
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTemperature-modulated selective C(sp3)–H or C(sp2)–H arylation through palladium catalysis†
Thirupathi
Gogula‡
,
Jinquan
Zhang‡
,
Madhava Reddy
Lonka
,
Shuaizhong
Zhang
and
Hongbin
Zou
 *
*
College of Pharmaceutical Sciences, Zhejiang University, Hangzhou, Zhejiang 310058, P. R. China. E-mail: zouhb@zju.edu.cn
First published on 24th September 2020
Abstract
Transition metal-catalysed C–H bond functionalisations have been extensively developed in organic and medicinal chemistry. Among these catalytic approaches, the selective activation of C(sp3)–H and C(sp2)–H bonds is particularly appealing for its remarkable synthetic versatility, yet it remains highly challenging. Herein, we demonstrate the first example of temperature-dependent selective C–H functionalisation of unactivated C(sp3)–H or C(sp2)–H bonds at remote positions through palladium catalysis using 7-pyridyl-pyrazolo[1,5-a]pyrimidine as a new directing group. At 120 °C, C(sp3)–H arylation was triggered by the chelation of a rare [6,5]-fused palladacycle, whereas at 140 °C, C(sp2)–H arylation proceeded instead through the formation of a 16-membered tetramer containing four 7-pyridyl-pyrazolo[1,5-a]pyrimidine–palladium chelation units. The subsequent mechanistic study revealed that both C–H activations shared a common 6-membered palladacycle intermediate, which was then directly transformed to either the [6,5]-fused palladacycle for C(sp3)–H activation at 120 °C or the tetramer for C(sp2)–H arylation at 140 °C with catalytic amounts of Pd(OAc)2 and AcOH. Raising the temperature from 120 °C to 140 °C can also convert the [6,5]-fused palladacycle to the tetramer with the above-mentioned catalysts, hence completing the C(sp2)–H arylation ultimately.
Introduction
Over the past few decades, C–H bond functionalisation catalysed by transition metals has emerged as a powerful and versatile synthetic method to construct novel carbon–carbon and carbon–heteroatom bonds.1 Complexes of various transition metals, such as Pd, Ru, Rh, Co, Ni and so on (Scheme 1a), have been successfully applied in C–H bond activation reactions,2 providing atom- and step-economical synthetic pathways to a myriad of organic compounds, including biologically active natural products and drug molecules.3 Meanwhile, a wide variety of directing groups have been designed and evaluated for regioselective transition metal-catalysed C(sp3)–H or C(sp2)–H functionalisations (Scheme 1a),4 most of which proceeded with either covalent or transient directing groups,5 involving an exogenous auxiliary and ligand in a bidentate- or monodentate-chelation system.6 The less-hindered and more-reactive C(sp2)–H is generally easier to activate than the C(sp3)–H bond in similar proximity to directing groups.7 Thus, most of the reported Pd-catalysed functionalisations of C(sp3)–H and C(sp2)–H bonds occurred separately on different substrates, and the selectivity between the two bonds was largely controlled by particular parameters, such as the choice of metal catalysts, directing groups, ligands or auxiliaries.8,9 To date, no single substrate has been reportedly able to initiate both C(sp3)–H and C(sp2)–H activations concurrently. A powerful synthetic strategy remains highly desirable to realize the selective direct functionalisation of unactivated C(sp3)–H and C(sp2)–H bonds.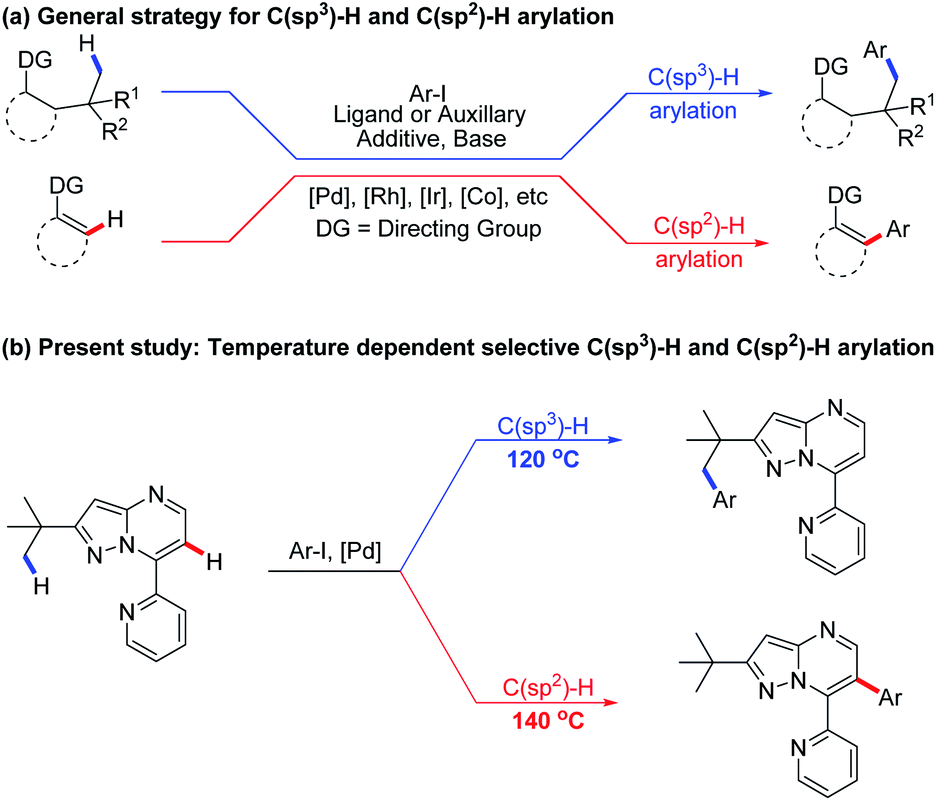 | ||
| Scheme 1 General strategy (a) and temperature dependent method (b) for C(sp3)–H and C(sp2)–H arylation. | ||
Pyrazolo[1,5-a]pyrimidines are a group of ubiquitous structures used in medicinal chemistry, materials science, and dyestuff industry.10,11 The ongoing research has given rise to diverse uses of pyrazolo[1,5-a]pyrimidines as functional groups.12 Very recently, we reported employing pyrazolo[1,5-a]pyrimidine as a novel directing group for rhodium-catalysed C(sp2)–H amidation under mild reaction conditions13 along with other C–H amidation reactions.14 Based on this and previous reports of substrate-directed C–H bond activation, we chose 7-pyridyl-pyrazolo[1,5-a]pyrimidine, a derivative of pyrazolo[1,5-a]pyrimidine, as a directing group in this study of palladium-catalysed C–H arylation with aryl iodides (Scheme 1b). The additional nitrogen atom on pyridine provides additional propulsion for the reaction by participating in cyclopalladation. To our excitement, selective arylation of 7-pyridyl-pyrazolo[1,5-a]pyrimidine was observed, resulting in the C(sp3)–H activation product at 120 °C and the C(sp2)–H arylation product at 140 °C under otherwise the same reaction conditions (Scheme 1b). To the best of our knowledge, this is the first report on a temperature-controlled selective C(sp3)–H or C(sp2)–H activation directed by a single substrate.
Results and discussion
We focused our initial efforts on seeking the optimal conditions for Pd(II)-catalysed C(sp3)–H arylation of 2-(tert-butyl)-7-(pyridin-2-yl)pyrazolo [1,5-a]pyrimidine (1a) using 4-iodoanisole (2a) as a model substrate. Treating 1a with 1.5 equiv. of 2a, 10 mol% of Pd(OAc)2, and 1.5 equiv. of AgTFA in AcOH at 120 °C for 24 h gave the desired C(sp3)–H arylation product 3aa in 18% yield, along with a small amount of unexpected 4aa, a product from C(sp2)–H arylation (Table 1, entry 1). Intrigued by this result, we next researched how varied conditions could affect the direction of the reaction. When AgOAc, AgF, and Ag2CO3 were attempted as additives, the yields of desired products 3aa and 4aa were both decreased, while the combination of AgBr with (NMe4)OC(CF3)3 (ref. 2h) failed to give products (see the ESI, Table S1†). And the screening result of HFIP, TFE, and DCE indicated HFIP as the best solvent choice (see the ESI, Table S1†). We then modified the solvent system with various ratios of AcOH and HFIP (Table 1, entries 2–5). The turnover of 3aa steadily grew with the increasing proportion of HFIP and reached the maximum of 58% in pure HFIP solvent (Table 1, entry 5). Next, we examined the production of 3aa under various reaction times and temperatures. By extending the reaction time to 36 h, we obtained 3aa in the highest yield of 74% (Table 1, entry 6), while further prolonging the reaction time reduced the product yield instead (Table 1, entry 7). On the other hand, both temperature decrease and increase from 120 °C attenuated the production of 3aa (Table 1, entries 8 and 9), whereas the yield of 4aa grew rapidly to 35% at a raised temperature of 130 °C (Table 1, entry 9). This phenomenon led us to further investigate the impact of temperature on the reaction (see the ESI, Table S1 and Fig. S1†). When we lowered the temperature to 110 °C, yields of both 3aa and 4aa were observed to decrease (Table 1, entry 10). In contrast, raising the temperature to 140 °C drastically hindered the C(sp3)–H arylation but promoted the C(sp2)–H activation as evidenced by the 3aa product decrease and 4aa yield increase (Table 1, entry 11). A further rise of the temperature to 150 °C and then to 160 °C only reduced the product yields (Table 1, entries 12 and 13). Furthermore, extending the reaction time to 48 h at 140 °C provided the highest yield of 4aa (Table 1, entry 14). Lastly, two alternative palladium catalysts were tested at optimal temperatures, which produced less impressive results than Pd(OAc)2 (see the ESI, Table S1†). We therefore concluded that both arylations share the same optimal reaction conditions in terms of catalyst (10 mol% of Pd(OAc)2), additive (1.5 equiv. of AgTFA) and solvent (HFIP), but the C(sp3)–H activation proceeded most potently at 120 °C given the reaction period of 36 h (Table 1, entry 6) and the C(sp2)–H arylation at 140 °C for 48 h (Table 1, entry 14).|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Temp (°C) | Time (h) | Yieldb (%) | |
| 3aa | 4aa | ||||
| a Reaction conditions: 1a (1.0 mmol), 2a (1.5 mmol), Pd(OAc)2 (10 mol%), AgTFA (1.5 mmol), solvent (3 mL), 100–160 °C, 24–48 h. b Isolated yield. | |||||
| 1 | AcOH | 120 | 24 | 18 | 6 |
| 2 | HFIP/AcOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
120 | 24 | 24 | 18 |
| 3 | HFIP/AcOH (6:4) |
120 | 24 | 33 | 14 |
| 4 | HFIP/AcOH (9:1) |
120 | 24 | 46 | 10 |
| 5 | HFIP | 120 | 24 | 58 | <5 |
| 6 | HFIP | 120 | 36 | 74 | <5 |
| 7 | HFIP | 120 | 48 | 61 | <5 |
| 8 | HFIP | 100 | 36 | 12 | <2 |
| 9 | HFIP | 130 | 36 | 20 | 35 |
| 10 | HFIP | 110 | 36 | 15 | <5 |
| 11 | HFIP | 140 | 36 | <5 | 42 |
| 12 | HFIP | 150 | 36 | <5 | 31 |
| 13 | HFIP | 160 | 36 | <5 | 26 |
| 14 | HFIP | 140 | 48 | <5 | 58 |
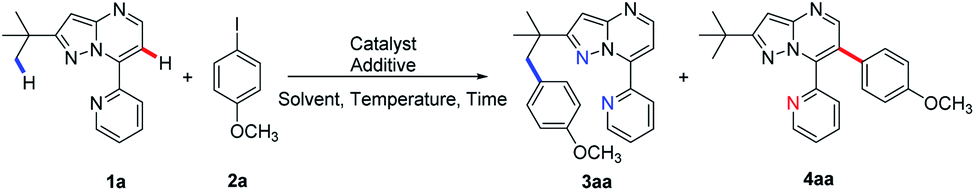
To expand the scope of the newly established protocol, we first subjected a series of structurally diverse aryl iodides (Scheme 2) to the optimal C(sp3)–H activation conditions (Table 1, entry 6). As a result, both electron-donating and -withdrawing aryl iodides were well tolerated by the reaction. For instance, aryl iodide harboring electron-donating methoxy (2a) at the para position underwent the reaction smoothly and gave 3aa in particularly good efficiency (Scheme 2, 74% yield). Aryl iodides with the halogen substituents (F, Cl, Br and I) also afforded desired products (3ab–3ae) in synthetically useful yields. Among them, the structure of 3ac was confirmed by X-ray diffraction analysis. Meanwhile, the presence of a strong electron-withdrawing group, such as a trifluoromethyl or nitro group at the para-position, was also compatible with the transformation (3af and 3ag). It is worth mentioning that bulky aryl partners, such as biphenyl and 4-iodo biphenyl, likewise underwent the reaction giving moderate to good yields (3ah and 3ai). Interestingly, aryl iodides with a methoxy substituent at the meta- or ortho-position gave a slightly less yield (3ak and 3al) than those with para-methoxy (3aa). Furthermore, aryl iodides hosting multiple substituents were also tolerated in moderate efficiency (3am and 3an). To further illustrate the potential application of this newly established protocol, a gram-scale experiment using 1a (1.008 g) and 4-phenyl iodobenzene (2h, 1.679 g) was conducted. To our delight, the desired product 3ah was furnished in 48% yield (0.775 g) under the standard C(sp3)–H arylation conditions.
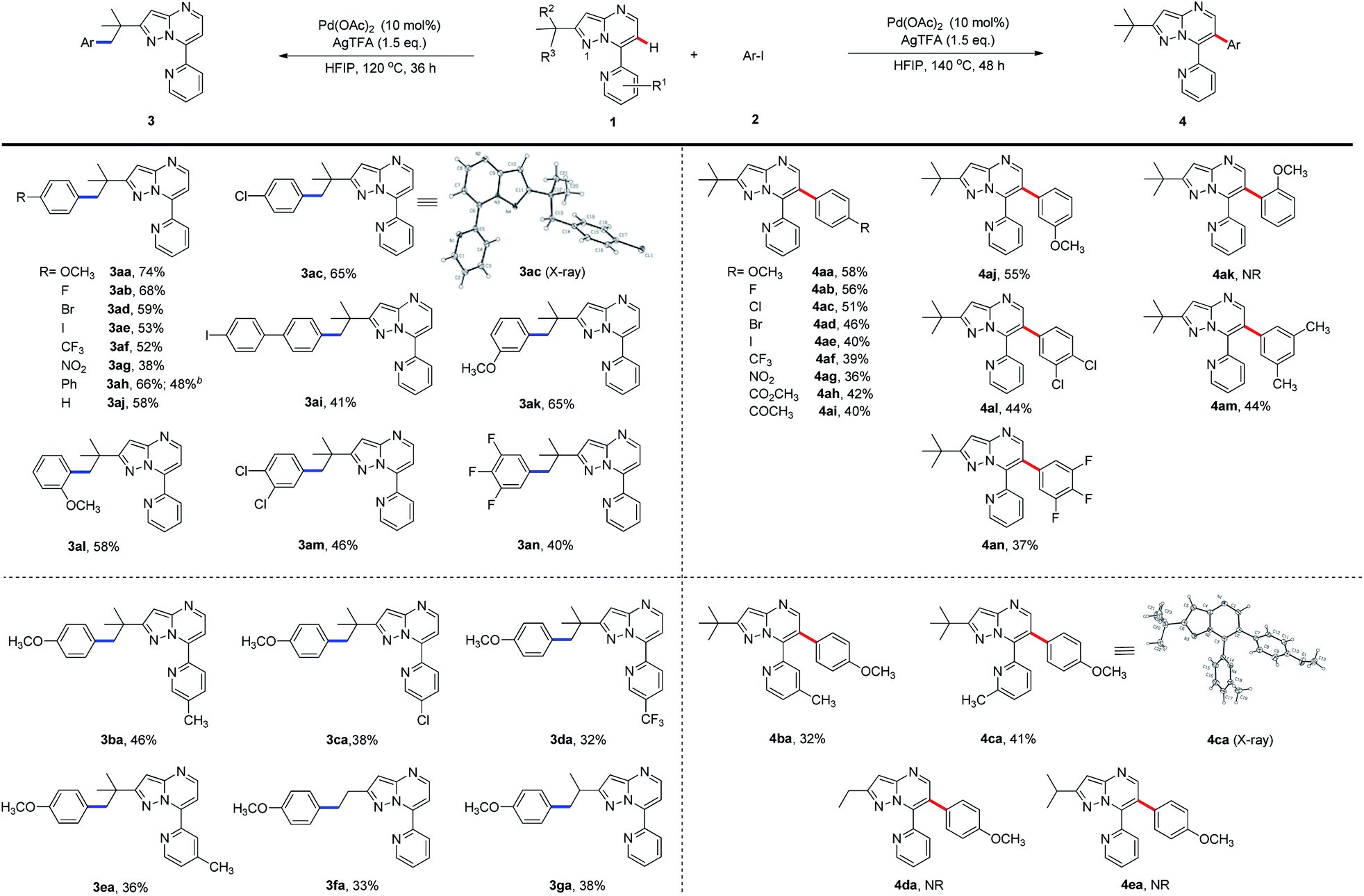 | ||
| Scheme 2 Scope of aryl iodides in Pd-catalysed C(sp3)–H and C(sp2)–H activations. (a) All reactions were carried out using 2-(tert-butyl)-7-pyridyl-pyrazolo[1,5-a]pyrimidine 1 (1.0 mmol), Ar–I (2, 1.5 mmol), Pd(OAc)2 (10 mol%), AgTFA (1.5 mmol), and HFIP (3.0 mL), at 120 °C, 36 h for C(sp3)–H arylation and at 140 °C, 48 h for C(sp2)–H arylation. (b) Yield of the scale-up reaction. | ||
As shown in Scheme 2, the generality of C(sp2)–H arylation with respect to aryl iodides was also examined under the optimal reaction conditions (Table 1, entry 14). Aryl iodide with the electron-donating para-methoxy group was well tolerated (4aa), so were those with electron-withdrawing halogen substituents (F, Cl, Br, and I) at the para-position, although giving slightly decreased yields (4ab–4ae). Strong electrophiles, such as trifluoromethyl and nitro groups, further reduced the reaction efficiency (4af, 4ag). Meanwhile, aryl iodide hosting electron-deficient ester or keto substituents at the para-position underwent the C(sp2)–H arylation in moderate yields (4ah and 4ai). Aryl iodide containing methoxy at the meta-position (4aj) was also well tolerated by the arylation. However, the ortho-methoxy-substituted aryl iodide failed to provide the product 4ak, presumably due to the steric hindrance of the methoxy group. Nonetheless, aryl iodides with multiple substituents went through the desired transformation in moderate yields (4al–4an).
We then sought to explore the scope of the C–H arylation system regarding 7-pyridyl-pyrazolo[1,5-a]pyrimidine (1a) containing various substituted pyridine rings using 4-iodoanisole (2a). Methyl substitution at the 4-, 5- or 6-position of the pyridine ring was tested for the production of C(sp3)–H and C(sp2)–H arylation products. Moderate efficiency was observed for C(sp3)–H arylated products (3ba and 3ea) with the methyl substituent at 4- and 5-positions and for C(sp2)–H arylated products (4ba and 4ca) with the methyl substituent at 4- and 6-positions, while very little (<5%) product was detected for the rest two cases. X-ray crystallography confirmed the structure of 4ca. However, 3-methyl-substituted 1a failed to furnish any arylated product, presumably because the 3-methyl group sterically hindered the planar conformation of pyridine and the pyrazolo[1,5-a]pyrimidine rings to form a palladacycle intermediate for C–H activation. Meanwhile, 7-pyridyl-pyrazolo[1,5-a]pyrimidines bearing electron-withdrawing chloro or trifluoromethyl substituents were tolerated by the reaction, giving the C(sp3)–H arylated products in relatively low yields (3ca and 3da) but no detectable C(sp2)–H arylated products. We next investigated the selectivity of the system using 2-substituted-7-pyridyl-pyrazolo[1,5-a]pyrimidines under standard reaction conditions. Exposing 2-ethyl- or isopropyl-substituted 1a to 2a afforded only moderate yields of C(sp3)–H arylated products (3fa and 3ga), while C(sp2)–H arylated products 4da and 4ea were not detected. Taken together, these results indicated that the C–H bond at the γ-position in relation to N1 is essential for C(sp3)–H arylation.
We have thus far realized the temperature-modulated selective C(sp3)–H and C(sp2)–H arylation of 7-pyridyl-pyrazolo[1,5-a]pyrimidines, as shown in Scheme 2. To the best of our knowledge, there is no report of such a palladium-catalysed diarylation of a single substrate. Next, we turned our attention to examine the sequential arylation processes of this catalytic system (Scheme 3). Interestingly, the reaction of C(sp2)–H arylated product 4af with 4-iodoanisole (2a) under standard conditions at 120 °C further led to the formation of C(sp3)–H arylated product 5 (Scheme 3a). On the other hand, the treatment of 3af from C(sp3)–H functionalisation with 4-(trifluoromethyl)-iodobenzene (2f) at 140 °C gave C(sp2)–H arylated product 6 (Scheme 3b). Containing both primary and methylene C–H bonds, compound 6 was then chosen for the subsequent experiment with 2a at 120 °C. Intriguingly, an unexpected regioselective arylation of C(sp3)–H occurred, as evidenced by a 31% yield of product 7 (Scheme 3c). Such a preference for the methylene C–H over the primary C–H bond is rare since the latter is usually more reactive than the former during the arylation reaction.15 Encouraged by this finding, we performed a one-pot sequential diarylation experiment by subjecting 1a to 2 equiv. of 2f and AgTFA at 120 °C for 36 h, followed by the treatment at 140 °C for another 48 h (Scheme 3d). Gratifyingly, the same product 6 obtained in Scheme 3b was once again furnished in a moderate yield of 26%, as a result of sequential C(sp3)–H and C(sp2)–H arylations of 1a. Deuterium-labeling experiments with 1a in the presence of HFIP-d2 were also performed (see the Mechanistic studies part of the ESI† for details). The results showed that 14% deuteration was observed at C(sp2)–H at 140 °C, while no deuteration was detected at C(sp2)–H or C(sp3)–H at 120 °C, which indicate that C(sp2)–H activation was more favored at 140 °C. These findings further confirmed that the selective C–H arylation of 7-pyridyl-pyrazolo[1,5-a]pyrimidines could be readily controlled by temperature, potentially opening new avenues for novel strategies to prepare functionally important polyarylates.16
 | ||
| Scheme 3 Sequential C(sp3)–H and C(sp2)–H arylation reactions. All reactions were performed using the 7-pyridyl-pyrazolo[1,5-a]pyrimidine derivative (1.0 mmol), aryl iodide (1.5 mmol), 10 mol% of Pd(OAc)2, and 1.5 equiv. of AgTFA in HFIP unless listed in the reaction equation. (a) Reaction of C(sp2)–H arylated product 4af with 4-iodoanisole (2a). (b) Reaction of C(sp3)–H arylated product 3af with 1-iodo-4-(trifluoromethyl)benzene (2f). (c) Reaction of C(sp2)–H and C(sp3)–H arylated product 6 with 2a. (d) Reaction of 1a with 2f under sequential C(sp3)–H and C(sp2)–H arylation conditions. | ||
Subsequently, we sought to uncover the mechanism of this selective C–H arylation system. We first synthesized and characterized a group of complexes which are plausibly related to the potential reaction intermediates (Scheme 4, see the Mechanistic studies part of the ESI† for details). The reaction of 2-(tert-butyl)-7-(pyridin-2-yl)pyrazolo[1,5-a]pyrimidine (1a) with Pd(OAc)2 in HFIP solvent at 120 °C afforded the corresponding [6,5]-fused palladacycle 8, whereas exposing 1a to Pd(OAc)2 in HFIP at 140 °C led to a possible intermediate 9 which was further recrystallized from chloroform and ethyl acetate to give a 16-membered tetramer 10 (Scheme 4a).8b It is an unexpected finding since the palladium chelation dimer is more favorable as the reaction intermediate, instead of the herein reported 16-membered tetramer, for C(sp2)–H activation in previous reports.17 The complexes 8 and 10 were unambiguously characterized by single-crystal X-ray diffraction analysis (Fig. 1). Intrigued by the complex structures, we then isolated a six-membered intermediate 11 through a mild reaction between 1a and Pd(OAc)2 in HFIP at 25 °C for 30 minutes, wherein Pd(OAc)2 was able to bond with N1 and the pyridine nitrogen. This intermediate could be readily transformed to complex 8 or 10 at 120 °C or 140 °C, respectively, indicating that 11 might be the shared intermediate for the C(sp3)–H and C(sp2)–H arylation reactions.
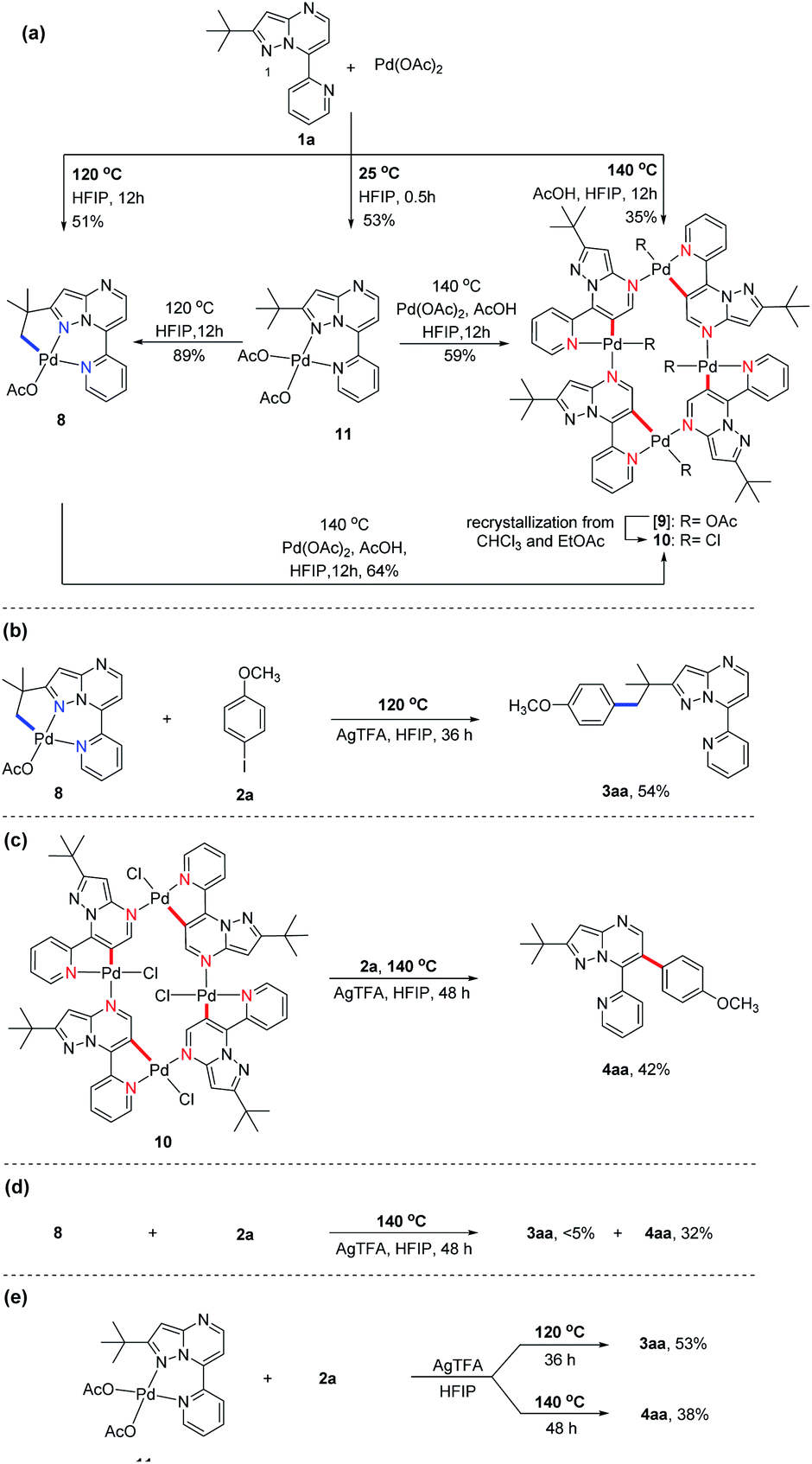 | ||
| Scheme 4 Mechanistic evidence for temperature-controlled selective C–H activation. (a) Preparation of intermediates 8, 10 and 11. (b) Reaction of 8 with 4-iodoanisole (2a) under C(sp3)–H arylation condition. (c) Reaction of 10 with 2a under C(sp2)–H arylation condition. (d) Reaction of 8 with 2a under C(sp2)–H arylation condition. (e) Reaction of 11 with 2a under C(sp3)–H or C(sp2)–H arylation condition. | ||
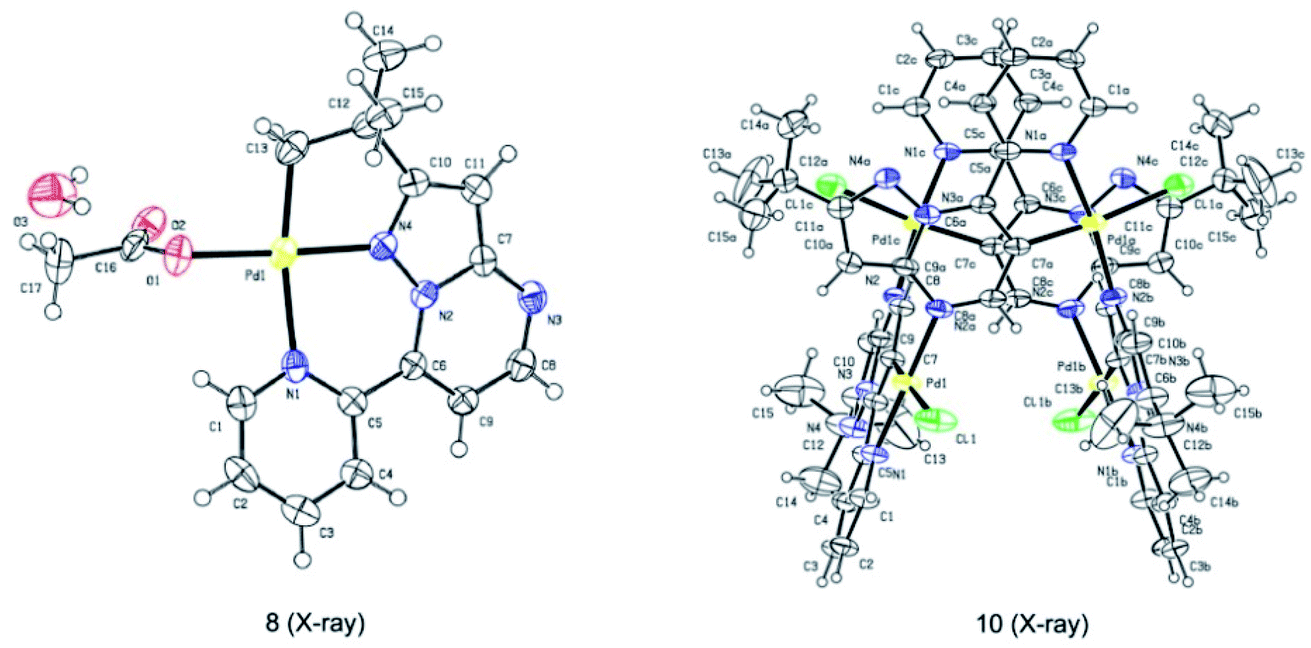 | ||
| Fig. 1 Molecular structure and atom numbering scheme for complexes 8 and 10. | ||
To confirm the relationship of newly isolated intermediates and their subsequent products, we carried out additional experiments (see the Mechanistic studies part of the ESI† for details). As expected, complex 8 reacted with 2a to give the C(sp3)–H arylated product 3aa at 120 °C (Scheme 4b) and complex 10 reacted with 2a to afford C(sp2)–H arylated product 4aa at 140 °C (Scheme 4c). Interestingly, the reaction of complex 8 with 2a at 140 °C furnished 4aa as the primary product (Scheme 4d), a result in accordance with the previous finding wherein the C(sp3)–H activation intermediate 8 could be directly converted to C(sp2)–H activation intermediate 10 (Scheme 4a). A trace amount of 3aa obtained from this reaction might be ascribed to the weak reaction between 8 and 2a as well as the slow conversion of 8 to 10. Meanwhile, direct treatment of the shared intermediate 11 with 2a provided 3aa at 120 °C and 4aa at 140 °C as expected (Scheme 4e).
Together, these results delineated the catalytic loop of the temperature-modulated selective C(sp3)–H and C(sp2)–H arylations, wherein the hanging pyridine nitrogen, appearing in each palladacycle intermediate, plays a key role. This result is in line with the outcome of another experiment, which employed an alternative substrate without the nitrogen atom of interest and produced no C–H arylated product under the otherwise same reaction conditions (see the ESI, Scheme S1†). Furthermore, the two nitrogen atoms, N1 and the pyridine nitrogen, act together as a bidentate ligand which coordinates to Pd(II) species at 120 °C to form a highly reactive complex that could effectively activate the proximately located tert-butyl C–H bonds. And the formation of the 16-membered complex from the tetrachelation of palladium and 1a also provided insights into the C(sp2)–H arylation cycle.
On the basis of our mechanistic studies and previous research reports,18,19 we proposed a plausible reaction mechanism for this temperature-controlled Pd-catalysed selective arylation of C(sp3)–H or C(sp2)–H bonds (Scheme 5). Initially, 2-(tert-butyl)-7-(pyridin-2-yl)pyrazolo[1,5-a]pyrimidine (1a) reacts with Pd(OAc)2 to form the six-membered palladacycle 11, which is subsequently converted to [6,5]-fused palladacycle 8 at 120 °C or a 16-membered tetrachelated complex 9 at 140 °C. These palladacycles then undergo oxidative addition with aryl iodide and give the intermediates A or B, respectively. Finally, compounds A and B go through reductive elimination and produce either the C(sp3)–H or C(sp2)–H arylated product 3 or 4, respectively, closing both catalytic cycles.
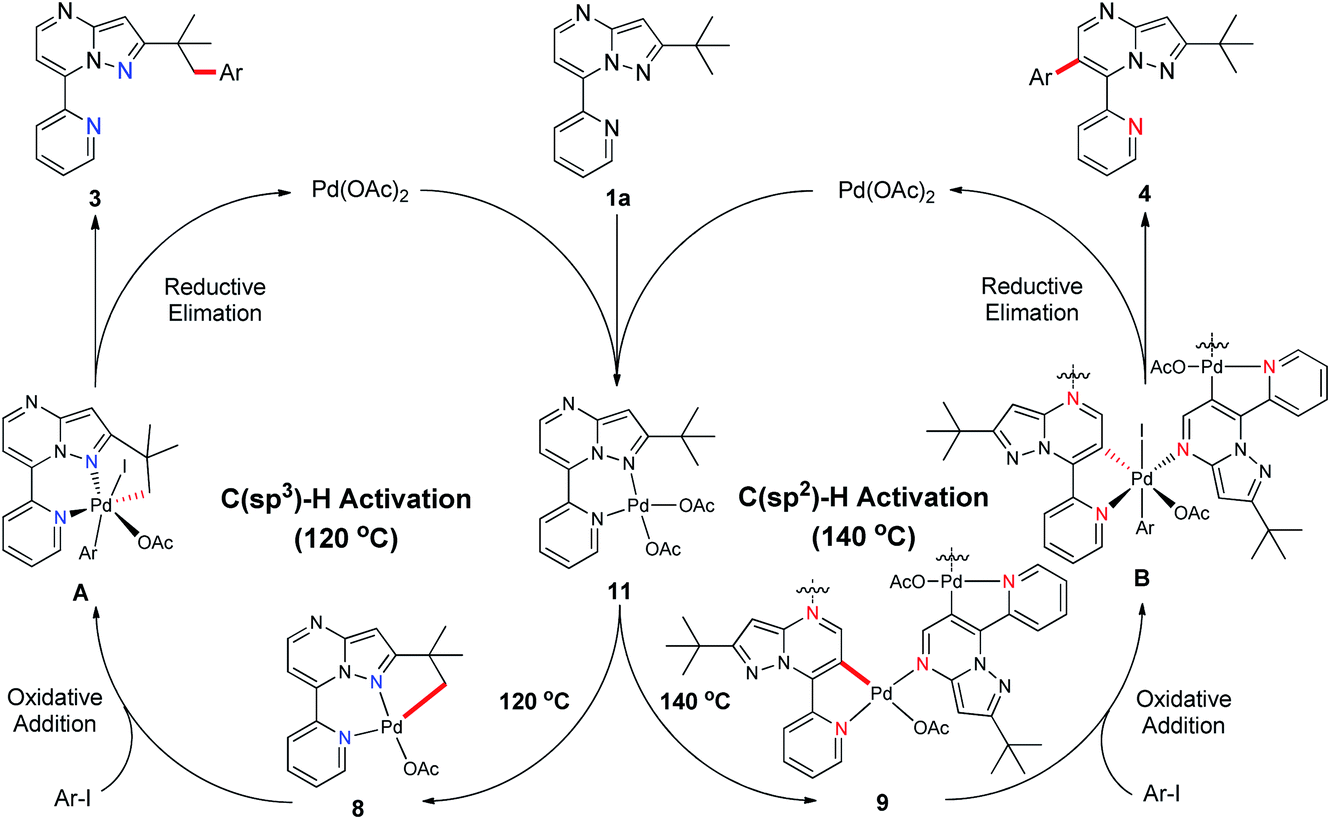 | ||
| Scheme 5 Proposed reaction pathways for C(sp3)–H and C(sp2)–H activation. | ||
Conclusions
In summary, we established an unprecedented temperature-controlled selective C(sp3)–H or C(sp2)–H activation under palladium catalysis. At 120 °C, a new 7-pyridyl-pyrazolo[1,5-a]pyrimidine acts as a bidentate ligand and forms a six-membered palladacycle to subsequently give C(sp3)–H arylated products. Alternatively, at 140 °C, the substrate serves as a monodentate ligand and undergoes the C(sp2)–H arylation reaction through the formation of a five-membered palladacycle in a 16-membered tetrachelation complex. These two direct palladacycle compounds were well characterized and their shared intermediate was also isolated to further illustrate the mutual transformation in the catalytic cycles. This novel temperature-dependent catalysed arylation approach provides promising potential to selectively produce diverse arylated C–H products from 7-pyridyl-pyrazolo[1,5-a]pyrimidines, which may propel the discovery of more promising directing groups as well as introduce new strategies for selective C–H bond functionalisation with precision, hence uncovering more possibilities for transition metal catalysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21472170) and the Zhejiang Provincial Fund for Distinguished Young Scholars (LR15B020001). We thank prof. Yongping Yu (Zhejiang University, China) for useful discussion.Notes and references
- (a) R. G. Bergman, Nature, 2007, 446, 391 CrossRef CAS; (b) H. M. L. Davies, J. Du Bois and J.-Q. Yu, Chem. Soc. Rev., 2011, 40, 1855 RSC; (c) L. McMurray, F. O'Hara and M. Gaunt, Chem. Soc. Rev., 2011, 40, 1885 RSC; (d) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS; (e) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef CAS; (f) H. M. Davies and D. J. Morton, J. Org. Chem., 2016, 81, 343 CrossRef CAS; (g) J. C. K. Chu and T. Rovis, Angew. Chem., Int. Ed., 2018, 57, 62 CrossRef CAS.
- (a) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS; (b) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS; (c) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS; (d) M. Moselage, J. Li and L. Ackermann, ACS Catal., 2016, 6, 498 CrossRef CAS; (e) R. Shang and L. I. E. Nakamura, Chem. Rev., 2017, 117, 9086 CrossRef CAS; (f) D. W. Ji, Y. C. Hu, H. Zheng, C. Y. Zhao, Q. A. Chen and V. M. Dong, Chem. Sci., 2019, 10, 6311 RSC; (g) H. Kim, R. S. Thombal, H. D. Khanal and Y. R. Lee, Chem. Commun., 2019, 55, 13402 RSC; (h) A. Panigrahi, D. Whitaker, I. J. Vitorica-Yrezabal and I. Larrosa, ACS Catal., 2020, 10, 2100 CrossRef CAS.
- (a) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792 CrossRef CAS; (b) Z. Huang, H. N. Lim, F. Mo, M. C. Young and G. Dong, Chem. Soc. Rev., 2015, 44, 7764 RSC; (c) R. S. Thombal and Y. R. Lee, Org. Lett., 2020, 22, 3397 CrossRef CAS.
- (a) Y. Xu, M. C. Young, C.-P. Wang, D. M. Magness and G. Dong, Angew. Chem., Int. Ed., 2016, 55, 9084 CrossRef CAS; (b) A. Yada, W. Liao, Y. Sato and M. Murakami, Angew. Chem., Int. Ed., 2017, 56, 1073 CrossRef CAS; (c) H. Lin, C. Wang, T. D. Bannister and T. M. Kamenecka, Chem.–Eur. J., 2018, 24, 9535 CrossRef CAS.
- (a) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236 CrossRef CAS; (b) F.-L. Zhang, K. Hong, T.-J. Li, H. Park and J.-Q. Yu, Science, 2016, 351, 252 CrossRef CAS.
- (a) K. M. Engle and J. Q. Yu, J. Org. Chem., 2013, 78, 8927 CrossRef CAS; (b) O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053 CrossRef CAS; (c) Y. L. Liu and H. Ge, Nat. Chem., 2017, 9, 26 CrossRef CAS.
- (a) Y. Wu, Y. Q. Chen, T. Liu, M. D. Eastgate and J. Q. Yu, J. Am. Chem. Soc., 2016, 138, 14554 CrossRef CAS; (b) Y.-J. Mao, S.-J. Lou, H.-Y. Hao and D. Xu, Angew. Chem., Int. Ed., 2018, 57, 14085 CrossRef CAS; (c) G. Xia, J. Weng, L. Liu, P. Verma, Z. Li and J.-Q. Yu, Nat. Chem., 2019, 11, 571 CrossRef CAS; (d) S. Rej, Y. Ano and N. Chatani, Chem. Rev., 2020, 120, 1788 CrossRef CAS.
- (a) X. Chen, C. E. Goodhue and J.-Q. Yu, J. Am. Chem. Soc., 2006, 128, 12634 CrossRef CAS; (b) H. Park, P. Verma, K. Hong and J. Q. Yu, Nat. Chem., 2018, 10, 755 CrossRef CAS; (c) Y. Q. Chen, Z. Wang, Y. Wu, S. R. Wisniewski, J. X. Qiao, W. R. Ewing, M. D. Eastgate and J. Q. Yu, J. Am. Chem. Soc., 2018, 140, 17884 CrossRef CAS; (d) A. Mondal, H. Chen, L. Flamig, P. Wedi and M. Van Gemmeren, J. Am. Chem. Soc., 2019, 141, 18662 CrossRef CAS.
- (a) B.-F. Shi, N. Maugel, Y.-H. Zhang and J.-Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 4882 CrossRef CAS; (b) H. Park, N. Chekshin, P.-X. Shen and J.-Q. Yu, ACS Catal., 2018, 8, 9292 CrossRef CAS; (c) W. Liu, Z. Ren, A. T. Bosse, K. Liao, E. L. Goldstein, J. Bacsa, D. G. Musaev, B. M. Stoltz and H. M. L. Davies, J. Am. Chem. Soc., 2018, 140, 12247 CrossRef CAS.
- (a) A. Damont, V. Médran-Navarrete, F. Cacheux, B. Kuhnast, G. Pottier, N. Bernards, F. Marguet, F. Puech, R. Boisgard and F. Dollé, J. Med. Chem., 2015, 58, 7449 CrossRef CAS; (b) S. Cherukupalli, R. Karpoormath, B. Chandrasekaran, G. A. Hampannavar, N. Thapliyal and V. N. Palakollu, Eur. J. Med. Chem., 2017, 126, 298 CrossRef CAS.
- (a) A. G. Al-Sehemi, A. Irfan and A. M. Fouda, Spectrochim. Acta, Part A, 2013, 111, 223 CrossRef CAS; (b) H. Kim, A. Jo, J. Ha, Y. Lee, Y. S. Hwang and S. B. A. Park, Chem. Commun., 2016, 52, 7822 RSC.
- (a) J. K. Jiang, X. Huang, K. Shamim, P. R. Patel, A. Lee, A. Q. Wang, K. Nguyen, G. Tawa, G. D. Cuny, P. B. Yu, W. Zheng, X. Xu, P. Sanderson and W. Huang, Bioorg. Med. Chem. Lett., 2018, 28, 3356 CrossRef CAS; (b) J.-C. Castillo, A. Tigreros and J. Portilla, J. Org. Chem., 2018, 83, 10887 CrossRef CAS.
- T. Gogula, J. Q. Zhang and H. B. Zou, Org. Lett., 2019, 21, 5933 CrossRef CAS.
- J. Q. Zhang, H. Xie, H. Zhu, S. Zhang, M. R. Lonka and H. B. Zou, ACS Catal., 2019, 9, 10233 CrossRef CAS.
- P. Dolui, J. Das, H. B. Chandrashekar, S. S. Anjana and D. Maiti, Angew. Chem., Int. Ed., 2019, 58, 13773 CrossRef CAS.
- T. Okazawa, T. Satoh, M. Miura and M. Nomura, J. Am. Chem. Soc., 2002, 124, 5286 CrossRef CAS.
- (a) N. R. Deprez and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 11234 CrossRef CAS; (b) T. Privalov, C. Linde, K. Zetterberg and C. Moberg, Organometallics, 2005, 24, 885 CrossRef CAS; (c) D. C. Powers, M. A. L. Geibel, J. E. M. N. Klein and T. Ritter, J. Am. Chem. Soc., 2009, 131, 17050 CrossRef CAS.
- (a) V. S. Thirunavukkarasu, K. Parthasarathy and C.-H. Cheng, Angew. Chem., Int. Ed., 2008, 47, 9462 CrossRef CAS; (b) P. Sehnal, R. J. K. Taylor and I. J. Fairlamb, Chem. Rev., 2010, 110, 824 CrossRef CAS.
- (a) R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242 RSC; (b) W. Li, Z. Yin, X. Jiang and P. Sun, J. Org. Chem., 2011, 76, 8543 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, synthetic procedures, characterization data and NMR spectra. CCDC 1987481–1987484. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc02328k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |