
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Transformation from helical to layered supramolecular organization of asymmetric perylene diimides via multiple intermolecular hydrogen bonding†
Mengmeng
Li‡
 abc,
Wojciech
Zajaczkowski‡
a,
Gangamallaiah
Velpula
d,
Daniel
Jänsch
a,
Robert
Graf
a,
Tomasz
Marszalek
af,
Sapun H.
Parekh
ae,
Yulian
Zagranyarski
a,
Kunal
Mali
d,
Manfred
Wagner
a,
Steven
De Feyter
d,
Chen
Li
a,
Klaus
Müllen
*a and
Wojciech
Pisula
*af
abc,
Wojciech
Zajaczkowski‡
a,
Gangamallaiah
Velpula
d,
Daniel
Jänsch
a,
Robert
Graf
a,
Tomasz
Marszalek
af,
Sapun H.
Parekh
ae,
Yulian
Zagranyarski
a,
Kunal
Mali
d,
Manfred
Wagner
a,
Steven
De Feyter
d,
Chen
Li
a,
Klaus
Müllen
*a and
Wojciech
Pisula
*af
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: muellen@mpip-mainz.mpg.de; pisula@mpip-mainz.mpg.de
bKey Laboratory of Microelectronic Devices and Integrated Technology, Institute of Microelectronics, Chinese Academy of Sciences, Beijing 100029, China
cUniversity of Chinese Academy of Sciences, Beijing, 100049, China
dDivision of Molecular Imaging and Photonics, Department of Chemistry, KU Leuven-University of Leuven, Celestijnenlaan 200 F, 3001 Leuven, Belgium
eDepartment of Biomedical Engineering, University of Texas at Austin, Austin, TX 78712, USA
fDepartment of Molecular Physics, Faculty of Chemistry, Lodz University of Technology, Zeromskiego 116, 90-924 Lodz, Poland
First published on 30th April 2020
Abstract
The self-assembly of an asymmetric perylene diimide (PDI) with a dove-tailed side chain is investigated after thermal annealing at various temperatures in solid-state. After annealing at low temperatures PDI dimers are formed through hydrogen bonding between the imide and carbonyl groups, together with π–stacking interactions leading to a helical packing of the molecules in supramolecular columnar structures. After annealing at higher temperatures a transformation into a layered organization with improved molecular order is observed. The driving force for this thermodynamically favorable reorganization and planarization of the PDI dimer is additional intermolecular hydrogen bonding between the carbonyl and aromatic H(Ar) that is also present in monolayers that are visualized by scanning tunneling microscopy. This study demonstrates the importance of hydrogen bonding on tuning the supramolecular organization of asymmetric PDI. Depending on the dominating interactions, a wide variety of complex supramolecular structures with unique properties can be initiated.
1. Introduction
The self-assembly of biological systems has inspired scientists to explore the complex overlap of intermolecular forces such as π-stacking, hydrogen bonding and dipole–dipole interactions for functional materials.1 A typical example are polycyclic aromatic hydrocarbons (PAHs) carrying flexible alkyl chains because they exhibit thermotropic properties due to self-assembly into columnar supramolecular structures.2 The resulting packing depends upon the nature of substituents, the size and symmetry of the core and the directed non-covalent interactions of hydrogen bonding.3 The lateral rotation between adjacent aromatic cores commonly results in a helical organization and affects the charge carrier transport along the columnar stacks due to changes in the orbital overlap.4 The adequate combination of π-stacking and hydrogen bonding in PAHs, thus, allows one to establish a desired liquid crystalline organization within a wide temperature range.A special case of PAHs are rylenes which consist of peri-fused naphthalenes.5 The most prominent molecular system are perylene tetracarboxydiimides (PDIs) which assemble into different helical structures depending on their substituents and functional groups.6 The substituents are commonly attached at the two imide positions of the PDI core in a symmetric fashion and, furnish, for instance, intermolecular hydrogen bonding and π-stacking interactions. In the case of PDI 1 helical nanofibers assemble due to hydrogen bonding between amide groups of adjacent molecules (Fig. 1).7 The helical pitch of few tens of nanometers of the fibers is attributed to the directed hydrogen bonding. Substituents with high steric demand at the two imide positions are also used to control the molecular packing. The dendritic groups of PDI 2 stimulate a lateral rotation of the molecules and induce complex helical columnar organizations depending on the length of the flexible spacer between the PDI core and the dendron. The helical columns can contain tetramers of PDI as basic repeat unit, which are based on two molecules lying side-by-side in each stratum.8 In another case, single-handed helical assemblies are generated following a cogwheel mechanism independent of the chirality of the molecules.9 Our group has found that simple, but space-demanding dove-tailed heptyloctyl side chains at the imides can also trigger a helical packing of PDI 3 containing 3 molecules per pitch.10 Interestingly, an identical packing has been observed for the larger terrylene and quaterrylene diimides substituted by the same side chains.11 When the hydrocarbon side chains are changed to longer triethyleneglycols, PDI 4 arranges in a staggered fashion.12
 | ||
| Fig. 1 Chemical structures of symmetrical and asymmetrical PDIs forming helical assemblies. | ||
An asymmetric substitution of the PDI core at the imide positions by alkyl side chains and hydrogen bonding functions offers further opportunities to tune the supramolecular self-assembly. Indeed, while the solid-state organization of symmetrically substituted PDIs has been well studied, little information is available for asymmetrically substituted PDIs in spite of few reported examples. The asymmetrical functionalization of PDI has great impact on the size, aspect ratio, and surface properties of the resulting self-assembled entities. A broad range of structures was observed for PDI 5 derivatives differing in the length of the alkyl spacer between the imide position and various pendant silylated alcohol functionalities.13 As another rare example of asymmetric PDIs, compound 6 bears a benzyl moiety with two chiral sec-butoxy substituents and a dodecyl side chain.14 The introduction of chiral substituents induces the formation of single-handed nano-coils and -fibers self-assembled after solution deposition.15 Introduction of side groups capable of non-covalent interactions can lead to further complex supramolecular structures of asymmetric PDIs as in the case of 7. This molecule contains a branched alkyl side chain and a carboxylic acid group leading to intermolecular hydrogen bonding in non-polar solvents and finally to catemers between molecules in neighboring stacks.16 Flash-photolysis time-resolved microwave conductivity measurements revealed a charge carrier mobility of 0.24 cm2 V−1 s−1 for this compound. In solution, the unsubstituted imide of the asymmetric PDI 8 is capable of two-fold hydrogen bonding with the carbonyl group of a neighboring core.17 The resulting organization consists of hydrogen-bonded dimers that are stacked in columns. The formation of hydrogen bonds and the self-assembly of asymmetric PDI can be adjusted by the polarity of the solvent.18 This dimer assembly significantly differs from the intracolumnar hydrogen bonding of the symmetrically substituted PDI 1 and resembles biological supramolecular structures such as DNA base pairing. While the solution self-assembly of asymmetrically substituted PDIs has been extensively studied, the solid-state organization remained less explored.
The structural study in this work reveals that mono-substituted PDI 9 with dove-tailed side chain arranges in hydrogen-bonded dimers in the bulk. However, the supramolecular organization strongly depends on the type of hydrogen bonding that is controlled by thermal annealing. The main structural information is derived from solid-state nuclear magnetic resonance (SSNMR) and wide-angle X-ray scattering measurements. After annealing at low temperatures, robust π-stacking interactions between aromatic PDI cores and hydrogen bonding between carbonyl and imide groups facilitate a helical self-organization of V-shaped dimers into columnar stacks. Higher annealing temperatures result in improved molecular order and planarization of the PDI dimers. Additional intermolecular hydrogen bonding between carbonyl and aromatic H(Ar) initiate a reorganization from the helical to a thermodynamically favorable layered supramolecular structure. The occurrence of multiple hydrogen bonding allows to further program and construct other supramolecular assemblies consisting of a high variety of aromatic building blocks.
2. Experiments
2.1 Differential scanning calorimetry (DSC)
DSC was measured using a Mettler DSC 30 setup at a scan rate of 5 °C min−1.2.2 UV-vis absorption
Thin films for UV-vis absorption were spin-coated from a 3 mg mL−1 chloroform solution onto cleaned quartz substrates, and then thermally annealed at 100 °C or 250 °C for 30 min in a nitrogen glovebox. The corresponding UV-vis spectra were recorded at room temperature through a PerkinElmer Lambda 950 spectrophotometer.2.3 Fourier transform infrared spectroscopy (FTIR)
FTIR spectra of spin-coated films was measured with a Nicolet 730 FTIR spectrometer. The FTIR spectra were averaged over three different separate films. The integration time for the IR measurements was 800 s. Average IR intensity values between the frequencies of 2120 cm−1 and 2140 cm−1 of each averaged FTIR spectra were used for the background subtraction.2.4 Solid-state nuclear magnetic resonance (SSNMR)
All SSNMR experiments were performed with a Bruker Avance III console operating at 850.27 MHz 1H Larmor frequency and ambient temperature using commercial double resonance MAS NMR probes designed for zirconia rotors with 2.5 mm out diameter. If not stated differently, 1H MAS NMR measurements were performed at 25 kHz MAS spinning frequency, whereas 13C CP-MAS NMR measurements were recorded at 18 kHz MAS spinning frequency, 3 ms Lee-Goldburg CP contact time and 100 kHz high power sweep-frequency TPPM dipolar decoupling. The 1H double quantum-single quantum correlation NMR spectra were acquired at 25 kHz MAS and at ambient temperature using the back-to-back double quantum recoupling scheme for 2 rotor periods double quantum excitation and reconversion.2.5 Two-dimensional wide-angle X-ray scattering (2DWAXS)
2DWAXS setup consists of the Siemens Kristalloflex X-ray source (copper anode X-ray tube, operated at 35 mV/20 mA), Osmic confocal MaxFlux optics, two collimating pinholes (1.0 and 0.5 mm Owis, Germany) and an antiscattering pinhole. 2DWAXS pattern was recorded on a MAR345 image plate detector (Marresearch, Germany). The sample was prepared by filament extrusion using a home-built mini-extruder, and then positioned perpendicular to the incident X-ray beam and vertical to the 2D detector.2.6 Film deposition by zone-casting
Zone-casting was utilized to deposit thin films of 9. The Si/SiO2 substrate was moved at a controlled speed and temperatures for both solution and substrate. The concentration of PDI 9 is 0.5 mg mL−1. The optimized processing parameters are 1,1,2,2-tetrachloroethane (TCE) as solvent, solution/substrate temperatures of 70/60 °C, and moving speed of 5 μm s−1. The details of optimization are shown in Fig. S7.† After film fabrication, thermal annealing at 100 °C for 0.5 h is applied to remove residual solvent.2.7 Topography characterizations
The topography of deposited films was characterized by a Digital Instruments Nanoscope IIIa atomic force microscopy (AFM) in tapping mode and transmission electron microscopy (TEM, FEI Tecnai F20). To gain information on the molecular organization of 9 in the zone-cast films, selected-area electron diffraction (SAED) patterns were recorded by a Philips CM 12 electron microscope.2.8 Scanning tunneling microscopy (STM)
Stock solution of PDI (8.3 × 10−4 M) was prepared by dissolving appropriate amount of solid in 1,2,4-trichlorobenzene (Sigma-Aldrich, >99%). The stock solution was diluted further to make concentration series. All STM experiments were performed at room temperature (21–23 °C) using a PicoLE (Agilent) machine operating in constant-current mode with the tip immersed in the supernatant liquid. STM tips were prepared by mechanically cutting a Pt/Ir wire (80%/20%, diameter 0.2 mm). Prior to imaging, a drop of warm PDI solution was placed onto a freshly cleaved surface of highly oriented pyrolytic graphite (HOPG, grade ZYB, Advanced Ceramics Inc., Cleveland, USA). The experiments were repeated in 2–3 sessions using different tips to check for reproducibility and to avoid experimental artifacts, if any. For analysis purposes, recording of a monolayer image was followed by imaging the graphite substrate underneath it under the same experimental conditions, except for increasing the current and the lowering the bias. The images were corrected for drift via Scanning Probe Image Processor (SPIP) software (Image Metrology ApS), using the recorded graphite images for calibration purposes, allowing a more accurate unit cell determination. The unit cell parameters were determined by examining at least 4 images and only the average values are reported. The images are Gaussian filtered. The imaging parameters are indicated in the figure caption: tunneling current (Iset), and sample bias (Vbias). The molecular models were built using Hyperchem™ 7.0 program.3. Results and discussion
3.1 Formation of hydrogen bonding
PDI 9 was synthesized according to literature,19 and the presence of its hydrogen bonding in solid-state is firstly confirmed by Fourier-transform infrared spectroscopy (FTIR) of films before and after annealing at 100 °C and 250 °C. The later annealing temperature was taken as maximum also for all other experiments, except SSNMR (see below), since evaporation of the material occurs above this point. The peak intensity of the vibration associated with hydrogen bonded carbonyl (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) groups slightly increases and is red shifted from 1690 cm−1 to 1680 cm−1 after annealing at 250 °C (Fig. 2a). Additionally, the low frequency part of the peak at 1680 cm−1 to 1695 cm−1 increases. These changes in peak position and intensity suggest increased hydrogen bonding mediated by carbonyl groups. Contrary to these observations, the intensity of the stretching mode related to hydrogen bonded imide (–NH) groups centered at 3200 cm−1 decreases and those attributed to non-hydrogen bonded (or free) imide groups at 3381 cm−1 increases after annealing the sample at 250 °C (Fig. 2b). These results indicate that the hydrogen bonding of the imide groups is weakened at higher annealing temperatures, while at the same time the non-covalent interaction by the carbonyl groups are enhanced. The moderate annealing temperature of 100 °C does not initiate any change in the sample. Large changes are also evident in the UV-vis absorption between films annealed at 100 °C and 250 °C (Fig. S1†). For 250 °C, the spectrum is red shifted and becomes less defined with broader features indicating more heterogeneous structures and improved electron coupling between the molecules. These observations will be confirmed in the structural analysis of Section 3.2 that also allows better understanding of the changes in hydrogen bonding after high temperature annealing.
O) groups slightly increases and is red shifted from 1690 cm−1 to 1680 cm−1 after annealing at 250 °C (Fig. 2a). Additionally, the low frequency part of the peak at 1680 cm−1 to 1695 cm−1 increases. These changes in peak position and intensity suggest increased hydrogen bonding mediated by carbonyl groups. Contrary to these observations, the intensity of the stretching mode related to hydrogen bonded imide (–NH) groups centered at 3200 cm−1 decreases and those attributed to non-hydrogen bonded (or free) imide groups at 3381 cm−1 increases after annealing the sample at 250 °C (Fig. 2b). These results indicate that the hydrogen bonding of the imide groups is weakened at higher annealing temperatures, while at the same time the non-covalent interaction by the carbonyl groups are enhanced. The moderate annealing temperature of 100 °C does not initiate any change in the sample. Large changes are also evident in the UV-vis absorption between films annealed at 100 °C and 250 °C (Fig. S1†). For 250 °C, the spectrum is red shifted and becomes less defined with broader features indicating more heterogeneous structures and improved electron coupling between the molecules. These observations will be confirmed in the structural analysis of Section 3.2 that also allows better understanding of the changes in hydrogen bonding after high temperature annealing.
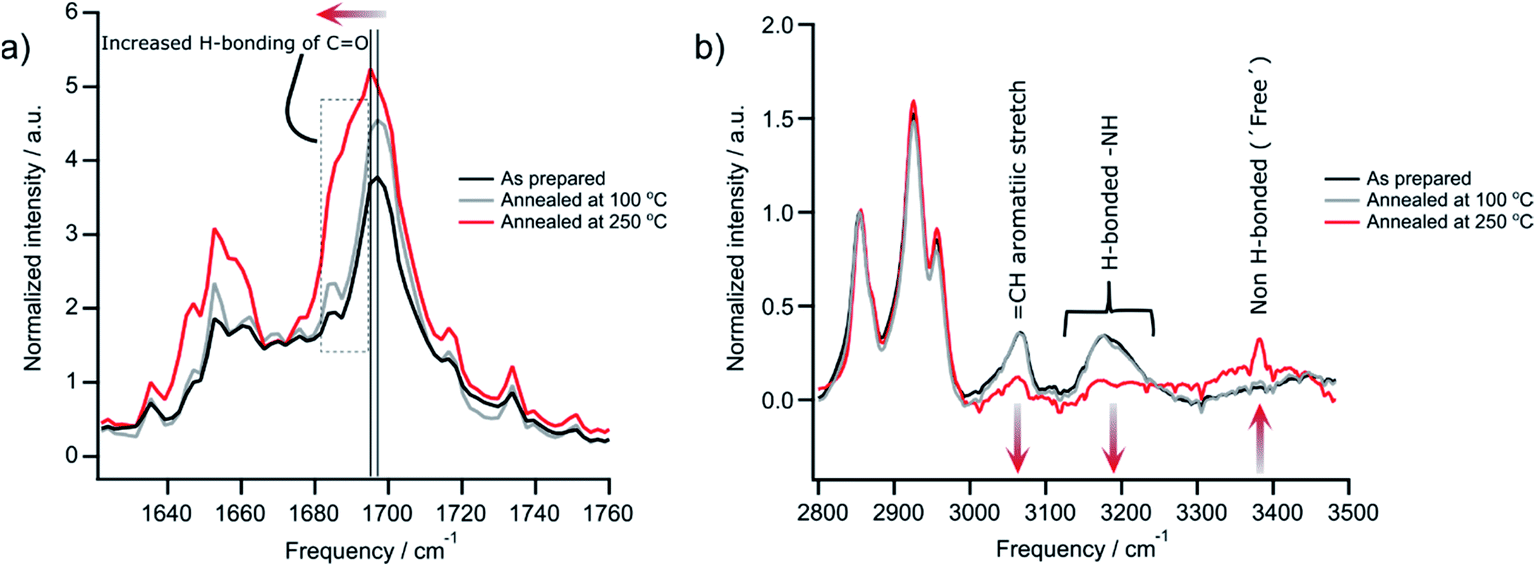 | ||
| Fig. 2 FTIR spectroscopy with (a) low and (b) high frequency of cast films of 9 after annealing at different temperatures. Spectra are averaged from three separate films on silicon substrates. All spectra were baseline subtracted and normalized by the CH2 vibration (2854 cm−1) to account for different film thickness prior to averaging. | ||
The direct visualization of the hydrogen bonding of PDI 9 is realized through scanning tunneling microscopy (STM) under ambient conditions. The molecular resolution STM image in Fig. 3a depicts that the bright features arise from rows of oval shaped structures and always appear in pairs. These oval features correspond to the aromatic backbones of 9 and their pairwise occurrence indicates the presence of hydrogen-bonded dimers on the surface as expected from the hydrogen bonding sites between carbonyl and imide groups on 9. Interestingly, adjacent dimers are shifted by almost half a molecule with respect to each other suggesting their additional interactions via carbonyl and aromatic H(Ar) (Fig. S2†). This type of hydrogen bonding has also been reported for unsubstituted for perylene–tetracarboxylic–dianhydride.20 The molecular rows run along one of the main symmetry axes of the HOPG substrate. The bright columns are separated by relatively darker narrow troughs where the alkyl chains are adsorbed. The alkyl chains could not be resolved, possibly in view of their mobility on the surface within the time scale of STM measurement. Molecular models based on the unit cell parameters indicate that there is not enough space in between adjacent molecules for the adsorption of both alkyl chains per molecule (Fig. 3b). The chains that are not adsorbed on the surface could be simply back-folded in the supernatant solution phase. A peculiar feature of the monolayer is the presence of defects in the form of kinks (green arrow, Fig. 3a) along the molecular rows. A careful inspection indicates that these defects do not interrupt the carbonyl–imide hydrogen bonding between individual PDI molecules (dashed white ovals, Fig. 3a). These insights provide important information between which groups of the PDI 9 molecules hydrogen bonding is formed.
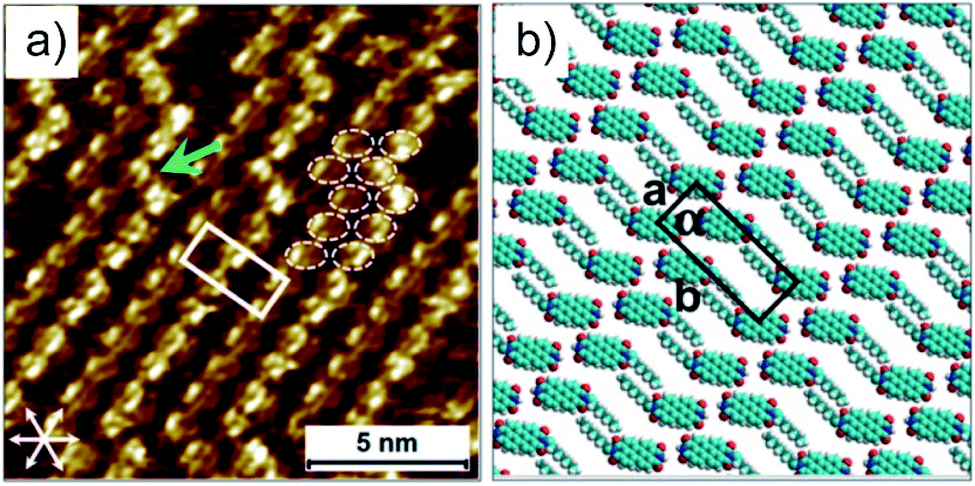 | ||
| Fig. 3 (a) STM image with molecular resolution (CPDI = 1.7 × 10−4 M). The green arrow shows a kink in the molecular row. The aromatic cores are highlighted by white ovals at a kink site. The graphite symmetry axes are given in the lower left corner. The unit cell is overlaid on the STM image and the cell parameters are: a = 1.2 ± 0.1 nm, b = 3.2 ± 0.1 nm and α = 90 ± 1°. Imaging parameters: Iset = 100 pA, Vbias = −400 mV. (b) Molecular model displaying the arrangement of 9 in the self-assembled monolayer. The molecules are drawn with only the octyl chain, while the heptyl chain is omitted for the sake of clarity. | ||
3.2 Structural analysis of supramolecular assembly
To correlate the results from FTIR and STM with the organization in bulk, the hydrogen bonding promoted self-organization of 9 is further explored by SSNMR. Since for these measurements the PDI samples were contained in a sealed holder, higher temperatures of 310 °C and 340 °C were additionally feasible being associated with phase transitions from DSC and annealing experiments (Fig. S3 and S4†). In the 1H MAS NMR spectra of the sample annealed at 100 °C, the aromatic signal is relatively broad (Fig. 4a). Upon further annealing, the aromatic 1H signal narrows gradually in the spectrum and shifts towards lower ppm values, indicating an improved π-stacking and intermolecular order. The 1H signal of the hydrogen bonded sites, seen as a weak shoulder around 11 ppm for the sample annealed at 100 °C, becomes better defined and shifts towards higher ppm values. This demonstrates the formation of better-ordered or additional hydrogen bonds after annealing at elevated temperatures. In the 13C CP-MAS NMR spectra the enhanced self-organization is also observed via the improved chemical shift resolution of the aromatic and carbonyl sites of the PDI cores (Fig. 4b). It should be pointed out, that the number of the observed 13C NMR signals exceeds the number of chemically distinct sites in the molecule, indicating the importance of the local packing for the chemical shifts in SSNMR. In contrast to the PDI core, the 13C signals of the aliphatic side chains show only a weak line narrowing with increasing annealing temperature, which might be attributed to the improved phase separation between the PDI cores and the aliphatic side chains due to the higher stacking order of the PDI cores.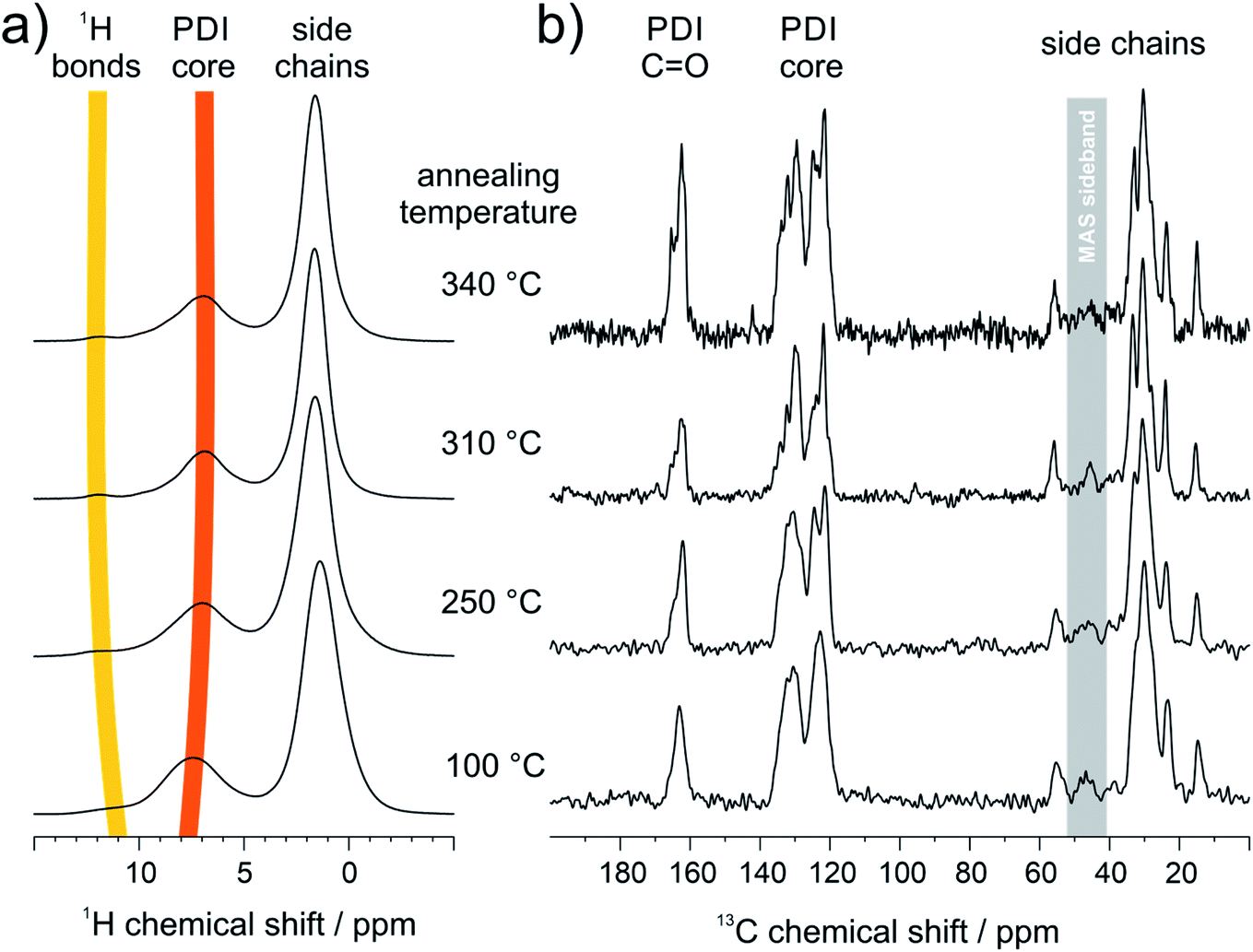 | ||
| Fig. 4 SSNMR results of PDI 9 annealed at different temperatures: (a) 1H MAS NMR spectra (850 MHz 1H Larmor frequency, 25 kHz MAS, ambient temperature) and (b) 13C CP-MAS NMR spectra (850 MHz 1H Larmor frequency, 18 kHz magic angle spinning frequency, 3 ms Lee-Goldburg CP and swf-TPPM decoupling, ambient temperature). | ||
Further information about the hydrogen bonding is obtained by two-dimensional 1H double quantum (DQ) correlation NMR spectra recorded at ambient temperature after the different annealing stages of the sample at ambient temperature. In Fig. 5a–d, pairwise correlations between 1H sites in close spatial proximity are monitored, so that the formation of hydrogen-bonded dimers can be distinguished from spatially isolated hydrogen bonding 1H sites. For perfectly symmetric hydrogen-bonded dimers with two equivalent hydrogen-bonded proton sites in close spatial proximity, a DQ correlation signal on the diagonal of the spectrum would be expected. On the other hand, a spatially isolated hydrogen bonding site would not be monitored, because DQ coherences can only be excited between spin ½ nuclei with significant dipolar couplings and all other signals are cancelled by an appropriate phase cycle. Weak distortions of the hydrogen-bonded dimers, where one of the hydrogen bonds is elongated while the second bond is slightly shortened, will cause a splitting of the signal symmetric to the diagonal. On the other hand, heterogeneous packing environment with neighboring PDI molecules will lead to a heterogeneous distribution of isotropic chemical shifts seen as an elongation of the DQ signal parallel to the diagonal of the spectrum. The 1H DQ correlation spectrum of the weakly annealed sample shows both features described above, the splitting due to distortions of the dimers, and an elongation along the diagonal due to columnar stacking heterogeneities resulting from variations of the helical pitch and π-stacking distances (Fig. 5a). Upon further annealing, the molecular order of the PDI dimers is substantially improved as seen by the refinement of spectral features in the 13C CP-MAS NMR spectrum assigned to the PDI core (110–180 ppm, Fig. 4b). The elongation along the diagonal as well as splitting of the NH proton signal vanishes with increasing annealing temperature, while at the same time a new DQ coherence involving hydrogen bonding protons at 12.2 and aromatic protons at 9.9 ppm (Fig. 5b–d) evolves. This observation confirms the formation of hydrogen bonding between aromatic proton and carbonyl sites of neighboring PDI cores and suggests that the planar packing arrangement (Fig. S2 and S5†), monitored by STM studies as surface structure, might resemble the bulk molecular packing after high temperature annealing. Moreover, it shows that the temperature dependent chemical shift of the hydrogen-bonded proton sites, discussed in Fig. 4, cannot be attributed to a thermal restructuring of the NH–carbonyl hydrogen bonding, but originates from the formation of hydrogen bonding between aromatic proton and carbonyl sites of the PDI cores as also found by FTIR.
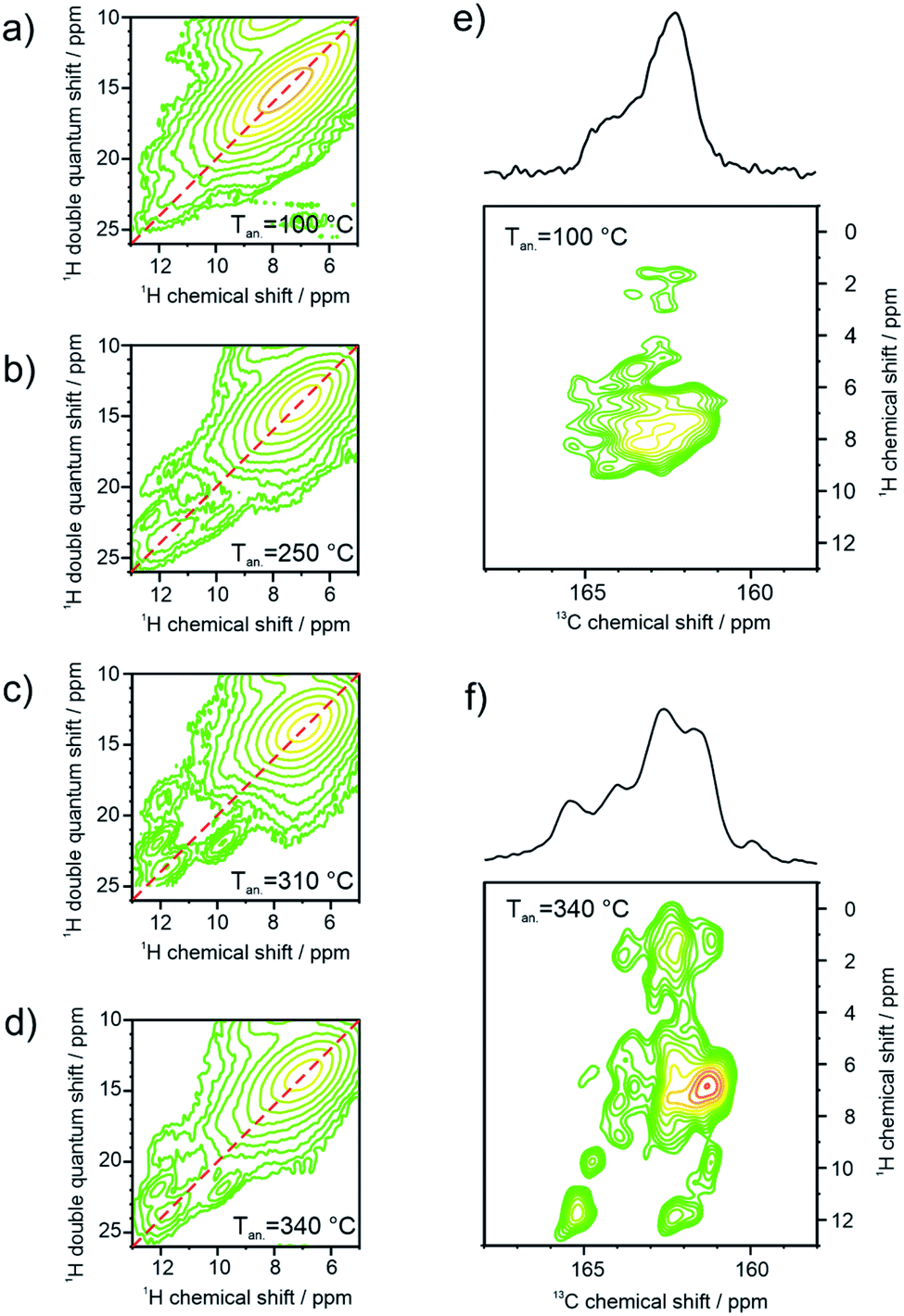 | ||
| Fig. 5 Two-dimensional SSNMR spectra of annealed 9 at different temperatures: (a–d) 1H double quantum correlation spectra (850 MHz 1H Larmor frequency, 25 kHz MAS, ambient temperature, 2 rotor periods BABA DQ excitation) and (e and f) 13C CP-MAS NMR spectra (850 MHz 1H Larmor frequency, 18 kHz magic angle spinning frequency, 3 ms LG-CP and swf-TPPM decoupling, ambient temperature). | ||
After annealing the sample at 340 °C, the intensity of the mixed coherence has approximately twice the intensity of the pure NH proton coherence, which is expected for the layered bulk structure. In Fig. 5e, the carbonyl region of a 13C–1H FSLG-CP correlation NMR spectrum of the 100 °C annealed sample reveals only ill-defined correlation signals to aromatic proton sites. In contrast, the same correlation spectrum of 9 after annealing at 340 °C, given in Fig. 5f, shows numerous well-defined correlations due to the improved molecular order. The new correlation signals at 13C chemical shifts above 165 ppm with hydrogen bonding protons, originate from carbonyl sites hydrogen-bonded to aromatic protons, which are not present in the initial helical packing of the sample. In summary, the solid-state NMR results indicate the formation of a new bulk structure on the molecular level driven by additional hydrogen bonding upon high temperature annealing consistent with the layered surface structure observed in the STM images and FTIR results.
To quantify the reorganization of PDI 9 upon annealing two-dimensional wide-angle X-ray scattering (2DWAXS) is performed for extruded fiber samples up to the annealing temperature of 250 °C. The patterns in Fig. 6 confirm a significant transition of the supramolecular structure with thermal annealing observed by SSNMR. Fig. 6a exhibits the 2DWAXS pattern after annealing at 100 °C. Equatorial reflections in the small-angle range (assigned as c1 in Fig. 6a and S6†) indicate columnar structures aligned along the fiber sample and arranged in a square-like lattice with the parameter of a = 4.18 nm. This value is in accordance with the calculated length of a PDI dimer with side chains of around 4.60 nm. Higher order reflections, up to the third order, are characteristic for a long-range molecular order. Interestingly, these scattering intensities exhibit an azimuthal splitting with maxima at γ = 11° (Fig. S6†) with respect to the equatorial plane suggesting a relative shift of 0.67 nm of the columns along the fiber axis towards each other. In the wide-angle range, reflections located at the off-meridional plane (assigned as a1 in Fig. 6a) correspond to the π-stacking distance of 0.34 nm of tilted PDI molecules. From the azimuthal position of the reflection a molecular tilt angle of β = 48° with respect to the columnar axis is determined. As illustrated in Fig. 7c, the dimers, formed with the assistance of hydrogen bonding dominated solely by interactions between –N–H and –CO groups, adopt a V-shape arrangement with titled PDI molecules. Additional low intensity meridional reflections (assigned as b1, Fig. 6a) in the small-angle region imply a helical organization of the dimers with a pitch of 1.41 nm containing three dimers.4,8a,21 Thereby, the dimers are rotated by around 60° to the next neighbor within the stack to ensure an identical positional order between each 4th dimer (Fig. 7b). The position and shape of the reflections in the pattern suggest a helix structure, similar to DNA, along the fiber direction. Note that the higher order reflections possess weak intensity characteristic to short-range order.
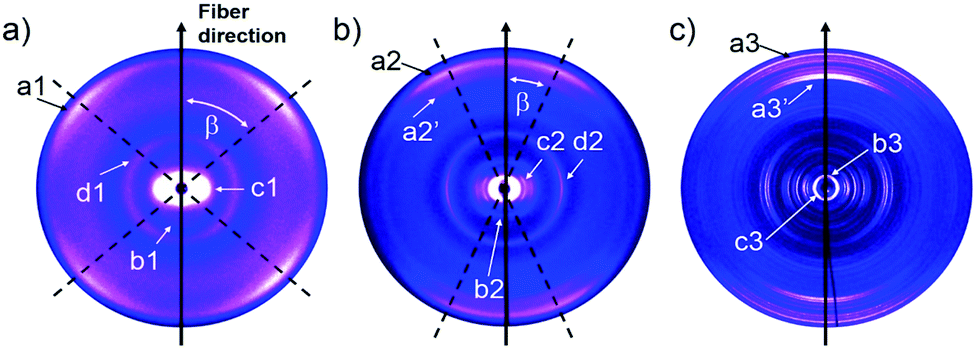 | ||
| Fig. 6 2DWAXS patterns for the extruded fiber of 9 after thermal annealing at (a) 100 °C, (b) 150 °C and (c) 250 °C. | ||
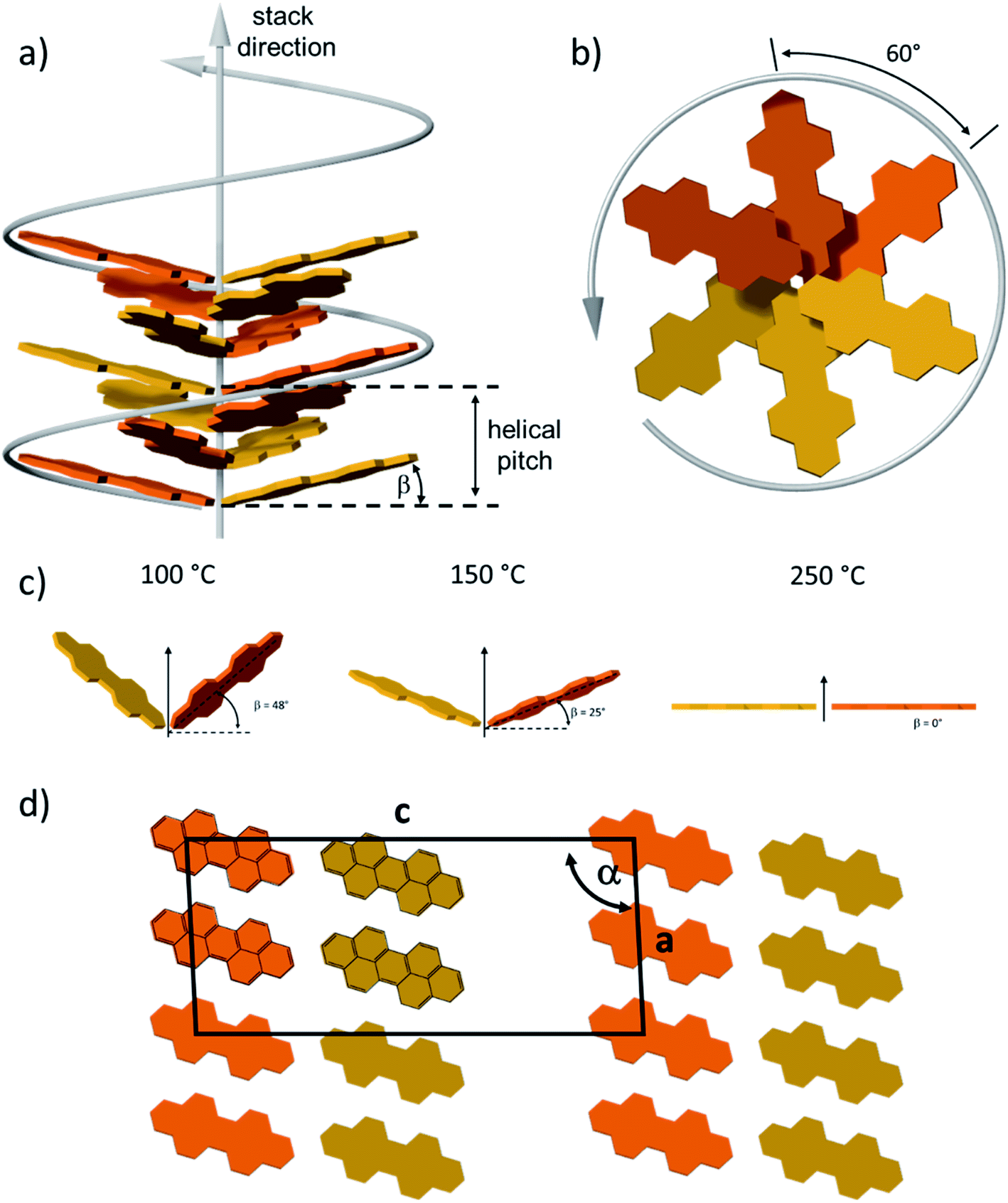 | ||
| Fig. 7 Model for the molecular arrangement of 9 in the (a) and (b) helical organization (in side and top view) after annealing at 150 °C and (d) layer structure (top view along the b crystal axis) after 250 °C. Different colors are used to schematically visualize the molecular rotation. Side chains are omitted. (c) Molecular tilting in the dimer after annealing at different temperatures. (d) Top view of the schematic unit cell of layer structure after 250 °C. | ||
After annealing at 150 °C, the molecular order is noticeably enhanced, as evident from the larger number of distinct and high intensity reflections. The annealing leads to a slight increase in the lattice parameter of the square-like unit cell to 4.26 nm. The azimuthal integration of the intercolumnar reflection (assigned as c2, Fig. 6b) reveals also a splitting with its maximum located at γ = 25° with respect to the equatorial plane. This might suggest that the columns remain aligned parallel along the fiber sample, but are translated by a larger value of 1.98 nm. From the split wide-angle reflections (a2, Fig. 6b) a π-stacking distance of 0.34 nm and molecular tilting of β = 25° are derived. This tilted molecular arrangement is confirmed by the additional reflection a2′ that is at the same azimuthal angle as a2 at a d-spacing of 0.37 nm for the intracolumnar period. The helical pitch of 1.46 nm remains almost unchanged as indicted by the small-angle scattering intensity b2 in Fig. 7b still including 3 dimers with a molecular rotation angle of 60°.
The sample annealed at 250 °C reveals an additional type of organization and significantly higher crystallinity in bulk as evident by the large number of distinct reflections (Fig. 6c). The shape of the main reflections in the small-angle range implies polymorphism with up to 4 different unit cells which vary only slightly from each other. The main identified unit cell is rectangular with a = 3.80 nm and b = 2.37 nm (c3, Fig. 6c). In this lattice the dimers are arranged in a layered order as shown in Fig. 7d analogous to the monolayer displayed by STM (Fig. 3). The driving force for this reorganization is additional intermolecular hydrogen bonding between carbonyl and aromatic H(Ar) which were found by FTIR and SSNMR. These interactions induce a planar molecular arrangement within the dimers (β = 0°). During this rearrangement, a certain amount of the hydrogen bonding between imide and carbonyl groups breaks causing an intensity decrease of the IR peak for hydrogen-bonded N–H. Since at the same time, new hydrogen bonded carbonyl is established, the intensity of this IR peak remains unchanged. The wide-angle range reflections, a3, located at the meridional plane are assigned to the molecular packing. However, the high number of reflections in this range does not allow to unambiguously distinguish the π-stacking distance but indicates a slight tilting of the entire dimer towards the stacking axis resulting in a herringbone structure (side view illustration of the lattice in Fig. S7†). Additionally, a minor phase of a hexagonal unit cell with ahex = 4.17 nm was also identified from the equatorial reflections of the 2DWAXS pattern. This hexagonal phase might be related to a not fully accomplished transition from helical to layered ordering as also revealed by SSNMR.
The good solubility of PDI 9 in common solvents allows the processing in solution to control the microstructure. Among solution processing techniques, zone-casting offers unique advantages such as a broad variation of the processing parameters for the uniaxial alignment of conjugated molecules.22 For PDI 9, the optimized zone-casting conditions are TCE as solvent, solution/substrate temperatures of 70/60 °C, and moving speed of 5 μm s−1, by which well-defined morphologies are achieved (Fig. S8†). As shown in Fig. 8a, the self-assembly of PDI 9 is directed by zone-casting with the formation of uniaxially aligned fiber-like superstructures along the casting direction. These oriented nanofibers reach few micrometers in length which might be attributed to intermolecular π–stacking interactions. A more detailed picture is obtained by TEM. Fig. 8b exhibits that individual nanofibers essentially consist of a bundle of smaller, ca. 3.8 nm thick columns that are also oriented in the zone-casting direction. This distance is in good agreement with the dimension of hydrogen-bonded dimer of PDI 9. Further structural information on these well-defined columns is obtained from selected area electron diffraction (SAED) in Fig. 8c. Two split reflections correspond to spacings of 0.34 nm and 0.37 nm and are related to the packing of the tilted PDI molecules in the stacks as illustrated in Fig. 7c. The reflection splitting is consistent with 2DWAXS results in Fig. 6a for the annealing temperature of 100 °C.
 | ||
| Fig. 8 High-resolution AFM phase image (a), TEM image with intracolumnar distance (b) and selected area electron diffraction (c) of PDI 9 thin film obtained by zone-casting. | ||
4. Conclusion
The supramolecular structure of monosubstituted PDI 9 with dove-tailed side chain is investigated in solid-state after thermal annealing at various temperatures. Due to hydrogen bonding between carbonyl and imide groups, the PDI 9 molecules form dimers. The initial organization consist of a helical columnar structure of π-stacked and V-shaped dimers. Annealing at high temperatures induces a transformation into a thermodynamically favorite state of a highly ordered layered structure in that the dimers are quasi planarized and are additionally hydrogen-bonded through carbonyl and aromatic H(Ar) interactions. This type of intermolecular hydrogen bonding is a rare example that can play a fundamental role in the solid-state supramolecular organization of perylene tetracarboxydiimides. The transformation between two distinct assemblies is driven by the occurrence of multiple interactions and might be accompanied by changes in the functionality of the material.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. Li has received funding from the National Key R&D Program of China (2019YFA0706100) and the Netherlands Organisation for Scientific Research (016.Veni.192.106). G. Velpula, K. Mali, and S. De Feyter thank Flemish Research Council (FWO) and KU Leuven – internal funds for financial support. T. Marszalek acknowledges the Foundation for Polish Science financed by the European Union under the European Regional Development Fund (POIR.04.04.00-00-3ED8/17-01). W. Pisula acknowledges the National Science Centre, Poland, through the grant UMO-2015/18/E/ST3/00322. Authors also thank Dr Yiran Zheng for the TEM measurement.References
- (a) J.-M. Lehn, Angew. Chem., Int. Ed. Engl., 1990, 29, 1304–1319 CrossRef; (b) T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed; (c) J. D. Badjić, A. Nelson, S. J. Cantrill, W. B. Turnbull and J. F. Stoddart, Acc. Chem. Res., 2005, 38, 723–732 CrossRef PubMed; (d) M. A. Mateos-Timoneda, M. Crego-Calama and D. N. Reinhoudt, Chem. Soc. Rev., 2004, 33, 363–372 RSC; (e) O. J. G. M. Goor, S. I. S. Hendrikse, P. Y. W. Dankers and E. W. Meijer, Chem. Soc. Rev., 2017, 46, 6621–6637 RSC; (f) M. Goel and M. Jayakannan, Chem.–Eur. J., 2012, 18, 11987–11993 CrossRef CAS PubMed.
- (a) W. Yuan, X.-K. Ren, M. Li, H. Guo, Y. Han, M. Wu, Q. Wang, M. Li and Y. Chen, Angew. Chem., Int. Ed., 2018, 57, 6161–6165 CrossRef CAS PubMed; (b) M. Ball, Y. Zhong, Y. Wu, C. Schenck, F. Ng, M. Steigerwald, S. Xiao and C. Nuckolls, Acc. Chem. Res., 2015, 48, 267–276 CrossRef CAS PubMed; (c) L. Chen, S. R. Puniredd, Y.-Z. Tan, M. Baumgarten, U. Zschieschang, V. Enkelmann, W. Pisula, X. Feng, H. Klauk and K. Müllen, J. Am. Chem. Soc., 2012, 134, 17869–17872 CrossRef CAS PubMed; (d) L. Chen, K. S. Mali, S. R. Puniredd, M. Baumgarten, K. Parvez, W. Pisula, S. De Feyter and K. Müllen, J. Am. Chem. Soc., 2013, 135, 13531–13537 CrossRef CAS PubMed; (e) B. Zhao, B. Liu, R. Q. Png, K. Zhang, K. A. Lim, J. Luo, J. Shao, P. K. H. Ho, C. Chi and J. Wu, Chem. Mater., 2010, 22, 435–449 CrossRef CAS.
- (a) W. Pisula, X. Feng and K. Müllen, Adv. Mater., 2010, 22, 3634–3649 CrossRef CAS PubMed; (b) D. Wasserfallen, I. Fischbach, N. Chebotareva, M. Kastler, W. Pisula, F. Jäckel, M. D. Watson, I. Schnell, J. P. Rabe, H. W. Spiess and K. Müllen, Adv. Funct. Mater., 2005, 15, 1585–1594 CrossRef CAS.
- (a) W. Pisula, Ž. Tomović, M. D. Watson, K. Müllen, J. Kussmann, C. Ochsenfeld, T. Metzroth and J. Gauss, J. Phys. Chem. B, 2007, 111, 7481–7487 CrossRef CAS PubMed; (b) X. Feng, W. Pisula, M. Takase, X. Dou, V. Enkelmann, M. Wagner, N. Ding and K. Müllen, Chem. Mater., 2008, 20, 2872–2874 CrossRef CAS.
- F. Wurthner and M. Stolte, Chem. Commun., 2011, 47, 5109–5115 RSC.
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef PubMed.
- X.-Q. Li, V. Stepanenko, Z. Chen, P. Prins, L. D. A. Siebbeles and F. Würthner, Chem. Commun., 2006, 3871–3873, 10.1039/b611422a.
- (a) V. Percec, S. D. Hudson, M. Peterca, P. Leowanawat, E. Aqad, R. Graf, H. W. Spiess, X. Zeng, G. Ungar and P. A. Heiney, J. Am. Chem. Soc., 2011, 133, 18479–18494 CrossRef CAS PubMed; (b) V. Percec, H.-J. Sun, P. Leowanawat, M. Peterca, R. Graf, H. W. Spiess, X. Zeng, G. Ungar and P. A. Heiney, J. Am. Chem. Soc., 2013, 135, 4129–4148 CrossRef CAS PubMed.
- (a) C. Roche, H.-J. Sun, P. Leowanawat, F. Araoka, B. E. Partridge, M. Peterca, D. A. Wilson, M. E. Prendergast, P. A. Heiney, R. Graf, H. W. Spiess, X. Zeng, G. Ungar and V. Percec, Nat. Chem., 2015, 8, 80 CrossRef PubMed; (b) B. E. Partridge, L. Wang, D. Sahoo, J. T. Olsen, P. Leowanawat, C. Roche, H. Ferreira, K. J. Reilly, X. Zeng, G. Ungar, P. A. Heiney, R. Graf, H. W. Spiess and V. Percec, J. Am. Chem. Soc., 2019, 141, 15761–15766 CrossRef CAS PubMed.
- F. Nolde, W. Pisula, S. Müller, C. Kohl and K. Müllen, Chem. Mater., 2006, 18, 3715–3725 CrossRef CAS.
- H. N. Tsao, W. Pisula, Z. Liu, W. Osikowicz, W. R. Salaneck and K. Müllen, Adv. Mater., 2008, 20, 2715–2719 CrossRef CAS PubMed.
- M. R. Hansen, T. Schnitzler, W. Pisula, R. Graf, K. Müllen and H. W. Spiess, Angew. Chem., Int. Ed., 2009, 48, 4621–4624 CrossRef CAS PubMed.
- R. Matthews, J. Swisher, K. M. Hutchins and E. B. Pentzer, Chem. Mater., 2018, 30, 3571–3577 CrossRef CAS.
- K. Hu, Y. Liu, W. Xiong, Y. Gong, Y. Che and J. Zhao, Chem. Mater., 2019, 31, 1403–1407 CrossRef CAS.
- X. Ma, Y. Zhang, Y. Zhang, Y. Liu, Y. Che and J. Zhao, Angew. Chem., Int. Ed., 2016, 55, 9539–9543 CrossRef CAS PubMed.
- D. Basak, D. S. Pal, T. Sakurai, S. Yoneda, S. Seki and S. Ghosh, Phys. Chem. Chem. Phys., 2017, 19, 31024–31029 RSC.
- R. van der Weegen, P. A. Korevaar, P. Voudouris, I. K. Voets, T. F. A. de Greef, J. A. J. M. Vekemans and E. W. Meijer, Chem. Commun., 2013, 49, 5532–5534 RSC.
- Z. Guo, K. Wang, P. Yu, S. Zhang, K. Sun and Z. Li, Phys. Chem. Chem. Phys., 2017, 19, 23007–23014 RSC.
- H. Langhals and S. Saulich, Chem.–Eur. J., 2002, 8, 5630–5643 CrossRef CAS.
- S. W. Cho, D. Newby Jr, A. DeMasi, K. E. Smith, L. F. J. Piper and T. S. Jones, J. Chem. Phys., 2013, 139, 184711 CrossRef CAS PubMed.
- V. Percec, M. Peterca, T. Tadjiev, X. Zeng, G. Ungar, P. Leowanawat, E. Aqad, M. R. Imam, B. M. Rosen, U. Akbey, R. Graf, S. Sekharan, D. Sebastiani, H. W. Spiess, P. A. Heiney and S. D. Hudson, J. Am. Chem. Soc., 2011, 133, 12197–12219 CrossRef CAS PubMed.
- (a) W. Pisula, A. Menon, M. Stepputat, I. Lieberwirth, U. Kolb, A. Tracz, H. Sirringhaus, T. Pakula and K. Müllen, Adv. Mater., 2005, 17, 684–689 CrossRef CAS; (b) A. Tracz, J. K. Jeszka, M. D. Watson, W. Pisula, K. Müllen and T. Pakula, J. Am. Chem. Soc., 2003, 125, 1682–1683 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc01911a |
| ‡ Contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |