
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegiocontrol in the oxidative Heck reaction of indole by ligand-enabled switch of the regioselectivity-determining step†
Yu-Jie
Wang
,
Chen-Hui
Yuan
,
De-Zhao
Chu
and
Lei
Jiao
 *
*
Center of Basic Molecular Science (CBMS), Department of Chemistry, Tsinghua University, Beijing 10084, China. E-mail: leijiao@mail.tsinghua.edu.cn
First published on 23rd September 2020
Abstract
Efficient control of regioselectivity is a key concern in transition-metal-catalyzed direct C–H functionalization reactions. Various strategies for regiocontrol have been established by tuning the selectivity of the C–H activation step as a common mode. Herein, we present our study on an alternative mode of regiocontrol, in which the selectivity of the C–H activation step is no longer a key concern. We found that, in a reaction where the C–H activation step exhibits a different regio-preference from the subsequent functionalization step, a ligand-enabled switch of the regioselectivity-determining step could provide efficient regiocontrol. This mode has been exemplified by the Pd(II)-catalyzed aerobic oxidative Heck reaction of indoles, in which a ligand-controlled C3-/C2-selectivity was achieved for the first time by the development of sulfoxide-2-hydroxypyridine (SOHP) ligands.
1. Introduction
Control of regioselectivity is a key concern in organic synthesis when the substrate bears more than one potential reactive site. The transition-metal-catalyzed C–H functionalization reactions represent an appealing approach to molecular modification,1 in which the substrates always contain more than one reactive C–H bonds, and efficient regiocontrol is of great significance. A variety of classical strategies have been developed to achieve this goal, generally focusing on the C–H activation step (Scheme 1a). For instance, in the C–H functionalization reactions of (hetero)arenes, a directing group (DG) strategy (for ortho-functionalization)2 and a template strategy (for remote functionalization)3 have been widely used for regiocontrol. For arenes without a suitable DG, many C–H functionalization reactions were reported to occur at the site prone to C–H activation (referred to as intrinsic regiocontrol).4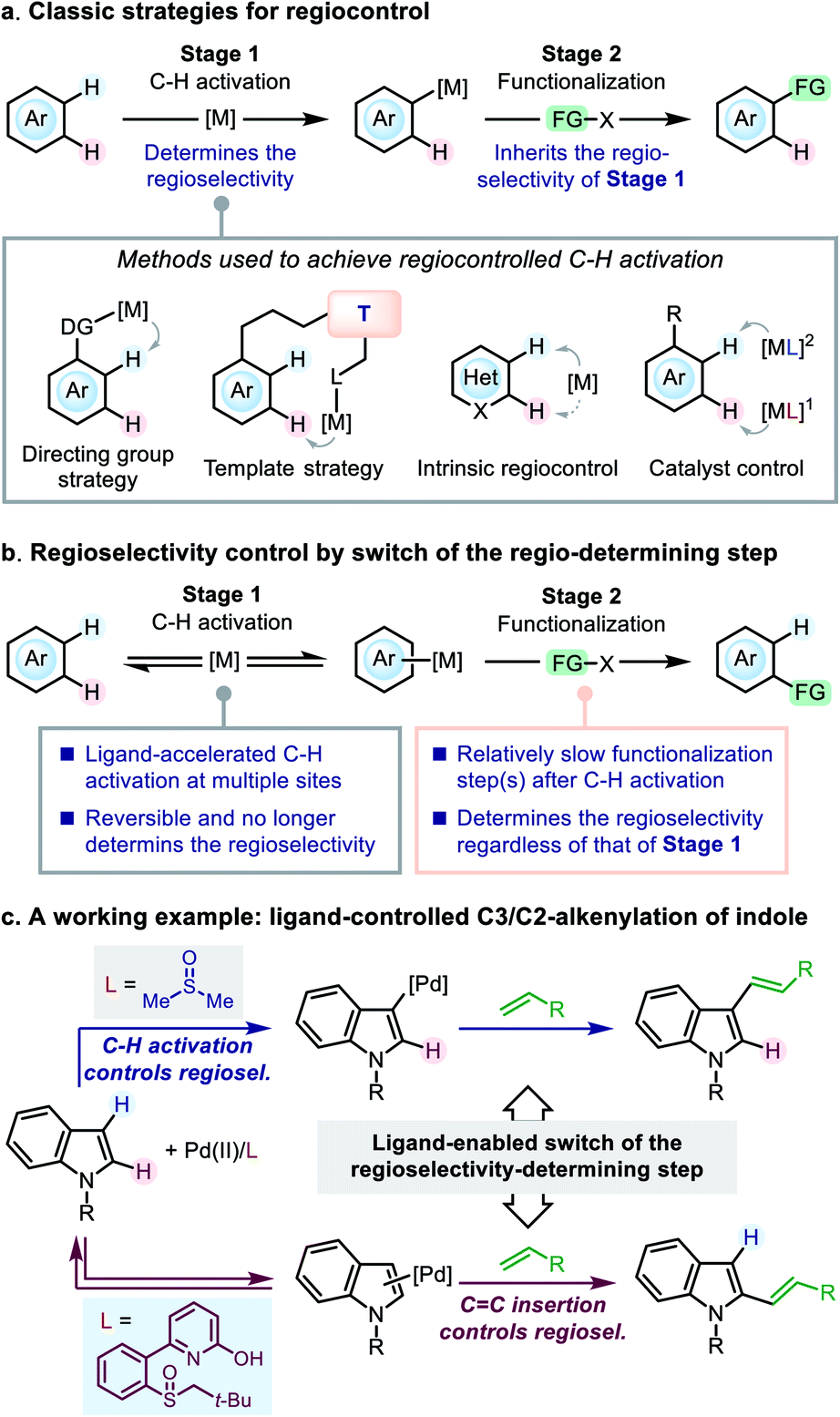 | ||
| Scheme 1 Regiocontrol in transition-metal-catalyzed C–H functionalization. | ||
Catalyst control is another useful strategy for tuning the regioselectivity. In particular, ligand development in the transition-metal-catalyzed C–H functionalization reactions has attracted significant attention in recent years,5–7 and ligand-enabled regiocontrol has emerged as a promising strategy. Representative advances in this direction arose from the field of Pd(II) catalysis,6 in which a few examples have been reported to achieve ligand-dictated regioselectivity in the C–H functionalization of arenes: Itami and co-workers reported ligand-controlled α-/β-arylation of thiophenes;8 the Yu group achieved regioselectivity control in the C–H alkenylation of arenes by using mono-protected amino acid (MPAA) ligands;6d the Stahl group utilized 4,5-diazafluorene (DAF) ligands to switch the C3-/C2-selectivity in the arylation of indole;6a the Sanford group demonstrated the role of pyridine as a ligand to dictate the regioselectivity of chloroarene acetoxylation;6k Carrow and co-workers developed a thioether-based ligand to tune the regioselectivity of the thiophene alkenylation.6y These studies highlighted the concept of ligand control in the C–H activation step; however, to date the principle of ligand design toward a desired regioselectivity on a given substrate remains ambiguous.
Herein, we present our study on an alternative mode of ligand-dictated regiocontrol, in which the selectivity of the C–H activation step is no longer a key concern (Scheme 1b). We envisioned that, when the functionalization step exhibits a different regio-preference from the C–H activation step, regiocontrol could be achieved by simply switching the regioselectivity-determining step of the reaction (i.e., the switch from scenario a to b described in Scheme 1), and suitably designed ancillary ligands could do this job by accelerating C–H metalation at multiple sites to make the C–H activation step reversible and no longer regioselectivity-determining. Although the principle is simple, such an example was rare in the literature and the development of such ligands remained challenging.9
In the present work, we take the Pd(II)-catalyzed oxidative C–H alkenylation reaction of indole derivatives as a working example to demonstrate this mode of regiocontrol (Scheme 1c). Owing to the development of a novel sulfoxide-2-hydroxypyridine (SOHP) ligand, the ligand-controlled C3-/C2-selectivity in this reaction was achieved for the first time by the switch of the regioselectivity-determining step. The in-depth mechanistic study has disclosed a clear picture of this control mode, which may provide inspirational insights into the ligand design and regiocontrol in related catalytic systems.
2. Results and discussion
The indole motif is the key substructure of many natural products, pharmacophores, and synthetic building blocks, and the methods for regioselective direct C–H functionalization of indole are highly demanded. Indole has long been employed as a typical substrate in various Pd(II)-catalyzed C–H functionalization reactions,10,11 and among them the oxidative C–H alkenylation (the oxidative Mizoroki–Heck reaction)12 of indole was selected as a suitable model reaction for the present study, because it has a well-established mechanistic picture, while the ligand-enabled regioselectivity control is unprecedented to date.It was established that the Pd(II)-catalyzed C–H alkenylation of indole took place at the C3-position with a high regioselectivity, which was attributed to a regioselectivity-determining C–H palladation step that preferentially occurs at the C3-position.13 The intrinsic C3-selectivity makes the selective C2-alkenylation of indole a difficult yet highly demanding task. While a number of protocols have been developed to achieve N1-, C2-, or C3-selective C–H arylation reaction of indole,10c,14 there are only a few methods to switch the site-selectivity of indole alkenylation from C3 to C2, such as the DG strategy15 or a solvent- or oxidant-controlled regioselectivity.13e,16 A ligand-enabled regiocontrol has not been worked out for this reaction. Aiming at developing a suitable catalytic system that allows for a ligand-controlled C3-/C2-alkenylation reaction of indole, we set out to investigate the origin of the intrinsic C3-alkenylation selectivity in the model reaction.
2.1 Understanding the intrinsic regioselectivity of the oxidative Heck reaction of indole
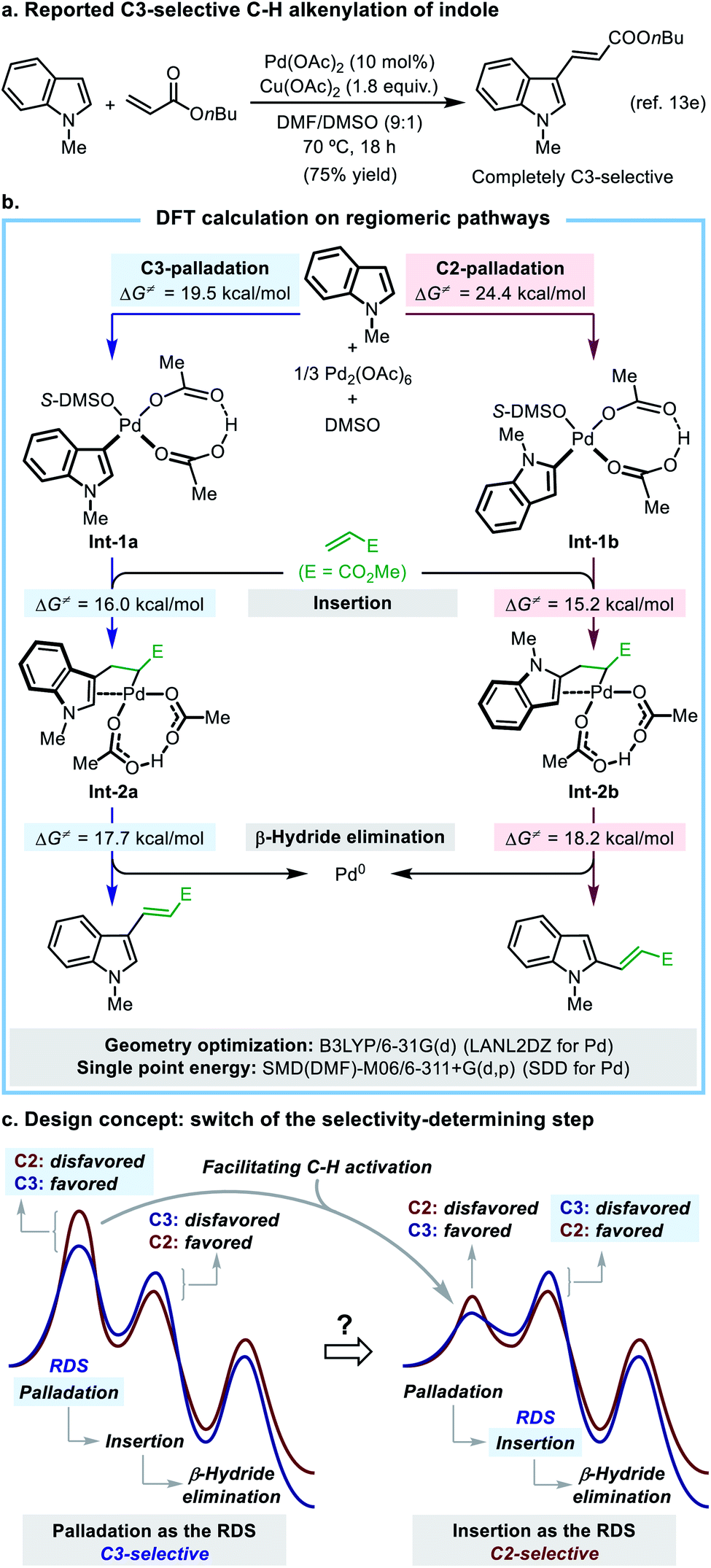 | ||
| Scheme 2 Understanding the origin of regioselectivity in the model reaction. | ||
In this mechanistic scheme, each regiomeric pathway is composed of three crucial steps including palladation, insertion, and β-hydride elimination (for full potential energy surfaces, see Fig. S12†). Palladation is an irreversible rate-determining step (RDS) in both pathways, which determines the overall regioselectivity of the reaction. The C3-palladation has a lower activation energy barrier (19.5 kcal mol−1) than that of the C2-palladation (24.4 kcal mol−1), which well accounts for the observed C3-selectivity. A simplified potential energy surface (Scheme 2c, left side) clearly illustrates the mode of regioselectivity control in this reaction. The key finding of the DFT study is that the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C insertion step after palladation exhibits a reversed regio-preference, with the C2-insertion (ΔG‡ = 15.2 kcal mol−1) being kinetically more favorable than the C3-insertion (ΔG‡ = 16.0 kcal mol−1). This is an important piece of mechanistic information that has not been disclosed in previous studies.
C insertion step after palladation exhibits a reversed regio-preference, with the C2-insertion (ΔG‡ = 15.2 kcal mol−1) being kinetically more favorable than the C3-insertion (ΔG‡ = 16.0 kcal mol−1). This is an important piece of mechanistic information that has not been disclosed in previous studies.
2.2 Ligand design and preliminary studies
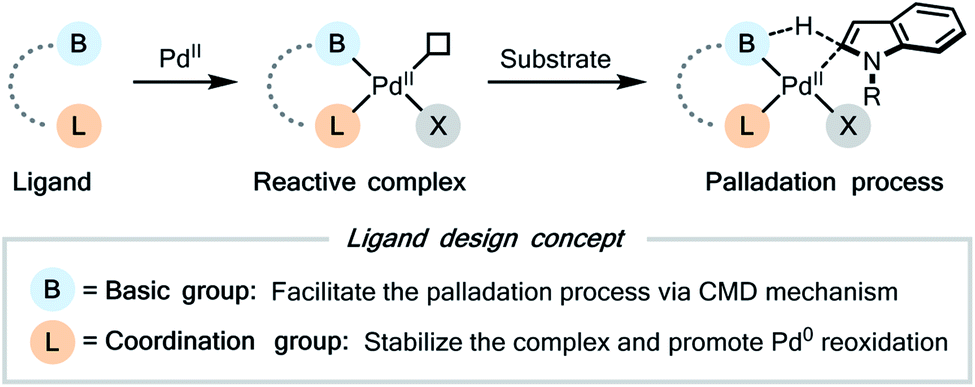 | ||
| Fig. 1 Ligand design. | ||
Then we further considered the detailed structure of the possible ligands. Both the acylamide (RCONH) group6c–g and the 2-hydroxypyridine (2-pyridone) group19 were known as efficient proton shuttle in the previous ligand design, thus we chose them as the potential basic group of the ligand. For the coordination group, a nitrogen- or oxygen-centered coordination site, such as pyridine, oxazoline, and carboxylic acid, was a typical choice. For the tether connecting two subunits of the ligand, either an alkyl chain or an aryl ring might be suitable.
We first searched in the established library of ligands that have been demonstrated to be efficient in Pd(II)-catalyzed C–H functionalization reactions. Among them, MPAAs match the above design, thus Boc-Ile-OH (L1) and Boc-Leu-OH (L2) were picked out as the representative members. DAF ligand L3, which was proved effective in promoting the C2-selective arylation of indole,6a was also selected. 6-Phenyl-2-pyridinol (L4) and 3-trifluoromethyl-2-pyridinol (L5)20 were chosen to evaluate the effect of the 2-hydroxypyridine motif alone. Finally, we added [2,2′-bipyridin]-6(1H)-one (bipy-6-OH, L6), a ligand that was reported to remarkably accelerate the CMD process in palladium catalysis.21 We intended to test the performance of these candidate ligands in the model oxidative Heck reaction of indole, with the hope that some of them could exhibit the desired C2-selectivity.
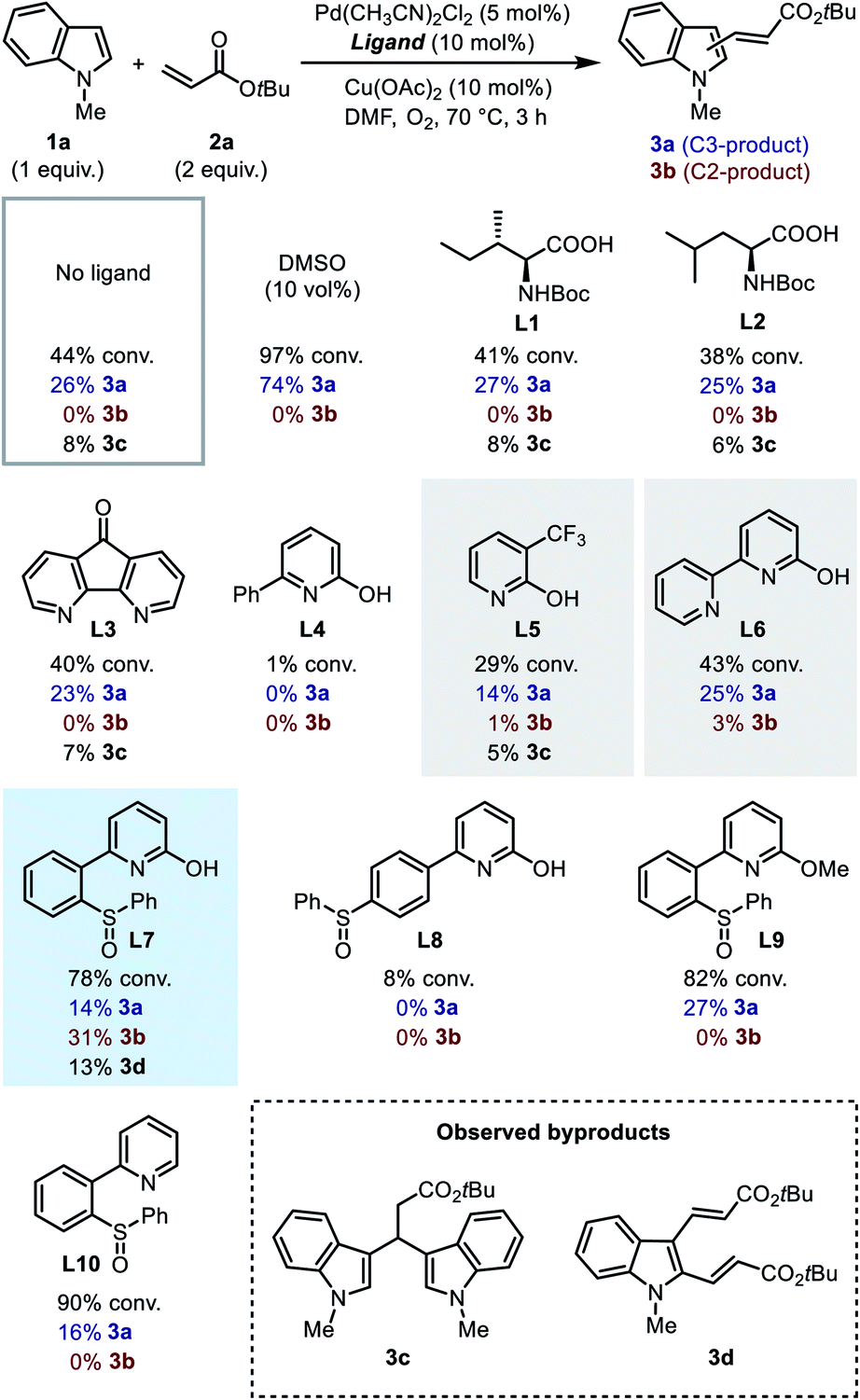 | ||
| Scheme 3 Ligand effects on the model reaction. Conversions were determined by GC analysis, and yields were determined by 1H NMR analysis. | ||
It was found that without a ligand the model reaction reached a moderate conversion to afford a low yield of C3-alkenylation product 3a, and palladium black formation was observed. With the addition of 10 vol% dimethylsulfoxide (DMSO) as a supporting ligand to prevent the aggregation of Pd(0), both the conversion of 1a and the yield of 3a increased dramatically, and the reaction remained completely C3-selective. To our disappointment, MPAA ligands L1 and L2 did not show an obvious promotive effect compared with the ligand-free conditions, nor did they alter the regioselectivity. In these reactions a minor amount of byproduct 3c resulting from Michael addition of indole to the C3-product 3a was obtained. DAF ligand L3 produced a similar result. For 2-hydroxypyridine-based ligands, ligand L4 had an inhibitory effect on the reaction, and both ligands L5 and L6 exhibited a low C3-reactivity. Interestingly, in the reactions with ligands L5 and L6, a trace amount of the C2-product was observed in the crude 1H NMR (<3%), which was never observed with other ligands. This observation indicated that the 2-hydroxypyridone motif might indeed be a key element in the ligand. Despite this, in general the established ligand library is not competent for regioselective C2-alkenylation of indole, which demonstrated the challenge for ligand design.
In order to probe the role of the two subunits in the SOHP ligand, partially modified ligands L8–L10 were tested (Scheme 3). When the sulfoxide moiety was moved from the ortho- to the para-position (L8), the catalytic activity lost completely, which supports the role of the sulfoxide as a coordination group rather than merely an electronic substituent. Blocking of the hydroxyl group by a methyl group (L9) or removal of the hydroxyl group (L10) in the ligand led to loss of C2-selectivity while maintaining a diminished reactivity. These results emphasized the important role of both 2-hydroxypyridine and sulfoxide subunits in dictating the C2-selectivity, as well as the cooperative effect of these two subunits.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of indole to acrylate were suitable reaction conditions for the desired C2-alkenylation (entries 13–16).
:1 ratio of indole to acrylate were suitable reaction conditions for the desired C2-alkenylation (entries 13–16).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Ligand | Conv. | Yields | |||
| 3a | 3b | 3d | Overall | |||
| a Conversions were determined by GC analysis, and yields were determined by 1H NMR analysis. b 10 vol% of DMSO was added. c 50 mol% of Cu2(OH)2CO3 was used instead of Cu(OTf)2. d 10 mol% PdCl2(CH3CN)2 was employed. e 2 equiv. of 1a and 1 equiv. of 2a were used, and the yields were calculated based on the amount of acrylate. f Reaction was performed at 50 °C for 12 h. | ||||||
| 1 | L7 | 86% | 12% | 41% | 10% | 63% |
| 2 | L11 | 42% | — | 9% | — | 9% |
| 3 | L12 | 81% | 10% | 42% | 10% | 62% |
| 4 | L13 | 23% | 1% | 13% | — | 14% |
| 5 | L14 | 34% | 4% | 24% | — | 28% |
| 6 | L15 | 43% | 6% | 26% | 2% | 34% |
| 7 | L16 | 59% | 4% | 34% | 2% | 40% |
| 8 | L17 | 83% | 6% | 46% | 6% | 58% |
| 9 | L18 | 65% | 6% | 40% | 5% | 51% |
| 10 | L19 | 36% | — | 17% | — | 17% |
| 11 | L20 | 82% | 10% | 37% | 4% | 51% |
| 12b | L17 | 53% | 12% | 14% | — | 26% |
| 13c | L17 | 58% | 10% | 43% | 4% | 54% |
| 14c,d | L17 | 96% | 6% | 50% | 15% | 71% |
| 15c,d,e | L17 | — | 7% | 46% | 12% | 65% |
| 16 , , , | L17 | — | 5% | 63% | 8% | 76% |
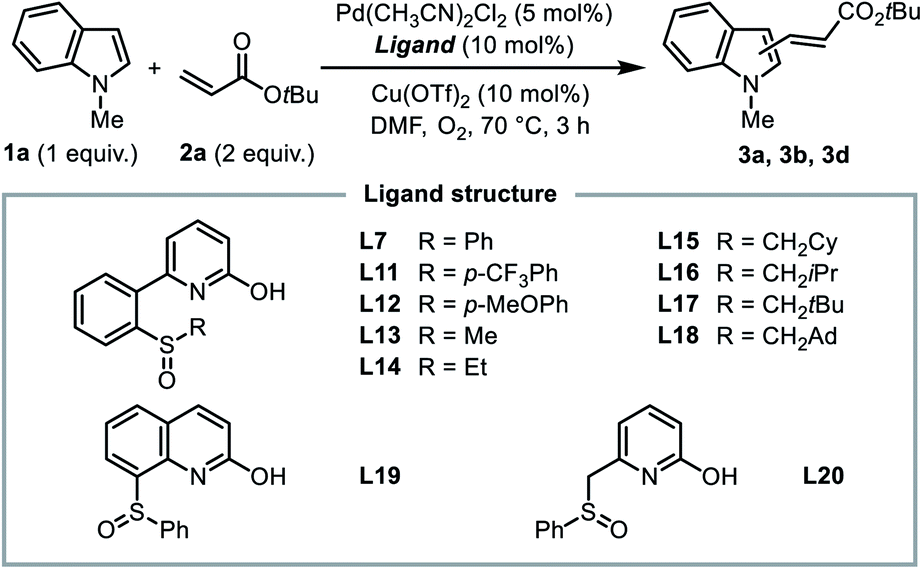
At this stage, we had identified a new ligand design that enables the unusual C2-selectivity in the oxidative Heck reaction of indole based on the concept of ligand-controlled switch of the rate-determining step. Our next goal was to draw a clear mechanistic picture of this control mode by a detailed mechanistic study.
2.3 Mechanistic studies
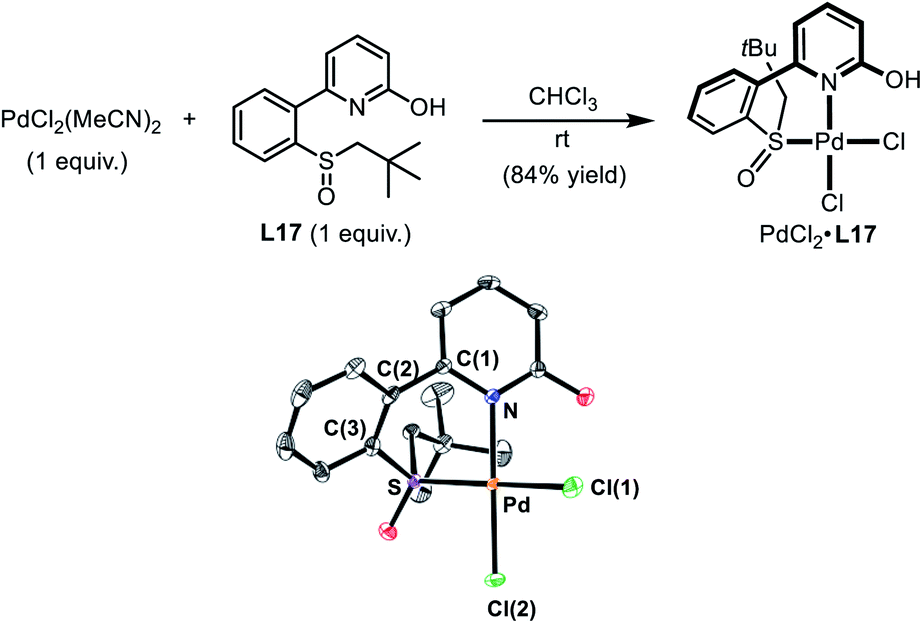 | ||
| Scheme 4 Preparation of the Pd(II)–SOHP Complex. In the ORTEP plot of PdCl2·L17 (CCDC 1978349), thermal ellipsoids are drawn at 50% probability level. The co-crystallized CHCl3 and all H atoms are omitted for clarity. Selected distances (Å) and a dihedral angle (deg): Pd–N 2.079(3), Pd–S 2.226(1), Pd–Cl(1) 2.324(1), Pd–Cl(2) 2.277 (1), and N–C(1)-C(2)-C(3) 44.9(5). | ||
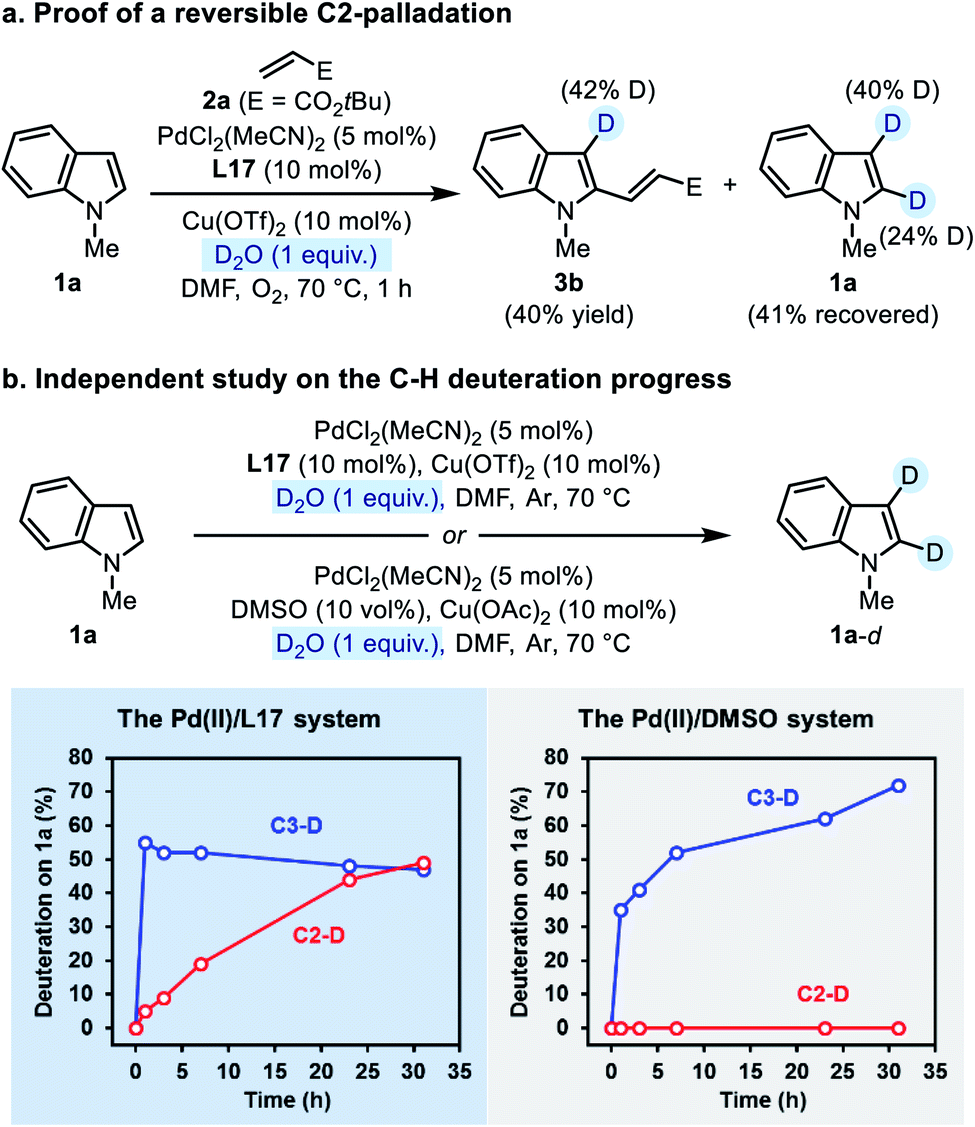 | ||
| Scheme 5 Probing the C2-palladation by deuteration. | ||
Then we performed an independent study on the deuteration of N-methylindole by running the reaction in the absence of 2a. The progress of both C3- and C2-deuteration was monitored as an indicator of C–H palladation at the corresponding site. In the Pd(II)/L17 system, the C3-deuteration occurred more rapidly than the C2-deuteration, but finally deuterium incorporation at both sites reached the same level (Scheme 5b, the left plot). In contrast, in the Pd(II)/DMSO system (the established C3-alkenylation conditions), only C3-deuteration was observed, which occurred slower than that in the Pd(II)/L17 system (Scheme 5b, the right plot). The contrast made clear that, compared with the Pd(II)/DMSO system, the Pd(II)/L17 system accelerates the C–H palladation process to make both C3- and C2-palladation kinetically accessible, while the regio-preference of the C–H activation step remains unchanged.
We noticed that the C2-deuteration of indole 1a proceeded more rapidly in the ligand-promoted oxidative Heck reaction than in the independent deuterium labeling experiment with Pd(II)/L17. Although a rigorous rationalization of this phenomenon has not been worked out at present, we tentatively attributed this difference to the different basicity of the two reaction systems.24 Nevertheless, the results from the deuterium labeling experiments revealed a reversible palladation at both C3- and C2-positions of indole enabled by the SOHP ligand, which confirmed our design concept that the ligand effect might render the C–H activation step reversible and no longer regioselectivity-determining.
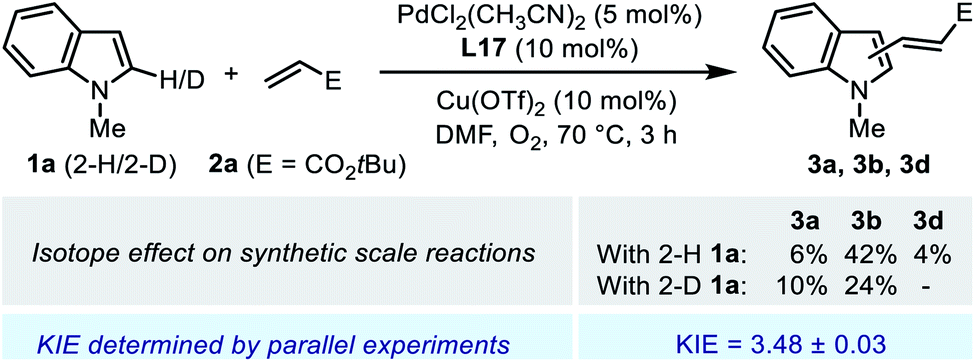 | ||
| Scheme 6 Study of isotope effect on C2-alkenylation. | ||
Parallel initial rate measurements were performed by employing 2-H and 2-D indole substrate 1a to determine the apparent kinetic isotope effect (KIE) of the reaction. The determined KIE of ca. 3.5 was consistent with the observation in the synthetic scale reaction (Scheme 6), and these results indicated a significant primary KIE for the C–H cleavage step. Normally, such a remarkable isotope effect indicates a rate-limiting irreversible C–H activation. However, it seemed contradictory to the reversible nature of the C–H palladation step revealed by the deuterium labeling experiment (Scheme 5a). We reasoned that, in the Pd(II)/SOHP reaction system, the activation energy barriers of the C–H palladation step and its following step were comparable to each other, thus the reaction could exhibit both a reversible C–H activation step and a significant apparent primary KIE, and this could be referred to as a quasi-reversible C–H activation. Interestingly, a similar circumstance has also been observed in the Pd(II)-catalyzed alkenylation reaction of thiophene derivatives, where a significant primary KIE was observed while the C–H activation step remained reversible.25 To figure out the rate-limiting step of the overall reaction, further experimental study was required.
C insertion step following C–H palladation is rate-limiting. The initial rate of the model reaction promoted by the Pd(II)/L17 system was measured with different acrylate concentrations (Scheme 7, the left plot). It was found that the C2-alkenylation reaction exhibited a clear positive dependence on [acrylate] within the range of 0.2–1.2 M, which seemed to follow a saturation kinetics. This implied that the migratory CC insertion step after C–H palladation played an important role in determining the rate of the reaction. Given the quasi-reversible C–H activation step revealed previously, we reasoned that this saturation-like kinetics did not imply a saturated pre-coordination equilibrium as usual, but instead reflected the result of the similar activation energy barriers of the C–H palladation step and the following insertion step. Taking these results together, it could be concluded that none of the two steps was a clear-cut rate-limiting step in the catalytic cycle, and both steps were important in determining the rate of the overall reaction.
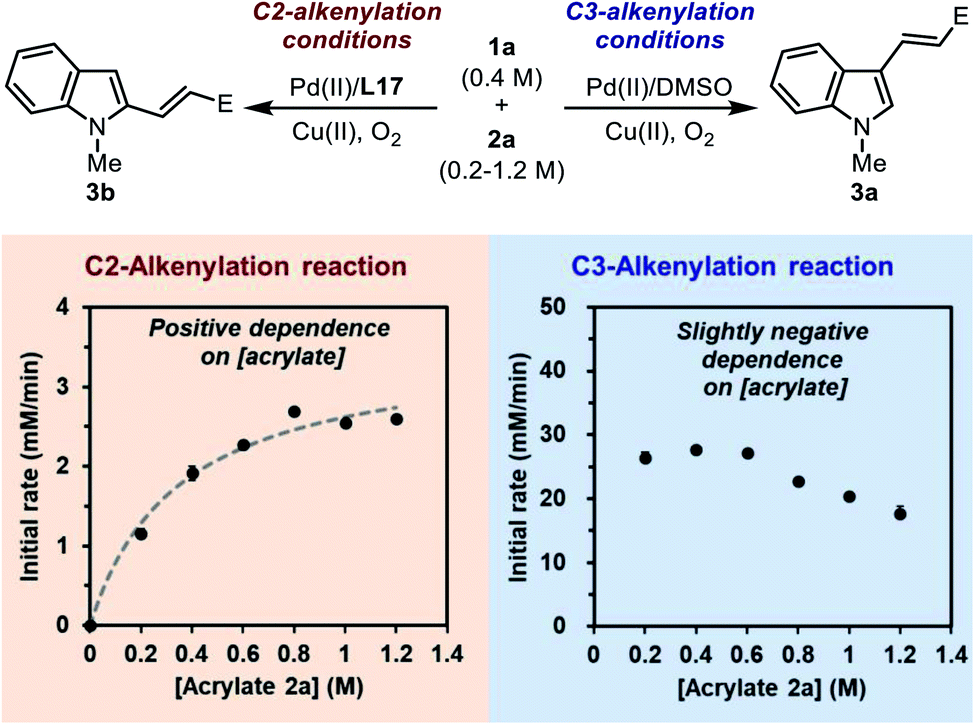 | ||
| Scheme 7 Effect of acrylate loading on the reaction. | ||
For comparison, the dependence of the initial rate on [acrylate] in the C3-alkenylation reaction catalyzed by the Pd(II)/DMSO system was also studied (Scheme 7, the right plot). Interestingly, a distinct dependence profile over the same range of [acrylate] was obtained, in which the initial rate was zero-order at low [acrylate] and gradually exhibited a negative-order at high [acrylate]. This result was consistent with a rate-limiting C–H activation step in the normal C3-alkenylation reaction of indole revealed by the DFT calculation (Scheme 2b). The negative order observed at high [acrylate] might be attributed to the competitive coordination of the acrylate substrate to the catalytic center that led to off-cycle species.
The results shown above manifested a change of the reaction profile from an irreversible rate-limiting C–H palladation (in the Pd(II)/DMSO system) to a complex rate-determining scenario with a quasi-reversible C–H palladation (in the Pd(II)/SOHP system). This supported our proposal that the ligand effect was able to switch the mode of rate- and selectivity-control in the catalytic cycle. The reversibility of the C–H palladation step implied that the CC migratory insertion step might play an important role in determining the regioselectivity, which was to be clarified by further studies.
To this end, two stoichiometric reactions between N-methylindole and norbornene were performed with both Pd(II)/DMSO (the C3-alkenylation conditions) and Pd(II)/L17 systems (the C2-alkenylation conditions). To our delight, the reaction could indeed stop after the insertion step, and upon treatment with NaBH4, the insertion intermediates afforded the corresponding norborylindole products (Scheme 8). It was found that, without the SOHP ligand, this reaction sequence only produced 3-norborylindole 5a, supporting an exclusive C3-palladation/insertion. In contrast, with the SOHP ligand, 2-norborylindole 5b was obtained as the major product together with a minor amount of 5a (C2:C3 = 3:1). Furthermore, if the crude C2-insertion intermediate formed in the Pd(II)/L17 system was treated with pyridinium hydrochloride (for anion exchange) and then triphenylphosphine (for coordination), the PPh3-coordinated C2-insertion complex 4 could be isolated in pure form by column chromatography, whose structure was confirmed by single crystal XRD analysis (Scheme 8).
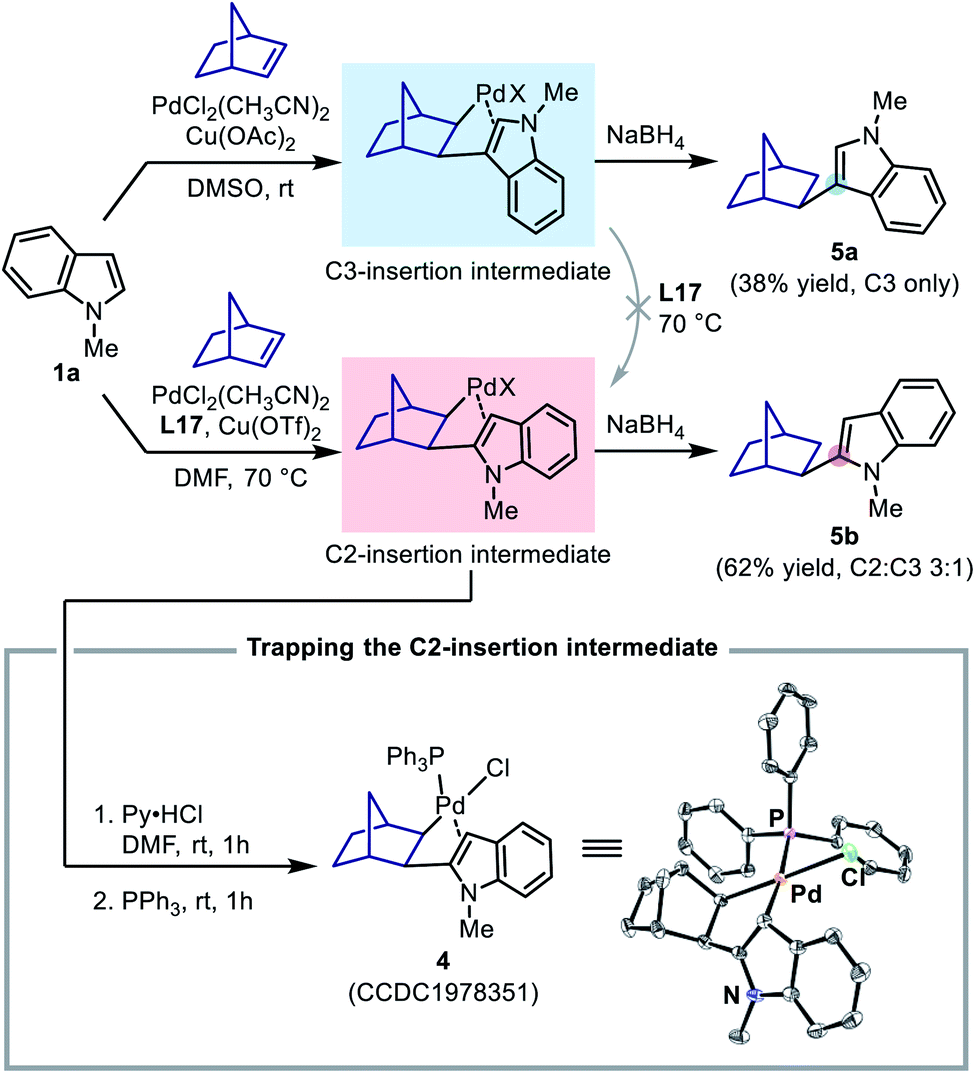 | ||
| Scheme 8 Study on the regioselectivity of the alkene insertion step. In the ORTEP plot of 4, thermal ellipsoids are drawn at the 50% probability level. The co-crystallized CH2Cl2 and all H atoms are omitted for clarity. | ||
The formation of the C2-insertion intermediate demonstrated the crucial role of the SOHP ligand in enabling the C2-palladation/C2-insertion pathway. However, because the insertion of norbornene into a C–Pd bond was found to be reversible in some cases, the formation of the C2-insertion intermediate could be rationalized either as a result of selective C2-insertion (kinetic control) or as an outcome of the equilibrium between insertion products (thermodynamic control). We found that, upon addition of stoichiometric ligand L17 to the generated C3-insertion intermediate and treatment at 70 °C, the C3-insertion intermediate remained intact and could not be transformed to its C2-counterpart, suggesting that the equilibrium between the regiomeric insertion products in the presence of the SOHP ligand was not viable. Therefore, the above results with norbornene served as a direct evidence for the C2-insertion process promoted by the SOHP ligand, and corroborated a regioselectivity-determining insertion step in the catalytic cycle.
C migratory insertion step played an important role; (3) the alkene insertion step exhibits a C2-selectivity and determines the overall regioselectivity.
In order to gain further details of the reaction mechanism, we performed a DFT computational study on the model reaction between 1a and 2b promoted by using the Pd(II)/SOHP ligand/Cu(OTf)2 system (Fig. 2).17 Methyl SOHP L13 was selected as a representative ligand for computational modeling to save computation time, because L13 exhibited similar C2-selectivity in the model reaction compared with the optimal ligand L17, though with an inferior reactivity.
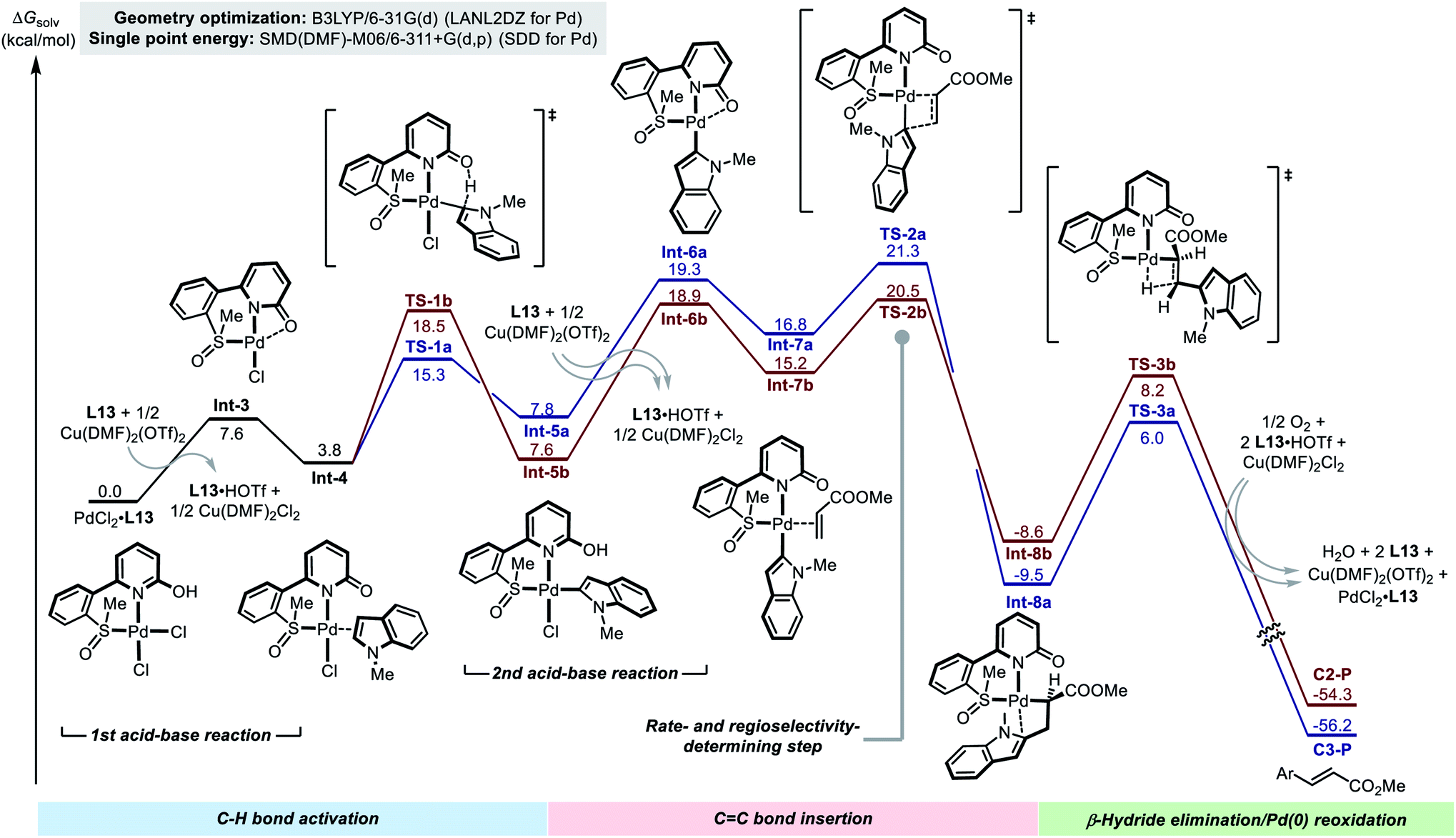 | ||
| Fig. 2 The potential energy surface of the C–H alkenylation reaction of indole 1a. Both C2- (red) and C3-alkenylation (blue) pathways are included, but only the intermediates and transition states in the C2-alkenylation pathway are shown for clarity. | ||
The reaction starts from the Pd(II)–ligand complex PdCl2·L13. In order to accommodate the indole substrate, a chloride anion should leave the Pd(II) center to make a vacant coordination site. This could be achieved by elimination of HCl from the complex to form Int-3, which is highly endergonic (ΔG > 20 kcal mol−1) according to the computational result (see the ESI† for details). We found that, when the HCl elimination process is coupled with the trapping of protons by the free SOHP ligand and the coordination of Cl− to Cu(II), its thermodynamic barrier becomes reasonable (ΔG = 7.6 kcal mol−1). Although there might still be some error in the calculated free energy of the deprotonation step due to the complexity of the reaction system, it was clear that the acid–base interaction facilitated the formation of Int-3 from the initial PdCl2–SOHP complex. In Int-3, the SOHP ligand adopts a κ3-S,N,O coordination mode, which brings about additional ring strain. The following substrate coordination releases this ring strain, affording complex Int-4.
Int-4 is the bifurcating point of the potential energy surface, from which two C–H activation transition states (TSs) evolve and lead to the C3- and C2-alkenylation pathways, respectively. In both C3- and C2-palladation transition states TS-1a and TS-1b, the 2-hydroxypyridine moiety in the SOHP ligand acts as an internal base to abstract a proton from the indole C–H bond. This CMD mechanism greatly facilitated the C–H palladation on both C3- and C2-sites of indole, as compared with the corresponding palladation pathways in the ligand-free system (Scheme 2b). As revealed by DFT calculation, the C3-palladation (ΔG‡ = 15.3 kcal mol−1) is still favored over C2-palladation (ΔG‡ = 18.5 kcal mol−1), which indicates that the palladation step remains C3-selective, as the experimental studies have indicated (Scheme 5b).
The C–H palladation steps lead to the formation of C3- and C2-Pd indole species Int-5a and Int-5b. Afterwards, another HCl elimination process has to occur to generate Int-6a and Int-6b, which allow for acrylate coordination and alkene insertion. These acid–base reactions were also found to be endergonic (ΔG > 10 kcal mol−1 for both pathways), even if the assistance of the base and the Cu(II) salt was considered. The alkene coordination occurs between these indolylpalladium species and the acrylate substrate to form Int-7a and Int-7b, and then migratory insertion of the C–Pd bond to the CC bond takes place viaTS-2a and TS-2b. Notably, similar to the “ligand-free” alkenylation reaction (Scheme 2), the insertion step in the ligand-promoted reaction also exhibits a C2-selectivity. In the C2-alkenylation pathway, the insertion step has the highest activation barrier, but the activation energy barrier of the C–H palladation step is close (TS1bvs.TS2b), which is in agreement with the conclusions drawn from the experimental study. The β-hydride elimination steps are facile in both pathways (viaTS-3a and TS-3b) and do not affect the rate or the regioselectivity.
The DFT calculation supported that the observed C2-alkenylation selectivity in the ligand-promoted reaction was a combined result of the quasi-reversible C–H activation and the regioselectivity-determining alkene insertion, which significantly differs from the original reaction without the SOHP ligand (Fig. 2vs. Fig. S12†). This ligand-enabled switch of the regioselectivity-control mode confirmed our design concept. Computations also revealed that the Cu(II) salt employed in the Pd(II)/SOHP reaction system is not only a redox catalyst, but also plays an important role in promoting the reaction by assisting the elimination of HCl from the key Pd(II) species.
2.4 Scope of the ligand-enabled regioselective oxidative Heck reactions
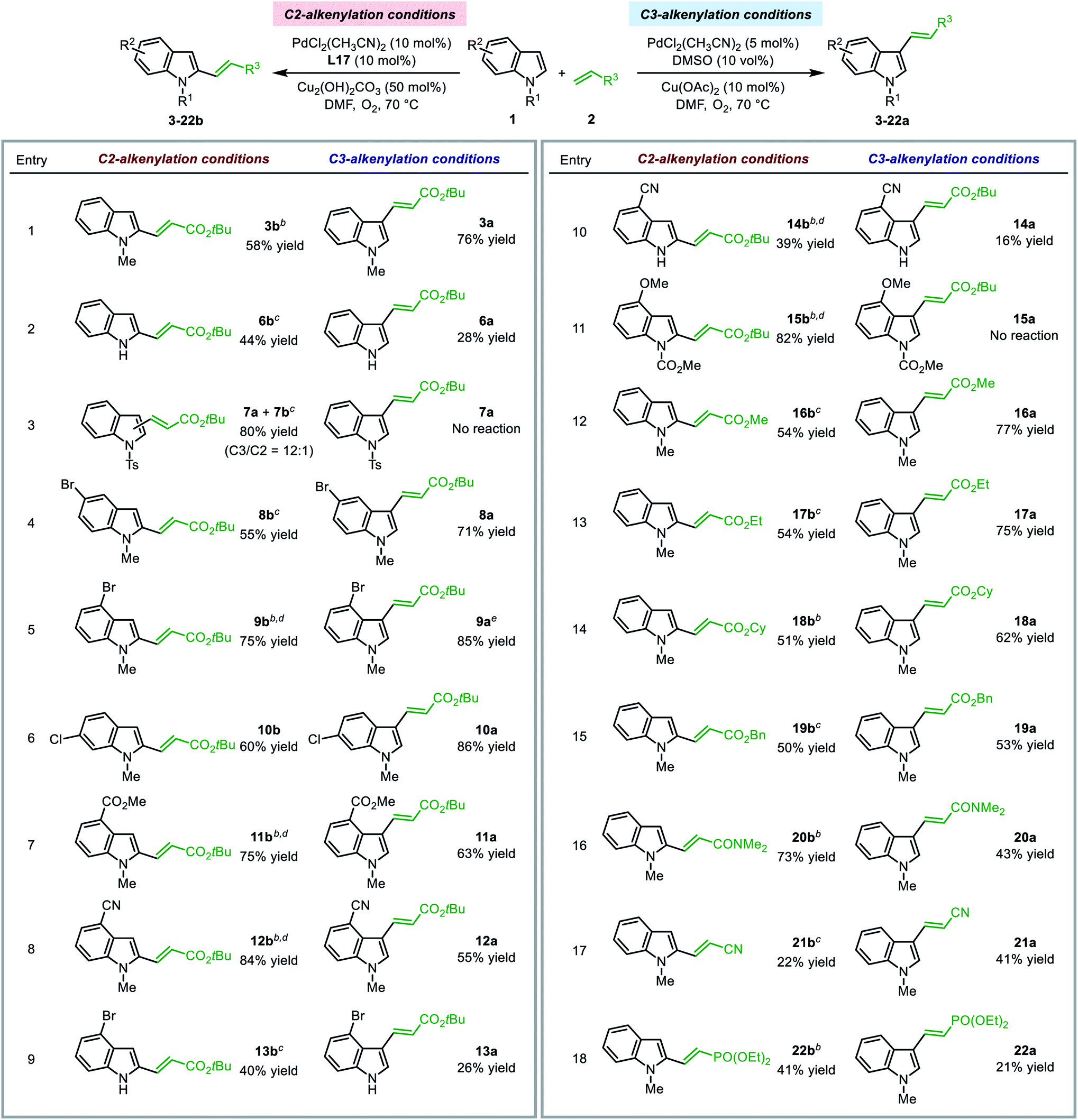
The optimal SOHP ligand enabled the regioselective synthesis of C2-alkenylated indole derivatives, while the established method utilizing DMSO as the ligand led to C3-alkenylation of indoles. The present method enriched the toolbox for regioselective functionalization of indoles under aerobic conditions. We performed the indole alkenylation reactions employing a variety of indole derivatives and electron deficient olefins under both conditions to explore the substrate scope.| a For C2-alkenylation reactions, 2 equiv. of 1 and 1 equiv. of 2 were used; for C3-alkenylation reactions, 1 equiv. of 1 and 2 equiv. of 2 were used. Yields of isolated products were reported (for C2-alkenylation reactions, only the C2-products were isolated). b Reaction was performed at 50 °C. c 5 mol% PdCl2(CH3CN)2 and 10 mol% Cu(OTf)2 was used instead of 10 mol% PdCl2(CH3CN)2 and 50 mol% Cu2(OH)2CO3. d 1 equiv. of 1 and 1.3 equiv. of 2 were used. e 10 mol% PdCl2(CH3CN)2 was used. |
|---|
|
Other electron-deficient olefins, such as methyl, ethyl, cyclopropyl, and benzyl acrylates, acrylonitrile, and diethyl vinylphosphonate, could be employed as the alkenylation reagent in the reactions to afford the C2- or C3-alkenylation products under regioselective alkenylation conditions (entries 13–18). Notably, for the substrates tested, no trace of the C2-product was observed under the traditional C3-alkenylation conditions, and C2-alkenylation could only be enabled by using the Pd(II)/SOHP system, which emphasized the key role of the designed ligand for C–H activation at the C2-position.
The limitation of the present C2-alkenylation reaction was that the olefin substrate should be electron-deficient and mono-substituted. Disubstituted olefins, such as crotonates and α-methyl acrylates, exhibited rather low reactivity and could not afford the alkenylation products in a reasonable yield. This might be caused by the high activation barrier of the alkene insertion step.
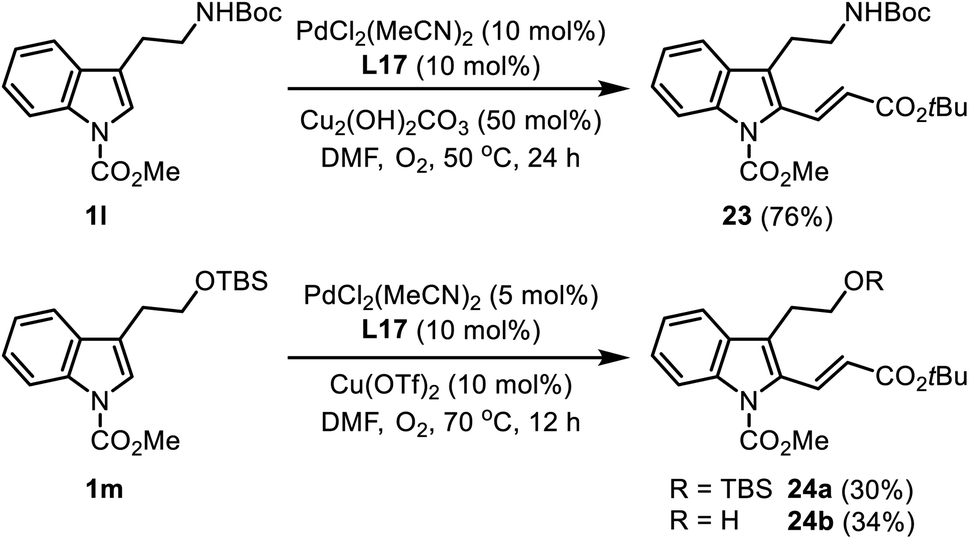 | ||
| Scheme 9 C2-alkenylation of tryptamine and tryptophol derivatives. | ||
3. Conclusion
In this work, we studied a new mode of regiocontrol in a transition-metal-catalyzed C–H functionalization reaction, which features a ligand-enabled switch of the regioselectivity-determining step. This mode has been demonstrated by the Pd(II)-catalyzed ligand-controlled C3/C2-selective aerobic oxidative Heck reaction of indole. A new type of ancillary ligand, sulfoxide-2-hydroxypyridine (SOHP), was developed based on a mechanistic understanding, which functioned as a key control factor in rendering the C2-selectivity of the reaction. This reaction represents a nice yet rare example of ligand-controlled regioselectivity in the direct C–H functionalization of heteroarenes.The mode of ligand-directed regioselectivity control in this reaction has been clearly elucidated through a detailed mechanistic study. It was found that the C–H activation and the CC insertion are two key steps in this oxidative Heck reaction, and under the traditional reaction conditions the C–H activation step with a C3-preference is rate- and regioselectivity-determining. The SOHP ligand facilitates C–H palladation by promoting proton abstraction via a CMD mechanism. As a result, the activation barriers of the C–H palladation step at both C3- and C2-sites are significantly decreased, rendering a quasi-reversible C–H activation step and a regioselectivity-determining CC insertion step. The reaction exhibits the regioselectivity determined by the insertion step, which is C2-selective. The present reaction system perfectly exemplified the ligand-enabled switch of regioselectivity-determining step as an efficient mode of regiocontrol.
The mechanistic information acquired in this work shed some light on the regioselectivity control in related reaction systems. It reminds chemists that for many C–H activation reactions, the regio-preferences of both the C–H metalation and the following functionalization steps should be considered with equal attention. In case there is a discrepancy, a rational ligand design that allows for a switch of regioselectivity-determining step between these two processes may open an avenue to a new ligand-controlled regioselective reaction. We hope that this mode of regiocontrol is extendable to more substrate types, and it could inspire the design of more regiocontrolled C–H functionalization reactions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The National Natural Science Foundation of China (Grant No. 21822304) is acknowledged for financial support. The technology platform of CBMS and the Tsinghua Xuetang Talents Program are acknowledged for providing instrumentation and computational resources.References
- For reviews, see: (a) J.-Q. Yu and Z. Shi, C−H Activation, Springer, 2010, vol. 292 Search PubMed; (b) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 Search PubMed; (c) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885–1898 Search PubMed; (d) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236–10254 Search PubMed; (e) S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936–946 Search PubMed; (f) P. H. Dixneuf and H. Doucet, C−H Bond Activation and Catalytic Functionalization I, Springer, 2016 Search PubMed; (g) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900–2936 Search PubMed; (h) J. Yamaguchi and K. Itami, Bull. Chem. Soc. Jpn., 2017, 90, 367–383 Search PubMed; (i) D. Wang, A. B. Weinstein, P. B. White and S. S. Stahl, Chem. Rev., 2018, 118, 2636–2679 Search PubMed; (j) P. Gandeepan, T. Muller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 Search PubMed.
- (a) M. Zhang, Y. Zhang, X. Jie, H. Zhao, G. Li and W. Su, Org. Chem. Front., 2014, 1, 843–895 Search PubMed; (b) Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107–1295 Search PubMed; (c) J. Maes and B. U. W. Maes, Adv. Heterocycl. Chem., 2016, 120, 137–194 Search PubMed; (d) G. Pototschnig, N. Maulide and M. Schnürch, Chem.–Eur. J., 2017, 23, 9206–9232 Search PubMed; (e) W. Ma, P. Gandeepan, J. Li and L. Ackermann, Org. Chem. Front., 2017, 4, 1435–1467 Search PubMed; (f) C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603–6743 Search PubMed.
- For selected examples of template-directed regioselective C–H functionalizations, see: (a) J. Spencer, B. Z. Chowdhry, A. I. Mallet, R. P. Rathnam, T. Adatia, A. Bashall and F. Rominger, Tetrahedron, 2008, 64, 6082–6089 Search PubMed; (b) D. Leow, G. Li, T.-S. Mei and J.-Q. Yu, Nature, 2012, 486, 518–522 Search PubMed; (c) L. Wan, N. Dastbaravardeh, G. Li and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 18056–18059 Search PubMed; (d) G. Yang, P. Lindovska, D. Zhu, J. Kim, P. Wang, R.-Y. Tang, M. Movassaghi and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 10807–10813 Search PubMed; (e) S. Bag, T. Patra, A. Modak, A. Deb, S. Maity, U. Dutta, A. Dey, R. Kancherla, A. Maji, A. Hazra, M. Bera and D. Maiti, J. Am. Chem. Soc., 2015, 137, 11888–11891 Search PubMed; (f) Z. Jin, L. Chu, Y.-Q. Chen and J.-Q. Yu, Org. Lett., 2018, 20, 425–428 Search PubMed.
- For selected examples of intrinsic regiocontrol in the C–H functionalization of heterocycles, see: (a) H. Choi, M. Min, Q. Peng, D. Kang, R. S. Paton and S. Hong, Chem. Sci., 2016, 7, 3900–3909 Search PubMed; (b) A. M. Prendergast and G. P. McGlacken, Eur. J. Org. Chem., 2018, 6068–6082 Search PubMed and references therein.
- For selected reviews, see: (a) K. M. Engle and J.-Q. Yu, J. Org. Chem., 2013, 78, 8927–8955 Search PubMed; (b) L. Ping, D. S. Chung, J. Bouffard and S. G. Lee, Chem. Soc. Rev., 2017, 46, 4299–4328 Search PubMed; (c) M. T. Mihai, G. R. Genov and R. J. Phipps, Chem. Soc. Rev., 2018, 47, 149–171 Search PubMed; (d) V. K. Tiwari and M. Kapur, Org. Biomol. Chem., 2019, 17, 1007–1026 Search PubMed.
- For selected examples of the ligand-promoted palladium-catalyzed C–H functionalization reactions, see: (a) A. N. Campbell, E. B. Meyer and S. S. Stahl, Chem. Commun., 2011, 47, 10257–10259 Search PubMed; (b) A. Vasseur, C. Laugel, D. Harakat, J. Muzart and J. Le Bras, Eur. J. Org. Chem., 2015, 944–948 Search PubMed; (c) B. F. Shi, N. Maugel, Y. H. Zhang and J. Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 4882–4886 Search PubMed; (d) D. H. Wang, K. M. Engle, B. F. Shi and J. Q. Yu, Science, 2010, 327, 315–319 Search PubMed; (e) L. Chu, K. J. Xiao and J. Q. Yu, Science, 2014, 346, 451–455 Search PubMed; (f) P. X. Shen, L. Hu, Q. Shao, K. Hong and J. Q. Yu, J. Am. Chem. Soc., 2018, 140, 6545–6549 Search PubMed; (g) Q.-F. Wu, X.-B. Wang, P.-X. Shen and J.-Q. Yu, ACS Catal., 2018, 8, 2577–2581 Search PubMed; (h) S. Ma and S. Yu, Tetrahedron Lett., 2004, 45, 8419–8422 Search PubMed; (i) Y. Izawa and S. S. Stahl, Adv. Synth. Catal., 2010, 352, 3223–3229 Search PubMed; (j) J. A. Schiffner and M. Oestreich, Eur. J. Org. Chem., 2011, 1148–1154 Search PubMed; (k) M. H. Emmert, A. K. Cook, Y. J. Xie and M. S. Sanford, Angew. Chem., Int. Ed., 2011, 50, 9409–9412 Search PubMed; (l) J. He, S. Li, Y. Deng, H. Fu, B. N. Laforteza, J. E. Spangler, A. Homs and J. Q. Yu, Science, 2014, 343, 1216–1220 Search PubMed; (m) W.-L. Jia and M. Á. Fernández-Ibáñez, Eur. J. Org. Chem., 2018, 6088–6091 Search PubMed; (n) H. Shi, P. Wang, S. Suzuki, M. E. Farmer and J. Q. Yu, J. Am. Chem. Soc., 2016, 138, 14876–14879 Search PubMed; (o) P. Wang, P. Verma, G. Xia, J. Shi, J. X. Qiao, S. Tao, P. T. W. Cheng, M. A. Poss, M. E. Farmer, K. S. Yeung and J. Q. Yu, Nature, 2017, 551, 489–493 Search PubMed; (p) R. Y. Zhu, Z. Q. Li, H. S. Park, C. H. Senanayake and J. Q. Yu, J. Am. Chem. Soc., 2018, 140, 3564–3568 Search PubMed; (q) M. S. Chen and M. C. White, J. Am. Chem. Soc., 2004, 126, 1346–1347 Search PubMed; (r) T. J. Osberger and M. C. White, J. Am. Chem. Soc., 2014, 136, 11176–11181 Search PubMed; (s) K. Yamaguchi, H. Kondo, J. Yamaguchi and K. Itami, Chem. Sci., 2013, 4, 3753–3757 Search PubMed; (t) H. Kondo, F. Yu, J. Yamaguchi, G. Liu and K. Itami, Org. Lett., 2014, 16, 4212–4215 Search PubMed; (u) W. Liu, S. Z. Ali, S. E. Ammann and M. C. White, J. Am. Chem. Soc., 2018, 140, 10658–10662 Search PubMed; (v) R. Ma and M. C. White, J. Am. Chem. Soc., 2018, 140, 3202–3205 Search PubMed; (w) W. H. Henderson, C. T. Check, N. Proust and J. P. Stambuli, Org. Lett., 2010, 12, 824–827 Search PubMed; (x) C. C. Le, K. Kunchithapatham, W. H. Henderson, C. T. Check and J. P. Stambuli, Chem.–Eur. J., 2013, 19, 11153–11157 Search PubMed; (y) B. J. Gorsline, L. Wang, P. Ren and B. P. Carrow, J. Am. Chem. Soc., 2017, 139, 9605–9614 Search PubMed.
- For selected examples of ligands used in other transition-metal catalyzed C–H functionalization reactions, see: (a) R. Bisht and B. Chattopadhyay, J. Am. Chem. Soc., 2016, 138, 84–87 Search PubMed; (b) S. Okumura, S. Tang, T. Saito, K. Semba, S. Sakaki and Y. Nakao, J. Am. Chem. Soc., 2016, 138, 14699–14704 Search PubMed; (c) Y. Nakao, K. S. Kanyiva and T. Hiyama, J. Am. Chem. Soc., 2008, 130, 2448–2449 Search PubMed; (d) Y. Nakao, Y. Yamada, N. Kashihara and T. Hiyama, J. Am. Chem. Soc., 2010, 132, 13666–13668 Search PubMed; (e) Y. Kuninobu, H. Ida, M. Nishi and M. Kanai, Nat. Chem., 2015, 7, 712–717 Search PubMed; (f) H. J. Davis and R. J. Phipps, Chem. Sci., 2017, 8, 864–877 Search PubMed; (g) H. J. Davis, M. T. Mihai and R. J. Phipps, J. Am. Chem. Soc., 2016, 138, 12759–12762 Search PubMed; (h) J. M. Alderson, A. M. Phelps, R. J. Scamp, N. S. Dolan and J. M. Schomaker, J. Am. Chem. Soc., 2014, 136, 16720–16723 Search PubMed.
- (a) S. Yanagisawa, K. Ueda, H. Sekizawa and K. Itami, J. Am. Chem. Soc., 2009, 131, 14622–14623 Search PubMed; (b) K. Ueda, S. Yanagisawa, J. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2010, 49, 8946–8949 Search PubMed.
- Regiocontrol by switch of the regioselectivity-determining step was rather rare in transition-metal catalysis (not limited to C–H functionalization reactions). For examples, see: (a) I. A. Sanhueza, A. M. Wagner, M. S. Sanford and F. Schoenebeck, Chem. Sci., 2013, 4, 2767–2775 Search PubMed; (b) E. P. Jackson and J. Montgomery, J. Am. Chem. Soc., 2015, 137, 958–963 Search PubMed.
- For selected reviews on C–H functionalization of indole, see: (a) M. Bandini and A. Eichholzer, Angew. Chem., Int. Ed., 2009, 48, 9608–9644 Search PubMed; (b) E. M. Beck and M. J. Gaunt, Top. Curr. Chem., 2010, 292, 85–121 Search PubMed; (c) L. Joucla and L. Djakovitch, Adv. Synth. Catal., 2009, 351, 673–714 Search PubMed; (d) A. H. Sandtorv, Adv. Synth. Catal., 2015, 357, 2403–2435 Search PubMed; (e) J. A. Leitch, Y. Bhonoah and C. G. Frost, ACS Catal., 2017, 7, 5618–5627 Search PubMed; (f) M. Petrini, Chem.–Eur. J., 2017, 23, 16115–16151 Search PubMed; (g) J.-B. Chen and Y.-X. Jia, Org. Biomol. Chem., 2017, 15, 3550–3567 Search PubMed; (h) J. Kalepu, P. Gandeepan, L. Ackermann and L. T. Pilarski, Chem. Sci., 2018, 9, 4203–4216 Search PubMed; (i) T. A. Shah, P. B. De, S. Pradhan and T. Punniyamurthy, Chem. Commun., 2019, 55, 572–587 Search PubMed.
- For selected recent examples on Pd-cataylzed functionalization of indole, see: (a) K.-Y. Jia, J.-B. Yu, Z.-J. Jiang and W.-K. Su, J. Org. Chem., 2016, 81, 6049–6055 Search PubMed; (b) Y. Yang, X. Qiu, Y. Zhao, Y. Mu and Z. Shi, J. Am. Chem. Soc., 2016, 138, 495–498 Search PubMed; (c) Y. Yang, P. Gao, Y. Zhao and Z. Shi, Angew. Chem., Int. Ed., 2017, 56, 3966–3971 Search PubMed; (d) A. J. Borah and Z. Shi, Chem. Commun., 2017, 53, 3945–3948 Search PubMed; (e) M. N. Godoi, F. de Azambuja, P. D. G. Martinez, N. H. Morgon, V. G. Santos, T. Regiani, D. Lesage, H. Dossmann, R. B. Cole, M. N. Eberlin and C. R. D. Correia, Eur. J. Org. Chem., 2017, 1794–1803 Search PubMed; (f) T. Hanakawa, K. Isa, S. Isobe, Y. Hoshino, B. Zhou and M. Kawatsura, J. Org. Chem., 2017, 82, 2281–2287 Search PubMed; (g) S. Zhao, W. Ma, M. Yasir, X. Zhou, J. Cheng and C. Jiang, ChemistrySelect, 2018, 3, 13319–13322 Search PubMed; (h) O. Moncea, D. Poinsot, A. A. Fokin, P. R. Schreiner and J.-C. Hierso, ChemCatChem, 2018, 10, 2915–2922 Search PubMed; (i) S. Fang, G. Jiang, M. Li, Z. Liu, H. Jiang and W. Wu, Chem. Commun., 2019, 55, 13769–13772 Search PubMed; (j) N. Thrimurtulu, A. Dey, A. Singh, K. Pal, D. Maiti and C. M. R. Volla, Adv. Synth. Catal., 2019, 361, 1441–1446 Search PubMed; (k) S. Guo, Z. Fang, B. Zhou, J. Hua, Z. Dai, Z. Yang, C. Liu, W. He and K. Guo, Org. Chem. Front., 2019, 6, 627–631 Search PubMed; (l) J. Zhang, M. Wu, J. Fan, Q. Xua and M. Xie, Chem. Commun., 2019, 55, 8102–8105 Search PubMed; (m) P.-G. Li, Y. Yang, S. Zhu, H.-X. Li and L.-H. Zou, Eur. J. Org. Chem., 2019, 73–76 Search PubMed.
- B. Karimi, H. Behzadnia, D. Elhamifar, P. F. Akhavan, F. K. Esfahani and A. Zamani, Synthesis, 2010, 1399–1427 Search PubMed.
- For selected examples on the oxidative Heck reaction of indole, see: (a) P. S. Baran and E. J. Corey, J. Am. Chem. Soc., 2002, 124, 7904–7905 Search PubMed; (b) G. Abbiati, E. M. Beccalli, G. Broggini and C. Zoni, J. Org. Chem., 2003, 68, 7625–7628 Search PubMed; (c) E. M. Ferreira and B. M. Stoltz, J. Am. Chem. Soc., 2003, 125, 9578–9579 Search PubMed; (d) S. Ma and S. Yu, Tetrahedron Lett., 2004, 45, 8419–8422 Search PubMed; (e) N. P. Grimster, C. Gauntlett, C. R. Godfrey and M. J. Gaunt, Angew. Chem., Int. Ed., 2005, 44, 3125–3129 Search PubMed; (f) L. Djakovitch and P. Rouge, Catal. Today, 2009, 140, 90–99 Search PubMed; (g) Q. Huang, Q. Song, J. Cai, X. Zhang and S. Lin, Adv. Synth. Catal., 2013, 355, 1512–1516 Search PubMed; (h) H. P. Gemoets, V. Hessel and T. Noel, Org. Lett., 2014, 16, 5800–5803 Search PubMed.
- For selected examples of regioselectivity switchable C–H arylation reactions of indole, see: (a) B. S. Lane, M. A. Brown and D. Sames, J. Am. Chem. Soc., 2005, 127, 8050–8057 Search PubMed; (b) D. R. Stuart, E. Villemure and K. Fagnou, J. Am. Chem. Soc., 2007, 129, 12072–12073 Search PubMed; (c) R. J. Phipps, N. P. Grimster and M. J. Gaunt, J. Am. Chem. Soc., 2008, 130(26), 8172–8174 Search PubMed.
- For selected examples of C2-alkenylation of indoles by the directing group strategy, see: (a) E. Capito, J. M. Brown and A. Ricci, Chem. Commun., 2005, 1854–1856 Search PubMed; (b) A. Maehara, H. Tsurugi, T. Satoh and M. Miura, Org. Lett., 2008, 10, 1159–1162 Search PubMed; (c) A. Garcia-Rubia, R. Gomez Arrayas and J. C. Carretero, Angew. Chem., Int. Ed., 2009, 48, 6511–6515 Search PubMed; (d) A. Garcia-Rubia, B. Urones, R. Gomez Arrayas and J. C. Carretero, Chem.–Eur. J., 2010, 16, 9676–9685 Search PubMed; (e) L. Wang, W. Guo, X. X. Zhang, X. D. Xia and W. J. Xiao, Org. Lett., 2012, 14, 740–743 Search PubMed; (f) Z.-L. Yan, W.-L. Chen, Y.-R. Gao, S. Mao, Y.-L. Zhang and Y.-Q. Wang, Adv. Synth. Catal., 2014, 356, 1085–1092 Search PubMed.
- For an example of solvent-controlled regioselective C–H alkenylation on pyrroles, see: Y. Su, H. Zhou, J. Chen, J. Xu, X. Wu, A. Lin and H. Yao, Org. Lett., 2014, 16, 4884–4887 Search PubMed.
- The calculations were performed using the Gaussian 09 software package. For computational details and the full citation of Gaussian 09, see the ESI.†.
- S. Zhang, Z. Chen, S. Qin, C. Lou, A. M. Senan, R. Z. Liao and G. Yin, Org. Biomol. Chem., 2016, 14, 4146–4157 Search PubMed.
- For selected reviews on 2-hydroxypyridine in ligand design, see: (a) J. M. Rawson and R. E. P. Winpenny, Coord. Chem. Rev., 1995, 139, 313–374 Search PubMed; (b) J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 Search PubMed; (c) C. M. Moore, E. W. Dahl and N. K. Szymczak, Curr. Opin. Chem. Biol., 2015, 25, 9–17 Search PubMed; (d) M. W. Drover, J. A. Love and L. L. Schafer, Chem. Soc. Rev., 2017, 46, 2913–2940 Search PubMed; (e) T. Higashi, S. Kusumoto and K. Nozaki, Chem. Rev., 2019, 119, 10393–10402 Search PubMed; (f) K. Fujita, Bull. Chem. Soc. Jpn., 2019, 92, 344–351 Search PubMed.
- P. Wang, M. E. Farmer and J. Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 5125–5129 Search PubMed.
- V. Salamanca, A. Toledo and A. C. Albeniz, J. Am. Chem. Soc., 2018, 140, 17851–17856 Search PubMed.
- For selected examples and reviews, see: (a) B. A. Steinhoff, S. R. Fix and S. S. Stahl, J. Am. Chem. Soc., 2002, 124, 766–767 Search PubMed; (b) W. Zierkiewicz and T. Privalov, Organometallics, 2005, 24, 6019–6028 Search PubMed; (c) B. A. Steinhoff and S. S. Stahl, J. Am. Chem. Soc., 2006, 128, 4348–4355 Search PubMed; (d) T. N. Diao, P. White, I. Guzei and S. S. Stahl, Inorg. Chem., 2012, 51, 11898–11909 Search PubMed; (e) G. Sipos, E. E. Drinkel and R. Dorta, Chem. Soc. Rev., 2015, 44, 3834–3860 Search PubMed.
- (a) A. H. Jackson and P. P. Lynch, J. Chem. Soc., Perkin Trans. 2, 1987, 1483–1488 Search PubMed; (b) X. Guo, S. Pan, J. Liu and Z. Li, J. Org. Chem., 2009, 74, 8848–8851 Search PubMed.
- We have found that the addition of a base (2,6-di-tert-butylpyridine) could accelerate the C2-deuteration reaction, which is consistent with the proposed reaction mechanism that deprotonation from the PdCl2·L17 complex is important for the reaction. Thus we hypothesized that the faster deuteration rate in the actual catalytic reaction was due to the existence of metal oxide species (generated from the aerobic re-oxidation of Pd(0)) as the base. For more details, see the ESI.†.
- L. Wang and B. P. Carrow, ACS Catal., 2019, 9, 6821–6836 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1978349 and 1978351. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc02246b |
| This journal is © The Royal Society of Chemistry 2020 |