
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Divergent uranium- versus phosphorus-based reduction of Me3SiN3 with steric modification of phosphido ligands†
Robert J.
Ward
a,
Pokpong
Rungthanaphatsophon
a,
Iker del
Rosal
b,
Steven P.
Kelley
 a,
Laurent
Maron
*b and
Justin R.
Walensky
*a
a,
Laurent
Maron
*b and
Justin R.
Walensky
*a
aDepartment of Chemistry, University of Missouri, Columbia, MO 65211, USA. E-mail: walenskyj@missouri.edu
bUniversite de Toulouse, CNRS, INSA, UPS, UMR, UMR 5215, LPCNO 135 Avenue de Ranguiel, 31077 Toulouse, France
First published on 27th May 2020
Abstract
We describe an example of a two-electron metal- and ligand-based reduction of Me3SiN3 using uranium(IV) complexes with varying steric properties. Reaction of (C5Me5)2U(CH3)[P(SiMe3)(Ph)] with Me3SiN3 produces the imidophosphorane complex, (C5Me5)2U(CH3)[N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P(SiMe3)2(Ph)] through oxidation of phosphorus. However, a similar reaction with a more sterically encumbering phosphido ligand, (C5Me5)2U(CH3)[P(SiMe3)(Mes)] forms the U(IV) complex, (C5Me5)2U[κ2-(N,N)–N(SiMe3)P(Mes)N(SiMe3)]. In probing the mechanism of this reaction, a U(VI) bis(imido) complex, (C5Me5)2U(NSiMe3){N[P(SiMe3)(Mes)]} was isolated. DFT calculations show an intramolecular reductive cycloaddition reaction leads to the formation of the U(IV) bis(amido)phosphane from the U(VI) bis(imido) complex. This is a rare example of the isolation of a reaction intermediate in f element chemistry.
P(SiMe3)2(Ph)] through oxidation of phosphorus. However, a similar reaction with a more sterically encumbering phosphido ligand, (C5Me5)2U(CH3)[P(SiMe3)(Mes)] forms the U(IV) complex, (C5Me5)2U[κ2-(N,N)–N(SiMe3)P(Mes)N(SiMe3)]. In probing the mechanism of this reaction, a U(VI) bis(imido) complex, (C5Me5)2U(NSiMe3){N[P(SiMe3)(Mes)]} was isolated. DFT calculations show an intramolecular reductive cycloaddition reaction leads to the formation of the U(IV) bis(amido)phosphane from the U(VI) bis(imido) complex. This is a rare example of the isolation of a reaction intermediate in f element chemistry.
Introduction
Many important reactions involve metal-based catalysis. Suzuki coupling,1 the Heck reaction,2 Wilkinson's catalyst,3 and many other catalytic cycles require oxidation and reduction reactions to work in tandem. These are all transition metal-based catalysts since two-electron redox couples are readily available. Within the 5f block,4 uranium is one of the only metals for which a two-electron redox couple is facile, and while oxidation is relatively easy to achieve, reduction is rarely observed without the use of an external reducing agent.5With uranium, two-electron metal-based oxidation is achieved most readily with U(III) either through using two equivalents of the U(III) starting material to form two U(IV) species, or direct oxidation to U(V). However, in all examples of oxidative chemistry with the actinides, a subsequent reductive step is rarely observed. Recently, the Liddle group reported the oxidation of U(III) to U(V) using azobenzene,6Scheme 1. Under reduced pressure and gentle heating, Liddle's U(V) dimer undergoes reduction to the U(III) starting material. This is the only example in which the oxidative and reductive steps have been isolated in which the oxidation state of the metal changes. The other reductive chemistry seen with the actinides is with redox-active ligands,7,8Scheme 1,9,10 with no observed change in the oxidation state of the metal. Since two-electron metal-based reactions are important in catalysis11,12 and small molecule activation,13,14 it is of interest to have a greater understanding of both the oxidative and reductive processes.
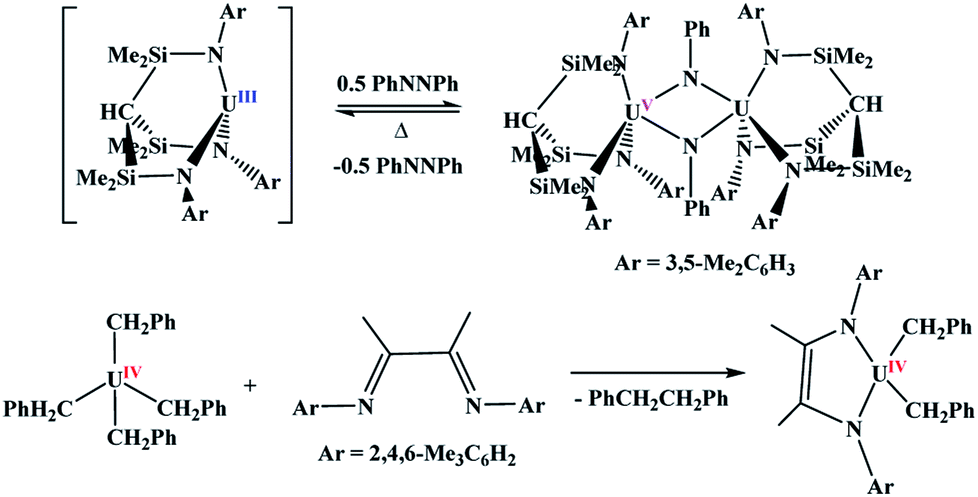 | ||
| Scheme 1 Examples of two-electron oxidative and reductive reactions in uranium chemistry. | ||
One of the most investigated substrates for interrogating U(III) reaction chemistry has been organic azides.15 This two-electron reduction typically is done by the metal centre to form a U(V) imido. There are limited examples of U(IV) oxidation to U(VI),16–23 all of them involving oxo- or imido-delivering agents. Our group has taken the approach of examining the reactivity of An(IV), An = Th, U, complexes with soft donor ligands, such as phosphorus, which also has a rich chemistry with organic azides.24 These complexes impart a mismatch between the hard, electropositive Lewis acidic actinide centre, and the soft Lewis basic nature of phosphorus, and have been shown to afford unpredictable and unusual chemistry.25–34 We recently showed that the U(III) complex, (C5Me5)2U(THF)[P(SiMe3)(Mes)] reacts with Me3SiN3 to form the U(VI) bis(imido) complex, (C5Me5)2U(NSiMe3)2.35 In order to prevent the formation of the bis(imido) complex, here, we used the mixed phosphido–methyl complexes (C5Me5)2U(CH3)[P(SiMe3)(R)], R = C6H5 (Ph), 1; 2,4,6-Me3C6H2 (Mes), 2. The mesityl complex was recently shown to react unusually with tBuNC through a series of cascade reactions to form an α-diimine,36 however, isocyanides do not undergo redox chemistry akin to organic azides such as Me3SiN3. Complexes 1 and 2 differ only in the steric properties of the R group associated with the phosphido ligand. Herein, we demonstrate that the steric properties of the aryl of the phosphido ligand play an integral role in product formation, including the isolation of a U(VI) intermediate, which subsequently reduces to U(IV) to form the final product. To our knowledge, there are no examples in f element chemistry in which an intermediate has been isolated and characterized.
Results and discussion
The phosphido–methyl complexes, (C5Me5)2U(CH3)[P(SiMe3)(Ph)], 1, and (C5Me5)2U(CH3)[P(SiMe3)(Mes)], 2,36 were prepared in high yield from the reaction of (C5Me5)2U(CH3)(I) with K[P(SiMe3)(R)], R = Ph or Mes, respectively, eqn (1). Both these complexes are brown-black in colour as compared to their dark red starting materials. No 31P NMR resonances were found from −5000 to +5000 ppm. While spectroscopic and analytical characterization of 1 was done, despite numerous attempts, the solid-state structure could not be obtained. However, due to the similarity in the 1H NMR spectra of 1 and 2, in addition to the reaction chemistry reported herein, we surmise that these two are structurally comparable.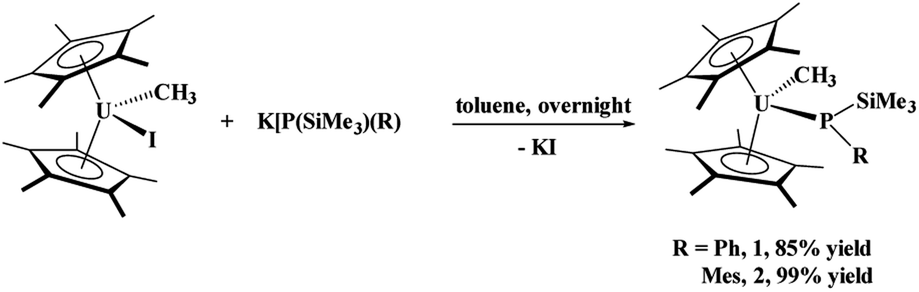 | (1) |
To probe the reactivity of these complexes, an organic azide was used since nitrogen is a hard Lewis base, compared to phosphorus, and organic azides have been shown to have both insertion37–40 and reductive reactivity.38,41 We specifically used Me3SiN3 since it does not typically insert into actinide–carbon bonds,37 and thus the reactivity should occur only at the uranium–phosphido bond. Reaction of (C5Me5)2U(CH3)[P(SiMe3)(Ph)], 1, with two equivalents of Me3SiN3, eqn (2), does not produce a colour change as the solution remains black, but effervescing was observed instantaneously. The low yield (18%) reported is based on the crystalline product, but the crude NMR spectrum shows the formation of one product. The low yield is attributed to high solubility of the complex in hydrocarbon solvents. Even with excess amount of Me3SiN3, the same product is obtained. The 1H NMR spectrum revealed a single product with a resonance at −194.4 ppm, indicative of a methyl group still coordinated to the paramagnetic uranium center.42–44 This resonance shifts slightly compared to the starting material at −190.2 ppm. In addition, the 1H NMR spectrum showed the (C5Me5)1− resonance at −1.35 ppm, but another resonance integrating to 18 protons was detected at 10.3 ppm. A signal in the 31P NMR spectrum at 518.0 ppm was located.
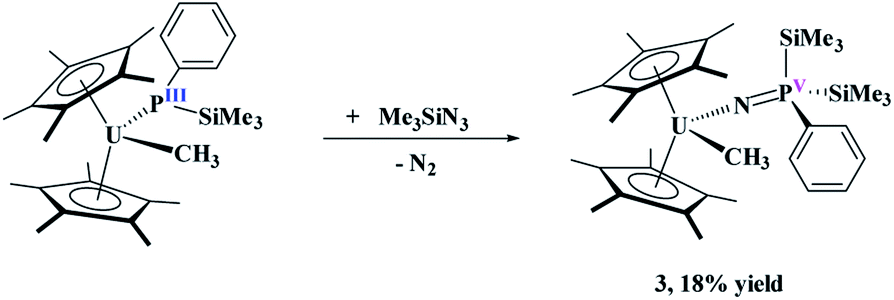 | (2) |
Dark yellow crystals, suitable for X-ray crystallography analysis, were grown from a saturated toluene solution at −25 °C. The solid-state structure, Fig. 1, identified the product as (C5Me5)2U(CH3)[NP(SiMe3)2(Ph)], 3. Complex 3 fits the NMR spectroscopy data with two trimethylsilyl groups coordinated to phosphorus and a methyl still bound to uranium. Complex 3 has a pseudo-tetrahedral arrangement with two (C5Me5)1− ligands as well as the methyl and the newly formed imidophosphorane ligand. The U(IV)-nitrogen bond distance of 2.098(3) Å is similar to uranium ketimide, (NCR2)1−, complexes. For example, (C5H5)3U[NC(Me)CHPMePh2]45 and (C5Me5)2U[NC(Ph)CH2Ph]2 (ref. 46) have U–N bond distances of 2.06(1) Å and 2.184(3) Å, respectively. A bond distance of 2.07(2) Å was observed in Gilje's imidophosphorane complex, (C5H5)3U(NPPh3).47 The U–N–P bond angle of 171.6(2)° in 3 is identical to the 172(1)° in Gilje's compound. Additionally, the U(IV) tetrakis(imidophosphorane) complex, U[NP(pip)3]4, pip = piperidinyl, was recently reported with average U–N bond lengths of 2.19(5) Å.18 A thorium complex, (1,2,4-tBu3C5H2)2Th(N3)[NP(2,4,6-tBu3C6H2)], with a similar Th–N (iminophosphino) bond length of 2.273(9) Å, has also been recently reported.25
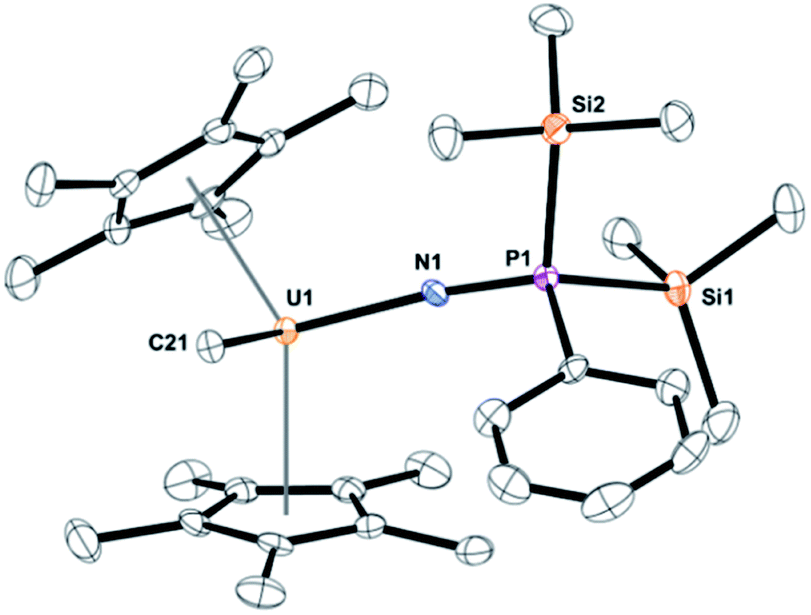 | ||
| Fig. 1 Thermal ellipsoid plot of 3 shown at the 50% probability level. The hydrogen atoms have been omitted for clarity. | ||
Formation of 3 involves the insertion of an imido unit into the uranium–phosphorus bond, with a silyl migration to phosphorus. Overall, this is a two-electron reduction of the azide to an imido moiety with simultaneous oxidation of the phosphorus from +3 to +5. We could find only one example of similar reactivity in the literature using a titanium phosphine complex with dinitrogen.48 In that case, the reduced dinitrogen oxidizes the phosphorus but without ligand addition to phosphorus. In fact, silylated iminophosphanes, (R3SiNPR3), are common starting materials to form transition metal complexes,49–52 however, in no case has the silyl been observed to migrate to phosphorus. We note that recently a mesityl ligand has been observed to migrate from nitrogen to phosphorus.53 To our knowledge, no examples of this imidophosphorane, with two trimethylsilyl groups and one phenyl, are known as mixed-substituted ligands are rare.
When the steric properties of the R group were increased from phenyl to mesityl, we expected the product to be the same, as was seen with the reactivity with tBuNC.15 Reaction of 2 with Me3SiN3 in 1,2-dimethoxyethane (DME) also had no colour change but effervescing was observed, eqn (3). The 1H NMR spectrum revealed two (C5Me5)1− resonances at 10.7 and 5.01 ppm and one SiMe3 resonance at −21.2 ppm. However, the resonance for a methyl group coordinated to uranium(IV) was not observed. A resonance in the 31P NMR spectrum was found at −180.8 ppm, shifted considerably from that observed in 3. Dark red crystals in 66% yield were grown from a saturated toluene solution at −25 °C, and the structure was unambiguously identified as (C5Me5)2U{κ2-(N,Nʹ)–[N(SiMe3)]2P(Mes)}, 4, Fig. 2.
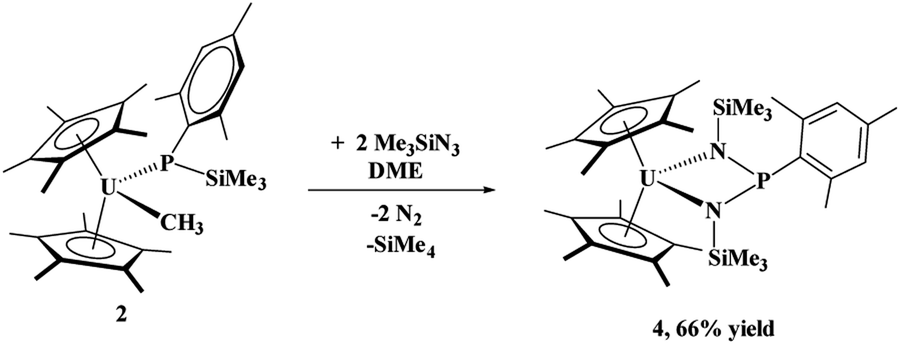 | (3) |
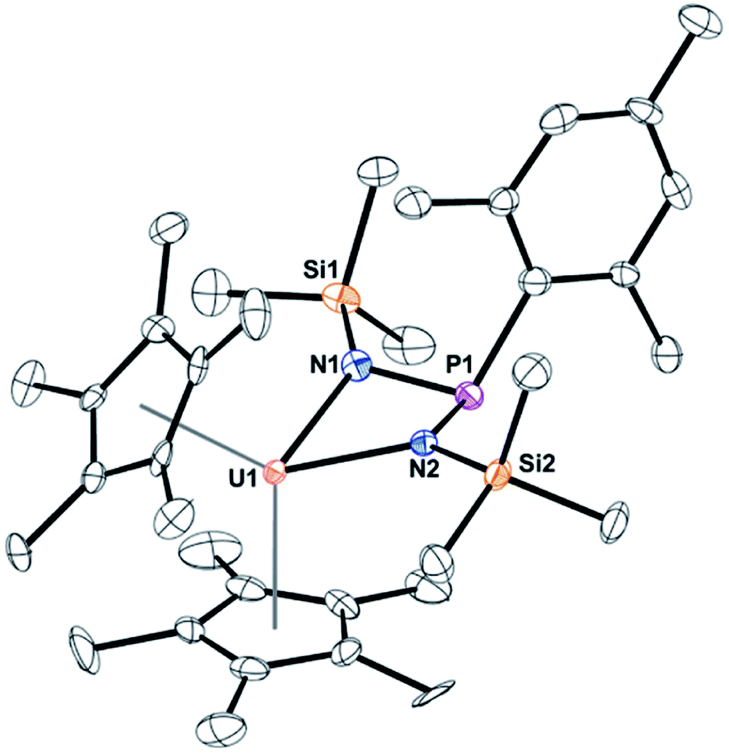 | ||
| Fig. 2 Thermal ellipsoid plot of 4 shown at the 50% probability level. The hydrogen atoms have been omitted for clarity. | ||
The structure of 4 is a uranium(IV) centre with the metallocene ligand framework and a bis(amido)phosphane ligand in a pseudo-tetrahedral environment.54–56 The uranium–nitrogen bond distances of 2.277(4) Å compare well with the U–N distances in U(IV) α-diimine complexes, (MesDABMe)2U(THF) of 2.251(4)–2.255(4) Å,57 or 2.273(2) and 2.3331(2) Å in (C5Me5)2U[κ2-(N,N)–N(tBu)CCN(tBu)CN(tBu)CH2].36 The P–N bond distances are 1.734(4) and 1.741(4) Å, much longer than the 1.600(3) Å found in 3, indicative of a P–N single bond.
The difference in reactivity of 1 and 2 with Me3SiN3 was examined using DFT calculations (see ESI†). Energetically, the formation of the mesityl analogue of 3, (C5Me5)2U(CH3)[NP(SiMe3)2(Mes)], 3Mes, is similar to the formation of 3, with the exception of the silyl transfer which is endothermic for 3Mes. Hence, this indicates the two reaction mechanisms are different from the initial step and do not share similarity. Thus, we then attempted to investigate the formation of 4 more closely. The reaction was conducted at −45 °C for 10 minutes in DME. To our surprise, a diamagnetic 1H NMR spectrum was obtained with one (C5Me5)1− resonance located at 4.67 ppm, and two SiMe3 groups at 1.03 and 1.17 ppm. The 31P NMR resonance was located at 157 ppm. Upon crystallization from a saturated diethyl ether solution at −45 °C, (C5Me5)2U{N[P(SiMe3)(Mes)]}[N(SiMe3)], 5, eqn (4), was identified as the product, but in very low yields (<10%). We have found that short reaction times are optimal for the isolation of 5, otherwise the product progresses to 4. Despite the low yield, crystalline material can be obtained from the reaction mixture in a reproducible manner. The byproduct, SiMe4, was observed in the crude NMR spectra for both 4 and 5. The formation of 5 using the UVI/IV redox couple is rare16,18–21 and nearly all examples involve forming bis(imido)17,22 complexes or uranyl functionalization23,58 as the UV/III redox is far more common,59 especially with azide reduction. Complex 5 is unusual as nearly all bis(imido) actinide complexes have a nitrogen–carbon, silicon, or hydrogen linkage.15 The 1H NMR spectrum of 5 shows temperature independent paramagnetism, a common feature of U(VI) bis(imido) complexes.60,61 When 5 is further stirred at room temperature, the conversion of 4 is observed, eqn (5). While there is precedent for oxidative chemistry with U(IV) complexes to form U(VI) bis(imido) complexes, those complexes do not reduce to U(IV) without addition of H2 (ref. 62) or an external reductant.63
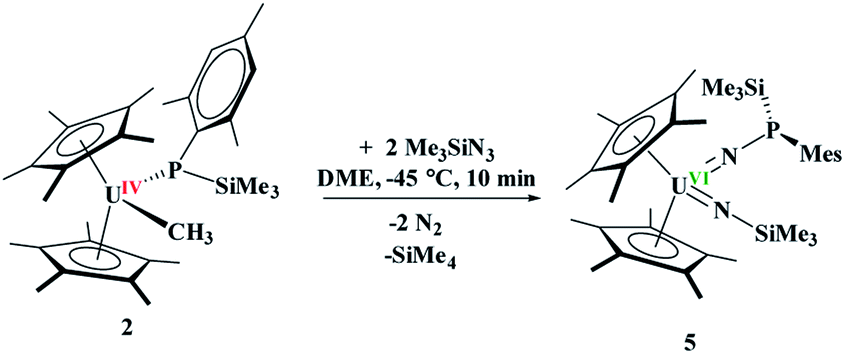 | (4) |
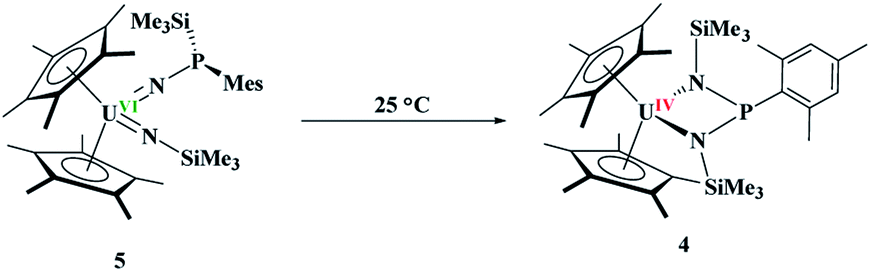 | (5) |
The solid-state structure of 5 was determined by X-ray crystallography, Fig. 3. Complex 5 is the first bis(imido) complex with a phosphorus coordinated to nitrogen, but reminiscent of the bis(imido) complex, (C5Me5)2U[NNCPh2][N(2,4,6-tBu3C6H2)].17 The U–N bond distances of 2.00(1) and 1.952(9) Å, compare well to those in (C5Me5)2U[NNCPh2][N(SiMe3)] of 2.031(6) and 1.987(5) Å. In both complexes, the longer U–N bond distance is the one associated with the phosphorus or nitrogen, respectively. The P(III)–N bond distance in 5 is 1.67(1) Å is identical to those observed in phosphanamides.64
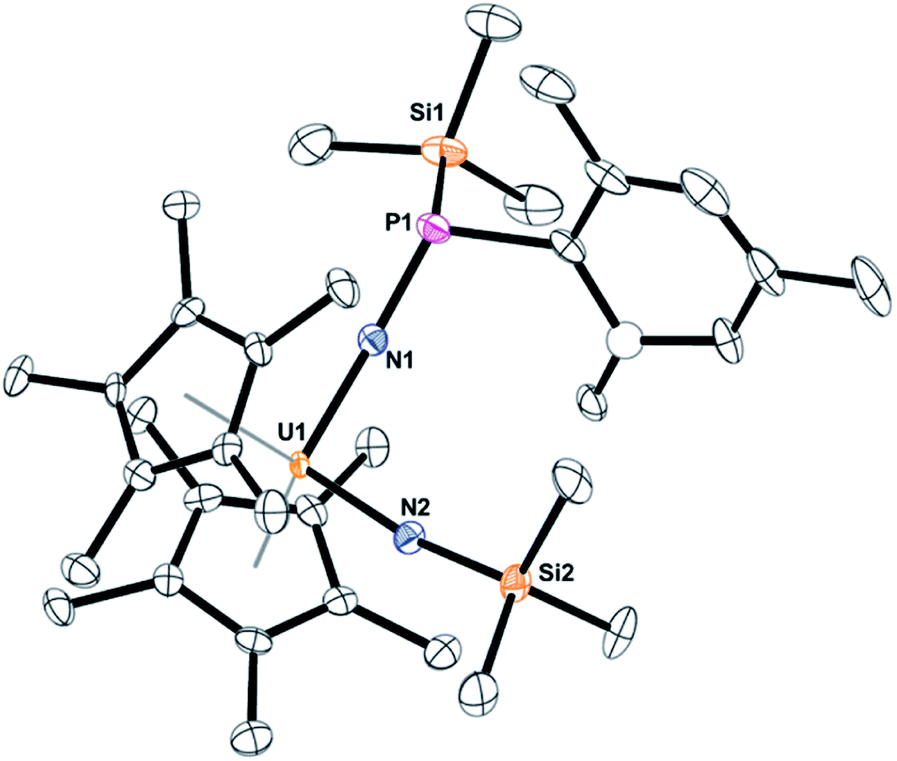 | ||
| Fig. 3 Thermal ellipsoid plot of 5 shown at the 50% probability level. The hydrogen atoms have been omitted for clarity. One carbon atom is shown anisotropically (see ESI†). | ||
Since 4 and 5 are structural isomers, the rearrangement must be intramolecular or solvent-assisted. Density functional theory calculations were performed to provide further insight into this transformation. A plausible reaction mechanism was obtained at the DFT level of theory (B3PW91, Fig. 4).
 | ||
| Fig. 4 Computed enthalpy profile for the transformation of complex 5 into complex 4. | ||
The reaction begins by the intramolecular transfer (1,2 shift) of the SiMe3 moiety on complex 5. The associated barrier is 31.0 kcal mol−1 meaning that the reaction is moderately fast, but high enough that 5 was able to be isolated. Interestingly, the TS is found on the triplet spin potential energy surface (PES), indicating that the reduction of the uranium centre already occurs during the silyl migration (the reaction on the singlet PES. The singlet PES for U(VI) was also computed and found to occur at a similar energy, 29.6 kcal mol−1 but yields a less stable intermediate, as shown in ESI†). Following the intrinsic reaction coordinates, the system evolves to the formation of a U(IV) imido-phospho-imino complex, that is only 1.7 kcal mol−1 less stable than complex 5. The latter complex readily undergoes a [2+2] cycloaddition (activation barrier of 1.8 kcal mol−1), yielding the final complex 4. Its formation is exothermic by 22.4 kcal mol−1.
With only a small change in the steric properties of phosphido ligands, Me3SiN3 was reduced by either P(III) or U(IV). The reasoning behind why one is favoured over another is not completely understood at present, but the calculations suggest that the imidophosphorane is not favourable with the mesityl group, hence an alternate, lower energy pathway, i.e. uranium oxidation, is performed instead of phosphorus oxidation. Until now, no examples of uranium oxidation and subsequent reduction were observed in the same reaction, and thus the conversion of 2 to 4via5 affords a snapshot of these processes working in concert. Finally, few cycloaddition reactions are known in f element chemistry,36,65–71 and none of them involve metal-based reduction.
Conclusions
In summary, we have examined the reactivity of Me3SiN3 with U(IV) metallocene complexes bearing mixed phosphido–methyl ligands. With a simple change of the aryl group on the phosphido ligand from phenyl to mesityl, the reactivity changed dramatically. With a smaller phenyl group, phosphorus oxidation is observed, and a silyl migration occurs from nitrogen to phosphorus. When the larger mesityl is present, a second azide is reduced. This was shown to proceed through a U(VI) bis(imido) complex followed by rearrangement through silyl transfer via a reductive intramolecular [2+2] cycloaddition. While the two-electron reduction of azides is well-known with phosphorus and uranium, we have demonstrated that either can reduce Me3SiN3 based on the steric properties of the phosphido ligand.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the Department of Energy, Office of Science Early Career Research Program under Award DE-SC-0014174 (JRW). Calmip is acknowledged for a generous grant of computing time. LM is member of the Institut Universitaire de France. The Humboldt Foundation and the Chinese Academy of Science are also acknowledged.Notes and references
- A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722–6737 CrossRef CAS PubMed.
- S. Jagtap, Catalysts, 2017, 7, 267 CrossRef.
- J. A. Osborn, F. H. Jardine, J. F. Young and G. Wilkinson, J. Chem. Soc., 1966, 1711–1732 RSC.
- R. R. Langeslay, M. E. Fieser, J. W. Ziller, F. Furche and W. J. Evans, Chem. Sci., 2015, 6, 517–521 RSC.
- E. Lu and S. T. Liddle, Dalton Trans., 2015, 44, 12924–12941 RSC.
- B. M. Gardner, C. E. Kefalidis, E. Lu, D. Patel, E. J. L. McInnes, F. Tuna, A. J. Wooles, L. Maron and S. T. Liddle, Nat. Commun., 2017, 8, 1898 CrossRef PubMed.
- M. E. Garner and J. Arnold, Organometallics, 2017, 36, 4511–4514 CrossRef CAS.
- W. J. Evans, K. A. Miller, S. A. Kozimor, J. W. Ziller, A. G. DiPasquale and A. L. Rheingold, Organometallics, 2007, 26, 3568–3576 CrossRef CAS.
- S. J. Kraft, P. E. Fanwick and S. C. Bart, J. Am. Chem. Soc., 2012, 134, 6160–6168 CrossRef CAS PubMed.
- E. M. Matson, S. M. Franke, N. H. Anderson, T. D. Cook, P. E. Fanwick and S. C. Bart, Organometallics, 2014, 33, 1964–1971 CrossRef CAS.
- A. R. Fox, S. C. Bart, K. Meyer and C. C. Cummins, Nature, 2008, 455, 341 CrossRef CAS PubMed.
- P. L. Cottingham and L. K. Barker, Ind. Eng. Chem. Prod. Res. Dev., 1973, 12, 41–47 CrossRef CAS.
- P. L. Arnold and Z. R. Turner, Nat. Rev. Chem., 2017, 1, 0002 CrossRef CAS.
- B. M. Gardner and S. T. Liddle, Eur. J. Inorg. Chem., 2013, 2013, 3753–3770 CrossRef CAS.
- D. Schädle and R. Anwander, Chem. Soc. Rev., 2019, 48, 5752 RSC.
- N. T. Rice, K. McCabe, J. Bacsa, L. Maron and H. S. La Pierre, J. Am. Chem. Soc., 2020, 142, 7368 CrossRef CAS PubMed.
- J. L. Kiplinger, D. E. Morris, B. L. Scott and C. J. Burns, Chem. Commun., 2002, 30–31 RSC.
- D. P. Mills, O. J. Cooper, F. Tuna, E. J. L. McInnes, E. S. Davies, J. McMaster, F. Moro, W. Lewis, A. J. Blake and S. T. Liddle, J. Am. Chem. Soc., 2012, 134, 10047–10054 CrossRef CAS PubMed.
- E. Lu, O. J. Cooper, J. McMaster, F. Tuna, E. J. L. McInnes, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2014, 53, 6696–6700 CrossRef CAS PubMed.
- A. J. Lewis, P. J. Carroll and E. J. Schelter, J. Am. Chem. Soc., 2013, 135, 13185–13192 CrossRef CAS PubMed.
- J. L. Brown, S. Fortier, G. Wu, N. Kaltsoyannis and T. W. Hayton, J. Am. Chem. Soc., 2013, 135, 5352–5355 CrossRef CAS PubMed.
- D. S. J. Arney and C. J. Burns, J. Am. Chem. Soc., 1995, 117, 9448–9460 CrossRef CAS.
- R. Faizova, F. Fadaei-Tirani, R. Bernier-Latmani and M. Mazzanti, Angew. Chem., Int. Ed., 2020, 59, 6756–6759 CrossRef CAS PubMed.
- H. Staudinger and J. Meyer, Helv. Chim. Acta, 1919, 2, 635–646 CrossRef CAS.
- C. Zhang, G. Hou, G. Zi, W. Ding and M. D. Walter, J. Am. Chem. Soc., 2018, 140, 14511–14525 CrossRef CAS PubMed.
- C. Zhang, G. Hou, G. Zi, W. Ding and M. D. Walter, Inorg. Chem., 2019, 58, 1571–1590 CrossRef CAS PubMed.
- C. Zhang, G. Hou, G. Zi and M. D. Walter, Dalton Trans., 2019, 48, 2377–2387 RSC.
- C. Zhang, Y. Wang, G. Hou, W. Ding, G. Zi and M. D. Walter, Dalton Trans., 2019, 48, 6921–6930 RSC.
- P. G. Edwards, M. B. Hursthouse, K. M. A. Malik and J. S. Parry, Chem. Commun., 1994, 1249–1250 RSC.
- S. P. Vilanova, M. L. Tarlton, C. L. Barnes and J. R. Walensky, J. Organomet. Chem., 2018, 857, 159–163 CrossRef CAS.
- S. P. Vilanova, I. del Rosal, M. L. Tarlton, L. Maron and J. R. Walensky, Angew. Chem., Int. Ed., 2018, 57, 16748–16753 CrossRef CAS PubMed.
- P. F. Rungthanaphatsophon, O. J. Fajen, S. P. Kelley and J. R. Walensky, Inorganics, 2019, 7, 105 CrossRef CAS.
- M. E. Garner, B. F. Parker, S. Hohloch, R. G. Bergman and J. Arnold, J. Am. Chem. Soc., 2017, 139, 12935–12938 CrossRef CAS PubMed.
- A. C. Behrle and J. R. Walensky, Dalton Trans., 2016, 45, 10042–10049 RSC.
- P. Rungthanaphatsophon, C. L. Barnes, S. P. Kelley and J. R. Walensky, Dalton Trans., 2018, 47, 8189 RSC.
- P. Rungthanaphatsophon, I. d. Rosal, R. J. Ward, S. P. Vilanova, S. P. Kelley, L. Maron and J. R. Walensky, Organometallics, 2019, 38, 1733–1740 CrossRef CAS.
- W. J. Evans, J. R. Walensky, J. W. Ziller and A. L. Rheingold, Organometallics, 2009, 28, 3350–3357 CrossRef CAS.
- S. J. Kraft, P. E. Fanwick and S. C. Bart, Organometallics, 2013, 32, 3279–3285 CrossRef CAS.
- Z. R. Turner, R. Bellabarba, R. P. Tooze and P. L. Arnold, J. Am. Chem. Soc., 2010, 132, 4050–4051 CrossRef CAS PubMed.
- N. S. Settineri and J. Arnold, Chem. Sci., 2018, 9, 2831–2841 RSC.
- A. Zalkin, J. G. Brennan and R. A. Andersen, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1988, 44, 1553–1554 CrossRef.
- P. J. Fagan, J. M. Manriquez, E. A. Maatta, A. M. Seyam and T. J. Marks, J. Am. Chem. Soc., 1981, 103, 6650–6667 CrossRef CAS.
- H. W. Turner, R. A. Andersen, A. Zalkin and D. H. Templeton, Inorg. Chem., 1979, 18, 1221–1224 CrossRef CAS.
- W. J. Evans, J. R. Walensky, F. Furche, J. W. Ziller, A. G. DiPasquale and A. L. Rheingold, Inorg. Chem., 2008, 47, 10169–10176 CrossRef CAS PubMed.
- R. E. Cramer, K. Panchanatheswaran and J. W. Gilje, J. Am. Chem. Soc., 1984, 106, 1853–1854 CrossRef CAS.
- K. C. Jantunen, C. J. Burns, I. Castro-Rodriguez, R. E. Da Re, J. T. Golden, D. E. Morris, B. L. Scott, F. L. Taw and J. L. Kiplinger, Organometallics, 2004, 23, 4682–4692 CrossRef CAS.
- R. E. Cramer, F. Edelmann, A. L. Mori, S. Roth, J. W. Gilje, K. Tatsumi and A. Nakamura, Organometallics, 1988, 7, 841–849 CrossRef CAS.
- L. Morello, P. Yu, C. D. Carmichael, B. O. Patrick and M. D. Fryzuk, J. Am. Chem. Soc., 2005, 127, 12796–12797 CrossRef CAS PubMed.
- H. J. Mai, S. Wocadlo, H. C. Kang, W. Massa, K. Dehnicke, C. Maichle-Mössmer, J. Strähle and D. Fenske, Z. Anorg. Allg. Chem., 1995, 621, 705–712 CrossRef CAS.
- T. Miekisch, H. J. Mai, R. M. Zu Köcker, K. Dehnicke, J. Magull and H. Goesmann, Z. Anorg. Allg. Chem., 1996, 622, 583–588 CrossRef CAS.
- H. J. Mai, S. Wocadlo, W. Massa, F. Weller and K. Dehnicke, Zeitschrift für Naturforschung B, 1995, 50, 1215–1221 CAS.
- Z. K. R. Meyer, F. Gerlinde, N. Bernhard, D. Kurt and M. Jörg, Z. Anorg. Allg. Chem., 1994, 620, 431–437 CrossRef.
- L. L. Liu, J. Zhou, L. L. Cao, R. Andrews, R. L. Falconer, C. A. Russell and D. W. Stephan, J. Am. Chem. Soc., 2018, 140, 147–150 CrossRef CAS PubMed.
- J. Vrana, R. Jambor, A. Ruzicka, M. Alonso, F. De Proft, A. Lycka and L. Dostal, Dalton Trans., 2015, 44, 4533–4545 RSC.
- K. Albahily, D. Al-Baldawi, S. Gambarotta, E. Koç and R. Duchateau, Organometallics, 2008, 27, 5943–5947 CrossRef CAS.
- K. Albahily, E. Koc, D. Al-Baldawi, S. Savard, S. Gambarotta, T. J. Burchell and R. Duchateau, Angew. Chem., Int. Ed., 2008, 47, 5816–5819 CrossRef CAS PubMed.
- S. J. Kraft, U. J. Williams, S. R. Daly, E. J. Schelter, S. A. Kozimor, K. S. Boland, J. M. Kikkawa, W. P. Forrest, C. N. Christensen, D. E. Schwarz, P. E. Fanwick, D. L. Clark, S. D. Conradson and S. C. Bart, Inorg. Chem., 2011, 50, 9838–9848 CrossRef CAS PubMed.
- B. E. Cowie, J. M. Purkis, J. Austin, J. B. Love and P. L. Arnold, Chem. Rev., 2019, 119, 10595–10637 CrossRef CAS PubMed.
- W. J. Evans and S. A. Kozimor, Coord. Chem. Rev., 2006, 250, 911–935 CrossRef CAS.
- L. P. Spencer, R. L. Gdula, T. W. Hayton, B. L. Scott and J. M. Boncella, Chem. Commun., 2008, 4986–4988 RSC.
- B. P. Warner, B. L. Scott and C. J. Burns, Angew. Chem., Int. Ed., 1998, 37, 959–960 CrossRef CAS PubMed.
- R. G. Peters, B. P. Warner and C. J. Burns, J. Am. Chem. Soc., 1999, 121, 5585–5586 CrossRef CAS.
- L. P. Spencer, E. J. Schelter, P. Yang, R. L. Gdula, B. L. Scott, J. D. Thompson, J. L. Kiplinger, E. R. Batista and J. M. Boncella, Angew. Chem., Int. Ed., 2009, 48, 3795–3798 CrossRef CAS PubMed.
- T. G. Wetzel, S. Dehnen and P. W. Roesky, Angew. Chem., Int. Ed., 1999, 38, 1086–1088 CrossRef CAS PubMed.
- O. P. Lam, P. L. Feng, F. W. Heinemann, J. M. O'Connor and K. Meyer, J. Am. Chem. Soc., 2008, 130, 2806–2816 CrossRef CAS PubMed.
- S. C. Bart, C. Anthon, F. W. Heinemann, E. Bill, N. M. Edelstein and K. Meyer, J. Am. Chem. Soc., 2008, 130, 12536–12546 CrossRef CAS PubMed.
- R. E. Jilek, N. C. Tomson, B. L. Scott and J. M. Boncella, Inorg. Chim. Acta, 2014, 422, 78–85 CrossRef CAS.
- A.-C. Schmidt, F. W. Heinemann, L. Maron and K. Meyer, Inorg. Chem., 2014, 53, 13142–13153 CrossRef CAS PubMed.
- C. J. Tatebe, M. Zeller and S. C. Bart, Inorg. Chem., 2017, 56, 1956–1965 CrossRef CAS PubMed.
- L. Maria, N. A. G. Bandeira, J. Marçalo, I. C. Santos and J. K. Gibson, Chem. Commun., 2020, 56, 431–434 RSC.
- L. P. Spencer, P. Yang, B. L. Scott, E. R. Batista and J. M. Boncella, J. Am. Chem. Soc., 2008, 130, 2930–2931 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1826642, 1826643 and 1995730. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc02261f |
| This journal is © The Royal Society of Chemistry 2020 |