
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ti-catalyzed ring-opening oxidative amination of methylenecyclopropanes with diazenes†
Evan P.
Beaumier
 ,
Amy A.
Ott
,
Xuelan
Wen
,
Zachary W.
Davis-Gilbert
,
T. Alexander
Wheeler
,
Joseph J.
Topczewski
,
Jason D.
Goodpaster
* and
Ian A.
Tonks
*
,
Amy A.
Ott
,
Xuelan
Wen
,
Zachary W.
Davis-Gilbert
,
T. Alexander
Wheeler
,
Joseph J.
Topczewski
,
Jason D.
Goodpaster
* and
Ian A.
Tonks
*
Department of Chemistry, University of Minnesota – Twin Cities, 207 Pleasant St SE, Minneapolis MN 55455, USA. E-mail: jgoodpas@umn.edu; itonks@umn.edu
First published on 23rd June 2020
Abstract
The ring-opening oxidative amination of methylenecyclopropanes (MCPs) with diazenes catalyzed by py3TiCl2(NR) complexes is reported. This reaction selectively generates branched α-methylene imines as opposed to linear α,β-unsaturated imines, which are difficult to access via other methods. Products can be isolated as the imine or hydrolyzed to the corresponding ketone in good yields. Mechanistic investigation via density functional theory suggests that the regioselectivity of these products results from a Curtin–Hammett kinetic scenario, where reversible β-carbon elimination of a spirocyclic [2 + 2] azatitanacyclobutene intermediate is followed by selectivity-determining β-hydrogen elimination of the resulting metallacycle. Further functionalizations of these branched α-methylene imine products are explored, demonstrating their utility as building blocks.
Introduction
Metal-catalyzed oxidative amination reactions are increasingly important methods of forming new carbon–nitrogen bonds.1 There are now many examples of metal-catalyzed oxidative C–H amination and alkene aziridination reactions, most commonly with late transition metals such as Fe, Rh, and Ag. In contrast, there are few examples with early transition metals such as Ti. Typically, Ti-catalyzed C–N bond forming reactions are redox neutral hydrofunctionalization reactions;2 the thermodynamic stability of TiIV makes catalytic transformations involving redox at the metal center more challenging.Recently, we found that it is possible to exploit the [2 + 2] cycloaddition reaction between Ti imidos (Ti![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) NR) and alkynes to perform oxidative amination reactions.3 Further insertion of unsaturated substrates into the azatitanacyclobutene resulting from [2 + 2] cycloaddition gave expanded metallacycles that underwent reductive processes to liberate aminated products. The resulting transient TiII species can be reoxidized to TiIV with azobenzene. Using this approach, we have designed new routes to pyrroles, α,β-unsaturated imines, α-iminocyclopropanes, carbodiimides, and pyrazoles.
NR) and alkynes to perform oxidative amination reactions.3 Further insertion of unsaturated substrates into the azatitanacyclobutene resulting from [2 + 2] cycloaddition gave expanded metallacycles that underwent reductive processes to liberate aminated products. The resulting transient TiII species can be reoxidized to TiIV with azobenzene. Using this approach, we have designed new routes to pyrroles, α,β-unsaturated imines, α-iminocyclopropanes, carbodiimides, and pyrazoles.
In light of these discoveries, we sought to expand on this oxidative amination beyond alkynes to other substrates capable of [2 + 2] cycloaddition with Ti imidos. In this regard, methylenecyclopropanes (MCPs) were targeted. Eisen demonstrated that Ti and Zr precatalysts can add primary amines across MCPs through a hydroamination mechanism that included a [2 + 2] cycloaddition with a group IV imido and subsequent β-carbon elimination of the corresponding spirocyclic azatitanacyclobutane, eventually providing linear or branched imines upon protonolysis (Fig. 1, top).4 Eisen suggested that the highly strained β-carbon in MCPs provides significant “sp-like” character which allows for accessible [2 + 2] cycloadditions with group IV imidos.4a,b Furthermore, the β-carbon elimination in MCPs can occur due to the release of ring strain from the cyclopropyl moiety.5 Based on this strategy, we report that Ti imido catalysts are capable of the ring-opening oxidative amination of terminal MCPs to yield unusual α-methylene substituted imines, which are challenging to access via other methods (Fig. 1, bottom).6 A full computational analysis of the regioselectivity toward forming the α-methylene imine product is presented, as well several examples of further functionalization of these useful building blocks.
 | ||
| Fig. 1 Top: Eisen's report of Ti-catalyzed ring-opening hydroamination of MCPs. Bottom: TI-catalyzed oxidative amination of MCPs (this work). | ||
Results and discussion
Reaction of 2.2 equiv. of 1-butyl-2-methylenecyclopropane (1a) with azobenzene and 5 mol% [py2TiCl2(NPh)]2 at 115 °C in PhCF3 for 62 hours cleanly provided the α-methylene imine product 3-methylene-N-phenylheptan-2-imine (2a) which was isolated in 68% yield on a >1 g scale (eqn (1)).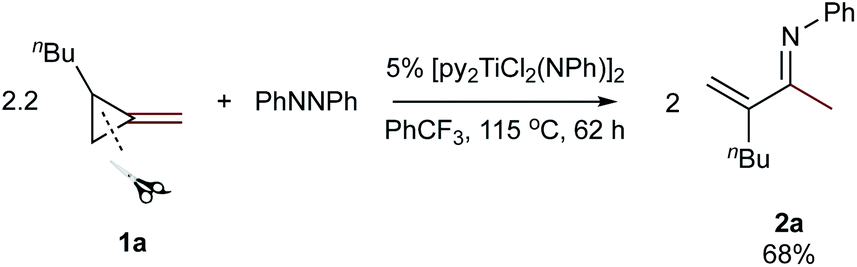 | (1) |
A full computational analysis of the catalytic reaction coordinate is presented in Fig. 2, as well as a simplified catalytic cycle in Fig. 3. As previously observed in [2 + 2 + 1] pyrrole synthesis,3d dative ligand speciation can impact the energetics of the overall catalytic cycle, and both mono-pyridine (Fig. 2, black) and no-pyridine (Fig. 2, grey) bound manifolds are possible, although the mono-pyridine bound Ti manifold is generally lower in energy. First, Markovnikov-like2a,4a,b [2 + 2] cycloaddition of a Ti imido (IM1) with 1a yields the spirocyclic azatitanacyclobutane IM3. Next, selective β-C elimination of the unsubstituted β-C-C bond generates an azatitana-methylenecyclopentane (IM4). The origin of selectivity will be discussed later (vide infra).
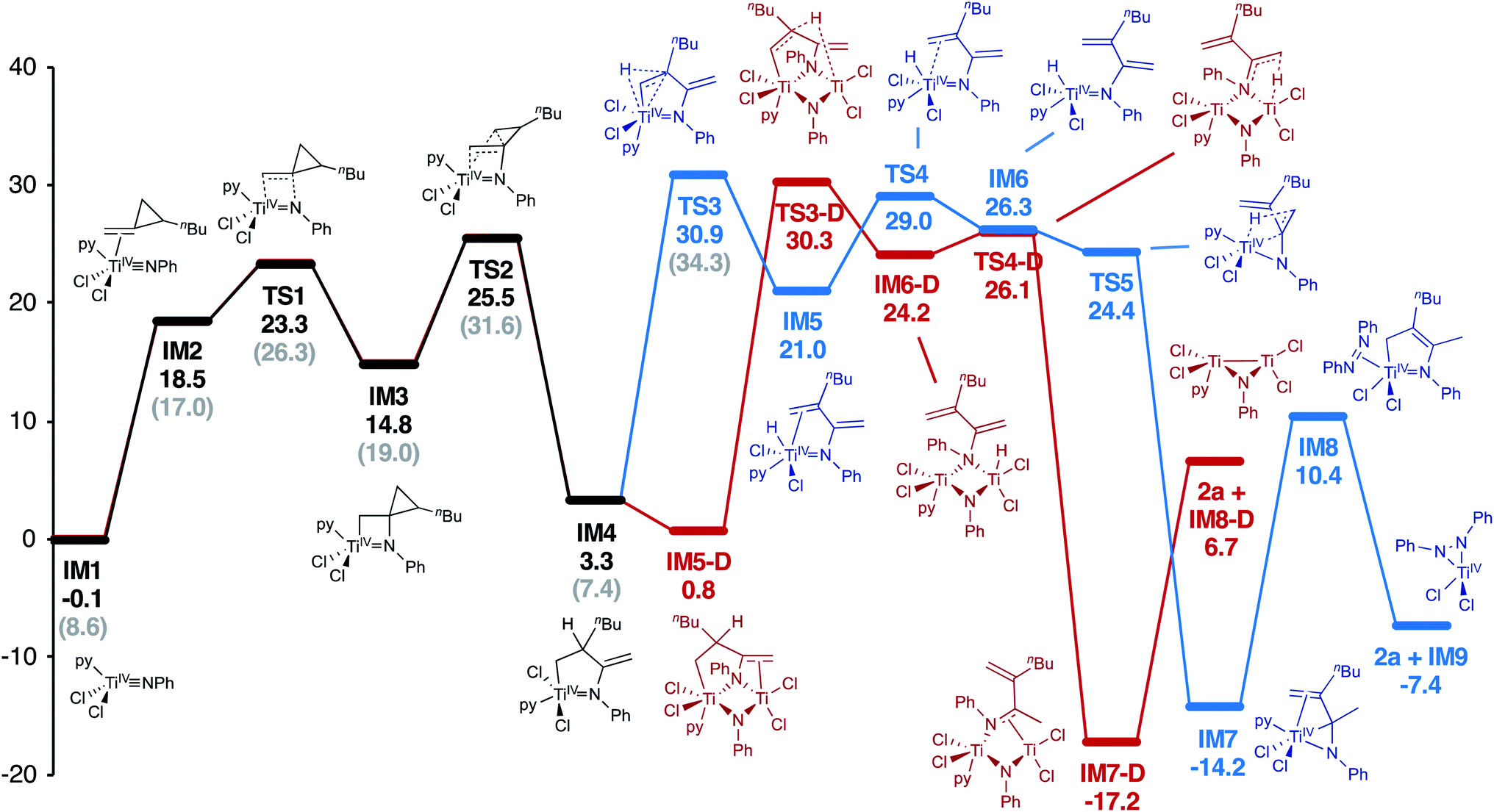 | ||
| Fig. 2 Computational analysis (M06/6-311G(d,p), SMD PhCF3, 388.15 K) of Ti-catalyzed oxidative amination of 1a. Free energies reported in kcal mol−1. Both a monometallic pathway (blue) and bimetallic pathway (red) are energetically feasible. Similarly, catalyst species with a ligated pyridine (black) or without ligated pyridine (grey) also possible, although the ligated pyridine pathway is lower in energy. | ||
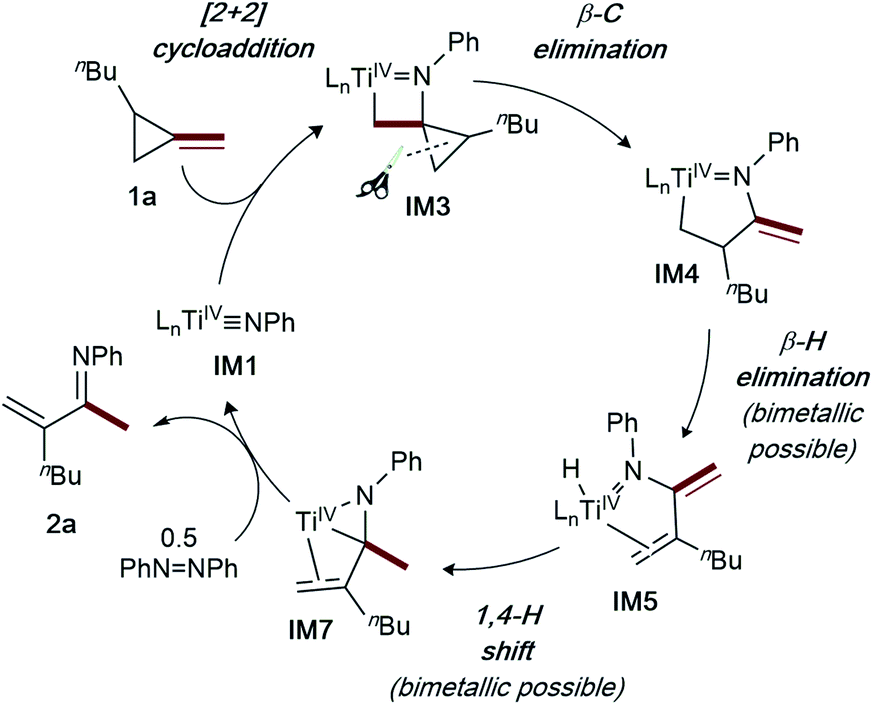 | ||
| Fig. 3 Simplified catalytic cycle for α-methylene imine formation via ring-opening oxidative amination of MCPs. | ||
From IM4, two feasible pathways diverge. First, a monometallic pathway in which IM4 undergoes rate-determining (TS3, 30.9 kcal mol−1) β-H elimination to form a Ti hydride (IM5). Next, ligand rearrangement (IM5 to IM6) and a 1,4-H migration7a yields the α,β-unsaturated imine adduct IM7. An N–H reductive elimination pathway was also considered from IM5, but it is significantly higher in energy (Fig. S58†). Associative displacement7a of the α,β-unsaturated imine by PhNNPh (IM8) results in charge transfer3g from the κ2-α,β-unsaturated imine to azobenzene, liberating the product and a TiIV η2-hydrazide (IM9) which can undergo disproportionation to close the catalytic cycle.3d Similar to a report on isocyanide oxidative amination,3g PhNNPh coordination is required for product release to avoid high-energy free TiII species.7
Alternately, IM4 can undergo roughly thermoneutral ligand exchange to form a bimetallic IM5-D, which similarly undergoes rate-determining (TS3-D, 30.3 kcal mol−1) β-H elimination to form IM6-D. Next, a 1,4-H migration generates IM7-D, which can undergo product release prior to PhNNPh reoxidation to close the catalytic cycle. Both pathways are very similar in overall energy; complex/intractable reaction kinetics are observed, indicating that both are likely contributing to the overall rate.
In this oxidative amination of 1a, β-C elimination occurs with opposite regioselectivity than in the previously-reported Ti-catalyzed hydroamination of 1-phenyl-2-methylenecyclopropane (1b) (Fig. 1, top). As a result, the regioselectivity of C–C ring-opening was investigated computationally (Fig. 4 and 5). For 1a, ring-opening of the substituted C–C bond to form IM4 or IM4′ is identical (TS2′ = TS2 = 25.5 kcal mol−1), but ultimately, IM4 (3.3 kcal mol−1) is significantly more stable than IM4′ (12.5 kcal mol−1) (Fig. 4).8 Since the subsequent β-H elimination (TS3/TS3′) is rate-determining, the C–C ring opening via β-C elimination is proposed to be reversible, leading to Curtin–Hammett control of the regioselectivity favoring reaction through TS3 instead of higher-energy TS3′. In contrast, for 1b ring-opening of the substituted benzylic C–C bond to form IM4′ is both kinetically and thermodynamically favored, and TS3′ is lower in energy than TS3, leading to the opposite regioselectivity compared to 1a (Fig. 5).9 Additional computations were performed considering initial pyridine dissociation before ring-opening (Fig. S59 and S60†) and lead to the same conclusion. Computational analysis of Eisen's hydroamination of 1b with Ti(NMe2)4 leads to the same conclusion (Fig. S61†).
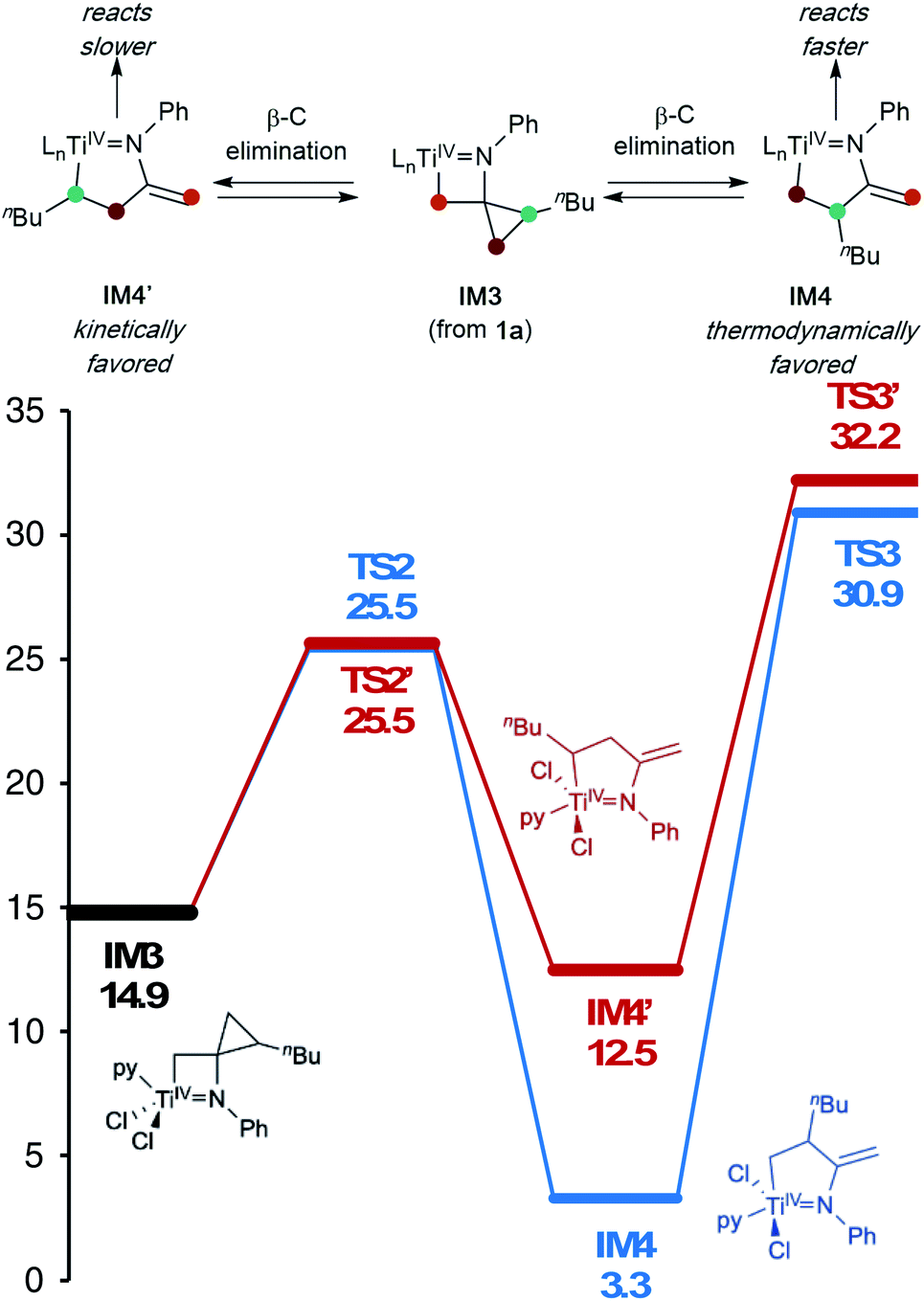 | ||
| Fig. 4 β-Carbon elimination from 1a thermodynamically favors formation of IM4 over IM4′, and kinetic formation of IM4′ is reversible (M06/6-311G(d,p), SMD PhCF3, 388.15 K). Ln = pyCl2. | ||
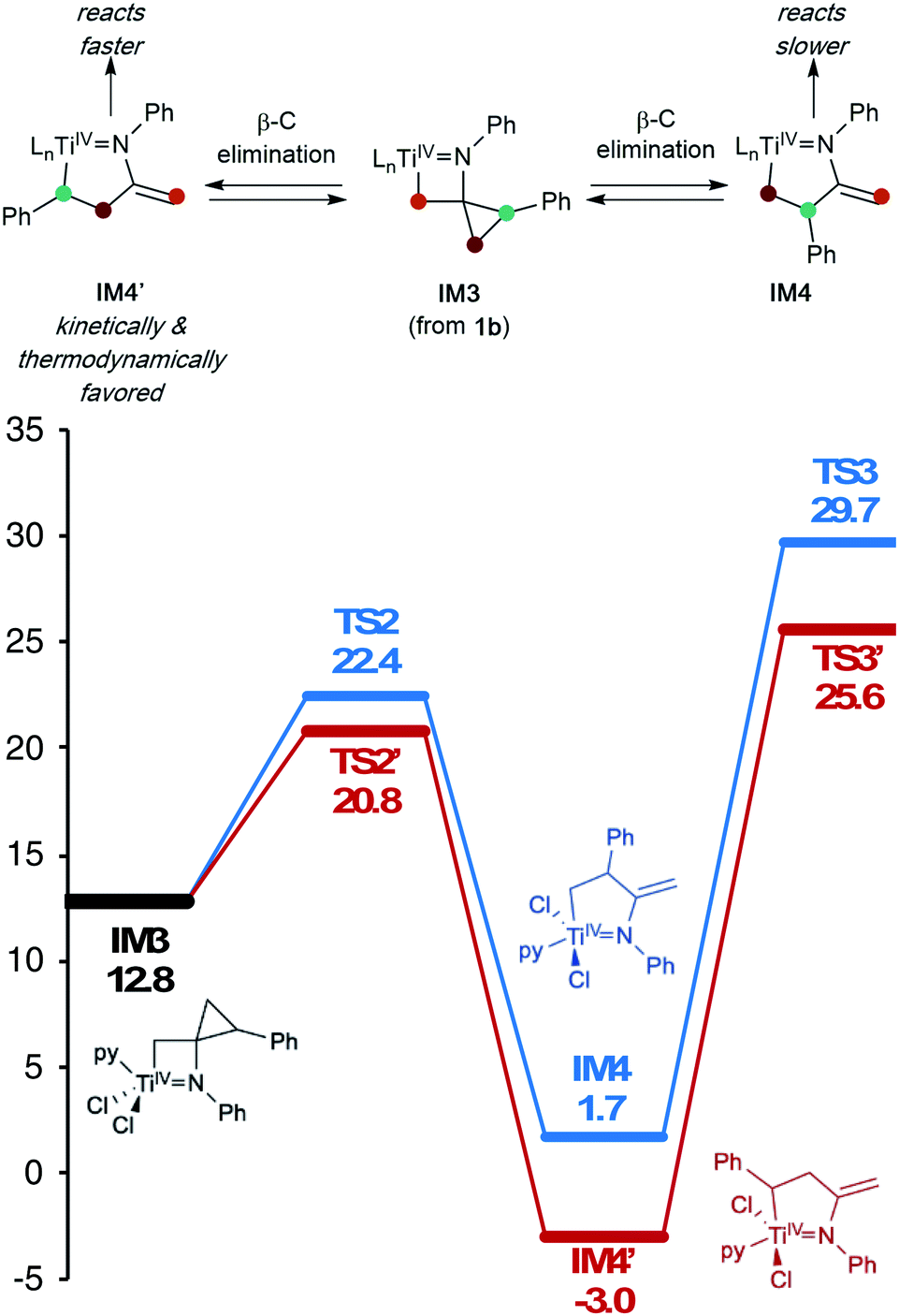 | ||
| Fig. 5 β-Carbon elimination from 1b both kinetically and thermodynamically favors formation of IM4′ over IM4 (M06/6-311G(d,p), SMD PhCF3, 388.15 K). Ln = pyCl2. | ||
Since protonolysis is often rate-determining in alkyne hydroamination,2a we hypothesized that similar manifolds of regiocontrol would also impact MCP hydroamination, and that hydroamination of 1a would lead to the formation of branched products, whereas 1b would lead to linear products (Fig. 6). In fact, reaction of 1a with PhNH2 catalyzed by [py2TiCl2(NPh)]2 under standard conditions leads to the formation of the branched product 2a-H2, while reaction of 1b leads to the linear product 2b′-H2. Attempts at oxidative amination of 1b led to an intractable mixture, although GC-MS analysis indicated the possible formation of a quinoline. Quinoline formation indicates that 2b′ had formed initially, as Mindiola had previously shown that α,β-unsaturated β-aryl imines underwent autoxidation to quinolines under similar reaction conditions (see ESI† for details).10 Nonetheless, these studies indicate that across multiple systems the regioselectivity of β-C elimination is under Curtin–Hammett control for alkyl and aryl-substituted cyclopropanes.
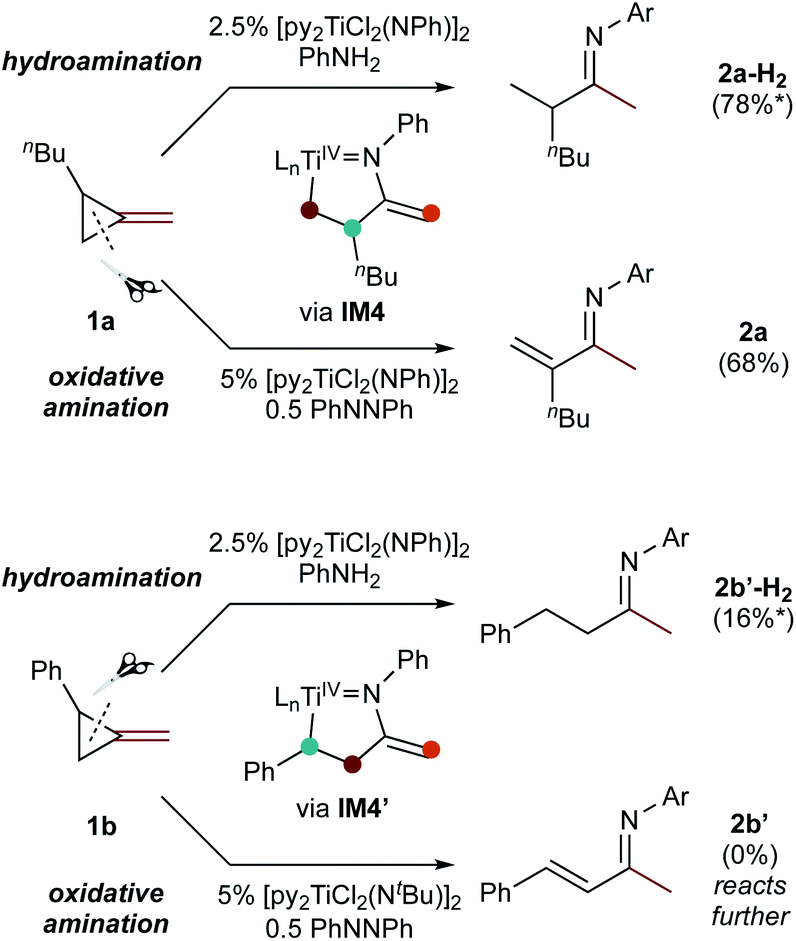 | ||
| Fig. 6 Regioselectivity of hydroamination and oxidative amination of 1a and 1b follow the same substrate control mechanisms. Conditions: 115 °C, 62 h (ox. amination) 20 h (hydroamination). *Yield determined via No-D 1H NMR spectroscopy. | ||
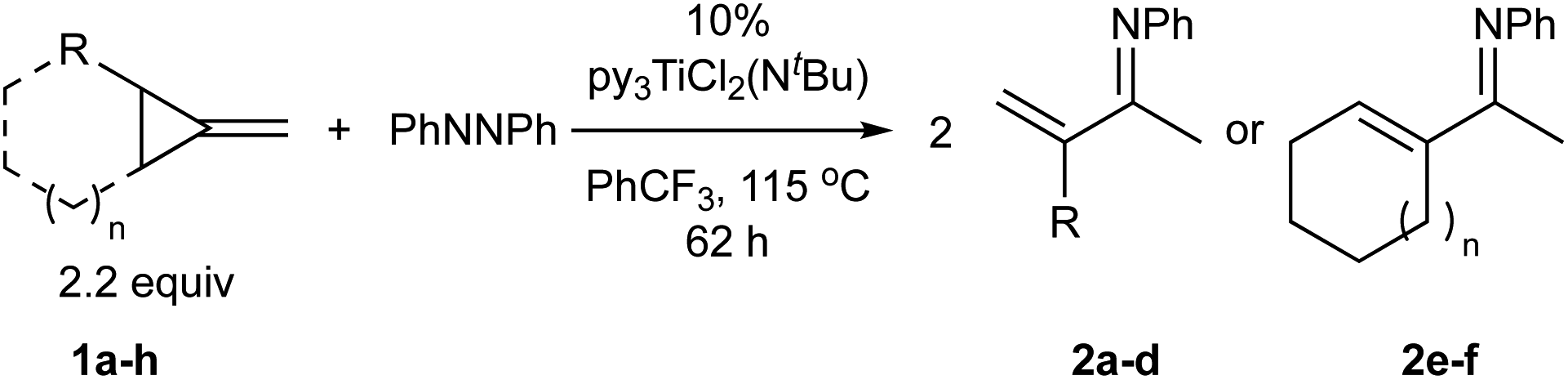
Table 1 depicts the initial scope of the MCP oxidative amination reaction. Monosubstituted aliphatic (1a, 1c), benzylic (1d), and bicyclic aliphatic (1e, 1f) MCPs all give good yields of the branched (or cyclic) α,β-unsaturated imines. Azobenzenes with electron donating and withdrawing groups also give good yields of 2a-tol and 2a-OCF3, respectively. Internally-substituted cyclopropylidene 1g and methylenecyclobutane 1h both did not react, presumably due to sterics (1g) or lack of sufficient ring strain (1h).11 Currently, the reaction scope is limited to monosubstituted or bicyclic aliphatic or benzylic MCPs. The substrate scope and practicality of this process are limited in large part by the difficulty in synthesizing MCPs via traditional routes.12 However, recent work from Uyeda on alkylidenecyclopropane synthesis indicates that more efficient access of these substrates may be possible in the future.13 Overall, this method represents an efficient route to generate α-methylene ketimines, which are often difficult to access via condensation of α,β-unsaturated ketones with primary amines because of competitive Michael addition.14
| a Isolated yields. b PhNNPh conversion. c Isolated as ketone after hydrolysis (2 M HCl, THF, 25 °C, 16 h). d 5% [py2TiCl2(NtBu)]2 as catalyst. e 5% [py2TiCl2(NPh)]2 as catalyst. f 145 °C, 110 h, 4 equiv. 1f. g 4 equiv. 1a. | |||
|---|---|---|---|
|
|||
|
1a |
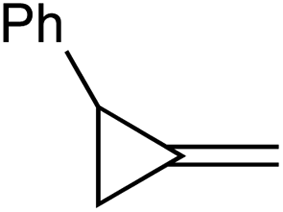
|
1b |
| 68% | 0% (98%)b | ||
|
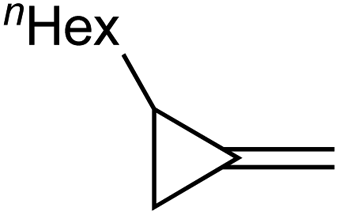
1c |
|
1d , |
| 77% | 59% | ||
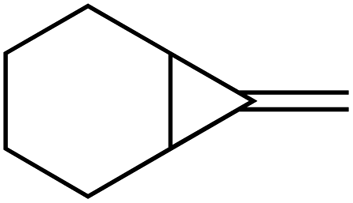
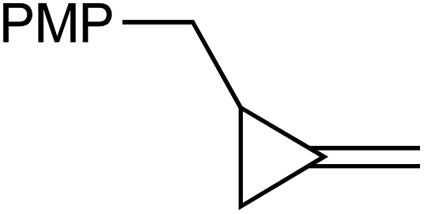
|
1e |
|
1f , |
| 67% | 35% | ||
|
1g |
|
1h |
| N.R. | N.R. | ||
| — | — | ||
|
1a |
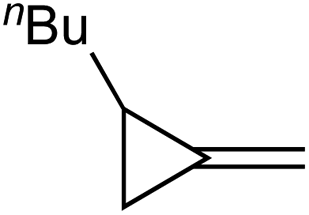
|
1a |
| Ph = p-tol (2a-tol) | Ph = p-CF3OPh (2a-OCF3) | ||
| 60% | 59% | ||
Given that there are few reports on the synthesis of α-methylene ketimines,6 we sought to explore the reactivity of products 2a and 2e (Fig. 7). Acidic hydrolysis of 2a and 2e generated ketones 3a and 3e, respectively. Intriguingly, the α-methylene moiety does not undergo acid-catalyzed isomerization to the internal alkene during the formation of 3a. Conjugate addition product 4a can be synthesized via reaction of CuCN and nBuLi with 2a, however isolation was complicated by enamine–imine tautomerization (see ESI† for details). β-Lactam 4e can be generated in modest yield from 2d using an in situ ketene formation, based on a procedure adapted from the flow system reported by Haftner and Ley.15 β-lactams are a key pharmacophore in antibiotics.16
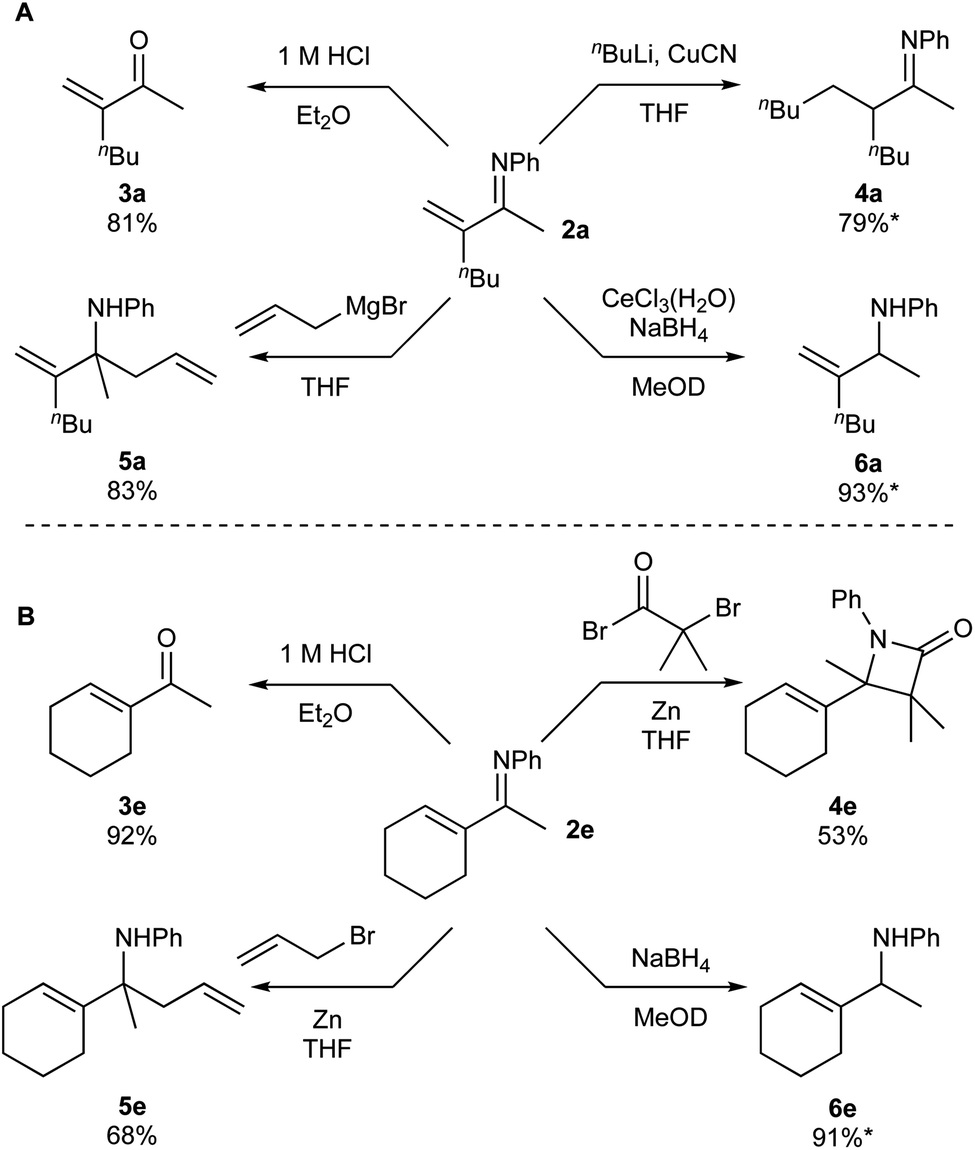 | ||
| Fig. 7 Further functionalizations of products 2a (A) and 2e (B). *Denotes yields that were determined via1H NMR spectroscopy. | ||
1,2-Allylations can also be accomplished via reaction of 2a with allyl magnesium bromide to form homoallylic amine 5a, or reaction of 2e with zinc and allyl bromide to form homoallylic amine 5e.17 Finally, both substrates can be reduced to allyl amines selectively, either via Luche reduction of 2a to form 6a or NaBH4 reduction of 2e to form 6e. Allyl amines are common precursors for nitrogen containing pharmaceuticals and natural products,18 and are functional groups present in some bioactive compounds, including naftifine and flunarizine.19 Thus, the reactivity of these unsaturated imines can be controlled in terms of selective 1,2 or 1,4-additions, depending on the reaction conditions. Additionally, cyclization reactions of these products present the potential for entry into heterocyclic moieties. With this reactivity in hand, we hope that these compounds can be incorporated toward the synthesis of useful nitrogen-containing compounds, including pharmaceuticals and natural products.
Conclusions
In conclusion, simple Ti imido halide complexes catalyze the ring-opening oxidative amination reaction of MCPs and diazenes to generate α-methylene imines in good yields. Importantly, this reaction further expands on the library of unsaturated substrates capable of undergoing Ti-catalyzed oxidative amination, indicating that aminations of more challenging substrates may be possible. The regioselectivity of ring-opening is dictated by the substituents on the cyclopropane ring, as determined by DFT calculations. For simple alkyl-substituted methylenecyclopropanes, branched products are obtained because rate-determining β-H elimination allows for equilibration to the more stable titanacycle that will ultimately yield the branched product. Reactivity of these α-methylene imine products has shown that diverse post-functionalizations can be achieved and hopefully may allow for access to important nitrogen-containing compounds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by the University of Minnesota (start-up funds, Wayland Noland Fellowship for AAO), the National Institutes of Health (1R35GM119457, 1R35GM124718) and the Alfred P. Sloan Foundation (Sloan Fellowship for IAT). NMR equipment was supported by the National Institutes of Health (S10OD011952) with matching funds from the University of Minnesota. Computational resources were provided by the Minnesota Supercomputing Institute (MSI) at the University of Minnesota and the National Energy Research Scientific Computing Center (NERSC), a DOE Office of Science User Facility supported by the U.S. Department of Energy under contract no. DE-AC02-05CH11231.References
- (a) Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247–9301 CrossRef CAS PubMed; (b) M.-L. Louillat and F. W. Patureau, Chem. Soc. Rev., 2014, 43, 901–910 RSC; (c) Y. Zhou, J. Yuan, Q. Yang, Q. Xiao and Y. Peng, ChemCatChem, 2016, 8, 2178–2192 CrossRef CAS; (d) B. J. Stokes and T. G. Driver, Eur. J. Org. Chem., 2011, 4071–4088 CrossRef CAS; (e) K. Shin, H. Kim and S. Chang, Acc. Chem. Res., 2015, 48, 1040–1052 CrossRef CAS PubMed; (f) M. M. Díaz-Requejo and P. J. Pérez, Chem. Rev., 2008, 108, 3379–3394 CrossRef PubMed; (g) H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417–424 CrossRef CAS PubMed; (h) P. Müller and C. Fruit, Chem. Rev., 2003, 103, 2905–2919 CrossRef PubMed; (i) H. Lu and X. P. Zhang, Chem. Soc. Rev., 2011, 40, 1899–1909 RSC; (j) F. Collet, C. Lescot and P. Dauban, Chem. Soc. Rev., 2011, 40, 1926–1936 RSC; (k) J. A. Halfen, Curr. Org. Chem., 2005, 9, 657–669 CrossRef CAS; (l) J. M. Alderson, J. R. Corbin and J. M. Schomaker, Acc. Chem. Res., 2017, 50, 2147–2158 CrossRef CAS PubMed.
- (a) T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795–3892 CrossRef PubMed; (b) A. L. Odom and T. J. McDaniel, Acc. Chem. Res., 2015, 48, 2822–2833 CrossRef CAS PubMed.
- (a) Z. W. Gilbert, R. J. Hue and I. A. Tonks, Nat. Chem., 2016, 8, 63–68 CrossRef CAS PubMed; (b) Z. W. Davis-Gilbert, L. J. Yao and I. A. Tonks, J. Am. Chem. Soc., 2016, 138, 14570–14573 CrossRef CAS PubMed; (c) H.-C. Chiu and I. A. Tonks, Angew. Chem., Int. Ed., 2018, 57, 6090–6094 CrossRef CAS PubMed; (d) Z. W. Davis-Gilbert, X. Wen, J. D. Goodpaster and I. A. Tonks, J. Am. Chem. Soc., 2018, 140, 7267–7281 CrossRef CAS PubMed; (e) Z. W. Davis-Gilbert, K. Kawakita, D. R. Blechschmidt, H. Tsurugi, K. Mashima and I. A. Tonks, Organometallics, 2018, 37, 4439–4445 CrossRef CAS PubMed; (f) H.-C. Chiu, X. Y. See and I. A. Tonks, ACS Catal., 2019, 9, 216–223 CrossRef CAS PubMed; (g) E. P. Beaumier, M. E. McGreal, A. R. Pancoast, R. H. Wilson, J. T. Moore, B. J. Graziano, J. D. Goodpaster and I. A. Tonks, ACS Catal., 2019, 9, 11753–11762 CrossRef CAS; (h) A. J. Pearce, R. P. Harkins, B. R. Reiner, A. C. Wotal, R. J. Dunscomb and I. A. Tonks, J. Am. Chem. Soc., 2020, 142, 4390–4399 CrossRef CAS PubMed; (i) E. P. Beaumier, A. J. Pearce, X. Y. See and I. A. Tonks, Nat. Rev. Chem., 2019, 3, 15–34 CrossRef PubMed; (j) Z. W. Davis-Gilbert and I. A. Tonks, Dalton Trans., 2017, 46, 11522–11528 RSC.
- (a) E. Smolensky, M. Kapon and M. S. Eisen, Organometallics, 2005, 24, 5495–5498 CrossRef CAS; (b) E. Smolensky, M. Kapon and M. S. Eisen, Organometallics, 2007, 26, 4510–4527 CrossRef CAS; For other examples of metal-catalyzed hydroamination of MCPs see (c) I. Nakamura, H. Itagaki and Y. Yamamoto, J. Org. Chem., 1998, 63, 6458–6459 CrossRef CAS; (d) I. Nakamura, H. Itagaki and Y. Yamamoto, Chem. Heterocycl. Compd., 2001, 37, 1532–1540 CrossRef CAS; (e) J.-S. Ryu, G. Y. Li and T. J. Marks, J. Am. Chem. Soc., 2003, 125, 12584–12605 CrossRef CAS PubMed; (f) M. Shi, L.-P. Liu and J. Tang, Org. Lett., 2006, 8, 4043–4046 CrossRef CAS PubMed; (g) D. Nishikawa, R. Sakae, Y. Miki, K. Hirano and M. Miura, J. Org. Chem., 2016, 81, 12128–12134 CrossRef CAS PubMed; (h) Y. Liu, A. A. Ogunlana and X. Bao, Dalton Trans., 2018, 47, 5660–5669 RSC; (i) J. C. Timmerman, B. D. Robertson and R. A. Widenhoefer, Angew. Chem., Int. Ed., 2015, 54, 2251–2254 CrossRef CAS PubMed; (j) J. C. Timmerman and R. A. Widenhoefer, Adv. Synth. Catal., 2015, 357, 3703–3706 CrossRef CAS; (k) M. Shi, Y. Chen and B. Xu, Org. Lett., 2003, 5, 1225–1228 CrossRef CAS PubMed.
- M. E. O'Reilly, S. Dutta and A. S. Veige, Chem. Rev., 2016, 116, 8105–8145 CrossRef PubMed.
- For some examples of α-methylene imine synthesis where the terminal alkene is favored over internal alkenes, see (a) N. De Kimpe, K. A. Tehrani and G. Fonck, J. Org. Chem., 1996, 61, 6500–6503 CrossRef CAS PubMed; (b) N. De Kimpe and W. Aelterman, Tetrahedron, 1996, 52, 12815–12820 CrossRef CAS.
- (a) J. Guo, Y. Lu, R. Zhao, Z. Liu, W. Menberu and Z.-X. Wang, Chem. –Eur. J., 2018, 24, 7010–7025 CrossRef CAS PubMed; (b) J. Guo, X. Deng, C. Song, Y. Lu, S. Qu, Y. Dang and Z.-X. Wang, Chem. Sci., 2017, 8, 2413–2425 RSC.
- J. F. Hartwig, in Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, Sausalito, 2010, ch. 3, p. 90 Search PubMed.
- Y. Jiao, M. E. Evans, J. Morris, W. W. Brennessel and W. D. Jones, J. Am. Chem. Soc., 2013, 135, 6994–7004 CrossRef CAS PubMed.
- (a) F. Basuli, H. Aneetha, J. C. Huffman and D. J. Mindiola, J. Am. Chem. Soc., 2005, 127, 17992–17993 CrossRef CAS PubMed; (b) D. J. Mindiola, Comments Inorg. Chem., 2008, 29, 73–92 CrossRef CAS.
- The strain energy is ca. 40.9 kcal mol−1 for methylenecyclopropane and ca. 26.9 kcal mol−1 for methylenecyclobutane. K. B. Wiberg, Angew. Chem., Int. Ed. Engl., 1986, 25, 312–322 CrossRef.
- K. Kitatani, T. Hiyama and H. Nozaki, Bull. Chem. Soc. Jpn., 1977, 50, 3288–3294 CrossRef CAS.
- S. Pal, Y.-Y. Zhou and C. Uyeda, J. Am. Chem. Soc., 2017, 139, 11686–11689 CrossRef CAS PubMed.
- (a) A. L. Odom, Dalton Trans., 2006, 225–233 Search PubMed; (b) For recent work on the reactivity of primary amines with α,β-unsaturated ketones and aldehydes see A. D. J. Calow, J. J. Carbó, J. Cid, E. Fernández and A. Whiting, J. Org. Chem., 2014, 79, 5163–5172 CrossRef CAS PubMed and references within.
- A. Hafnter and S. V. Ley, Synlett, 2015, 26, 1470–1474 CrossRef.
- K.-F. Kong, L. Schneper and K. Mathee, APMIS, 2010, 118, 1–36 CrossRef CAS PubMed.
- L.-M. Zhao, S.-Q. Zhang, H.-S. Jin, L.-J. Wan and F. Dou, Org. Lett., 2012, 14, 886–889 CrossRef CAS PubMed.
- M. Johannsen and K. A. Jørgensen, Chem. Rev., 1998, 98, 1689–1708 CrossRef CAS PubMed.
- (a) G. Petranyi, N. S. Ryder and A. Stütz, Science, 1984, 224, 1239–1241 CrossRef CAS PubMed; (b) A. Stütz, Angew. Chem., Int. Ed. Engl., 1987, 26, 320–328 CrossRef; (c) A. Stütz, A. Georgopoulos, W. Granitzer, G. Petranyi and D. Berney, J. Med. Chem., 1986, 29, 112–125 CrossRef PubMed; (d) J. Olesen, J. Neurol., 1991, 238, S23–S27 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental and computational details. See DOI: 10.1039/d0sc01998d |
| This journal is © The Royal Society of Chemistry 2020 |