
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesigning a highly stable coordination-driven metallacycle for imaging-guided photodynamic cancer theranostics†
Lizhen
He‡
b,
Li-Xuan
Cai‡
 a,
Meng-Hua
Li
c,
Guang-Lu
Zhang
a,
Li-Peng
Zhou
a,
Tianfeng
Chen
*b,
Mei-Jin
Lin
*c and
Qing-Fu
Sun
*a
a,
Meng-Hua
Li
c,
Guang-Lu
Zhang
a,
Li-Peng
Zhou
a,
Tianfeng
Chen
*b,
Mei-Jin
Lin
*c and
Qing-Fu
Sun
*a
aState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou 350002, P. R. China. E-mail: qfsun@fjirsm.ac.cn
bDepartment of Chemistry, Jinan University, Guangzhou 510632, P. R. China. E-mail: tchentf@jnu.edu.cn
cState Key Laboratory of Photocatalysis on Energy and Environment College of Chemistry, Fuzhou University, Fuzhou 350116, P. R. China. E-mail: meijin_lin@fzu.edu.cn
First published on 30th June 2020
Abstract
Coordination-driven self-assembly features good predictability and directionality in the construction of discrete metallacycles and metallacages with well-defined sizes and shapes, but their medicinal application has been limited by their low stability and solubility. Herein, we have designed and synthesized a highly stable coordination-driven metallacycle with desired functionality derived from a perylene-diimide ligand via a spontaneous deprotonation self-assembly process. Brilliant chemical stability and singlet oxygen production ability of this emissive octanuclear organopalladium macrocycle make it a good candidate toward biological studies. After cellular uptake by endocytosis, the metallacycle exhibits potent fluorescence cell imaging properties and cancer photodynamic therapeutic ability through enhancing ROS production, with high biocompatibility and safety. This study not only provides a rational design strategy for highly stable luminescent organopalladium metallacycles, but also sheds light on their application in imaging-guided photodynamic cancer therapy.
Introduction
Coordination-driven self-assembly features good predictability and directionality in the construction of discrete metallacycles and metallacages with well-defined sizes and shapes,1–10 which have shown great potential in diverse fields.11–19 Since the assembly of metallic molecular squares by Fujita and Stang in the early 1990s,20,21 more efforts have been dedicated to design of metallacycles possessing increased structural complexity and functionality.22–30 Especially, luminescent metallacycles have attracted intense attention due to their important and burgeoning biomedical applications, such as drug delivery,31 sensing,22,32 bio-imaging,33,34 and cancer therapy.35–37Photodynamic therapy (PDT) is now considered as a promising treatment modality for a variety of solid tumors, and in general relies on the singlet oxygen production ability of photosensitive dyes.38–41 Perylene tetracarboxylic acid diimides, in short perylene-diimides (PDIs), with characteristics of exceptional high thermal and photochemical stabilities, chemical inertness, outstanding optoelectronic properties and high biocompatibility,42,43 have been extensively investigated as well-known fluorescent dyes. Due to their easy modification in structures, derivative PDI dyes with efficient singlet oxygen generation ability warrant themselves as potent imaging and PDT photosensitizers.44,45 However, most bay-unsubstituted PDIs possess low singlet oxygen production ability due to their weak S1 → T1 intersystem crossing (ISC) processes. Common strategies to achieve a strong ISC for a PDI dye include incorporation of heavy atoms into the π-scaffolds and/or distortion of the PDI cores into nonplanarity by bay-substitution.46,47
Previously, Würthner and co-workers have developed very interesting metallosupramolecular macrocycles with PDI-based ligands, which exhibited unique fluorescence and redox properties.48–50 Possibly due to the low stability of the known PDI complexes, their bio-imaging and PDT applications have never been explored so far, though such applications have been demonstrated with water-soluble organic PDI-derivatives.51 Inspired by fluorescent self-assembled metallacycles with anticancer activity reported recently,31,35,36 we report here a highly luminescent metallosquare Pd8L4 (metallacycle 2) from four N,N′-3,5-di-methyl-pyrazole substituted PDI ligands (1) and eight 2,2-bipyridine-blocked palladium corners [BpyPd(NO3)2] (Fig. 1A). Due to the formation of dipalladium clips during a spontaneous deprotonation assembly process,52,53 metallacycle 2 exhibits unusual chemical stability toward acidic conditions. Photophysical properties, singlet oxygen production and the bio-imaging and intracellular translocation of metallacycle 2in vitro and vivo have been thoroughly studied. Our results demonstrate that this new organopalladium macrocycle possesses excellent PDT activity towards HeLa cervical cancer cells. In addition, we have synthesized a control mono-substituted PDI-pyrazole ligand 3 and its dinuclear complex Pd2L2 (complex 4) in order to probe whether the simpler metallo-organic complex is similarly effective in PDT (Fig. 2A). It is worth noting that metallacycle 2 with expanded size and complicated structure exhibited better growth inhibitory effects on HeLa cervical cancer cells than complex 4. Therefore, this study not only provides a rational design strategy for highly stable luminescent organopalladium metallacycles, but also sheds light on their application in imaging-guided photodynamic cancer therapy.
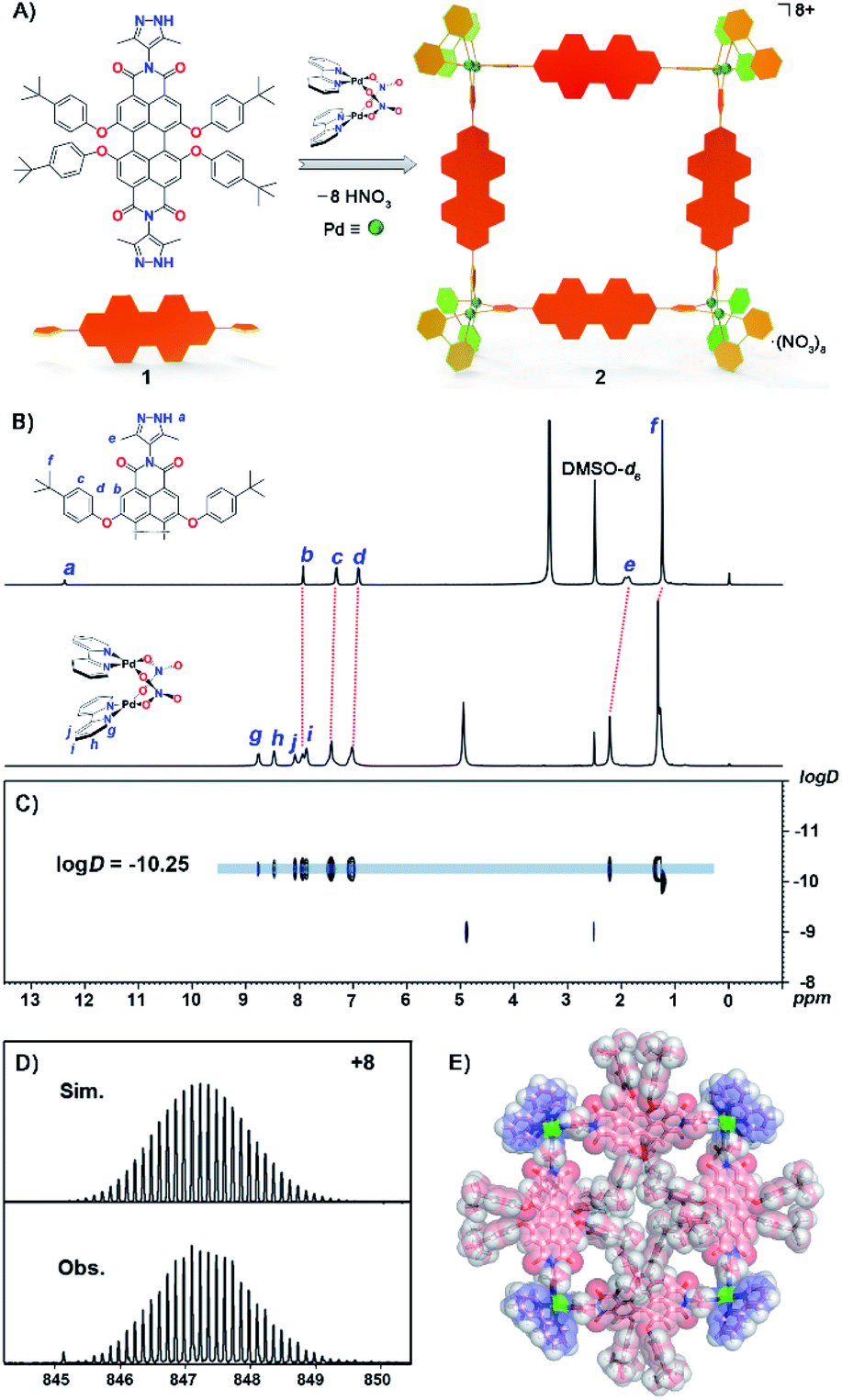 | ||
| Fig. 1 (A) Construction of Pd8L4 metallacycle 2 from a PDI-based ligand 1via a spontaneous deprotonation self-assembly process. (B) 1H NMR spectra (400 MHz, DMSO-d6, 298 K) of ligand 1 and metallacycle 2 with signals assigned to Ha–Hf on ligand 1 and Hg–Hj on the Bpy group, (C) 1H DOSY spectrum of metallacycle 2. (D) ESI-TOF-MS spectrum of metallacycle 2 showing the observed and simulated isotopic patterns of the 8+ peak. (E) The X-ray structure of metallacycle 2. For clarity, anions and solvent molecules have been omitted. | ||
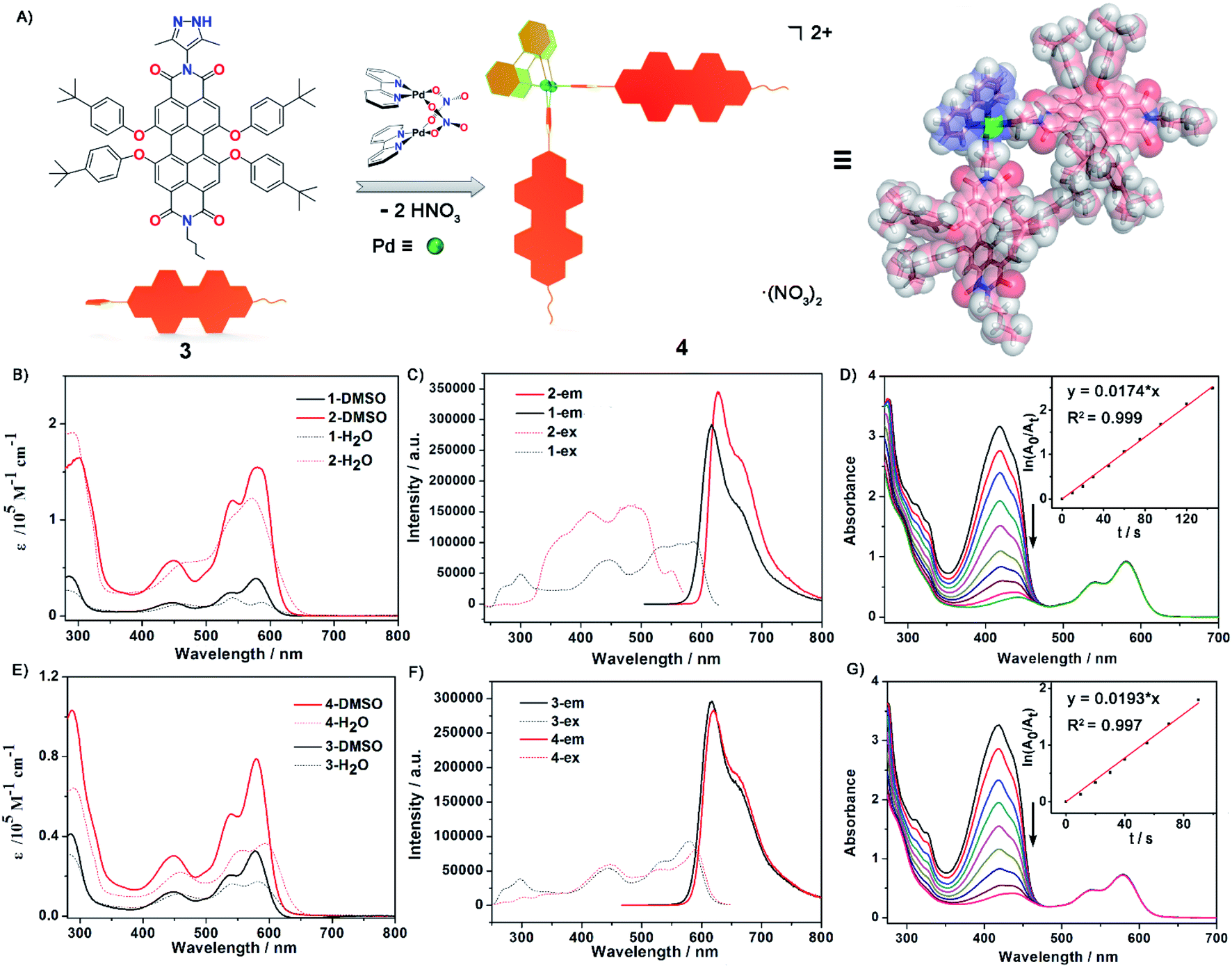 | ||
| Fig. 2 (A) Construction and optimized structure of Pd2L2 complex 4 from a PDI-based ligand 3via a spontaneous deprotonation self-assembly process. (B) and (E) The UV-Vis absorption spectra in DMSO and H2O solution and (C) and (F) the excitation and emission spectra of ligand 1, metallacycle 2, ligand 3 and complex 4 in DMSO (c = 2.5 × 10−5 M). (D) and (G) Measurement of 1O2 generation efficiency via decrease in absorption maxima of DPBF at 418 nm versus irradiation time in the presence of metallacycle 2 and complex 4 with the inset showing plots for the absorption decays of DPBF (150 μM) (for 2: ln(A0/At) versus time, where y = 0.0174x, R2: 0.999; for 4: ln(A0/At) versus time, where y = 0.0193x, R2: 0.997). | ||
Results and discussion
Self-assembly and X-ray crystal structure of metallacycle 2
The synthesis of ligand 1 starts from 1,6,7,12-tetrachloroperylene-3,4,9,10-tetracarboxylic dianhydride and 4-amino-3,5-dimethyl-pyrazole, both of which were obtained according to previous literature.54,55 Then intermediate tetrachloroperylene bisimide was reacted with 2,6-di-tert-butylphenol in dry N-methylpyrrolidinone (NMP). Following chromatographic purification, ligand 1 was obtained in a high overall yield (55% in two steps; see ESI† for details). The mono-pyrazole substituted control ligand 3 is synthesized by a similar method to 1 (Scheme S1†). However, it has to be pointed out that synthesis of 3 was much more challenging as the asymmetric precursor S2 was obtained in a much lower yield (23%) compared to that of S1 (80% yield). Molecular formulae of precursors S1 and S2, and ligands 1 and 3 have been clearly confirmed by NMR and ESI-TOF mass spectroscopy (Fig. S1–S18†). Subsequently, the octanuclear metallacycle Pd8L4 was quantitatively prepared by vigorous stirring of ligand 1 with (bpy)Pd(NO3)2 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio in deuterated dimethylsulfoxide (DMSO-d6) at 90 °C for 10 h. The formation of the octametallic square was first confirmed by the broadening and splitting of the 1H NMR spectrum of 2 when compared to that of ligand 1 (Fig. 1B and S19†). All the proton signals were fully assigned according to a 1H–1H COSY experiment (Fig. S20†). The singlet signal (12.38 ppm) assigned to Ha on the pyrazole groups of ligand 1 disappeared during the spontaneous deprotonation self-assembly process, which was accompanied by the generation of HNO3. The doublet signals on the t-butylphenol substituents of PDI displayed slight downfield shifts as compared to that of free ligand 1. The singlet peak for the methyl group on pyrazole (He) showed a downfield shift upon coordination of the N atoms to Pd(II). Diffusion-ordered spectroscopy (DOSY) NMR of metallacycle 2 also provided clear evidence for the formation of a single species with a single diffusion band at logD = −10.25 (Fig. 1C). Similarly, complex 4 was self-assembled from ligand 3 according to the same procedure of metallacycle 2, except in a ligand/metal ratio of 1:1 (Fig. S23†). The formulae of complexes 2 and 4 were then confirmed by high-resolution electrospray ionization mass spectrometry (ESI-TOF-MS, Fig. S27 and S28†). For example, the highly-resolved 8+ and 2+ peaks observed at m/z = 847.2290 and 1394.5089 correspond very well to the simulated isotopic patterns of the charged molecular complexes of [Pd8(bpy)814]8+ and [Pd2(bpy)232]2+ (Fig. 1D, S27 and S28†), respectively.
:2 ratio in deuterated dimethylsulfoxide (DMSO-d6) at 90 °C for 10 h. The formation of the octametallic square was first confirmed by the broadening and splitting of the 1H NMR spectrum of 2 when compared to that of ligand 1 (Fig. 1B and S19†). All the proton signals were fully assigned according to a 1H–1H COSY experiment (Fig. S20†). The singlet signal (12.38 ppm) assigned to Ha on the pyrazole groups of ligand 1 disappeared during the spontaneous deprotonation self-assembly process, which was accompanied by the generation of HNO3. The doublet signals on the t-butylphenol substituents of PDI displayed slight downfield shifts as compared to that of free ligand 1. The singlet peak for the methyl group on pyrazole (He) showed a downfield shift upon coordination of the N atoms to Pd(II). Diffusion-ordered spectroscopy (DOSY) NMR of metallacycle 2 also provided clear evidence for the formation of a single species with a single diffusion band at logD = −10.25 (Fig. 1C). Similarly, complex 4 was self-assembled from ligand 3 according to the same procedure of metallacycle 2, except in a ligand/metal ratio of 1:1 (Fig. S23†). The formulae of complexes 2 and 4 were then confirmed by high-resolution electrospray ionization mass spectrometry (ESI-TOF-MS, Fig. S27 and S28†). For example, the highly-resolved 8+ and 2+ peaks observed at m/z = 847.2290 and 1394.5089 correspond very well to the simulated isotopic patterns of the charged molecular complexes of [Pd8(bpy)814]8+ and [Pd2(bpy)232]2+ (Fig. 1D, S27 and S28†), respectively.
The structure of complex 2 was also unambiguously determined by X-ray crystallography (Table S1†). Crystallization was achieved by slow vapor diffusion of tetrahydrofuran into a DMSO solution of 2 over three weeks. Dark red single crystals were obtained and subsequent crystallographic analysis clearly confirmed the formation of an octanuclear metallacycle. The complex crystallized in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group, and thus two unique ligands and dipalladium clips are found in the asymmetric unit. In the crystal structure (Fig. 1E and S29†), four PDI ligands are connected by four dipalladium clips to form a metallic square, with a dimension of 21.8 Å × 21.6 Å (the distances between adjacent dipalladium corners). The spontaneous deprotonation of the 3,5-dimethylpyrazole group occurred upon coordination to the dipalladium center during the self-assembly process in solution, which is consistent with previous reports.52,53,56 The average Pd⋯Pd separation in the dimetallic clips was 3.16 Å, indicative of the existence of a weak PdII–PdII interaction (in the range of 2.60–3.30 Å). The dihedral angles of two pyrazolate planes in the dipalladium corners are 88.1° and 89.0° respectively. Among eight t-butylphenol substituents toward the inner cavity of the metallic square, four lie on the equatorial plane while the other four stretch outside the square plane due to steric hindrance. Four PDI aromatic backbones are also severely twisted out of the square plane with significant twist angles between the two naphthalene units being 30.5° and 30.8° (Fig. S30†). It is worth pointing out that only a few tetraphenoxy-PDIs have been characterized by single crystal analysis and most of them like here show a breaking of symmetry.57,58 We infer that the steric congestion evoked by tetraphenoxy substituents in the bay positions and the crowded square cavity leads to the core distortion.59 Such a nonplanar conformation of the ligands and the highly charged nature of the complex cooperatively suppress the aggregation of the central perylene diimide chromophores toward face-to-face π–π stacking (Fig. S31†).
space group, and thus two unique ligands and dipalladium clips are found in the asymmetric unit. In the crystal structure (Fig. 1E and S29†), four PDI ligands are connected by four dipalladium clips to form a metallic square, with a dimension of 21.8 Å × 21.6 Å (the distances between adjacent dipalladium corners). The spontaneous deprotonation of the 3,5-dimethylpyrazole group occurred upon coordination to the dipalladium center during the self-assembly process in solution, which is consistent with previous reports.52,53,56 The average Pd⋯Pd separation in the dimetallic clips was 3.16 Å, indicative of the existence of a weak PdII–PdII interaction (in the range of 2.60–3.30 Å). The dihedral angles of two pyrazolate planes in the dipalladium corners are 88.1° and 89.0° respectively. Among eight t-butylphenol substituents toward the inner cavity of the metallic square, four lie on the equatorial plane while the other four stretch outside the square plane due to steric hindrance. Four PDI aromatic backbones are also severely twisted out of the square plane with significant twist angles between the two naphthalene units being 30.5° and 30.8° (Fig. S30†). It is worth pointing out that only a few tetraphenoxy-PDIs have been characterized by single crystal analysis and most of them like here show a breaking of symmetry.57,58 We infer that the steric congestion evoked by tetraphenoxy substituents in the bay positions and the crowded square cavity leads to the core distortion.59 Such a nonplanar conformation of the ligands and the highly charged nature of the complex cooperatively suppress the aggregation of the central perylene diimide chromophores toward face-to-face π–π stacking (Fig. S31†).
Photophysical properties and chemical stability
The UV-Vis absorption and emission spectra of PDI ligand 1 and the metallic square 2 were explored in DMSO solution (Fig. 2B and C). The photophysical data for both ligands, metallacycle 2 and complex 4 were measured in detail and are summarized in the ESI (Fig. 2E and F, S32–S42 and Table S2).† Ligand 1 exhibited characteristic PDI absorption bands in the range of 400–630 nm, with the intense 578 nm band assigned to the S0–S1 electronic transition of the perylene chromophore and high-energy absorption bands centered at 534 and 446 nm attributed to the transition from the ground state to a higher excitation state. A similar absorption pattern for metallacycle 2 was observed with the absorption maxima at 449 nm, 540 nm, and 580 nm respectively. Based on the crystal structure of metallacycle 2 discussed above, we infer that the coordination-induced bathochromic-shift was counter-balanced by the increased HOMO–LUMO band-gap resulting from the twisted ligand conformation on the complex. The extinction coefficient of metallacycle 2 in DMSO was as high as 1.6 × 105 M−1 cm−1, roughly four times that of ligand 1 (Fig. 2B). When excited on the PDI chromophore at 486 nm, both ligand 1 and metallacycle 2 showed a strong red emission band tailing up to the NIR region, with λem maxima centered at 617 and 627 nm respectively. The photoluminescence quantum yields (PLQY) of ligand 1 (λex = 447 nm) and metallacycle 2 (λex = 486 nm) in solution were determined to be 57.7% and 50.7%, respectively. It is worth noting that negligible fluorescence quenching of Pd(II)-perylene bisimide metallacycles has been observed in previous studies.48,50 Photophysical properties of ligand 3 and complex 4 were also explored in order to compare with those of ligands 1 and 2. Similar UV-Vis and emission spectra of 3 and 4 in DMSO were observed, except smaller molar extinction coefficients (0.33 × 105 M−1 cm−1 for 3 and 0.79 × 105 M−1 cm−1 for 4, Fig. 2E). Compared to ligand 1 and complex 2, both 3 and 4 showed lower photo-luminescent quantum yields in DMSO (44.7% for 3 and 46.1% for 4), suggesting that the introduction of the second pyrazole chelate and the rigidified PDI chromophores on metallacycle 2 have dramatically enhanced its photoluminescence (50.7% for 2). Based on these photophysical investigations, we conclude that: (1) metallacycle 2 based on the PDI photosensitizer possesses high molar extinction coefficients and long wavelength absorptions, which make it a good candidate for photodynamic therapy application; (2) the increased Stokes shift in metallacycle 2 compared to free ligand 1 reduces the energy loss of the excited-states through the self-quenching process; (3) the 50.7% luminescent quantum yield of 2 is quite nice as half of the excited states lead to emission as needed for imaging and the other half presumably to triplet states as needed for singlet oxygen generation.60,61Water solubility and chemical stability of the supramolecular complexes are the prerequisite for their biological applications. We then examined the water solubility of both ligand 1 and metallacycle 2 by UV-Vis and fluorescence spectroscopy. The absorptions of ligand 1 start to level-off when its concentration reaches 3 × 10−5 M in water. By clear contrast, no obvious aggregation was observed for metallacycle 2 until up to 2 × 10−4 M (Fig. S43†). Enhanced water solubility has thus been achieved by formation of the 8+ charge on the complex balanced by strongly solvating nitrate counter-ions. The luminescence properties were also investigated in aqueous media. The emission spectra of ligand 1 and the metallacycle 2 were measured in H2O/DMSO mixed solvent systems (Fig. S44†). With the addition of water, the emission intensity for the solution of ligand 1 decreased sharply, with a decay rate much faster than metallacycle 2. The emission intensity of metallacycle 2 remains high in 50% H2O/DMSO solution. Enhanced steric congestion attributed to four aryloxyl groups stretching outside the square plane of the metallacycle may contribute to less π–π stacking aggregation, which leads to stronger luminescence of metallacycle 2 than that of ligand 1 in the aqueous phase.
Evaluation of the stability of Pd8L4 metallacycle 2 in aqueous solutions under different pH conditions has been conducted by monitoring the changes in the UV-Vis absorption spectrum over time. Different pH values adjusted by phosphate-buffered saline (PBS) were used to simulate the physiological condition (pH = 7.4), tumor microenvironment (pH = 6.8) and lysosome environment (pH = 5.3), respectively. No obvious changes (less than 8% of intensity decrease) were observed, indicative of good stability of metallacycle 2 (Fig. S49–S51†). We also examined the emission spectra of complex 2 in different solutions. The emission shapes are consistent with those in water, along with a slight increase in the emission intensity of metallacycle 2.
Singlet oxygen (1O2) generation studies
Singlet molecular oxygen 1O2, as one of the cytotoxic reactive oxygen species (ROS) responsible for the tumor cell death and damage, is crucial and beneficial for photodynamic therapy (PDT). To evaluate 1O2 generation capacity by photosensitization with ligand 1, ligand 3, metallacycle 2 and complex 4, the singlet oxygen production rate was examined by measuring the degradation of 1,3-diphenyl-isobenzofuran (DPBF, an 1O2 scavenger62) upon photoexcitation of all compounds at an excitation wavelength of 510 nm. The optical density values at 510 nm of all compound solutions were kept the same (0.25) to guarantee the absorption of the same number of photons. In the presence of metallacycle 2, a sharp decrease of the strong absorption band centered at 418 nm for DPBF was observed with prolonged irradiation time, as shown in Fig. 2D. The 1O2 generation rate constants of the sensitizer can be calculated from ln(A0/At) = kt, where A0 and At denote the initial and final absorbance of DPBF at 418 nm, and k is the slope of the corresponding linear curves after plotting ln(A0/At) vs. time. The rate constant of singlet oxygen generation by metallacycle 2 was determined to be 0.0179 (±0.0006) s−1 (Fig. S46†), about 3 times larger than that produced by PDI ligand 1 (0.0069 (±0.0005) s−1, Fig. S45†). More significant decrease in absorption maxima of DPBF was also observed in the presence of metallacycle 2 than ligand 1, indicating better 1O2 generation ability of metallacycle 2. The rate constants of 1O2 generation for 3 and 4 were determined to be 0.0113 (±0.0006) s−1 and 0.0191 (±0.0009) s−1, respectively (Fig. 2G, S47 and S48†). The larger rate constants observed for complex 4 were reasonable considering that it exhibited lower fluorescence quantum yields.PDT-based anticancer activity and in vitro1O2 overproduction
Studies were carried out to compare the PDT-based anticancer activities of both ligands and complexes, as well as BpyPd(NO3)2. As shown in Fig. 3A, ligands 1 and 3 and complex 4 (Pd2L2) didn't exhibit obvious anticancer activity and PDT enhancement effects. In sharp contrast, metallacycle 2 (Pd8L4) exhibited significant growth inhibitory effects on HeLa cervical cancer cells. The IC50 values of complex 4 and metallacycle 2 against HeLa cells were about 29.7 μM and 9.7 μM, respectively, while IC50 for BpyPd(NO3)2 was 9.0 μM, suggesting the important contribution of Pd(II) to the anticancer activity of the complex. After irradiation at 0.15 W cm−2 for 2 min, the IC50 of metallacycle 2 against HeLa cells decreased significantly, indicating the enhancement of anticancer activities in combination with PDT therapy. However, no PDT enhancement effects could be found in BpyPd(NO3)2 and cisplatin, due to their lack of absorbance upon laser irradiation at 560 nm. It is worth noting that metallacycle 2 with expanded size and complicated structure exhibited better growth inhibitory effects on HeLa cervical cancer cells than complex 4, indicating its functional role in construction this metallacycle structure. We next investigated the cytotoxic effects of metallacycle 2 on a series of cancer cells, and found that metallacycle 2 exhibited broad-spectrum antitumor activity (Fig. 3B). Based on the photo-activation properties, we further confirmed the PDT efficiency of metallacycle 2 against SiHa cervical cancer cells at grade II (squamous cell carcinoma). As shown in Fig. 3C, S52 and S53,† after incubation with metallacycle 2 for 6 h followed by laser irradiation, the cell viability decreased more significantly than metallacycle 2 and laser irradiation alone, especially the HeLa cells treated with 20 μM of metallacycle and 0.15 W cm−2 irradiation. In contrast, ligand 1, ligand 3 and complex 4 exhibited much lower PDT activities, while no PDT effect was observed for BpyPd(NO3)2 and cisplatin. More importantly, we compared the cytotoxicity of 1 equiv. metallacycle 2 and 8 equiv. BpyPd(NO3)2 against Ect1/E6E7 human normal cervical immortalized squamous cells (Fig. 3D and S54†), where metallacycle 2 exhibited much lower cytotoxicity toward normal cells than BpyPd(NO3)2. These results demonstrate the safety advantage of metallacycle 2 compared to BpyPd(NO3)2. Following studies were carried out to examine the modes of cell death induced by metallacycle-based PDT. As shown in Fig. 3E and F and S55,† co-treatment with metallacycle 2 and irradiation lead to a significantly higher G0/G1 cell population than those of individual treatment, indicating that metallacycle 2 synergistically enhances PDT-induced cell cycle arrest to inhibit cancer cell growth.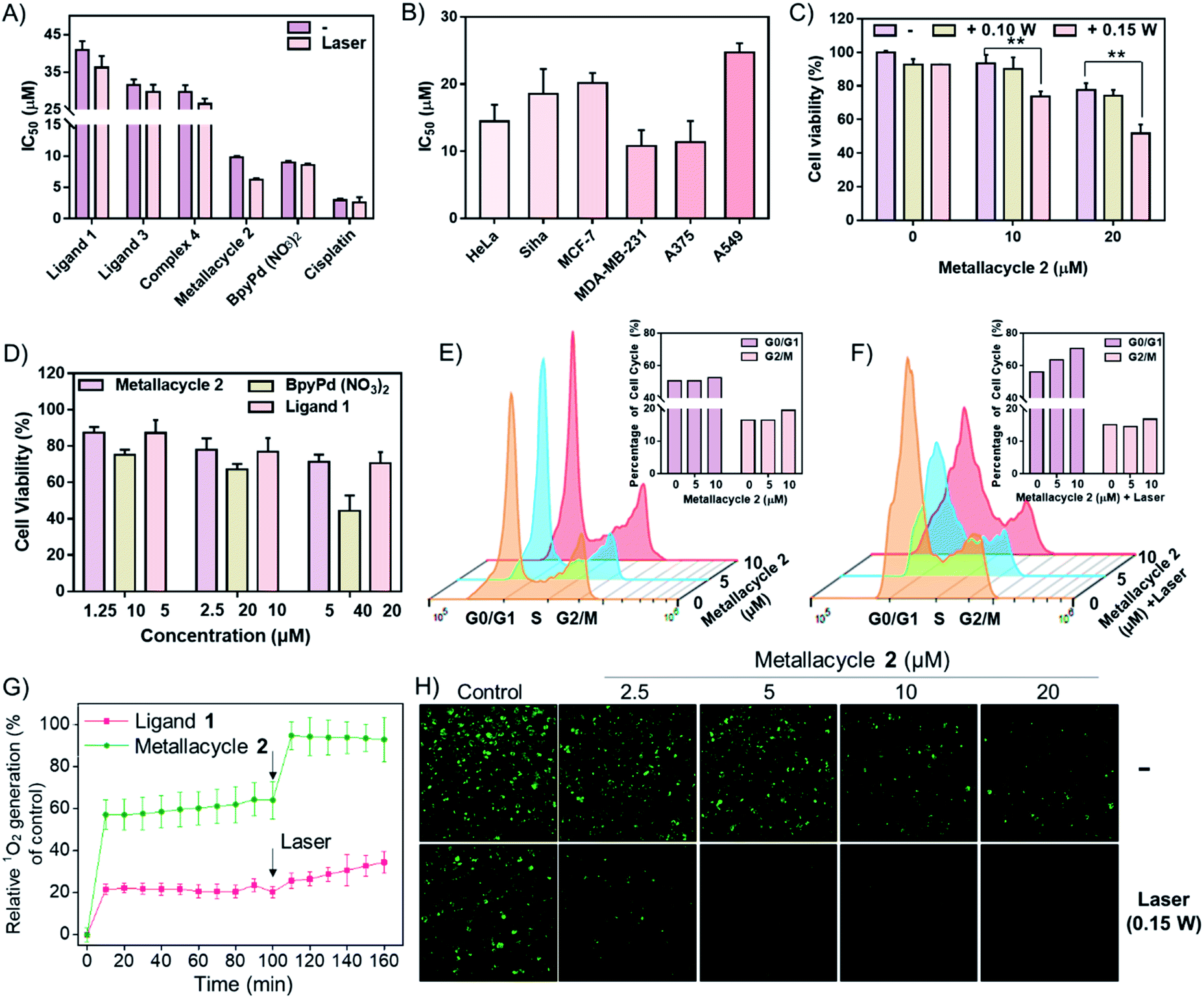 | ||
| Fig. 3 Anticancer action and induction of 1O2 overproduction by metallacycle-based PDT. (A) IC50 values for various molecules against HeLa cells with or without PDT (0.15 W cm−2 irradiation for 2 min). The cells were treated with these molecules for 72 h. (B) Cytotoxic effects of metallacycle 2 on various human cancer cells after 72 h incubation. Values expressed are means ± SD of triplicates. (C) HeLa cell viability of metallacycle 2 combined with PDT for 24 h-incubation. (D) Ect1/E6E7 human normal cervical immortalized squamous cell viability induced by ligand 1, BpyPd(NO3)2 and metallacycle 2. The cells were treated with these molecules for 72 h. (E and F) Metallacycle 2 enhances PDT-induced cell cycle arrest in HeLa cells for 24 h-incubation. (G) Comparison between metallacycle 2 (2.5 μM) and ligand 1 (10 μM) in PDT-induced singlet oxygen overproduction in HeLa cells. (H) Fluorescence imaging technique to examine the fluorescence intensity of 1O2 overproduction in HeLa cells treated with metallacycle 2 and PDT. The decrease in fluorescence intensity of DPBF reflects the level of 1O2 production. | ||
Due to the important role of 1O2 in PDT, we have examined the in vitro1O2 generation in HeLa cells promoted by metallacycle 2 and laser irradiation by the DPBF assay. Since metallacycle 2 is a highly stable molecule with 4 PDI cores, we have compared the effects of metallacycle 2 and 4-times concentrations of ligand 1. As shown in Fig. 3G and S56,† under LED irradiation at 0.15 W cm−2 for 2 min, 2.5 μM of metallacycle 2 produced an 1O2 generation increase of 85.1% at 160 min, which was significantly higher than that of 10 μM ligand 1 (44.5%). This finding was also confirmed by fluorescence imaging. As expected, metallacycle 2 promoted 1O2 generation in a dose-dependent manner, as evidenced by the significant decrease in green fluorescence, which was further enhanced by LED irradiation (Fig. 3H). These results suggest that LED irradiation activates metallacycle 2 to enhance 1O2 overproduction in HeLa cells and enhance anticancer efficacy.
Fluorescence imaging and localization
Fluorescence imaging and intracellular translocation of metallacycle 2in vitro and vivo were then investigated, with the aim to realize its future application as a diagnostic agent during cancer therapy. As shown in Fig. 4A, ligand 1 and ligand 3 show extremely weak fluorescence after 24 h incubation with HeLa cells, while complex 4 exhibits week fluorescence intensity inside the cells. BpyPd(NO3)2 shows almost no fluorescence. Interestingly, metallacycle 2 exhibited much higher red fluorescence intensity inside the cells. Based on this fluorescence signal, we then investigated the internalization process of metallacycle 2 in HeLa cells. As shown in Fig. S57,† metallacycle 2 (red fluorescence) accumulated in the cell membrane after 6 h incubation, which enhanced gradually and then started to enter the cytoplasm after 12 h treatment, and finally dispersed in the whole cytoplasm after 24 h. In contrast, no fluorescent signal was detected for ligand 1 treatment at 12 h, and only a negligible red fluorescent signal was determined at 24 h (Fig. S58†). As can be expected, no fluorescence signal was detected for BpyPd(NO3)2 even after 24 h (Fig. S59†). The cellular uptake of metallacycle 2 in HeLa cells was then quantitatively examined by flow cytometry, which showed that, intracellular metallacycles increase in a time-dependent manner (Fig. 4B). This time-dependent intracellular translocation process was further confirmed using a high-magnification confocal fluorescence microscope (Fig. 4C). The images in z-stack mode by three-dimensional (3D) visualization revealed that metallacycle 2 with red fluorescence not only binds to the cell membrane, but is also internalized inside HeLa cells (Fig. 4D).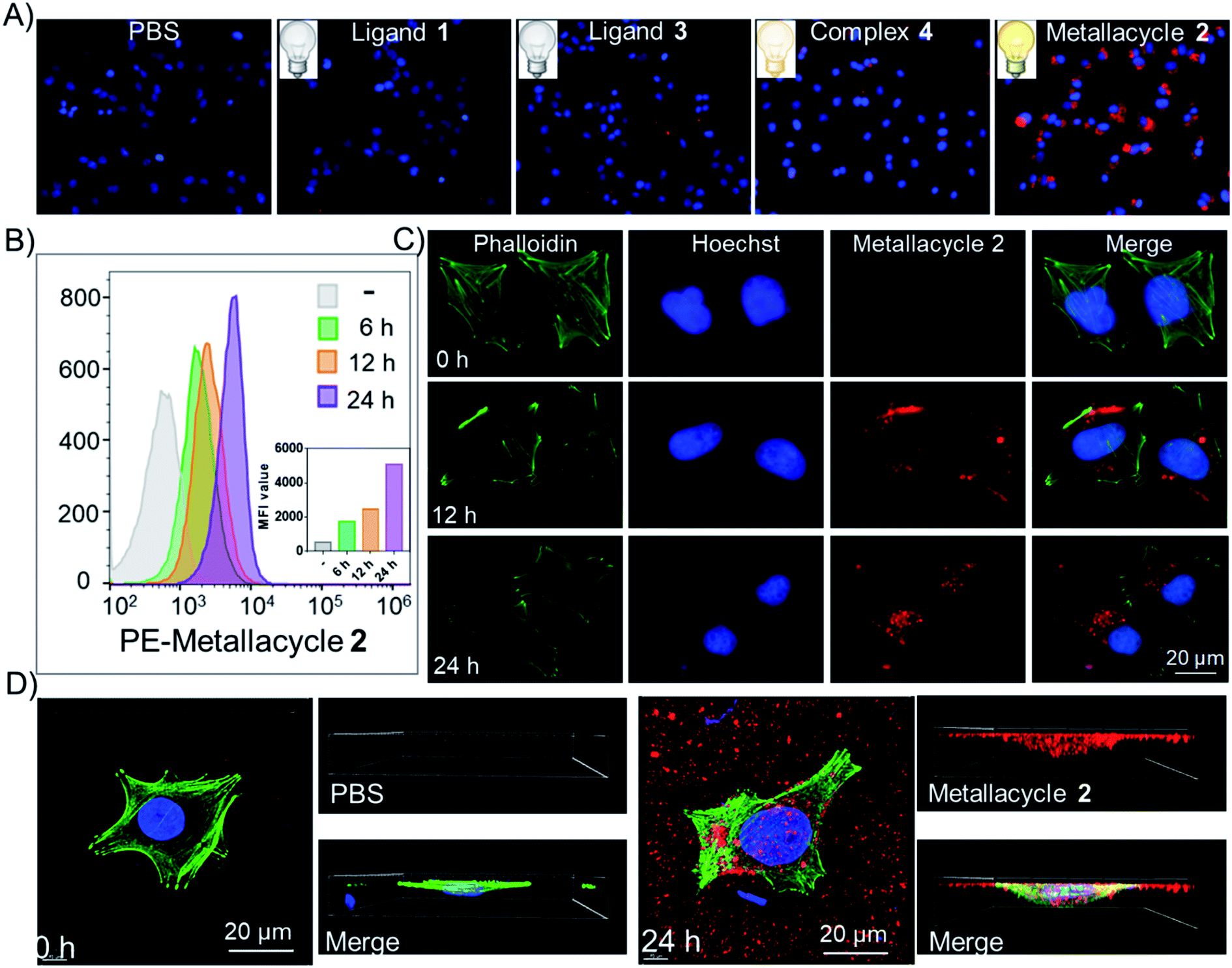 | ||
| Fig. 4 Fluorescence imaging by metallacycle 2in vitro. (A) The fluorescence imaging properties of ligands and complexes in HeLa cells. The cells were incubated with 10 μM of these molecules for 24 h. (B) Cellular uptake of metallacycle 2 (10 μM) in HeLa cells at different times examined by flow cytometry. Inset is quantitative mean fluorescence intensity. (C) The distribution of metallacycle 2 in HeLa cells examined using a high magnification confocal fluorescence microscope. (D) Three-dimensional (3D) fluorescence images of HeLa cells after incubation with 10 μM of metallacycle 2 for 24 h. | ||
We examined the in vivo fluorescence imaging and biodistribution of metallacycle 2 in HeLa xenograft nude mice. As shown in Fig. 5A, metallacycle 2 distributed in the whole body of tumor-bearing nude mice after intravenous injection, which gathered around the tumor sites after 48 h and the fluorescence intensity increased after 72 h. We also investigated the biodistribution of metallacycle 2 in the main organs. As expected, metallacycle 2 was mainly distributed in the liver and tumor tissues, and a small amount was found in the kidneys after 48 h. While after 72 h, the accumulation of metallacycle 2 in the liver decreased, but increased in the kidneys and tumor tissues (Fig. 5B). These results suggest that metallacycle 2 may exhibit a long residence time in the tumor sites, and the possible metabolism pathways of 2 may be through clearance by the liver and excretion by the kidneys. Consistently, we also found that the red fluorescence of metallacycle 2 increased in a time-dependent manner in the tumor section (Fig. 5C and D). Furthermore, we also examined the biocompatibility and cytotoxicity of metallacycle 2 or complex 4in vivo. As shown in Table S3,† after being intravenously injected with 2 mg kg−1 of metallacycle 2 or complex 4 for 3 days, the results of blood routine examination were both in the normal range compared with the control group and have no significant changes. Furthermore, we also performed hematological analysis and H&E staining to examine the effects of each treatment on the functions of the main organs. As shown in Fig. S60,† the blood indexes of liver and kidney functions also fluctuate within the normal range after treatment with metallacycle 2 or complex 4. The H&E staining results showed that no obvious pathological changes are observed in the main organs (including the heart, liver, spleen, lungs, and kidneys) with different treatments (Fig. S61†). These results confirm the high biocompatibility and safety of metallacycle 2 and complex 4. Moreover, metallacycle 2 exhibits strong in vivo fluorescence properties, and thus could be further developed as diagnostic agents during cancer therapy.
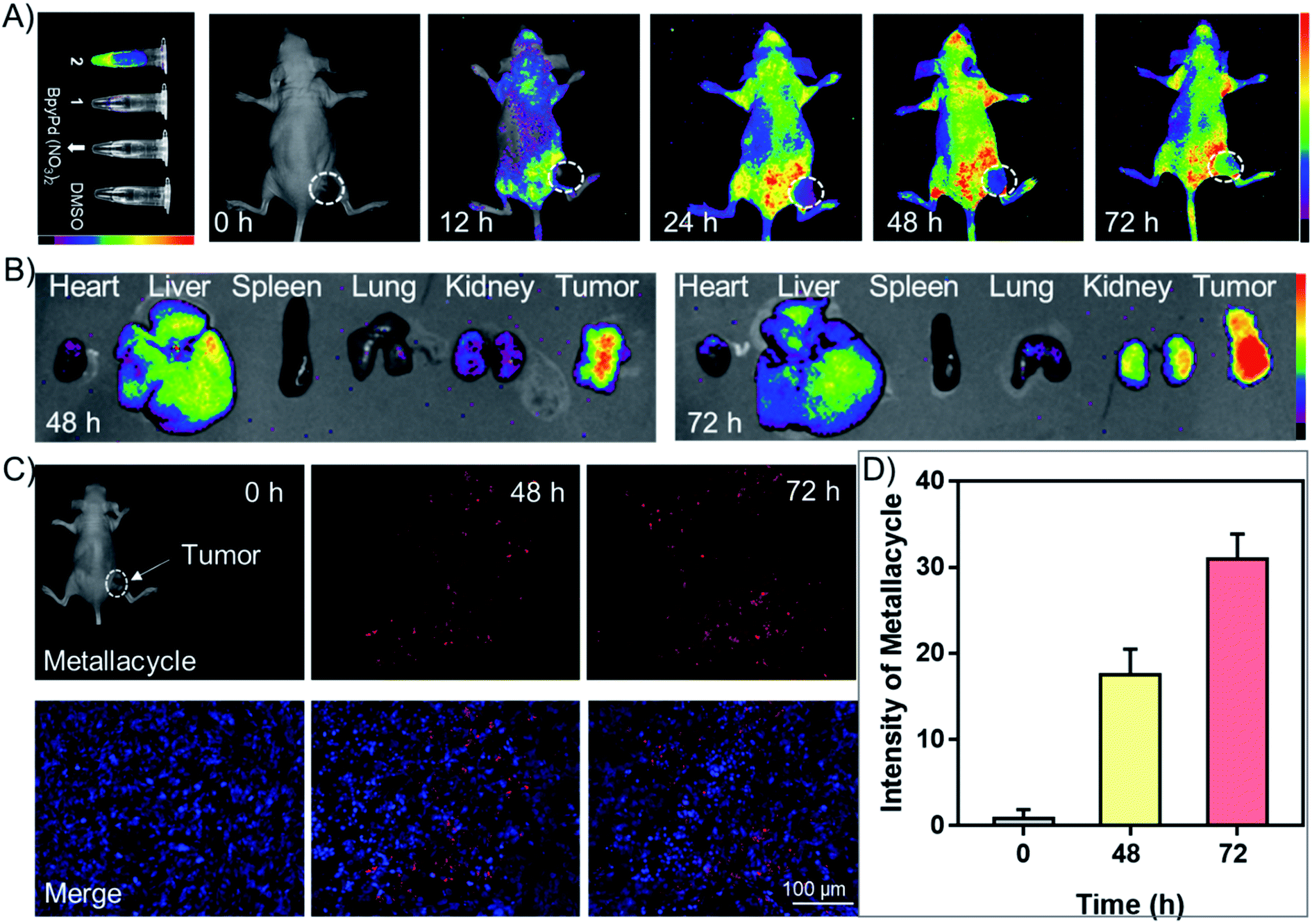 | ||
| Fig. 5 Fluorescence imaging by metallacycle 2in vivo. (A) Fluorescence imaging monitors the accumulation of metallacycle 2 in HeLa xenograft nude mice at different time points. (B) The biodistribution of metallacycle 2 in the main organs after 48 h and 72 h intravenous injection. (C) The distribution of metallacycle 2 (2 mg kg−1) in HeLa xenografts nude mice and 8 μm sections of tumor tissues. (D) Fluorescence intensity of metallacycle 2 in tumor sections analyzed by Image J. The HeLa xenograft nude mice were intravenously injected with 2 mg kg−1 of metallacycle 2. | ||
Studies were also carried out to investigate the cellular uptake process and localization of metallacycle 2 in HeLa cells. As shown in Fig. 6A, after incubation for 12 h, the red fluorescence of metallacycle 2 was found around the lysosome and entered lysosomes (green fluorescence from lysotracker), and the colocalization ratio reached 31–41%. After incubation for 24 h, metallacycle 2 matched well with lysosomes, and the colocalization ratio reached up to 75–86%, indicating that metallacycle 2 is internalized into cancer cells through lysosome-mediated endocytosis. Furthermore, different chemical endocytosis inhibitors were employed to clarify the internalization pathway. Among them, sodium azide (NaN3), chlorpromazine and low temperature (4 °C) are the inhibitors of energy-dependent endocytosis. As shown in Fig. 6B, after pretreatment with NaN3, chlorpromazine and low temperature (4 °C) for 1 h, the cellular uptake of metallacycle 2 significantly decreased, especially with chlorpromazine and low temperature pretreatment. In contrast, sucrose, a specific inhibitor of clathrin-mediated endocytosis, didn't inhibit the internalization of metallacycle 2. Meanwhile, dynasore and nystatin markedly decreased the internalization of metallacycle 2, suggesting that dynamin-mediated lipid raft endocytosis may also be the main pathway. In addition, after combination of low temperature and NaN3 with the inhibitors of chlorpromazine and dynasore, the cellular uptake of metallacycle 2 decreased (Fig. S62†). Taken together, we infer that both energy-dependent and dynamin-mediated lipid raft endocytosis work for the internalization of metallacycle 2 in cancer cells.
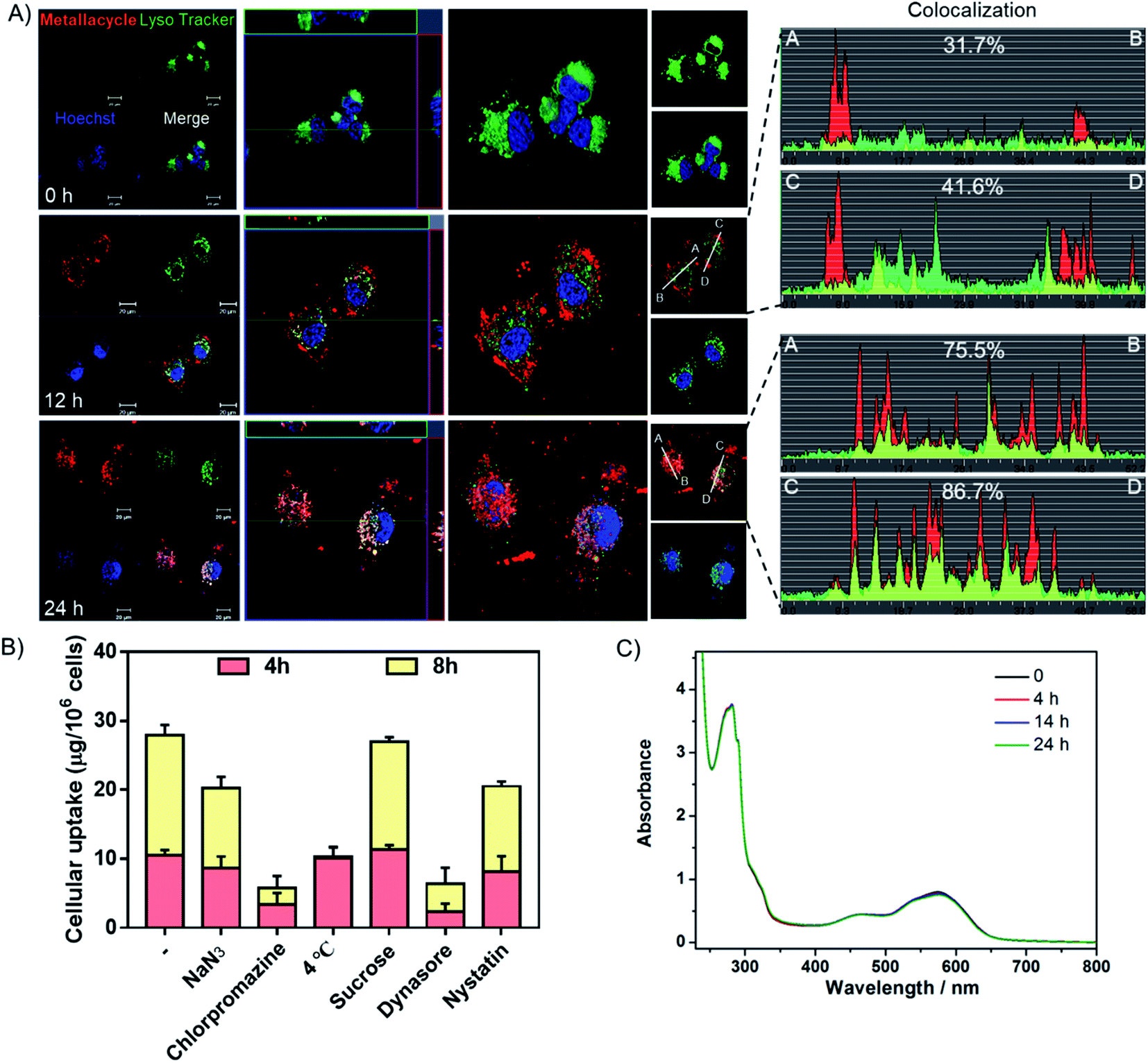 | ||
| Fig. 6 Translocation of metallacycle 2. (A) Subcellular localization of metallacycle 2 (red) in HeLa cells by staining with the special fluorescent tracers, lysotracker (green) and Hoechst 33342 (blue). Metallacycle 2 localization analysis (the first column); z-stack mode analysis (the second column); three-dimensional (3D) reconstruction and co-localization analysis (the third column). (B) Intracellular uptake of the metallacycle in HeLa cells after pretreatment with different endocytosis inhibitors. HeLa cells were pretreated with various endocytosis inhibitors for 1 h and then incubated with 10 μM of metallacycle for different periods of time at 37 °C or 4 °C. The control group was incubated with metallacycle 2 only at 37 °C. (C) Changes in the UV-Vis spectrum of metallacycle 2versus time in PBS buffer at pH = 5.3 containing lysozymes (c = 10 μM). | ||
Finally, the chemical stability of metallacycle 2 was confirmed under physiological conditions. First of all, no obvious changes were observed in the absorption (Fig. 6C) spectra of metallacycle 2 in PBS buffer at pH = 5.3 containing lysozymes (to simulate the environment of lysosomes), indicating the good stability of metallacycle 2 under physiological conditions. We then also evaluated the absorption and emission spectra of metallacycle 2 extracted from the cell lysate after incubation with HeLa cells for 24 h. As shown in Fig. S63,† no obvious changes in the UV-Vis spectrum further indicated the stability of metallacycle 2 even in HeLa cells. It is worth pointing out that a slight increase in emission intensity was observed for metallacycle 2 (Fig. S64†). The successful observation of the m/z signals assignable to [2–n(NO3−)]n+ (n = 6, 7, 8) in the ESI-TOF-MS spectrum for metallacycle 2 in water at pH = 5.3 containing lysozymes also indicates the good stability of 2 (Fig. S65†). For comparison, the stability of ligand 1, metallacycle 2, ligand 3 and complex 4 in PBS buffer at pH = 5.3 containing lysozymes was evaluated by monitoring their UV-Vis absorptions and emission spectra (Fig. S66–S69†). In the UV-Vis spectrum, roughly 78.6% and 77.2% of intensity decreases for ligand 1 and 3 were observed after 19 h, suggesting that they both tend to aggregate even in dilute solutions. While complex 4 also shows obvious 83.9% of absorbance decrease, only 5.6% of absorbance drop for metallacycle 2 was noticed after 24 h, indicating the best stability of metallacycle 2 under physiological conditions. Moreover, a time-dependent decrease in the emission intensity of both ligands and complex 4 was also observed, but a slight increase in emission intensity was found for metallacycle 2 (Fig. S67†). This observation indicated the possible internalization of metallacycle 2 into the confined space of the small cellular organelle, leading to the enhancement of luminescence imaging properties, which allows us to trace the in vitro and in vivo translocalization and enables its application in future cancer diagnosis.
Conclusion
In summary, a novel metallacycle with desired functionality derived from a new PDI-based ligand was developed via coordination-driven self-assembly, and was fully characterized by NMR, ESI-MS, and X-ray analysis. The use of the “dual-edge” Pd–Pd clipping unit leads to the unusual chemical stability of metallacycle 2. High extinction coefficients, long wavelength absorptions and strong luminescent properties of metallacycle 2 have been demonstrated, as well as good chemical stability and singlet oxygen production ability. More interestingly, based on the synergistic effect of four ligands, this metallocycle with expanded size and complicated structure exhibits good fluorescence imaging power in vitro and vivo, along with much higher anticancer efficacy using PDT compared with ligand 1, control ligand 3 and its dinuclear complex 4. Overall, the current study not only provides a facile design strategy for highly stable luminescent organo-palladium species, but also sheds light on the potential roles of photo-active metallacycles in efficient cancer therapy.Ethical statement
This study was performed in strict accordance with the national guidelines for the care and use of laboratory animals (Laboratory animal-Guideline for ethical review of animal welfare. GB/T 35892-2018) and was approved by the Animal Experimentation Ethics Committee of Jinan University (Guangzhou, China).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of China (Grant No. 21825107, 21877049, 21801241 and 21971041), the Natural Science Foundation of Fujian Province (Grant No. 2017J05037), the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB20000000), the Major Program for Tackling Key Problems of Industrial Technology in Guangzhou (201902020013), the Dedicated Fund for Promoting High-Quality Marine Economic Development in Guangdong Province (GDOE-2019-A31), the Guangzhou Key Laboratory of Molecular and Functional Imaging for Clinical Translation (201905010003), and the Innovation Team Project in Guangdong Colleges and Universities (2019KCXTD008).Notes and references
- T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001–7045 CrossRef CAS PubMed.
- S. Mukherjee and P. S. Mukherjee, Chem. Commun., 2014, 50, 2239–2248 RSC.
- H.-T. Feng, Y.-X. Yuan, J.-B. Xiong, Y.-S. Zheng and B. Z. Tang, Chem. Soc. Rev., 2018, 47, 7452–7476 RSC.
- L. Xu, Y.-X. Wang, L.-J. Chen and H.-B. Yang, Chem. Soc. Rev., 2015, 44, 2148–2167 RSC.
- M. Han, D. M. Engelhard and G. H. Clever, Chem. Soc. Rev., 2014, 43, 1848–1860 RSC.
- J.-C. Chang, S.-H. Tseng, C.-C. Lai, Y.-H. Liu, S.-M. Peng and S.-H. Chiu, Nat. Chem., 2016, 9, 128–134 CrossRef.
- A. Baba, T. Kojima and S. Hiraoka, J. Am. Chem. Soc., 2015, 137, 7664–7667 CrossRef CAS PubMed.
- L.-X. Cai, S.-C. Li, D.-N. Yan, L.-P. Zhou, F. Guo and Q.-F. Sun, J. Am. Chem. Soc., 2018, 140, 4869–4876 CrossRef CAS PubMed.
- A. V. Zhukhovitskiy, M. Zhong, E. G. Keeler, V. K. Michaelis, J. E. Sun, M. J. Hore, D. J. Pochan, R. G. Griffin, A. P. Willard and J. A. Johnson, Nat. Chem., 2016, 8, 33–41 CrossRef CAS PubMed.
- T. Zhang, L.-P. Zhou, X.-Q. Guo, L.-X. Cai and Q.-F. Sun, Nat. Commun., 2017, 8, 15898 CrossRef CAS PubMed.
- M. Yamashina, S. Kusaba, M. Akita, T. Kikuchi and M. Yoshizawa, Nat. Commun., 2018, 9, 4227 CrossRef PubMed.
- D. Preston, J. E. Lewis and J. D. Crowley, J. Am. Chem. Soc., 2017, 139, 2379–2386 CrossRef CAS PubMed.
- D. Zhang, T. K. Ronson and J. R. Nitschke, Acc. Chem. Res., 2018, 51, 2423–2436 CrossRef CAS PubMed.
- C. M. Hong, R. G. Bergman, K. N. Raymond and F. D. Toste, Acc. Chem. Res., 2018, 51, 2447–2455 CrossRef CAS PubMed.
- X. Jing, C. He, L. Zhao and C. Duan, Acc. Chem. Res., 2019, 52, 100–109 CrossRef CAS.
- J. Jiao, C. Tan, Z. Li, Y. Liu, X. Han and Y. Cui, J. Am. Chem. Soc., 2018, 140, 2251–2259 CrossRef CAS PubMed.
- B. P. Burke, W. Grantham, M. J. Burke, G. S. Nichol, D. Roberts, I. Renard, R. Hargreaves, C. Cawthorne, S. J. Archibald and P. J. Lusby, J. Am. Chem. Soc., 2018, 140, 16877–16881 CrossRef CAS PubMed.
- C.-Y. Zhu, M. Pan and C.-Y. Su, Isr. J. Chem., 2019, 59, 209–219 CrossRef CAS.
- S. K. Samanta, D. Moncelet, V. Briken and L. Isaacs, J. Am. Chem. Soc., 2016, 138, 14488–14496 CrossRef CAS PubMed.
- M. Fujita, J. Yazaki and K. Ogura, J. Am. Chem. Soc., 1990, 112, 5645–5647 CrossRef CAS.
- P. J. Stang and D. H. Cao, J. Am. Chem. Soc., 1994, 116, 4981–4982 CrossRef CAS.
- M. L. Saha, X. Yan and P. J. Stang, Acc. Chem. Res., 2016, 49, 2527–2539 CrossRef CAS PubMed.
- C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Chem. Rev., 2015, 115, 3012–3035 CrossRef CAS PubMed.
- H. Xue, F. Jiang, Q. Chen, D. Yuan, J. Pang, G. Lv, X. Wan, L. Liang and M. Hong, Chem. Commun., 2015, 51, 13706–13709 RSC.
- M. Zhang, S. Yin, J. Zhang, Z. Zhou, M. L. Saha, C. Lu and P. J. Stang, PNAS, 2017, 114, 3044–3049 CrossRef CAS PubMed.
- Z. Zhou, X. Yan, M. L. Saha, M. Zhang, M. Wang, X. Li and P. J. Stang, J. Am. Chem. Soc., 2016, 138, 13131 CrossRef CAS PubMed.
- M. L. Saha and M. Schmittel, Inorg. Chem., 2016, 55, 12366–12375 CrossRef CAS.
- L. Zhao, J. Wei, J. Lu, C. He and C. Duan, Angew. Chem., Int. Ed. Engl., 2017, 56, 8692–8696 CrossRef CAS.
- P. Wei, T. R. Cook, X. Yan, F. Huang and P. J. Stang, J. Am. Chem. Soc., 2014, 136, 15497–15500 CrossRef CAS.
- Y. Ai, M. Ng, E. Y.-H. Hong, A. K.-W. Chan, Z.-W. Wei, Y. Li and V. W.-W. Yam, Chem.–Eur. J., 2018, 24, 11611–11618 CrossRef CAS.
- G. Yu, M. Zhang, M. L. Saha, Z. Mao, J. Chen, Y. Yao, Z. Zhou, Y. Liu, C. Gao, F. Huang, X. Chen and P. J. Stang, J. Am. Chem. Soc., 2017, 139, 15940–15949 CrossRef CAS PubMed.
- M. Zhang, M. L. Saha, M. Wang, Z. Zhou, B. Song, C. Lu, X. Yan, X. Li, F. Huang, S. Yin and P. J. Stang, J. Am. Chem. Soc., 2017, 5067–5074 CrossRef CAS PubMed.
- M. Zhang, S. Li, X. Yan, Z. Zhou, M. L. Saha, Y.-C. Wang and P. J. Stang, PNAS, 2016, 113, 11100–11105 CrossRef CAS PubMed.
- J. Liu, H. Lai, Z. Xiong, B. Chen and T. Chen, Chem. Commun., 2019, 55, 9904–9914 RSC.
- Z. Zhou, J. Liu, T. W. Rees, H. Wang, X. Li, H. Chao and P. J. Stang, PNAS, 2018, 115, 5664–5669 CrossRef CAS PubMed.
- J. Zhou, Y. Zhang, G. Yu, M. R. Crawley, C. R. P. Fulong, A. E. Friedman, S. Sengupta, J. Sun, Q. Li, F. Huang and T. R. Cook, J. Am. Chem. Soc., 2018, 140, 7730–7736 CrossRef CAS PubMed.
- Z. Chen, H. Lai, L. Hou and T. Chen, Chem. Commun., 2019, 56, 179–196 RSC.
- L. Zhang, Z. Huang, D. Dai, Y. Xiao, K. Lei, S. Tan, J. Cheng, Y. Xu, J. Liu and X. Qian, Org. Lett., 2016, 18, 5664–5667 CrossRef CAS PubMed.
- J. Zhou, G. Yu and F. Huang, Chem. Soc. Rev., 2017, 46, 7021–7053 RSC.
- N. Kong, X. Ji, J. Wang, X. Sun, G. Chen, T. Fan, W. Liang, H. Zhang, A. Xie, O. C. Farokhzad and W. Tao, Nano Lett., 2020, 20, 3943–3955 CrossRef CAS PubMed.
- Z. Tang, N. Kong, J. Ouyang, C. Feng, Na Y. Kim, X. Ji, C. Wang, O. C. Farokhzad, H. Zhang and W. Tao, Matter, 2020, 2, 297–322 CrossRef.
- F. Wurthner, C. R. Saha-Moller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef PubMed.
- S. K. M. Nalluri, J. Zhou, T. Cheng, Z. Liu, M. T. Nguyen, T. Chen, H. A. Patel, M. D. Krzyaniak, W. A. Goddard, M. R. Wasielewski and J. F. Stoddart, J. Am. Chem. Soc., 2019, 141, 1290 CrossRef PubMed.
- Q. Fan, K. Cheng, Z. Yang, R. Zhang, M. Yang, X. Hu, X. Ma, L. Bu, X. Lu, X. Xiong, W. Huang, H. Zhao and Z. Cheng, Adv. Mater., 2015, 27, 843–847 CrossRef CAS PubMed.
- J. Zhang, F. An, Y. Li, C. Zheng, Y. Yang, X. Zhang and X. Zhang, Chem. Commun., 2013, 49, 8072–8074 RSC.
- M. Schulze, A. Steffen and F. Wurthner, Angew. Chem., Int. Ed. Engl., 2015, 54, 1570–1573 CrossRef CAS PubMed.
- Z. Zhao, X. Zhang, C. E. Li and T. Chen, Biomaterials, 2019, 192, 579–589 CrossRef CAS PubMed.
- Y. Wu, R. M. Young, M. Frasconi, S. T. Schneebeli, P. Spenst, D. M. Gardner, K. E. Brown, F. Würthner, J. F. Stoddart and M. R. Wasielewski, J. Am. Chem. Soc., 2015, 137, 13236–13239 CrossRef CAS PubMed.
- F. Schlosser, J. Sung, P. Kim, D. Kim and F. Würthner, Chem. Sci., 2012, 3, 2778–2785 RSC.
- S. Peter and F. Würthner, J. Photochem. Photobiol., C, 2017, 31, 114–138 CrossRef.
- M. M. Sun, W. Y. Yin, X. H. Dong, W. T. Yang, Y. L. Zhao and M. Z. Yin, Nanoscale, 2016, 8, 5302–5312 RSC.
- S.-Y. Yu and H.-L. Lu, Isr. J. Chem., 2019, 59, 166–183 CrossRef CAS.
- G. L. Zhang, L. P. Zhou, D. Q. Yuan and Q. F. Sun, Angew. Chem., Int. Ed. Engl., 2015, 54, 9844–9848 CrossRef CAS PubMed.
- L. Li, Y.-J. Hong, D.-Y. Chen and M.-J. Lin, Chem.–Eur. J., 2017, 23, 16612–16620 CrossRef CAS PubMed.
- H. Ke, L.-J. Weng, S.-Y. Chen, J.-Z. Chen and M.-J. Lin, Dyes Pigm., 2015, 113, 318–324 CrossRef CAS.
- Z.-M. Chen, Y. Cui, X.-F. Jiang, J. Tong and S.-Y. Yu, Chem. Commun., 2017, 53, 4238–4241 RSC.
- H. X. Gong, Z. Cao, M. H. Li, S. H. Liao and M. J. Lin, Org. Chem. Front., 2018, 5, 2296–2302 RSC.
- R. M. Pinto, E. M. Macoas, A. I. Neves, S. Raja, C. Baleizao, I. C. Santos and H. Alves, J. Am. Chem. Soc., 2015, 137, 7104–7110 CrossRef CAS PubMed.
- M. J. Lin, M. Schulze, K. Radacki and F. Wurthner, Chem. Commun., 2013, 49, 9107–9109 RSC.
- K. Nagarajan, A. R. Mallia, K. Muraleedharan and M. Hariharan, Chem. Sci., 2017, 8, 1776–1782 RSC.
- K. Nagarajan, A. R. Mallia, V. S. Reddy and M. Hariharan, J. Phys. Chem. C, 2016, 120, 8443–8450 CrossRef CAS.
- R. H. Young, K. Wehrly and R. L. Martin, J. Am. Chem. Soc., 1971, 93, 5774–5779 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1893884. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc02236e |
| ‡ Lizhen He and Li-Xuan Cai contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |