
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA practical catalytic reductive amination of carboxylic acids†
Emma L.
Stoll
 a,
Thomas
Tongue
a,
Keith G.
Andrews
a,
Damien
Valette
b,
David J.
Hirst
b and
Ross M.
Denton
*a
a,
Thomas
Tongue
a,
Keith G.
Andrews
a,
Damien
Valette
b,
David J.
Hirst
b and
Ross M.
Denton
*a
aSchool of Chemistry, GlaxoSmithKline Carbon Neutral Laboratories for Sustainable Chemistry, University of Nottingham, 6 Triumph Road, Nottingham, NG7 2GA, UK. E-mail: ross.denton@nottingham.ac.uk
bGlaxoSmithKline, Gunnels Wood Road, Stevenage, SG1 2NY, UK
First published on 12th August 2020
Abstract
We report reductive alkylation reactions of amines using carboxylic acids as nominal electrophiles. The two-step reaction exploits the dual reactivity of phenylsilane and involves a silane-mediated amidation followed by a Zn(OAc)2-catalyzed amide reduction. The reaction is applicable to a wide range of amines and carboxylic acids and has been demonstrated on a large scale (305 mmol of amine). The rate differential between the reduction of tertiary and secondary amide intermediates is exemplified in a convergent synthesis of the antiretroviral medicine maraviroc. Mechanistic studies demonstrate that a residual 0.5 equivalents of carboxylic acid from the amidation step is responsible for the generation of silane reductants with augmented reactivity, which allow secondary amides, previously unreactive in zinc/phenylsilane systems, to be reduced.
Introduction
Amines and their derivatives are found in a wide range of fundamentally and commercially important molecules. As a result, there are numerous methods for the construction of carbon–nitrogen bonds, such as N-alkylation, Buchwald–Hartwig coupling,1 the Ullman reaction2 and hydroamination.3 In the context of N-alkylation, the most important and widely used methods involve either nucleophilic substitution with alkyl halides/pseudohalides or reductive amination using aldehydes.4 In both cases, the required electrophiles are typically generated from alcohols through either stoichiometric activation or oxidation. Alternatively, alcohols can be directly used in the more atom economical “borrowing hydrogen” approach.5,6 A much less explored but potentially powerful method of N-alkylation involves the use of carboxylic acids as nominal electrophiles. Carboxylic acids are relatively abundant, inexpensive, easier to handle than aldehydes and avoid the toxicity issues associated with alkyl halides.7–10 For these reasons a general reductive amination protocol using carboxylic acids would be of significant value.The first reductive alkylation reactions of amines with carboxylic acids were reported in the 1970s using super-stoichiometric metal borohydrides.11–13 In general, these alkylations were narrow in scope and employed liquid carboxylic acids as the reaction solvent or were limited to aniline substrates. More recently, Beller and co-workers disclosed a catalytic N-alkylation of primary and secondary amines with carboxylic acids, using Karstedt's catalyst and phenylsilane as the terminal reductant (Fig. 1A).14
 | ||
| Fig. 1 Reductive amination of amines using carboxylic acids exploiting the dual reactivity of phenylsilane. | ||
Whilst this method represents a clear breakthrough, large excesses of phenylsilane (up to 10 equiv.) and carboxylic acid (up to 5.5 equiv.) are required to mitigate the non-productive reduction of aldehyde intermediates (Fig. 1A). Further work by Fu and co-workers15 used polymethylhydrosiloxane (PMHS) as the reducing agent in the presence of substoichiometric tris(perfluorophenyl)borane. Again, large excesses of the carboxylic acid (up to 5 equiv.) and PMHS (up to 8 equiv.) are required for the reaction, which is also performed under Schlenk conditions. Following on from these two examples, further catalytic methods have been documented which involve Ru,16 Rh,17 Ir,18 Cu19 and Pt20 catalysts in combination with silane terminal reductants. Additionally, systems involving dihydrogen as the terminal reductant have also been described.21,22 Despite these reports a practical method that is applicable to primary and secondary amines and mediated by an inexpensive catalyst is yet to be realized. Here we report a general reductive amination protocol of carboxylic acids using Zn(OAc)2 in combination with phenylsilane, which has been demonstrated on a 305 mmol scale (Fig. 1B). The reductive amination exhibits predictable selectivity that allows the chemistry to be deployed in sequential amination processes as well as in API synthesis. Mechanistic studies demonstrate that excess carboxylic acid from the amidation stage of the reaction is responsible for modification of the silane reductant in situ, which allows previously unreactive substrates to be reduced.
Results and discussion
In contrast to previous work (Fig. 1A) our reaction design (Fig. 1B) is based upon the dual reactivity of hydrosilanes, which mediate direct amide coupling23–26 and amide reduction in the presence of a metal catalyst.27–30Combining these processes gives a reductive amination reaction in which carbon–nitrogen bond formation is complete before reduction begins, thereby avoiding unwanted reduction of the carboxylic acid, leading to alcohol by-products. Although we had established proof-of-concept for this general approach using iridium catalysis,18 the synthesis of secondary amines was complicated by relatively high iridium loadings and the fact that different silanes were required for the amidation and reduction step. In developing a general and more practical catalytic reductive amination we were attracted to the Zn(OAc)2 because of its low cost31 and reported activity for tertiary amide reduction in combination with triethoxysilane.32 Unfortunately, the reduction of secondary amides, required for the synthesis of secondary amines, was known to be poor with Zn(OTf)2/phenylsilane.33 Therefore, in order to realize a general catalytic reductive amination, we began by developing a new Zn(OAc)2-catalyzed reduction protocol using phenylsilane as the terminal reductant.We first examined the reduction step of the proposed reductive amination reaction in isolation using a model secondary amide (Table 1). An initial experiment with 1 equivalent of phenylsilane and 10 mol% Zn(OAc)2 (entry 1) gave a very poor conversion of 1% which was increased to a maximum of 22% as the amount of phenylsilane was increased (entries 2 and 3). Since the remainder of the mass balance in these reactions was the unreacted amide, the reaction time was increased to 24 hours (entry 4). However, this resulted in only a very modest improvement and gave 27% of the amine product. These initial results were in agreement with related observations of Beller and co-workers.33
We had previously observed that Brønsted acids react rapidly with phenylsilane to generate modified silanes with enhanced reducing properties34 and, remarkably, the addition of 50 mol% of benzoic acid (entry 5) resulted in a substantial improvement in the yield to 65%. More significantly, we found that the carboxylic acid could be changed to match that from which the secondary amide was derived (entry 6). This result implies that a 0.5 equivalent excess of carboxylic acid (i.e. 1.5 equiv. at the beginning of the reaction) should result in the catalytic reductive amination of primary amines that we sought, in which phenylsilane functions as an amide coupling reagent and then as the terminal reductant. Gratifyingly, this proved to be the case and we therefore surveyed the scope of the carboxylic acid enhanced Zn(OAc)2/phenylsilane reductive amination (Table 2). Beginning with secondary amines, previously unattainable from secondary amides in zinc/phenylsilane systems, it can be seen that a range of potentially reducible functional groups are tolerated in both the acid and amine component, including aryl halides (entries 1, 5 and 8), acetals (entry 2), nitro (entry 4) and alkenes (entries 3 and 7). Free hydroxyl groups are also tolerated but additional silane is required in order to compensate for silyl ether formation (entry 8). Alternatively, standard silyl ether protected substrates can be used without the requirement for additional silane (entry 10). Finally, with respect to the carboxylic acid, the scope includes electron rich aromatic carboxylic acids (entries 2 and 3), electron deficient aromatic carboxylic acids (entries 4 and 6) as well as aliphatic carboxylic acids (entries 7 and 10). With respect to tertiary amine synthesis (Table 2) we note that the reduction of the amide intermediate in this case is faster and complete within 2 hours compared to 6 hours for secondary amide intermediates. Amination was efficient for a wide range of substituted benzoic acids containing electron donating groups (entries 12, 15, 16 and 17), electron withdrawing groups (entries 13 and 20), halides (entries 14, 18 and 19) and a boronic acid ester (entry 22). Disappointingly aniline substrates (not depicted) showed poor reactivity in the reaction.
| a Reaction conditions: secondary amine products – PhSiH3 (0.75 equiv.), toluene, reflux, 16 h then PhSiH3 (3.0 equiv.), Zn(OAc)2 (10 mol%), toluene, reflux, 6 h; tertiary amine products – PhSiH3 (0.75 equiv.), toluene, reflux, 16 h then PhSiH3 (2.0 equiv.), Zn(OAc)2 (10 mol%), toluene, reflux, 2 h. b PhSiH3 (1.75 equiv.), toluene, reflux, 16 h then PhSiH3 (3.0 equiv.), Zn(OAc)2 (10 mol%), toluene, reflux, 6 h. |
|---|

|
The latter example provides a good illustration of how the reductive amination can be deployed for the construction of a tertiary amine building block with two versatile functional groups for further derivatization. Pyridines are tolerated in both the acid and amine component (entries 21, 24 and 25) and aliphatic carboxylic acids also undergo efficient coupling with both cyclic and acyclic amines (entries 23, 24 and 25). In all but 4 cases, the tertiary amine products were obtained by acid–base work-up without recourse to chromatography.
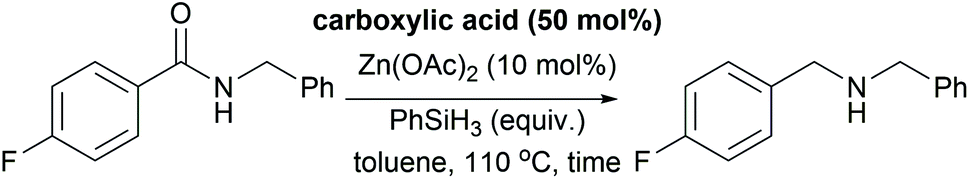
We next sought to explore further the utility and practicality of the reductive amination reaction and began by examining a large-scale reaction (Scheme 1A). Reductive amination between benzylamine (305 mmol) and benzoic acid was carried out in a one litre controllable lab reactor equipped with overhead stirring and a heating jacket. Given that hydrogen gas is generated during the amidation-phase of the process, the silane was added by syringe pump at a rate of 2 mL min−1. Following completion of the reductive amination reaction, the crude reaction mixture was decanted from the reactor and zinc precipitate was removed by filtration through Celite. The product was obtained in an isolated yield of 92% (with an HPLC purity of 93%) as the hydrochloride monohydrate after treatment of the filtrate with concentrated hydrochloric acid.
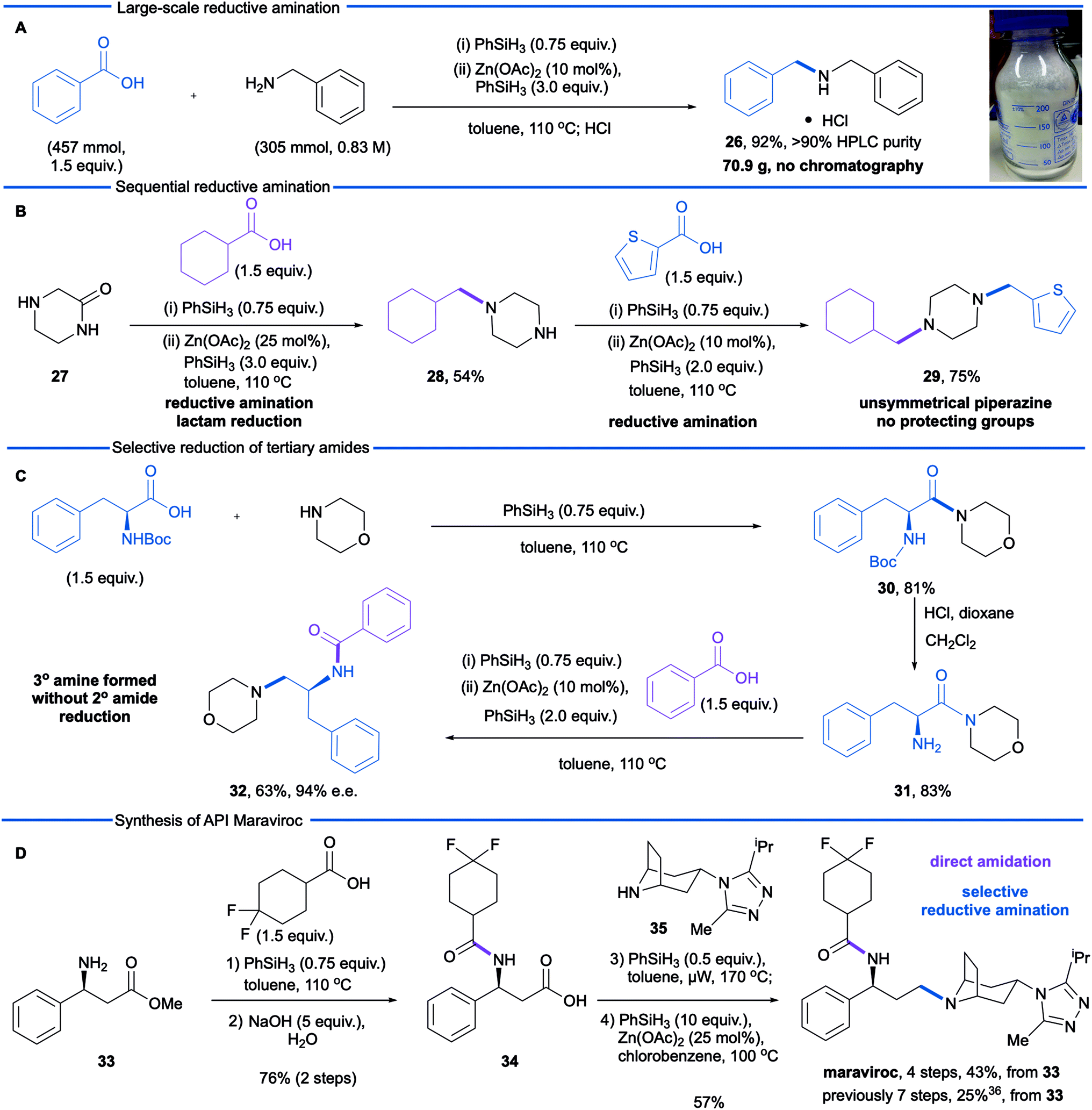 | ||
| Scheme 1 Large-scale reductive amination and synthesis applications. | ||
In a second study, we examined the construction of differentially N,N-disubstituted piperazines (Scheme 1B). These are a privileged class of compounds with biological activities including antifungals, antidepressants, antivirals, and serotonin receptor (5-HT) antagonists/agonists.35 The conventional approach to N,N-disubstituted piperazines involves mono Boc-protection of piperazine followed by reductive amination or alkylation, carbamate cleavage and a second reductive amination or alkylation. Depicted in Scheme 1B is a more efficient approach using 2-oxopiperazine 27 as a building block in lieu of N-Boc piperazine. Reductive amination of 2-oxopiperazine 27 affords the corresponding amine 28, resulting from N-alkylation and lactam reduction. A second reductive amination with 2-thiophenecarboxylic acid afforded the differentially substituted product 29, which demonstrates a modular synthesis of privileged diamine structures.
During our optimization studies we established that catalytic reduction of secondary amides is significantly slower than tertiary substrates. With target synthesis applications in mind, we carried out a study to explore selective reductions (Scheme 1C). Silane-mediated direct amidation of morpholine with N-Boc-phenylalanine gave amide 30 which was deprotected to give amine 31. Treatment of this primary amine with benzoic acid formed the secondary amide intermediate and subsequent addition of Zn(OAc)2/phenylsilane resulted in reduction of only the tertiary amide to afford tertiary amine 32 in good yield and excellent stereochemical integrity (63%, 97% e.e.). Reduction of the secondary amide was not observed.
In a final study, we made use of the reductive amination protocol in a short, convergent synthesis of the anti-retroviral medicine maraviroc. The construction of the tertiary amine (Scheme 1D) has proven to be challenging and, in the process developed by Pfizer,36 was accomplished by conventional reductive amination of an aldehyde. The aldehyde intermediate was derived from β-phenylalanine methyl ester 33, by benzoyl protection of the amine, ester hydrolysis, reduction of the derived acid to alcohol and, finally, oxidation of the alcohol. This gives a total of three redox steps associated with the construction of the carbon–nitrogen bond. We envisioned a more redox economical route37 based on the reductive amination of carboxylic acid 34 and tropane 35.36 The required carboxylic acid was prepared from β-aminoester 33 in excellent yield (76% over two steps) by phenylsilane-mediated direct amidation38 with 4,4-difluorocyclohexanecarboxylic acid, followed by saponification of the methyl ester. Reductive amination was then performed by treatment of the carboxylic acid and amine 35 with phenylsilane to effect the difficult amidation reaction. Selective reduction of the tertiary amide using modified conditions (25 mol% Zn(OAc)2) gave the active pharmaceutical ingredient in 57% yield. This synthesis demonstrates the advantages of a carboxylic acid reductive amination in terms of step and redox economy37 and compares favourably to the Pfizer synthesis (4 steps versus 7 steps from the common intermediate 33).
Mechanism: role of the carboxylic acid
Having explored the scope and utility of the Zn(OAc)2-catalyzed reductive amination reaction, we next carried out experiments in order to probe the role played by residual carboxylic acid in the amide reduction step. Beller and co-workers had previously reported that the combination of Zn(OTf)2 and phenylsilane33 was poor for the reduction of secondary amides and our own experiments (Table 1) demonstrated that the combination of Zn(OAc)2 and phenylsilane gave similar results.However, the addition of a carboxylic acid resulted in a significantly improved reduction process (Table 1, entries 5 and 6). We reasoned that the carboxylic acid was modifying the silane in situ generating a more reactive species, which, in combination with Zn(OAc)2, was able to reduce secondary amides at enhanced rates. In order to investigate this possibility, we carried out a 19F NMR study using carboxylic acid 36. We first examined the 19F NMR spectrum of the crude reaction mixture at the end of the amidation phase of the reaction (Scheme 2A). As expected, amide 37 was visible (19F δ = −107.9) along with the remaining 0.5 equivalents of carboxylic acid 36 (19F δ = −105.6).39 Subsequent addition of Zn(OAc)2 and additional phenylsilane resulted in the generation of hydrogen gas and a broad range of new 19F environments downfield of the carboxylic acid (19F δ = −103.3 to −105.2).39 These signals we attribute to multiple silyl esters that are generated by a dehydrogenative silylation process which could be catalyzed by residual amine from the amidation step or, as the reaction progresses, by the amine product.34
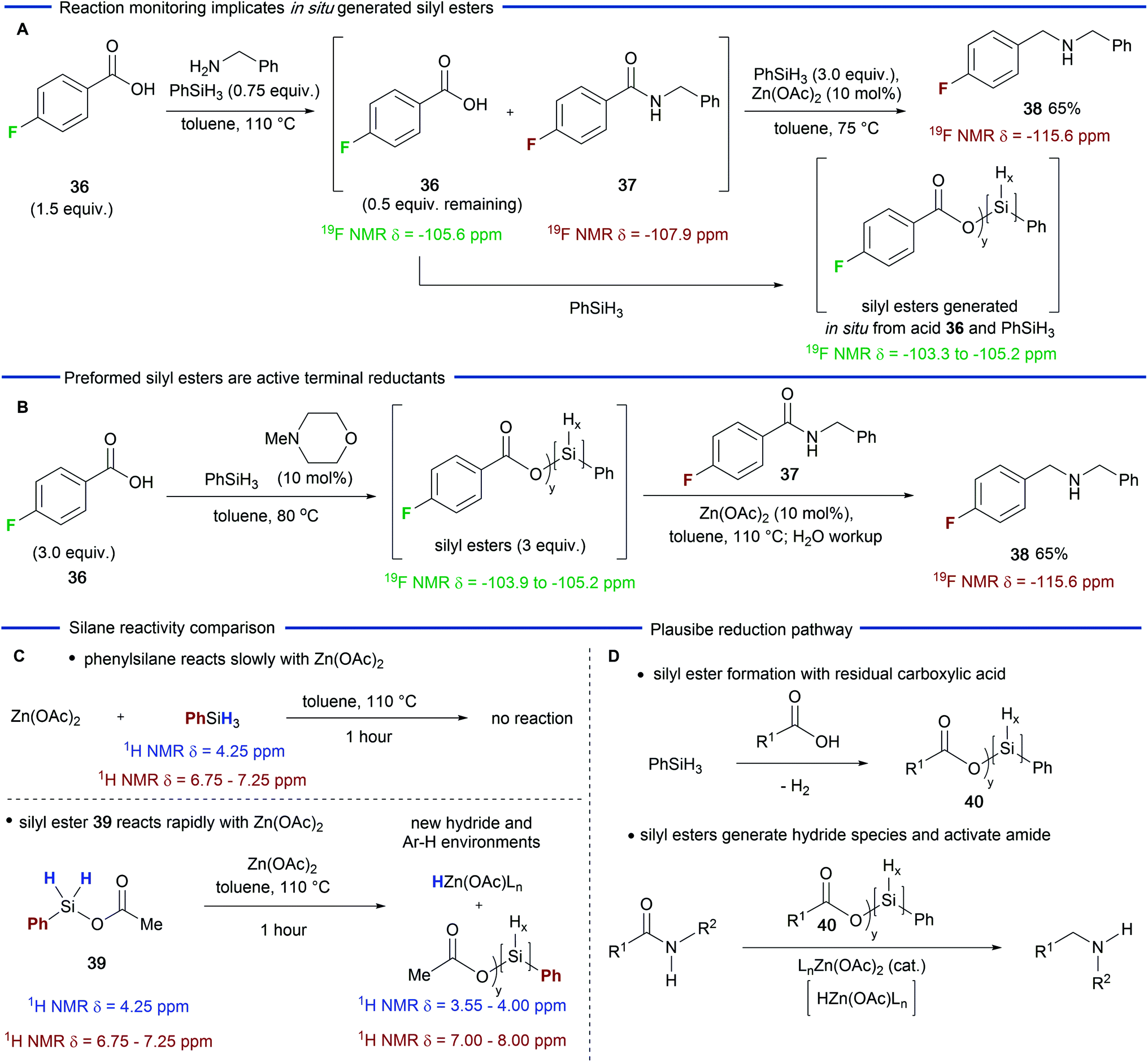 | ||
| Scheme 2 The role of the carboxylic acid in the reduction phase of the amination reaction. | ||
Having observed species that we believed to be silyl ester intermediates in the reduction phase of the reaction, we carried out a further experiment in which carboxylic acid 36 was treated with phenylsilane and catalytic N-methylmorpholine (Scheme 2B) – conditions which will generate a mixture of silyl esters.34 Gratifyingly, a similar 19F NMR profile was observed (19F δ = −103.9 to −105.2) along with some residual carboxylic acid.39 Subsequent addition of secondary amide 37 and Zn(OAc)2 to this mixture gave amine 38 in 65% yield. This experiment provides further evidence that silyl esters participate in the amide reduction.
In a final experiment we sought to compare the reactivity of phenylsilane with an independently prepared silyl ester 39 (Scheme 2C).39 Refluxing phenylsilane with Zn(OAc)2 (1 equiv.) for one hour resulted in no observable reaction as judged by 1H NMR spectroscopy. However, the reaction of silyl ester 39 with Zn(OAc)2 resulted in a range of new aromatic environments (1H δ = 8.15 to 7.45), the disappearance of species 39 and new broad hydride environments (1H δ = 4.15 to 3.55) upfield of phenylsilane. These observations are consistent with silane modification, and, possibly, the formation of a zinc hydride species.40 On the basis of these observations we can derive a plausible pathway for the catalytic amide reduction (Scheme 2D). We speculate that the residual carboxylic acid from the amidation step of the reaction undergoes dehydrogenative silylation to afford a mixture of silyl esters depicted as the general structure 40. Once formed this mixture reacts with Zn(OAc)2 more rapidly than the parent silane and this combination is responsible for activation and reduction of the amide. The nature of the activation and reduction process is obscured by the complex speciation of the silyl esters. However, the formation of a zinc hydride species is feasible40 and they have been invoked by Mlynarski and co-workers in Zn(OAc)2-catalzyed reduction of imines41 and, more recently, by Beller and co-workers32 in Zn(OAc)2/triethoxysilane reductions of tertiary amides. The initial reduction would lead to a silylated tetrahedral intermediate (not depicted) which is then likely to be further activated and then reduced to the amine product. Finally, we note that although zinc has been depicted with acetate ligands, exchange with the residual carboxylic acid is possible but has not been depicted for the sake of clarity.
Conclusions
We have demonstrated a practical Zn(OAc)2-catalyzed N-alkylation reaction of amines that allows carboxylic acids to be used directly in lieu of aldehydes and alkyl halides. The reactions are conducted in conventional laboratory glassware, are tolerant of other potentially reducible functional groups and have been demonstrated on a large scale (305 mmol with respect to the amine component). Finally, the elucidation of the role played by the residual carboxylic acid provides mechanistic insight that can potentially be transferred to the design of new reduction reactions in which silane reactivity is augmented in situ using Brønsted acids.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Prof Hon Wai Lam for the use of chiral HPLC equipment and Mr Andrew Payne for assistance with process safety studies. The authors gratefully acknowledge funding (CASE studentship E. S. L. and PDRA funding T. T.) from GlaxoSmithKline.Notes and references
- P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS PubMed.
- S. V. Ley and A. W. Thomas, Angew. Chem., Int. Ed., 2003, 42, 5400–5449 CrossRef CAS PubMed.
- L. J. Gooßen, L. Huang, M. Arndt, K. Gooßen and H. Heydt, Chem. Rev., 2015, 115, 2596–2697 CrossRef PubMed.
- A. F. Abdel-Magid, K. G. Carson, B. D. Harris, C. A. Maryanoff and R. D. Shah, J. Org. Chem., 1996, 61, 3849–3862 CrossRef CAS PubMed.
- M. H. S. A. Hamid, P. A. Slatford and J. M. J. Williams, Adv. Synth. Catal., 2007, 349, 1555–1575 CrossRef CAS.
- B. G. Reed-Berendt, K. Polidano and L. C. Morrill, Org. Biomol. Chem., 2019, 17, 1595–1607 RSC.
- P. S. Baran, J. Wang, M. Shang, H. Lundberg, K. S. Feu, S. J. Hecker, T. Qin and D. G. Blackmond, ACS Catal., 2018, 8, 9537–9542 CrossRef PubMed.
- A. Fawcett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Science, 2017, 357, 283–286 CrossRef CAS PubMed.
- C. P. Johnston, R. T. Smith, S. Allmendinger and D. W. C. MacMillan, Nature, 2016, 536, 322–325 CrossRef CAS PubMed.
- E. R. Welin, C. Le, D. M. Arias-Rotondo, J. K. McCusker and D. W. C. MacMillan, Science, 2017, 355, 380–385 CrossRef CAS PubMed.
- G. W. Gribble and P. W. Heald, Synthesis, 1978, 10, 766–768 CrossRef.
- P. Marchini, G. Liso, A. Reho, F. Liberatore and F. M. Moracci, J. Org. Chem., 1975, 40, 3453–3456 CrossRef CAS.
- G. W. Gribble, P. D. Lord, J. Skotnicki, S. E. Dietz, J. T. Eaton and J. Johnson, J. Am. Chem. Soc., 1974, 96, 7812–7814 CrossRef CAS.
- I. Sorribes, K. Junge and M. Beller, J. Am. Chem. Soc., 2014, 136, 14314–14319 CrossRef CAS PubMed.
- M. C. Fu, R. Shang, W. M. Cheng and Y. Fu, Angew. Chem., Int. Ed., 2015, 54, 9042–9046 CrossRef CAS PubMed.
- M. Minakawa, M. Okubo and M. Kawatsura, Tetrahedron Lett., 2016, 57, 4187–4190 CrossRef CAS.
- T. V. Q. Nguyen, W.-J. Yoo and S. Kobayashi, Adv. Synth. Catal., 2016, 358, 452–458 CrossRef CAS.
- K. G. Andrews, D. M. Summers, L. J. Donnelly and R. M. Denton, Chem. Commun., 2016, 52, 1855–1858 RSC.
- C. Qiao, X. Liu, X. Liu and L. He, Org. Lett., 2017, 19, 1490–1493 CrossRef CAS PubMed.
- L. Zhu, L.-S. Wang, B. Li and B. Fu, Catal. Sci. Technol., 2016, 6, 6172–6176 RSC.
- I. Sorribes, J. R. Cabrero-Antonino, C. Vicent, K. Junge and M. Beller, J. Am. Chem. Soc., 2015, 137, 13580–13587 CrossRef CAS PubMed.
- A. A. Nunez Margo, G. Eastham and D. J. Cole-Hamilton, Chem. Commun., 2007, 3154–3156 RSC.
- Z. Ruan, R. M. Lawrence and C. B. Cooper, Tetrahedron Lett., 2006, 47, 7649–7651 CrossRef CAS.
- M. Sayes and A. B. Charette, Green Chem., 2017, 19, 5060–5064 RSC.
- D. C. Braddock, P. D. Lickiss, B. C. Rowley, D. Pugh, T. Purnomo, G. Santhakumar and S. J. Fussell, Org. Lett., 2018, 20, 950–953 CrossRef CAS PubMed.
- E. Morisset, A. Chardon, J. Rouden and J. Blanchet, Eur. J. Org. Chem., 2020, 388–392 CrossRef CAS.
- L. G. Xie and D. J. Dixon, Nat. Commun., 2018, 9, 1–8 CrossRef PubMed.
- For related work involving amine synthesis by silane-mediated reduction followed by nucleophilic addition see L. G. Xie and D. J. Dixon, Chem. Sci., 2017, 8, 7492–7497 RSC.
- P. Trillo and H. Adolfsson, ACS Catal., 2019, 9, 7588–7595 CrossRef CAS.
- B. J. Simmons, M. Hoffmann, J. Hwang, M. K. Jackl and N. K. Garg, Org. Lett., 2017, 19, 1910–1913 CrossRef CAS PubMed.
- A recent comparison of metal chlorides which eliminates cost of any associated ligands, designated zinc as inexpensive and less than $100 per mol whilst platinum and iridium were classed as extremely expensive with prices over $10
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 per mol. See O. Berger, K. R. Winters, A. Sabourin, S. V. Dzyuba and J. L. Montchamp, Inorg. Chem. Front., 2019, 6, 2095–2108 CAS.
000 per mol. See O. Berger, K. R. Winters, A. Sabourin, S. V. Dzyuba and J. L. Montchamp, Inorg. Chem. Front., 2019, 6, 2095–2108 CAS. - S. Das, D. Addis, S. Zhou, K. Junge and M. Beller, J. Am. Chem. Soc., 2010, 132, 1770–1771 CrossRef CAS PubMed.
- S. Das, D. Addis, K. Junge and M. Beller, Chem.–Eur. J., 2011, 17, 12186–12192 CrossRef CAS PubMed.
- K. G. Andrews, R. Faizova and R. M. Denton, Nat. Commun., 2017, 8, 15913 CrossRef PubMed.
- A. Mordini, G. Reginato, M. Calamante and L. Zani, Curr. Top. Med. Chem., 2014, 14, 1308–1316 CrossRef CAS PubMed.
- S. J. Haycock-Lewandowski, A. Wilder and J. Åhman, Org. Process Res. Dev., 2008, 12, 1094–1103 CrossRef CAS.
- N. Z. Burns, P. S. Baran and R. W. Hoffmann, Angew. Chem., Int. Ed., 2009, 48, 2854–2867 CAS.
- Full details will be reported elsewhere..
- See the ESI† for further details..
- V. Bette, A. Mortreux, D. Savoia and J. F. Carpentier, Adv. Synth. Catal., 2005, 347, 289–302 CAS.
- A. Bezłada, M. Szewczyk and J. Mlynarski, J. Org. Chem., 2016, 81, 336–342 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of experimental procedures and characterisation of compounds. See DOI: 10.1039/d0sc02271c |
| This journal is © The Royal Society of Chemistry 2020 |