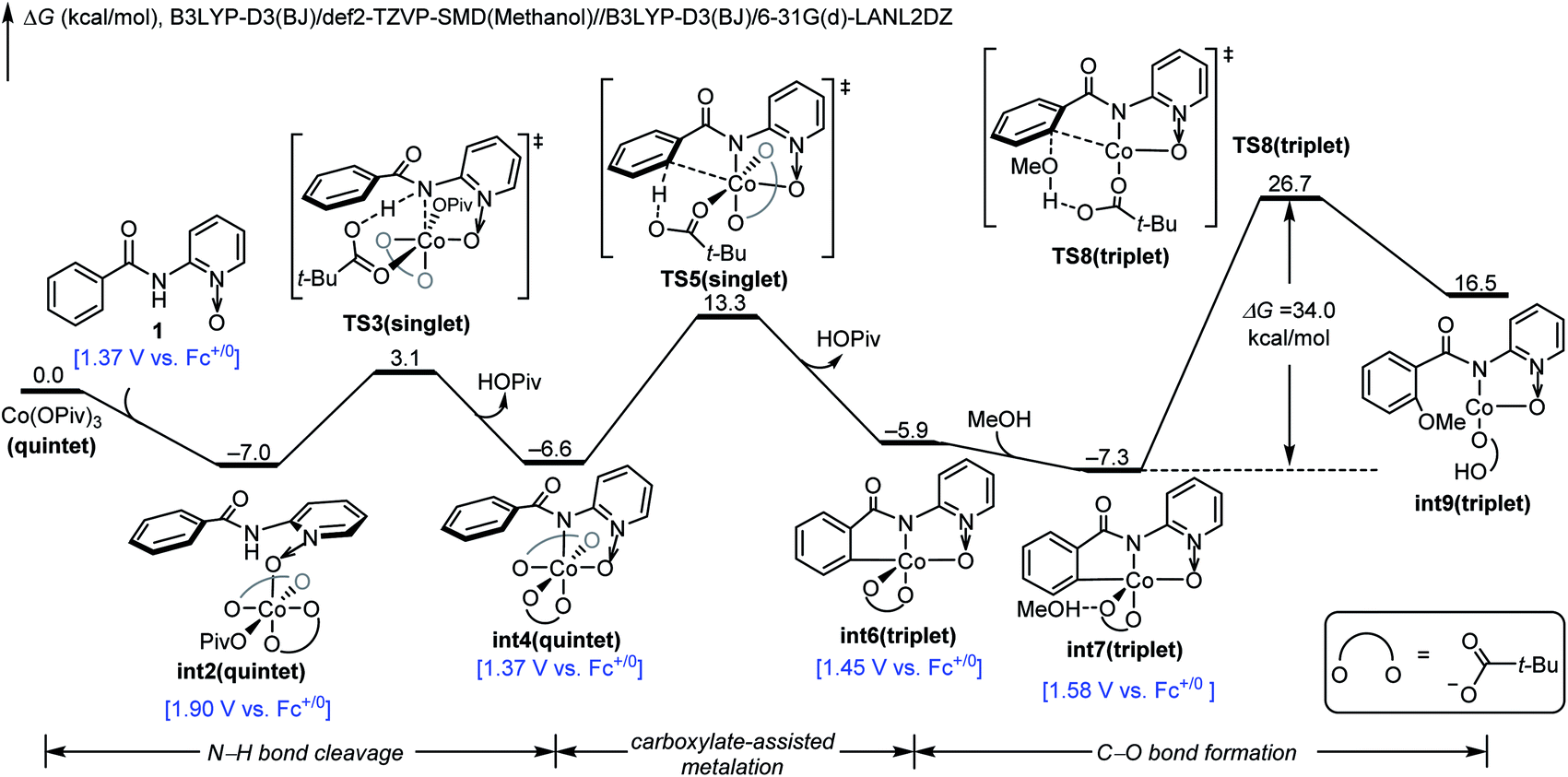 | ||
| Fig. 1 DFT-computed free energy changes of mechanism A and computed oxidation potentials of intermediates before C–O bond formation. | ||
From int7, the C–O bond formation viaTS8 requires an unsurmountable barrier of 34.0 kcal mol−1. We also considered the C–O bond formation process via reductive elimination of arylcobalt(III)(OMe) species, and this alternative process is even less favourable (Fig. S1†). Therefore, mechanism A is not operative due to the high C–O bond formation barrier of the arylcobalt(III) intermediate for the room-temperature catalytic C–H oxygenation. Only the most stable spin states are presented in Fig. 1; the detailed free energy profile with minimum-energy crossing point information and IRC confirmations of each transition state are included in the ESI (Fig. S2–S6†).
Our above computations suggest that the oxidation of Co(III) species prior to C–O bond formation is necessary. Based on the computed oxidation potentials of the involved organocobalt intermediates (Fig. 1), the amide substrate 1 and amidylcobalt(III) intermediate int4 have the lowest oxidation potentials. These two species have the same oxidation potentials because the oxidation of int4 mainly occurs on the coordinating amidyl fragment (Fig. S7†). The oxidation of int4 leads to a Co(III) complex with a coordinating amidyl radical, a formal Co(IV) species int10 (Fig. 2). If the electrochemical oxidation occurs on int4, Co(IV) generation involves sequential electrochemical oxidation of Co(II) pivalate and a chemical process (EC process, Fig. 2). Alternatively, the electrochemical oxidation of the amide substrate leads to two independent electrochemical oxidations (EE process, Fig. 2).11
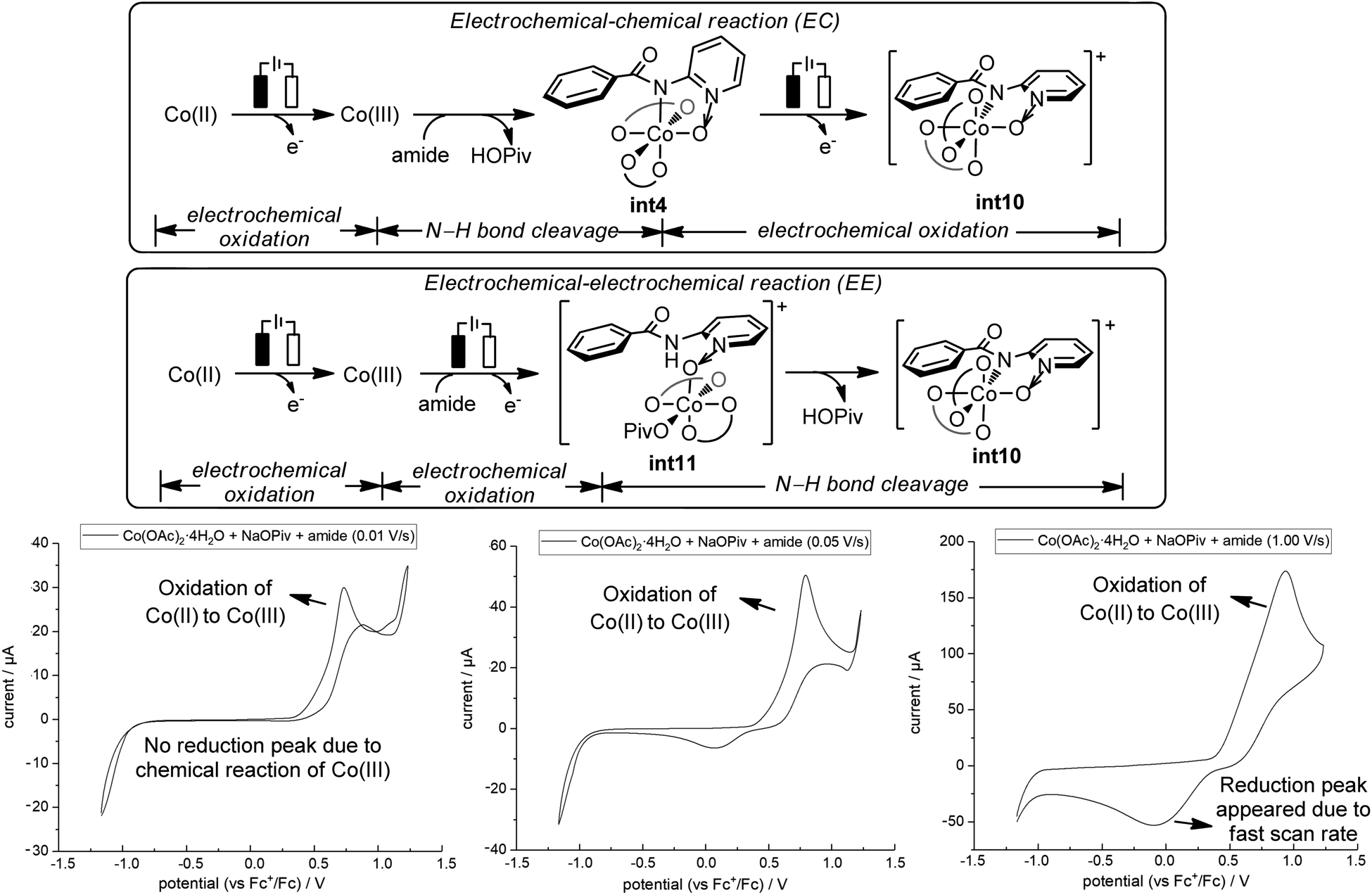 | ||
Fig. 2 Cyclic voltammetry studies with varying scan rates. The measurements were carried out at scan rates of 0.01 V s−1, 0.05 V s−1, and 1.00 V s−1 respectively in a solvent mixture of DCM and MeOH (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), with electrolyte n-Bu4NPF6 (0.05 M). Amide (substrate 1) (0.5 mM), NaOPiv·H2O (2 mM), and Co(OAc)2·4H2O (0.5 mM). :1), with electrolyte n-Bu4NPF6 (0.05 M). Amide (substrate 1) (0.5 mM), NaOPiv·H2O (2 mM), and Co(OAc)2·4H2O (0.5 mM). | ||
The EC process can be identified by varying the scan rate in the cyclic voltammetry (CV) study.11 The mixture of Co(OAc)2·4H2O, NaOPiv·H2O and amide 1 at a scan rate of 0.01 V s−1 showed an oxidation peak which corresponds to the oxidation of Co(II) to Co(III).12 No reduction peak was identified at this slow scan rate, suggesting that the oxidized Co(III) species underwent a chemical transformation and the reduction of Co(III) did not occur. With increasing scan rate, the rate of the chemical transformation did not match the fast scan rate, and a reduction peak appeared (0.05 V s−1 and 1.00 V s−1). CV studies with additional scan rates are included in the ESI (Fig. S16†). This observation is strongly suggestive of an EC process, and the key chemical process has a number of possibilities, including the coordination of the amide substrate, or the amide N–H bond cleavage.
The preparation, characterization and stoichiometric transformations of the proposed amidylcobalt(III) species revealed that the key chemical transformation that connects the two electro-oxidations is the amide N–H bond cleavage. The proposed amidylcobalt(III) species A was synthesized by reacting the amide substrate 1 with cobalt triacetoacetate, in the presence of silver acetate and sodium pivalate hydrate in chloroform (Scheme 3a). By the ESI-MS study of the synthesized A, a consistent molecular weight was found (Scheme 3a), and the structure of A was unambiguously determined by X-ray diffraction (Scheme 3b). Additional NMR and UV-Vis characterization of A is included in the ESI.† The CV study of the amidylcobalt(III) species A is shown in Scheme 3c (red curve). The oxidation peak of A matched well with the second oxidation peak of the reaction mixture involving the amide substrate 1, Co(acac)2 and NaOPiv·H2O. This further supports the mechanistic proposal that the second electro-oxidation occurs on the amidylcobalt(III) intermediate with the arene C–H bonds being intact. A is stable in basic solution without additional oxidants (Scheme S3†). This is consistent with the above calculations that the Co(III)-mediated C–O bond formation is unfeasible (Fig. 1). Under electro-oxidizing conditions, A is smoothly transformed into the oxygenation product 12 (Scheme 3d). This corroborated the mechanistic proposal that a second on-cycle electro-oxidation is necessary for C–O bond formation. It should be noted that the chemical oxidation with Ag2O also allowed the desired oxygenation of A to occur (Scheme S4†), which suggests that the amidylcobalt(IV) intermediate could be involved in both electro-chemical and chemical oxidation processes.
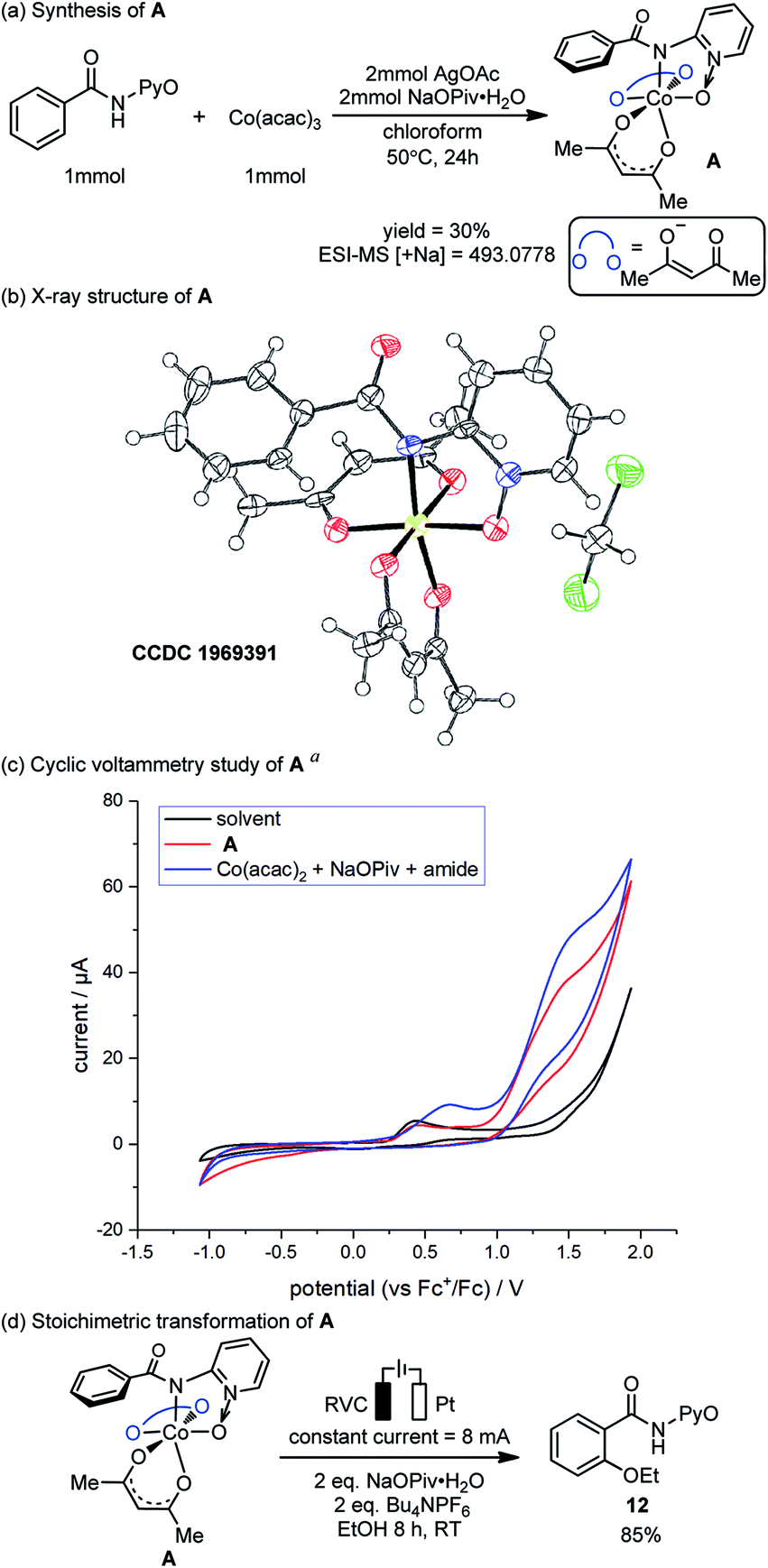 | ||
| Scheme 3 Synthesis, characterization, cyclic voltammetry study and stoichiometric reaction of amidylcobalt(III) species. aThe measurements were carried out at a scan rate of 0.10 V s−1 in a solvent mixture of DCM and MeOH (4:1), with electrolyte n-Bu4NPF6 (0.05 M). Amide (substrate 1) (0.5 mM), NaOPiv·H2O (2 mM), and Co(acac)2 (0.5 mM). | ||
We next investigated the mechanistic pathway from the formal Co(IV) intermediate, and the DFT-computed free energy changes are included in Fig. 3. A complexation of int10 with the pivalate anion leads to the neutral intermediate int13. This intermediate undergoes a carboxylate-assisted HAT process to cleave the arene C–H bond viaTS14 (vide infra), generating a high-energy intermediate int15 with a phenyl radical. The alternative proton transfer process for the arene C–H bond cleavage is much less favorable (Fig. S8†), and the CMD transition state cannot be located despite extensive efforts. The phenyl radical of int15 is intramolecularly trapped by cobalt to form the arylcobalt intermediate int16. This exergonic cobalt–aryl bond formation compensates the endergonicity of the HAT step. From int16, the coordinating pivalic acid dissociates to generate int17. Subsequently, methanol complexes through hydrogen-bonding in int18, and the C–O bond formation proceeds viaTS19. This C–O bond formation only requires a barrier of 16.0 kcal mol−1 and is significantly more efficient than the corresponding process viaTS8 (Fig. 1). Information on further exergonic proto-demetalation, additional spin states, and IRC confirmations of each transition state are included in the ESI (Fig. S9–S12†).
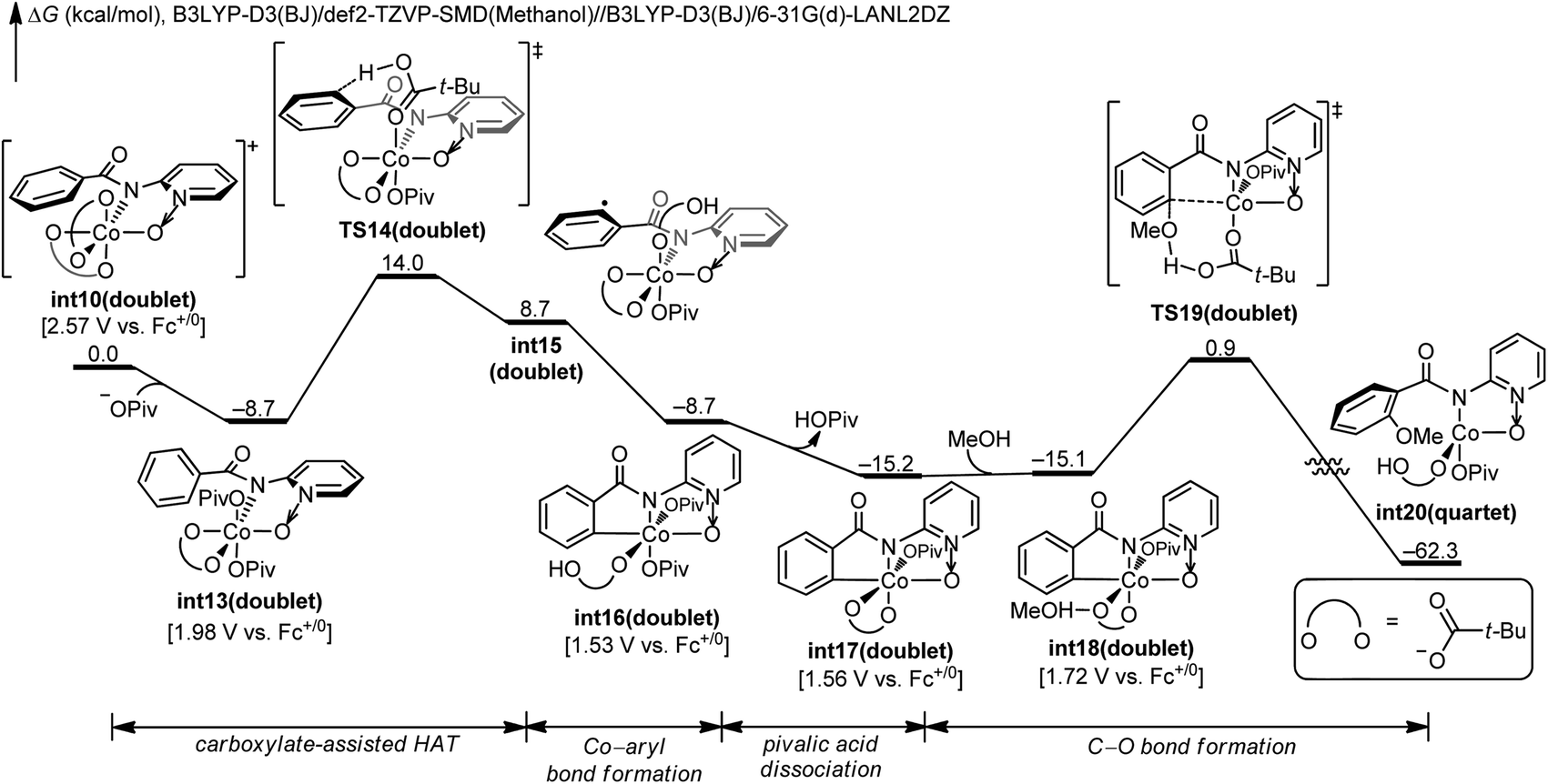 | ||
| Fig. 3 DFT-computed free energy changes of the pathway from formal Co(IV) intermediate int10 and computed oxidation potentials of intermediates. | ||
The nature of hydrogen-atom-transfer of the carboxylate-assisted C–H bond activation was further characterized by the natural spin density distribution (Fig. 4). In the doublet int13, the radical spin is mainly located in O1, N and O2. The radical character of the pivalate oxygen O1 allows the hydrogen-atom-transfer though TS14, and a significant radical distribution exists on the forming phenyl radical in this transition state. The overall hydrogen-atom-transfer produces the phenyl radical in int15, and the spin density is now mainly distributed in the carbon radical atom. This change of spin distribution is consistent with previous theoretical studies on HAT,13 and our computational Hammett analysis also confirmed that the radical-stabilizing substituents can facilitate this HAT process (Scheme S1†).14
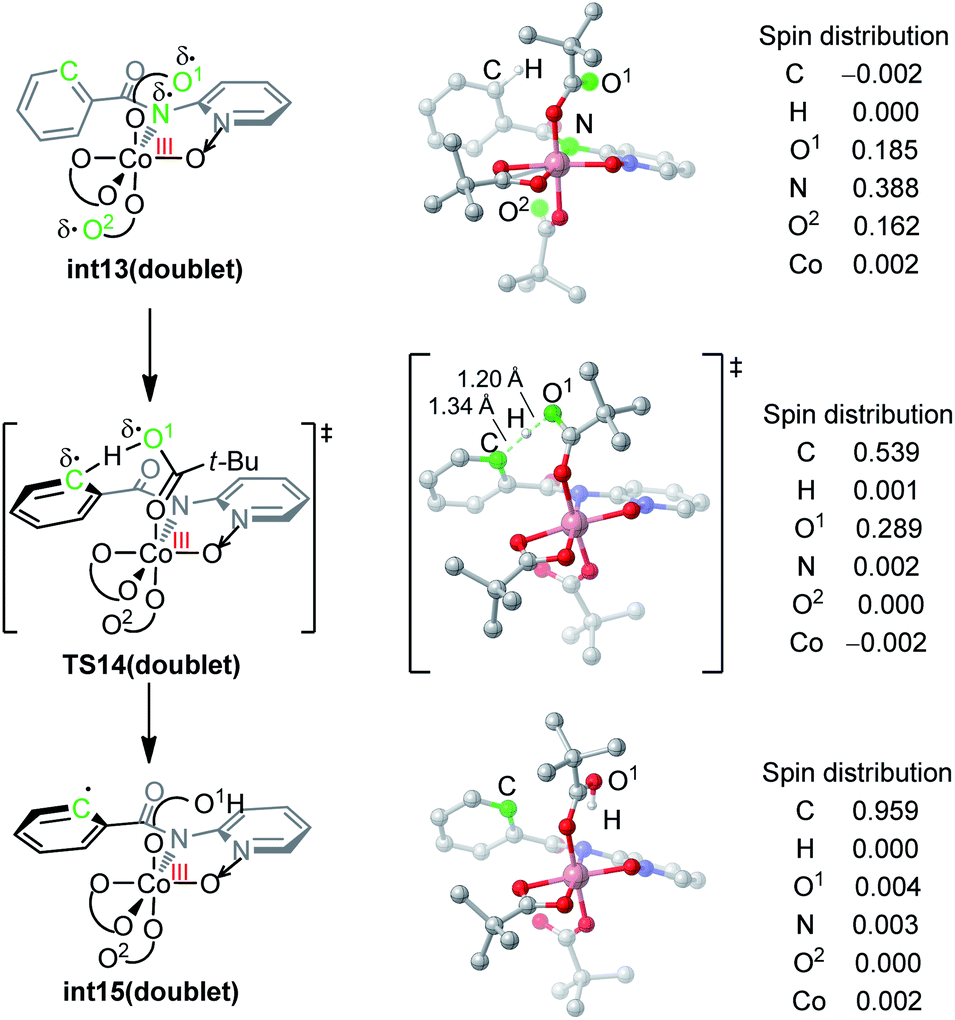 | ||
| Fig. 4 DFT-optimized structures and the spin density distribution of the C–H activation transition state and intermediates. | ||
We want to emphasize that the radical character of the coordinating pivalate is crucial for the HAT mechanism. Pivalate is generally considered as a basic and anionic ligand instead of a radical ligand. This is why the carboxylate-assisted arene C–H bond activation generally proceeds via a concerted-metalation-deprotonation mechanism in which the hydrogen transfers as a proton.7c,g In the electro-oxidized formal Co(IV) intermediate int13, considerable radical distribution exists on the coordinating heteroatoms (Fig. 4). This allows the coordinating pivalate to behave as a radical-type hydrogen atom acceptor, leading to the HAT mechanism for arene C–H bond cleavage.
The mechanistic elucidation indicates that the anodic oxidation is not simply responsible for the regeneration of the active transition metal catalyst at the end of the catalytic cycle,15 or the oxidation of an organometallic intermediate to a high-valent species for efficient reductive elimination.16 In the case of the studied electrochemical cobalt-catalyzed C–H oxygenation with the pyridine-N-oxide directing group, the anodic oxidation plays two important roles in transforming cobalt(II) into cobalt(III) and oxidation of the amidylcobalt(III) species to the corresponding formal cobalt(IV) intermediate. These insights emphasized the mechanistic importance of a redox non-innocent ligand17 in 3d transition metal catalysis, which requires cautious mechanistic proposals. In addition, the change of the directing group may also alter the mechanistic picture, which is currently under investigation in our laboratories. It should be noted that the related C–H activated cyclometallated cobalt(III) species was recently discovered under the electro-oxidative conditions, whose transformations followed the sequence of C–H activation and a following oxidatively induced reductive elimination upon anodic oxidation.18
Conclusions
In summary, the potential mechanism of electrochemical cobalt-catalyzed C–H oxygenation was revealed by combined computational and experimental studies (Scheme 4). An initial electrochemical oxidation generates Co(III) pivalate, and subsequent N–H bond cleavage leads to the amidylcobalt(III) intermediate. This intermediate undergoes a second electrochemical oxidation, which oxidizes the coordinating amidyl fragment to the corresponding radical in the formal Co(IV) intermediate. In the formal Co(IV) intermediate, the radical character of the coordinating carboxylate allows a HAT process to cleave the arene C–H bond. Subsequent C–O bond formation proceeds in a concerted fashion, and the formed amidylcobalt(II) intermediate undergoes a proto-demetalation to release the oxygenated product as well as regenerate the Co(II) catalyst. A Co(II/III/I) catalytic cycle involving a CMD-type mechanism could be excluded by computational studies due to the unfeasible Co(III)-mediated C–O bond formation. This HAT-type C–H bond activation is distinctive from the general CMD mechanism in thermal catalysis, which could provide advantages for C(sp3)–H bond activation. Further studies on the controlling factors of the carboxylate-assisted HAT and mechanism-based design of electrochemical C–H functionalizations are ongoing in our laboratories and will be reported in due course.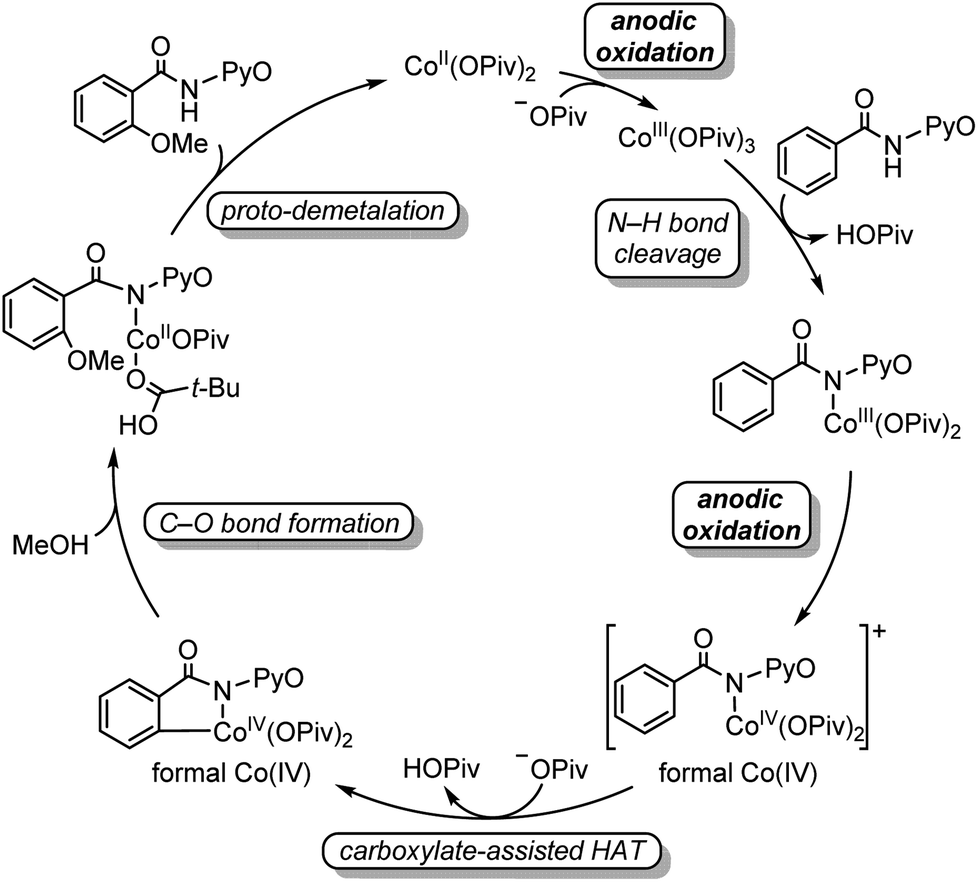 | ||
| Scheme 4 Overall catalytic cycle of electrochemical cobalt-catalyzed C–H oxygenation. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the NSFC (21702182 and 21873081 for X. H. 51771174 for F.-H. C.), DFG (Gottfried-Wilhelm-Leibniz prize) (L. A.), “Fundamental Research Funds for the Central Universities” (2-2050205-19-361 for X. H. and H.-J. X.) and China Postdoctoral Science Foundation (2018M640546 for S.-Q. Z.) is gratefully acknowledged. Calculations were performed on a high-performance computing system at the Department of Chemistry, Zhejiang University.Notes and references
- For selected reviews of electrochemical C–H activation, see: (a) L. Ackermann, Acc. Chem. Res., 2020, 53, 84 CrossRef CAS PubMed; (b) T. H. Meyer, L. H. Finger, P. Gandeepan and L. Ackermann, Trends in Chemistry, 2019, 1, 63 CrossRef; (c) Z. Ye and F. Zhang, Chin. J. Chem., 2019, 37, 51 CrossRef; (d) C. Song, K. Liu, X. Dong, C. Chiang and A. Lei, Synlett, 2019, 30, 1149 CrossRef CAS; (e) M. Kärkäs, Chem. Soc. Rev., 2018, 47, 578 RSC; (f) K. Liu, C. Song and A. Lei, Org. Biomol. Chem., 2018, 16, 2375 RSC; (g) S. Tang, Y. Liu and A. Lei, Chem, 2018, 4, 27 CrossRef CAS; (h) Q. Yang, P. Fang and T. Mei, Chin. J. Chem., 2018, 36, 338 CrossRef CAS. For general reviews on organic electrosynthesis see: (i) A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594 CrossRef CAS PubMed; (j) M. Yan, Y. Kawamata and P. Baran, Chem. Rev., 2017, 117, 13230 CrossRef CAS PubMed; (k) E. Horn, B. Rosen and P. Baran, ACS Cent. Sci., 2016, 2, 302 CrossRef CAS PubMed; (l) R. Francke and R. Little, Chem. Soc. Rev., 2014, 43, 2492 RSC.
- For selected reviews of electrochemical transition metal-catalyzed C–H functionalization, see: (a) H. Wang, X. Gao, Z. Lv, T. Abdelilah and A. Lei, Chem. Rev., 2019, 119, 6769 CrossRef CAS PubMed; (b) N. Sauermann, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Catal., 2018, 8, 7086 CrossRef CAS; (c) C. Ma, P. Fang and T. Mei, ACS Catal., 2018, 8, 7179 CrossRef CAS; (d) K. Jiao, C. Zhao, P. Fang and T. Mei, Tetrahedron Lett., 2017, 58, 797 CrossRef CAS; (e) F. Kakiuchi and T. Kochi, Isr. J. Chem., 2017, 57, 953 CrossRef CAS. For a review on cobalt-catalyzed electrochemical C–H functionalization, see: (f) N. Sauermann, T. H. Meyer and L. Ackermann, Chem.–Eur. J., 2018, 24, 16209 CrossRef CAS PubMed.
- (a) C. Tian, U. Dhawa, J. Struwe and L. Ackermann, Chin. J. Chem., 2019, 37, 552 CrossRef CAS; (b) N. Sauermann, T. H. Meyer, C. Tian and L. Ackermann, J. Am. Chem. Soc., 2017, 139, 18452 CrossRef CAS PubMed.
- (a) N. Sauermann, R. Mei and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 5090 CrossRef CAS; (b) X. Gao, P. Wang, L. Zeng, S. Tang and A. Lei, J. Am. Chem. Soc., 2018, 140, 4195 CrossRef CAS PubMed.
- (a) R. Mei, W. Ma, Y. Zhang, X. Guo and L. Ackermann, Org. Lett., 2019, 21, 6534 CrossRef CAS PubMed; (b) J. Chen, L. Jin, J. Zhou, X. Jiang and C. Yu, Tetrahedron Lett., 2019, 60, 2054 CrossRef CAS; (c) C. Tian, L. Massignan, T. H. Meyer and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 2383 CrossRef CAS PubMed; (d) R. Mei, N. Sauermann, C. Joao, A. Oliveira and L. Ackermann, J. Am. Chem. Soc., 2018, 140, 7913 CrossRef CAS PubMed; (e) T. H. Meyer, J. C. A. Oliveira, S. Sau, N. Ang and L. Ackermann, ACS Catal., 2018, 8, 9140 CrossRef CAS; (f) S. Tang, D. Wang, Y. Liu, L. Zeng and A. Lei, Nat. Commun., 2018, 9, 798 CrossRef PubMed.
- (a) S. Sau, R. Mei, J. Struwe and L. Ackermann, ChemSusChem, 2019, 12, 1 CrossRef PubMed; (b) L. Zeng, H. Li, S. Tang, X. Gao, Y. Deng, G. Zhang, C. Pao, J. Chen, J. Lee and A. Lei, ACS Catal., 2018, 8, 5448 CrossRef CAS.
- For selected reviews of mechanistic studies on transition metal-catalyzed C–H bond activation, see: (a) X. Qi, Y. Li, R. Bai and Y. Lan, Acc. Chem. Res., 2017, 50, 2799 CrossRef CAS PubMed; (b) Y. Yang, X. Hong, J. Yu and K. N. Houk, Acc. Chem. Res., 2017, 50, 2853 CrossRef CAS PubMed; (c) D. Davies, S. Macgregor and C. McMullin, Chem. Rev., 2017, 117, 8649 CrossRef CAS PubMed; (d) X. Zhang, L. Chung and T. Wu, Acc. Chem. Res., 2016, 49, 1302 CrossRef CAS PubMed; (e) T. Sperger, I. Sanhueza, I. Kalvet and F. Schoenebeck, Chem. Rev., 2015, 115, 9532 CrossRef CAS PubMed; (f) D. Musaev, T. Figg and A. Kaledin, Chem. Soc. Rev., 2014, 43, 5009 RSC; (g) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed; (h) T. Lyons and M. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed.
- For a review on mechanistic studies of electrochemical transition-metal catalysis, see: A. Jutand, Chem. Rev., 2008, 108, 2300 CrossRef CAS PubMed.
- For selected mechanistic studies on cobalt-catalyzed C–H bond activation, see: (a) P. Ma and H. Chen, ACS Catal., 2019, 9, 1962 CrossRef CAS; (b) Y. W. C. Du, Y. Wang, X. Guo, L. Fang, M. Song, J. Niu and D. Wei, Adv. Synth. Catal., 2018, 360, 2668 CrossRef; (c) M. Li and J. Wang, Org. Lett., 2018, 20, 6490 CrossRef CAS PubMed; (d) R. Mei, H. Wang, S. Warratz, S. Macgregor and L. Ackermann, Chem.–Eur. J., 2016, 22, 6759 CrossRef CAS PubMed; (e) S. Maity, R. Kancherla, U. Dhawa, E. Hoque, S. Pimparpar and D. Maiti, ACS Catal., 2016, 6, 5493 CrossRef CAS; (f) O. Planas, C. Whiteoak, V. Martin-Diaconescu, I. Gamba, J. Luis, T. Parella, A. Company and X. Ribas, J. Am. Chem. Soc., 2016, 138, 14388 CrossRef CAS PubMed; (g) X. Guo, L. Zhang, D. Wei and J. Niu, Chem. Sci., 2015, 6, 7059 RSC.
- Computations are performed with the Gaussian 09 program, reference: Frisch, M. J., et al., Gaussian 09, revision C.01; Gaussian Inc., Wallingford, CT, 2016, Computational details are provided in the ESI.†.
- (a) A. Bard and L. Faulkner, Electrochemical Methods Fundamentals and Applications, John Wiley & Sons, 2001 Search PubMed; (b) For a recent study involving mechanistic understading of electrochemical organic synthesis, see: B. Peters, K. Rodriguez, S. Reisberg, S. Beil, D. Hickey, Y. Kawamata, M. Collins, J. Starr, L. Chen, S. Udyavara, K. Klunder, T. Gorey, S. Anderson, M. Neurock, S. Minteer and P. S. Baran, Science, 2019, 363, 838 CrossRef CAS PubMed.
- The CV studies on individual species confirmed the oxidation potential of Co(II), details are included in the ESI (Fig. S15†).
- (a) J. Li, S. Zhou, J. Zhang, M. Schlangen, T. Weiske, D. Usharani, S. Shaik and H. Schwarz, J. Am. Chem. Soc., 2016, 138, 7973 CrossRef CAS; (b) J. Li, S. Zhou, J. Zhang, M. Schlangen, D. Usharani, S. Shaik and H. Schwarz, J. Am. Chem. Soc., 2016, 138, 11368 CrossRef CAS PubMed; (c) H. Schwarz, Chem. Phys. Lett., 2015, 629, 91 CrossRef CAS.
- For a consistent Hammett analysis of the HAT process, see: J. Paulo, T. Zaragoza, M. Siegler and D. Goldberg, J. Am. Chem. Soc., 2018, 140, 4380 CrossRef.
- (a) Y. Qiu, A. Scheremetjew and L. Ackermann, J. Am. Chem. Soc., 2019, 141, 2731 CrossRef CAS; (b) Q. Yang, Y. Xing, X. Wang, H. Ma, X. Weng, X. Yang, H. Guo and T. Mei, J. Am. Chem. Soc., 2019, 141, 18970 CrossRef CAS; (c) T. Koyanagi, A. Herath, A. Chong, M. Ratnikov, A. Valiere, J. Chang, V. Molteni and J. Loren, Org. Lett., 2019, 21, 816 CrossRef CAS; (d) X. Gao, P. Wang, L. Zeng, S. Tang and A. Lei, J. Am. Chem. Soc., 2018, 140, 4195 CrossRef CAS PubMed; (e) Y. Qiu, W. Kong, J. Struwe, N. Sauermann, T. Rogge, A. Scheremetjew and L. Akermann, Angew. Chem., Int. Ed., 2018, 57, 5828 CrossRef CAS PubMed.
- (a) A. Shrestha, M. Lee, A. Dunn and M. Sanford, Org. Lett., 2018, 20, 204 CrossRef CAS PubMed; (b) S. Zhang, R. Samanta, N. Sauermann and L. Ackermann, Chem.–Eur. J., 2018, 24, 19166 CrossRef CAS PubMed; (c) Y. Li, Q. Yang, P. Fang, T. Mei and D. Zhang, Org. Lett., 2017, 19, 2905 CrossRef CAS PubMed; (d) C. Ma, C. Zhao, Y. Li, L. Zhang, X. Xu, K. Zhang and T. Mei, Chem. Commun., 2017, 53, 12189 RSC; (e) Q. Yang, Y. Li, C. Ma, P. Fang, X. Zhang and T. Mei, J. Am. Chem. Soc., 2017, 139, 3293 CrossRef CAS PubMed.
- For selected reviews of redox non-innocent ligands, see: (a) J. I. Vlugt, Chem.–Eur. J., 2019, 25, 2651 CrossRef PubMed; (b) V. Lyaskovskyy and B. Bruin, ACS Catal., 2012, 2, 270 CrossRef CAS.
- T. H. Meyer, J. C. A. Oliveira, D. Ghorai and L. Ackermann, Angew. Chem., Int. Ed., 2020, 59, 1 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1969391. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc01898h |
| This journal is © The Royal Society of Chemistry 2020 |