
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMethylbismuth: an organometallic bismuthinidene biradical†
Deb Pratim
Mukhopadhyay
 a,
Domenik
Schleier
a,
Sara
Wirsing
a,
Jacqueline
Ramler
b,
Dustin
Kaiser
a,
Engelbert
Reusch
a,
Patrick
Hemberger
c,
Tobias
Preitschopf
a,
Ivo
Krummenacher
b,
Bernd
Engels
*a,
Ingo
Fischer
*a and
Crispin
Lichtenberg
*b
a,
Domenik
Schleier
a,
Sara
Wirsing
a,
Jacqueline
Ramler
b,
Dustin
Kaiser
a,
Engelbert
Reusch
a,
Patrick
Hemberger
c,
Tobias
Preitschopf
a,
Ivo
Krummenacher
b,
Bernd
Engels
*a,
Ingo
Fischer
*a and
Crispin
Lichtenberg
*b
aInstitute of Physical and Theoretical Chemistry, University of Würzburg, Am Hubland, D-97074 Würzburg, Germany. E-mail: bernd.engels@uni-wuerzburg.de; ingo.fischer@uni-wuerzburg.de
bInstitute of Inorganic Chemistry, University of Würzburg, Am Hubland, D-97074 Würzburg, Germany. E-mail: crispin.lichtenberg@uni-wuerzburg.de
cLaboratory for Femtochemistry and Synchrotron Radiation, Paul Scherrer Institut (PSI), CH-5232 Villigen, Switzerland. E-mail: patrick.hemberger@psi.ch
First published on 3rd June 2020
Abstract
We report the generation, spectroscopic characterization, and computational analysis of the first free (non-stabilized) organometallic bismuthinidene, BiMe. The title compound was generated in situ from BiMe3 by controlled homolytic Bi–C bond cleavage in the gas phase. Its electronic structure was characterized by a combination of photoion mass-selected threshold photoelectron spectroscopy and DFT as well as multi-reference computations. A triplet ground state was identified and an ionization energy (IE) of 7.88 eV was experimentally determined. Methyl abstraction from BiMe3 to give [BiMe2]• is a key step in the generation of BiMe. We reaveal a bond dissociation energy of 210 ± 7 kJ mol−1, which is substantially higher than the previously accepted value. Nevertheless, the homolytic cleavage of Me–BiMe2 bonds could be achieved at moderate temperatures (60–120 °C) in the condensed phase, suggesting that [BiMe2]• and BiMe are accessible as reactive intermediates under these conditions.
Introduction
Low-valent molecular compounds of group 15 elements, E–R, with the central atom E in the oxidation state of +1 are highly reactive, electron-deficient species (E = N–Bi; R = monoanionic ligand).1 In principle, they can adopt either singlet or triplet electronic ground states, which strongly influences their physical and chemical properties (Scheme 1). Thus, understanding their (electronic) structures is key to rationalizing the reactivity of these species, which represent important intermediates in fundamental transformations such as insertion and ring expansion reactions.2 The isolation of such compounds, in which the group 15 atom is bound to only one additional atom, is extremely challenging and has only recently been achieved for the lightest congeners in landmark contributions (E = N, P, Scheme 1a).2,3 Access to the heavier homologues is increasingly difficult due to their tendency to undergo degradation reactions such as oligomerization, disproportionation, and bond activation processes. For the heaviest group 15 element bismuth, mononuclear organometallic compounds with the metal atom in the oxidation state of +1 (so-called bismuthinidenes) could so far only be accessed by stabilization through adduct formation with Lewis bases (Scheme 1b).4–9 Very recently, the intriguing properties of such bismuthinidenes have been exploited for (electro-)catalytic and photophysical applications: a BiI/BiIII redox couple has been proposed to be a key component in bismuth-catalyzed transfer hydrogenation reactions with ammonia borane.10 In addition, BiI oxalates have been suggested to be involved in the electrochemical reduction of carbon dioxide at bismuth electrodes.11 When embedded in host materials such as zeolites, [BiI]+ centers show intriguing photophysical properties such as ultrabroad near-infrared emission.12,13 Without any exceptions, the isolable, Lewis-base-stabilized bismuthinidenes reported to date show singlet ground states. This situation changes, when non-stabilized, (so far) non-isolable, electron-deficient molecular bismuthinidenes, Bi–R, are targeted. To date, only inorganic species Bi–X have been reported and were accessed by comproportionation (e.g. at high temperature in the melt) or by reaction of Bi atoms with reactive species such as F2 in the gas phase (X = H, F–I, AlCl4).14–18 Importantly, these synthetic routes did not allow to study the reactivity of bismuthinidenes, they did not allow to work in conventional solvents at moderate temperature, and they did not allow to access compounds with tunable substituents R, such as organometallic bismuthinidenes Bi–R (R = alkyl, aryl; Fig. 1c).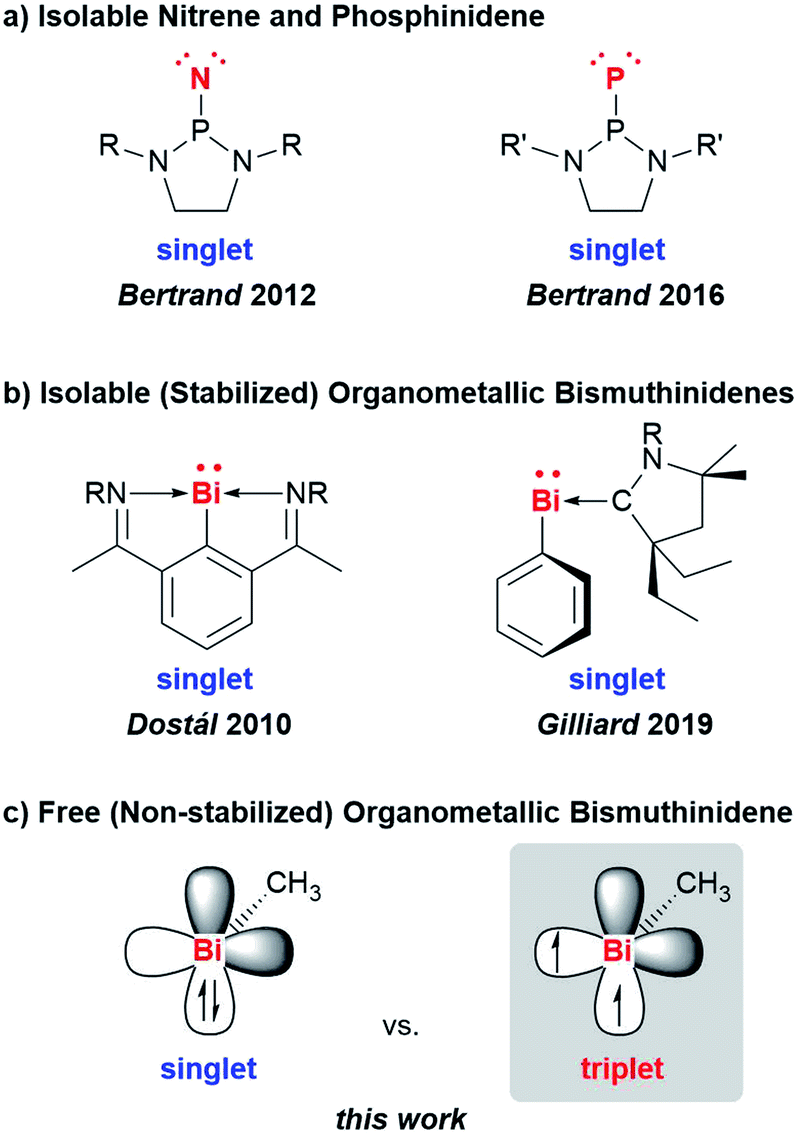 | ||
| Scheme 1 Low-valent group 15 compounds: (a) isolable nitrene and phosphinidene (singlet ground state). (b) stabilized organometallic bismuthinidenes (singlet ground state; occupied bismuth 6s-orbital omitted for clarity); (c) free (non-stabilized) singlet and triplet species that may be envisaged for BiMe (only two bismuth p-orbitals shown; p-orbital used for Bi–Me bonding and occupied s-orbital are omitted for clarity). R = 2,6-Me2-C6H3; 2,6-iPr2-C6H3; R′ = 2,6-[(4-tBu-C6H4)2CH]2-4-Me-C6H3. | ||
 | ||
| Fig. 1 Photoionization mass spectra recorded at 8.5 eV without pyrolysis (top trace), low pyrolysis power (T ≈ 470 K, center trace) and medium pyrolysis power (T ≈ 600 K, bottom trace). The signal at m/z = 91 is due to a background signal from previous experiments. | ||
Here we report the generation and characterization of the first free (i.e. non-stabilized) organometallic bismuthinidene methylbismuth, BiMe, in a gas-phase reaction with implications for related reactions in condensed phase at moderate temperature. In addition, dimethylbismuth, [BiMe2]• was studied to determine the Bi–CH3 bond dissociation energy in BiMe3.
Results and discussion
In order to access the elusive class of non-stabilized organometallic bismuthinidenes, we aimed at a “top-down approach”, more specifically the homolytic cleavage of Bi–C bonds from the well-defined organometallic precursor BiMe3 (1) as shown in Scheme 2. The long-known BiMe3 has been investigated from many different perspectives,19–21 including its application in chemical vapor deposition, where the abstraction of all its methyl groups is targeted.22,46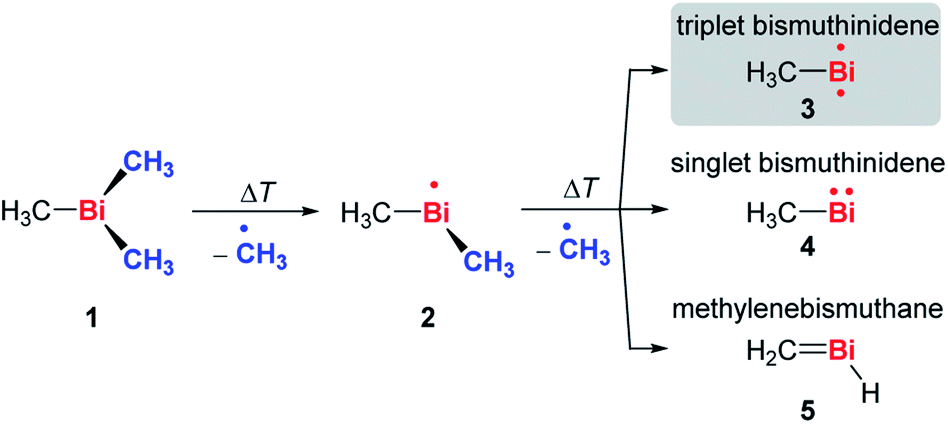 | ||
| Scheme 2 Controlled, stepwise abstraction of CH3 radicals from 1 in the gas phase by flash pyrolysis. | ||
In a recent study, it was demonstrated that the abstraction of a single methyl group can be achieved by dissociative photoionization of 1, i.e. the Bi–C bond cleavage of the [BiMe3]•+ cation in the gas phase.23 Under these conditions, only a single methyl group was abstracted, yielding the cation [BiMe2]+. Here we attempt a thermally-induced, controlled and stepwise abstraction of methyl groups from neutral BiMe3. Thus, a sample of BiMe3 diluted in Ar was pyrolysed in a microreactor and analyzed by photoelectron–photoion coincidence spectroscopy (PEPICO) using synchrotron radiation.24 This method permits to record photoion mass-selected threshold photoelectron spectra (ms-TPE) for each species by correlating ions and electrons produced in a single photoionization event. Isomer-selective information is then obtained from an analysis of the photoelectron spectrum based on computations.
Fig. 1 shows mass spectra under various pyrolysis conditions. Without pyrolysis (top trace) only the parent ion 1•+ is visible, thus dissociative photoionization is irrelevant under our experimental conditions. Already at low pyrolysis power (center trace) a stepwise methyl loss down to atomic Bi occurs, associated with formation of 2 and one of the products 3–5. The small intensity of m/z = 239 ([Bi(CH3)2]+) compared to m/z = 224 ([BiCH3]•+) indicates that cleavage of the second methyl group is more facile than the first. In addition, Bi2 is visible at m/z = 418, due to dimerization of bismuth atoms (see ESI, Fig. S4†). In some experiments a further peak appeared at m/z = 478 and is most likely due to Me2Bi–BiMe2. Note that CH3 is not observed due to its ionization energy of 9.83891 eV.25,26 Traces of BiI from the synthesis are also present in the spectrum.
When the pyrolysis power is further increased (bottom trace) the precursor is fully converted and experimental conditions are suitable for studying the molecule formed after loss of two methyl groups. Three structures are possible for m/z = 224, the bismuthinidenes 3 (triplet)/4 (singlet) or the methylenebismuthane 5. Threshold photoelectron spectroscopy provides structural isomer-selective information through comparison with Franck–Condon-simulated or reference spectra. Fig. 2 represents the ms-TPE spectrum of a species with the composition BiCH3 at m/z = 224. The first major band at 7.88 eV is assigned to the adiabatic ionization energy (IEad). It is followed by several smaller bands that are ca. 40–50 meV apart. Simulations based on DFT and multi-reference calculations were carried out for 3, 4 and 5. While DFT often provides very accurate geometries and frequencies even for molecules with complicated electronic structures,48 it is in many cases less accurate for the computation of energy surfaces or excitation energies.49 Hence, the ωB97X-D3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 50 functional was employed for frequency computations, but the multi-reference NEVPT251 approach was used to determine geometrical changes and to compute ionization energies as well as the energy difference between the two relevant states of the [BiCH3]•+ cation (vide infra). Both methods (DFT and multi-reference) were combined with a scalar relativistic approach and with the SARC-ZORA-TZVP basis set which allows for an all-electron treatment of bismuth52 (for computational details, see ESI†). Bismuthinidene 3 with a C3v symmetry and an X 3E (T0) triplet ground state is the lowest-energy structure (ΔE = 0 kJ mol−1). The computations on the NEVPT2-level show a very good agreement with the experimentally determined IE of 7.88 eV (IEcalc = 7.98 eV for the X+ 2A′′ ← X 3E transition (vide infra)), as compared to singlet bismuthinidene 4 (ΔE = +0.78 eV/+75 kJ mol−1; IEcalc = 7.21 eV) and methylenebismuthane 5 (ΔE = +0.91 eV/+88 kJ mol−1; IEcalc = 8.68 eV; for energy values obtained through DFT calculations see ESI†). This shows, that the ground state (electronic) structure of species with the sum formula ECH3 are fundamentally different, depending on the choice of the element E. For E = Bi the triplet bismuthinidene 3 is energetically favored and observed (vide supra), whereas for the lighter congeners (E = N, P), the formation of the methylene species HN
50 functional was employed for frequency computations, but the multi-reference NEVPT251 approach was used to determine geometrical changes and to compute ionization energies as well as the energy difference between the two relevant states of the [BiCH3]•+ cation (vide infra). Both methods (DFT and multi-reference) were combined with a scalar relativistic approach and with the SARC-ZORA-TZVP basis set which allows for an all-electron treatment of bismuth52 (for computational details, see ESI†). Bismuthinidene 3 with a C3v symmetry and an X 3E (T0) triplet ground state is the lowest-energy structure (ΔE = 0 kJ mol−1). The computations on the NEVPT2-level show a very good agreement with the experimentally determined IE of 7.88 eV (IEcalc = 7.98 eV for the X+ 2A′′ ← X 3E transition (vide infra)), as compared to singlet bismuthinidene 4 (ΔE = +0.78 eV/+75 kJ mol−1; IEcalc = 7.21 eV) and methylenebismuthane 5 (ΔE = +0.91 eV/+88 kJ mol−1; IEcalc = 8.68 eV; for energy values obtained through DFT calculations see ESI†). This shows, that the ground state (electronic) structure of species with the sum formula ECH3 are fundamentally different, depending on the choice of the element E. For E = Bi the triplet bismuthinidene 3 is energetically favored and observed (vide supra), whereas for the lighter congeners (E = N, P), the formation of the methylene species HN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 and HPCH2 has been determined to be more favorable.27–32,53
CH2 and HPCH2 has been determined to be more favorable.27–32,53
 | ||
| Fig. 2 Mass-selected threshold photoelectron spectrum of m/z = 224. The simulation based on 3 as the carrier (blue line) fits the experiment well. Transitions into the X+ 2A′′ ground state of the ion are given as grey bars, transitions into the A+ 2A′ state as red bars. | ||
Upon photoionization of 3, one electron is removed from either of the two degenerate SOMOs, which correspond in first approximation to the px and py orbital on the Bi center. The computations for the ionic ground state, [BiCH3]•+ (3•+), yielded a shortening of the Bi–C bond (from 2.27 Å in 3 to 2.21 Å in 3•+) and a deformation of the methyl group with a tilt angle of 4° relative to the Bi–C axis. This was ascribed to antibonding interactions between the unpaired electron and the bonding electrons of the two C–H groups, which are approximately aligned with the singly occupied p-type orbital of bismuth (Table S18†). This leads to a loss of the C3 axis and a reduction to Cs symmetry. As a consequence of this Jahn–Teller distortion, the 2E state in the C3v symmetric cation splits into a X+ 2A′′ and a A+ 2A′ component. The computations indicate an energy difference of only 50 meV between the two states at the geometry of the X+ 2A′′. Thus, transitions into both states contribute to the spectrum and have to be included in the simulation in Fig. 2 (blue curve). Transitions from the T0 ground state of neutral 3 into the X+ 2A′′ ionic ground state are given as grey bars, while transitions into the A+ 2A′ excited state are shown as red bars. In addition, there is a vibrational structure evident in the spectrum with a spacing of around 50 meV, including a hot band transition at 7.83 eV, which is assigned to the Bi–C stretching motion (corresponding to ν′′ ≈ 50 meV). Vibrational activity is expected due to the reduction of the Bi–C bond lengths in the cation by > 0.06 Å (see ESI† for all geometry parameters). Franck–Condon simulations based on computations of isomer 5 further support the triplet bismuthinidene 3 as the carrier of the spectrum (Fig. S3†). First, the computed IE of 5 is 0.80 eV higher than the experimental value, and second, a more pronounced vibrational progression with a maximum intensity for a transition into an excited vibrational state would be expected for 5. Thus a contribution of 5 to the spectrum can be ruled out.
The computed IE of 3 (for the lowest state of 3•+) and the relative energies for the two states of the cation 3•+ are in excellent agreement with the corresponding experimental data. This indicates that the neglected spin–orbit effects33 do not play a key role for the determination of the ionization energies, possibly because they are similar in magnitude for all involved states. While the experimental and calculated IEs nicely agree, deviations were observed in the shape of the spectra and were ascribed to the flatness of the potential energy surface (PES) of the cation. Two factors mainly contribute to the flatness of this PES: (i) the X+ 2A′′ state is threefold degenerate due to facile rotation around the Bi–C bond. (ii) The shape of the PES going from the equilibrium geometry of the X+ 2A′′ towards the equilibrium geometry of the A+ 2A′ state is expected to be non-harmonic. Efforts to obtain a better description of the surface were so far unsuccessful due to strong correlations of the various internal coordinates, so that high dimensional surfaces would be necessary for an appropriate description.
To gain additional information on the formation of BiMe (3) by stepwise abstraction of methyl groups from BiMe3 (1), an ms-TPE spectrum of [BiMe2]• (2) (m/z(2+) = 239) was recorded at a low pyrolysis temperature (Fig. 3; cf. Fig. 1). Simulations of the spectrum based on DFT calculations indicate C2v symmetry for both 2 and 2+ as well as a X+ 1A1 ← X 2B1 transition in [BiMe2]• (2). The first major band at 7.27 eV is assigned to the IE, in excellent agreement with the computed value of 7.35 eV at the NEVTP2 level of theory. A vibrational progression with a spacing of 60 meV is visible and is dominated by the symmetric Bi–C stretching mode in the cation. Additional torsional modes of the CH3 groups may lead to a broadening of the bands. While there is a good agreement between simulation and experiment, the vibrational intensities (including hot bands) are somewhat underestimated in the simulations. This indicates a slightly larger change of the Bi–C bond length upon ionization than the computed shortening of 0.05 Å (see ESI† for all geometry parameters).
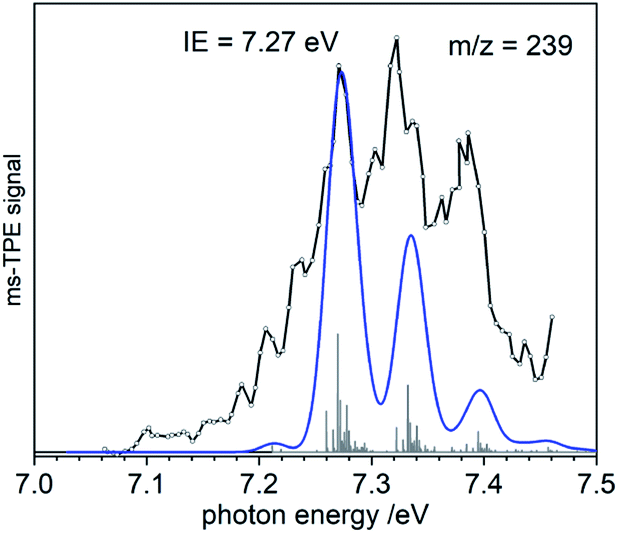 | ||
| Fig. 3 Ms-TPE spectrum of Bi(CH3)2, m/z = 239. The vibrational progression is due to the symmetric bismuth–carbon stretch and its combination with torsional motion. The blue line represents the simulated spectrum based on 2. | ||
Bond dissociation energies (BDEs) can be determined via thermochemical cycles that combine appearance energies (AE) and ionization energies. The zero Kelvin appearance energy AE0K for the abstraction of the first methyl radical from 1 has been determined with very high accuracy, AE0K(Bi(CH3)3, Bi(CH3)2+) = 9.445 ± 0.064 eV.23 According to computations, the methyl loss in the cation is a simple homolytic bond cleavage without a reverse barrier. Combined with the IE of 2, the Me2Bi–CH3 bond dissociation energy in 1, BDE(Me2Bi–CH3), can be derived:
| BDE(Me2Bi–CH3) = AE0K(BiMe3, [BiMe2]•+) − IE([BiMe2]•) | (1) |
From eqn (1) a value of 210 ± 7 kJ mol−1 is obtained for BDE(Me2Bi–CH3). The bond dissociation energy of the first Bi–CH3 bond in BiMe3 (BDE(Me2Bi–CH3)) can be expected to be the highest of the three Bi–CH3 BDEs in this molecule34 and is thus crucial for any type of reaction initiation via Bi–CH3 homolysis. However, the value of BDE(Me2Bi–CH3) has never been determined explicitly in the primary literature. Based on previous investigations into the thermal decomposition of 1,34–36 an estimation of BDE(Me2Bi–CH3) can be made, which yields a value of 182 kJ mol−1 as the upper limit that would be possible for this parameter.37 Thus, our results substantially revise the bond dissociation energy of the first Bi–Me bond in 1, which is key to the radical chemistry of 1 and related bismuth compounds. The calculated isodesmic reaction provides a value of 226 kJ mol−1 in good agreement with the experimental findings.
Our correction of the BDE in a fundamentally important organometallic compound such as BiMe3 raised the question, whether homolytic Bi–C bond cleavage is possible at moderate reaction temperatures in the condensed phase, making this process relevant for synthetic chemistry under conventional experimental conditions. To test for methyl radical abstraction from BiMe3, a benzene solution of BiMe3 and the radical trap 6 was heated to 60 °C and subsequently analyzed by EPR spectroscopy (Fig. 4). Indeed, a resonance was detected with giso = 2.006 and a(14N) = 41.6 MHz, a(1H) = 9.63 MHz, a(13C) = 12.9 MHz, indicating the formation of 7 by methyl radical transfer.38,54,55
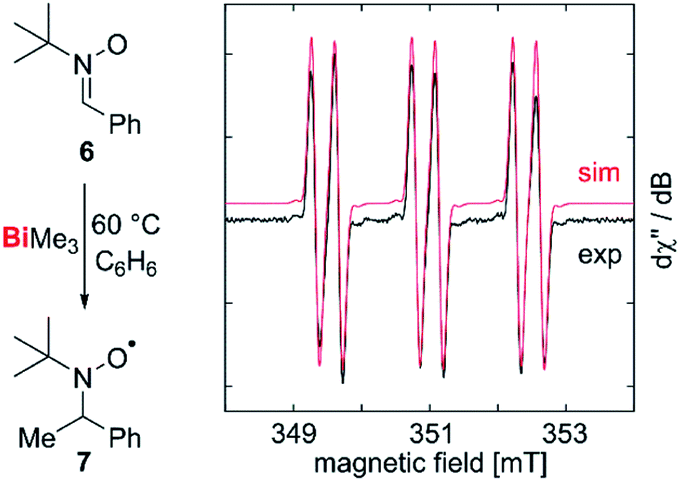 | ||
| Fig. 4 Abstraction and spin trapping of methyl radicals from BiMe3. | ||
In order to gain further hints at the generation and subsequent trapping of BiMe in the condensed phase, neat BiMe3 was reacted with stoichiometric amounts of (PhS)2 at 120 °C (Fig. 5a). BiMe(SPh)2 (8)39–41 was isolated as the product of this reaction in 41% yield and fully characterized (Fig. 5b and c; for details see ESI†). Methane was detected in the headspace of the reaction by IR spectroscopy (Fig. S5†), suggesting the appearance of methyl radicals in this reaction.
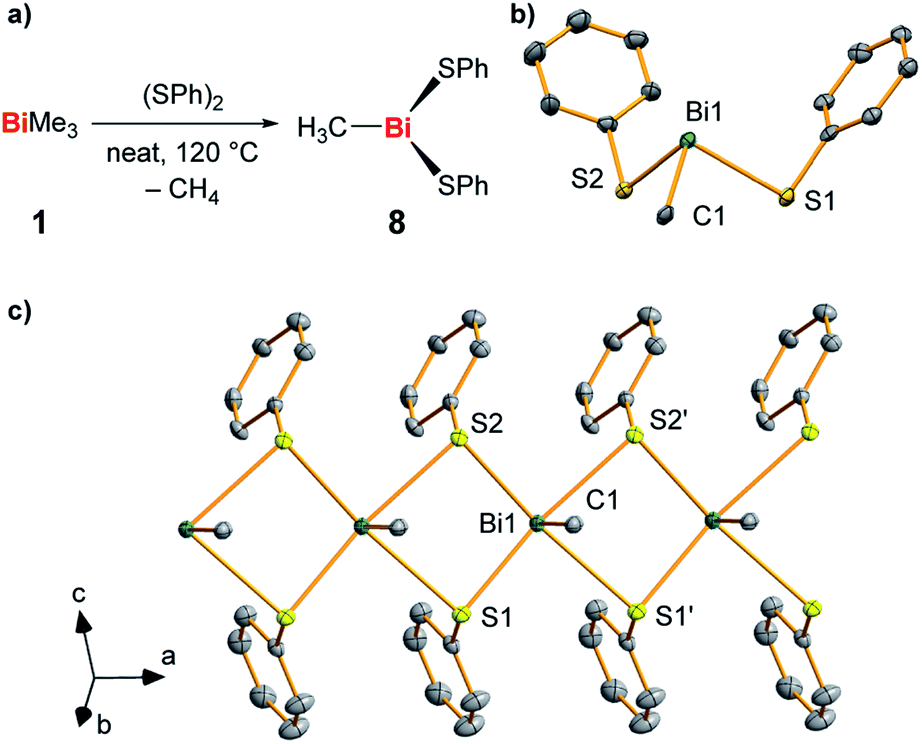 | ||
| Fig. 5 (a) Synthesis of BiMe(SPh)2 (8) from BiMe3 and (SPh)2 with methane as a detected by-product. (b and c) Molecular structure of 8 in the solid state with one formula unit shown in (b) and a cutout of the coordination polymer shown in (c). Displacement ellipsoids are shown at the 50% probability level. Hydrogen atoms and one set of split positions of disordered atoms are omitted for clarity. For detailed discussion of structural parameters see ESI.† Selected bond lengths (Å) and angles (°): Bi1–C1, 2.208(10); Bi1–S1, 2.736(2); Bi1–S2, 2.699(2); C1–Bi1–S1, 89.6(3); C1–Bi1–S2, 89.4(3); S1–Bi1–S2, 93.18(7). | ||
This – together with literature reports42 – supports the potential of BiMe to act as a transient reactive species in this reaction. The thioether MeSPh was also detected, suggesting that methyl radical attack at sulfur or σ-bond metathesis/disproportionation sequences may also be operative as a parallel reaction pathway.41 In the solid state, compound 8 forms a coordination polymer through bridging coordination modes of the thiolate ligands (Fig. 5c). This is a unique structural feature within the small number of literature-known compounds BiR(SR′)2 (R, R′ = aryl, alkyl; for details see ESI†).42–45
Conclusions
In conclusion, we have generated methylbismuth (BiMe), a fundamental organometallic compound and the first example of an organometallic non-stabilized bismuthinidene. BiMe was accessed via controlled thermal homolysis in the gas phase. The title compounds shows a triplet (biradical) ground state and an ionization energy of 7.88 eV, as revealed by combination of photoelectron spectroscopy and computations. The homolytic dissociation of the first Me2Bi–CH3 bond in BiMe3 is crucial to the radical chemistry of this compound (and related species) and is the initiating step in the formation of BiMe. Our results reveal a Me2Bi–CH3 homolytic bond dissociation energy of 210 ± 7 kJ mol−1, i.e. the previously reported value is revised by more than +15% (+28 kJ mol−1). Nevertheless, reactions in the condensed phase demonstrate that the abstraction of methyl radicals from BiMe3 is possible at moderate reaction conditions and suggest that BiMe may act as an intermediate in reactions with suitable trapping reagents. Future research will be directed towards the generation of non-stabilized bismuthinidenes and their exploitation in synthetic chemistry.Methods and experimental
Details of experimental conditions for synchrotron experiments, for calculations with DFT and multi-reference methods, for the preparation of compounds 1 and 8 (including two methods for the preparation of 119), and for IR and EPR spectroscopic experiments are given in the ESI.†
All calculations were performed with the ORCA program package, version 4.1.1 and 4.2.56
The spectroscopic experiments were carried out at the VUV beamline of the Swiss Light Source (SLS) at the Paul-Scherrer Institute, Villigen/CH. In most experiments the photon energy was scanned in 5 meV steps and calibrated using autoionization resonances in Ar. The ionization energies reported in the main paper are accurate to within ±20 meV and were corrected for the Stark-shift by the extraction field (8–9 meV). Note that in some experiments 10 meV steps were used. A detailed description of the beamline is given in the literature.47
[MeBi(SPh)2] (8).39
Neat trimethyl bismuth (100 mg, 0.394 mmol) and diphenyl disulfide (85.9 mg, 0.393 mmol) were heated to 120 °C for 3 d. After cooling to ambient temperature, a green solid was obtained, which was dissolved in benzene (1.5 mL) and layered with n-pentane (1.5 mL). After 16 h at ambient temperature, the product could be isolated by filtration and dried in vacuo as yellow needles. Yield: 70.8 mg, 0.160 mmol, 41%.1H NMR (500 MHz, C6D6): δ = 0.94 (s, 3H, CH3), 6.86 (dd, 2H, 3JHH = 7.4 Hz, 3JHH = 7.5 Hz, p-C6H5), 7.03 (t, 4H, 3JHH = 7.6 Hz, m-C6H5), 7.48 (d, 4H, 3JHH = 7.8 Hz, o-C6H5) ppm.
13C NMR (126 MHz, C6D6): δ = 40.40 (br, CH3), 127.29 (s, p-C6H5), 128.60 (s, m-C6H5), 135.62 (s, o-C6H5), 136.06 (s, ipso-C6H5) ppm.
Elemental analysis: anal. calc. for: [C13H13BiS2] (442.35 g mol−1): C 35.30, H 2.96, S 14.50; found: C: 35.12, H 2.90, S 14.40.
Conflicts of interest
There are no conflicts to declare.Author contributions
DPM, DS, ER and PH carried out the synchrotron radiation experiments, JR carried out all synthetic work, SW and DK performed the computations, IK performed the ESR and TP the IR spectroscopy. IF, BE and CL planned the project, contributed to the interpretation of the results and wrote the manuscript.Acknowledgements
Funding by the DFG (funding to CL, IF (FI575/13-1) and through GRK2112), by the Swiss Federal Office for Energy (BFE Contract Number SI/501269-01), and the FCI (PhD and Liebig scholarships to DK and CL) are gratefully acknowledged. CL thanks Prof. Holger Braunschweig for continuous support. The spectroscopic experiments were performed at the VUV beamline of the Swiss Light Source, located at the Paul Scherrer Institute (PSI).Notes and references
- L. Dostál, Coord. Chem. Rev., 2017, 353, 142–158 CrossRef.
- F. Dielmann, O. Back, M. Henry-Ellinger, P. Jerabek, G. Frenking and G. Bertrand, Science, 2012, 337, 1526–1528 CrossRef CAS PubMed.
- L. Liu, D. A. Ruiz, D. Munz and G. Bertrand, Chem, 2016, 1, 147–153 CAS.
- P. Šimon, F. de Proft, R. Jambor, A. Růžička and L. Dostál, Angew. Chem., Int. Ed., 2010, 49, 5468–5471 CrossRef PubMed.
- I. Vranova, M. Alonso, R. Lo, R. Sedlak, R. Jambor, A. Ruzicka, F. De Proft, P. Hobza and L. Dostál, Chem.–Eur. J., 2015, 21, 16917–16928 CrossRef CAS PubMed.
- I. Vranova, M. Alonso, R. Jambor, A. Ruzicka, M. Erben and L. Dostál, Chem.–Eur. J., 2016, 22, 7376–7380 CrossRef CAS PubMed.
- G. C. Wang, L. A. Freeman, D. A. Dickie, R. Mokrai, Z. Benkő and R. J. Gilliard, Chem.–Eur. J., 2019, 25, 4335–4339 CrossRef CAS PubMed.
- C. Lichtenberg, Angew. Chem., Int. Ed., 2016, 55, 484–486 CrossRef CAS PubMed.
- A. M. Arif, A. H. Cowley, N. C. Norman and M. Pakulski, J. Am. Chem. Soc., 1985, 107, 1062–1063 CrossRef CAS.
- F. Wang, O. Planas and J. Cornella, J. Am. Chem. Soc., 2019, 141, 4235–4240 CrossRef CAS PubMed.
- M. C. Thompson, J. Ramsay and J. M. Weber, Angew. Chem., Int. Ed., 2016, 55, 15171–15174 CrossRef CAS PubMed.
- H. T. Sun, Y. Matsushita, Y. Sakka, N. Shirahata, M. Tanaka, Y. Katsuya, H. Gao and K. Kobayashi, J. Am. Chem. Soc., 2012, 134, 2918–2921 CrossRef CAS PubMed.
- H. T. Sun, Y. Sakka, N. Shirahata, Y. Matsushita, K. Deguchi and T. Shimizu, J. Phys. Chem. C, 2013, 117, 6399–6408 CrossRef CAS.
- E. H. Fink, K. D. Setzer, D. A. Ramsay, M. Vervloet and J. M. Brown, J. Mol. Spectrosc., 1990, 142, 108–116 CrossRef CAS.
- J. D. Corbett, J. Am. Chem. Soc., 1958, 80, 4257–4260 Search PubMed.
- P. Kuijpers and A. Dymanus, Chem. Phys. Lett., 1976, 39, 217–220 CrossRef CAS.
- E. H. Fink, K. D. Setzer, D. A. Ramsay and M. Vervloet, Chem. Phys. Lett., 1991, 179, 95–102 CrossRef CAS.
- R. A. Lynde and J. D. Corbett, Inorg. Chem., 1971, 10, 1746–1749 CrossRef.
- K. Schäfer and F. Hein, Z. Anorg. Allg. Chem., 1917, 100, 249–303 CrossRef.
- S. Schulz, A. Kuczkowski, D. Bläser, C. Wolper, G. Jansen and R. Haack, Organometallics, 2013, 32, 5445–5450 CrossRef CAS.
- J. M. Herbelin, R. Klingberg, D. J. Spencer, M. A. Kwok, H. Bixler, R. Ueunten, R. Cook and W. Hansen, Opt. Commun., 1981, 36, 475–476 CrossRef CAS.
- S. W. Kang, K. M. Jeon, J. S. Shin, J. R. Chun, Y. H. Kim, S. J. Lee and J. Y. Yun, Chem. Vap. Deposition, 2013, 19, 61–67 CrossRef CAS.
- B. Hornung, A. Bodi, C. I. Pongor, Z. Gengeliczki, T. Baer and B. Sztáray, J. Phys. Chem. A, 2009, 113, 8091–8098 CrossRef CAS PubMed.
- T. Baer and R. P. Tuckett, Phys. Chem. Chem. Phys., 2017, 19, 9698–9723 RSC.
- B. K. Cunha de Miranda, C. Alcaraz, M. Elhanine, B. Noller, P. Hemberger, I. Fischer, G. Garcia, H. Soldi-Lose, B. Gans, L. A. Viera Mendez, S. Boye-Peronne, S. Douin, J. Zabka and P. Botschwina, J. Phys. Chem. A, 2010, 114, 4818–4830 CrossRef CAS PubMed.
- A. M. Schulenburg, C. Alcaraz, G. Grassi and F. Merkt, J. Chem. Phys., 2006, 125, 104310 CrossRef CAS PubMed.
- H. Bock and R. Dammel, Chem. Ber., 1987, 120, 1961–1970 CrossRef CAS.
- F. Holzmeier, M. Lang, K. Hader, P. Hemberger and I. Fischer, J. Chem. Phys., 2013, 138, 214310 CrossRef PubMed.
- S.-J. Kim, T. P. Hamilton and H. F. Schaefer III, J. Phys. Chem., 1993, 97, 1872–1877 CrossRef CAS.
- S. Lacombe, D. Gonbeau, J.-L. Cabioch, B. Pellerin, J.-M. Denis and G. Pfister-Guillouzo, J. Am. Chem. Soc., 1988, 110, 6964–6967 CrossRef CAS.
- S. Y. Liang, P. Hemberger, J. Levalois-Grützmacher, H. Grützmacher and S. Gaan, Chem.–Eur. J., 2017, 23, 5595–5601 CrossRef CAS PubMed.
- S. Y. Liang, P. Hemberger, N. M. Neisius, A. Bodi, H. Grützmacher, J. Levalois-Grützmacher and S. Gaan, Chem.–Eur. J., 2015, 21, 1073–1080 CrossRef CAS PubMed.
- G. Herzberg, Molecular Spectra and Molecular Structure, Krieger, Malabar/FL, 1966 Search PubMed.
- S. J. W. Price and A. F. Trotman-Dickenson, Trans. Faraday Soc., 1958, 54, 1630–1637 RSC.
- L. H. Long and J. F. Sackman, Trans. Faraday Soc., 1954, 50, 1177–1182 RSC.
- S. J. W. Price, in Comprehensive Chemical Kinetics, ed. C. H. Bamford and C. F. H. Tipper, Elsevier, 1972, vol. 4, pp. 197–257 Search PubMed.
- The average bond dissociation energy for a Bi–CH3 bond in 1 has been reported to be 33.8 kcal mol−1 (141 kJ mol−1) (ref. 35). The sum of the bond dissociation energies associated with the cleavage of the second and the third Bi–CH3 bond in 1 (BDE(MeBi–CH3) plus BDE(Bi–CH3)) was estimated to be at least 57.8 kcal mol−1 (242 kJ mol−1) (ref. 34). This gives a maximum value of 43.6 kcal mol−1 (182 kJ mol−1) for the bond dissociation energy of the first Bi–CH3 bond in 1.
- D. L. Haire, U. M. Oehler, P. H. Krygsman and E. G. Janzen, J. Org. Chem., 1988, 53, 4535–4542 CrossRef.
- In a different synthetic approach, compound 8 has previously been obtained from reaction of in situ generated [MeBi(OEt)2] with two equiv. HSPh: M. Wieber and U. Baudis, Z. Anorg. Allg. Chem., 1976, 423, 40–46 CrossRef CAS.
- A. G. Davies and S. C. W. Hook, J. Chem. Soc. B, 1970, 735–737 RSC.
- M. Wieber and I. Sauer, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1984, 39, 1668–1670 Search PubMed.
- P. Simon, R. Jambor, A. Ruzicka and L. Dostál, Organometallics, 2013, 32, 239–248 CrossRef CAS.
- M. Dräger and B. M. Schmidt, J. Organomet. Chem., 1985, 290, 133–145 CrossRef.
- K. M. Anderson, C. J. Baylies, A. H. M. Monowar Jahan, N. C. Norman, A. G. Orpen and J. Starbuck, Dalton Trans., 2003, 3270–3277 RSC.
- P. Šimon, R. Jambor, A. Růžička and L. Dostál, J. Organomet. Chem., 2013, 740, 98–103 CrossRef.
- For reactions of the related precursor Sb2Me4 under near-chemical vapor deposition conditions see: N. Bahlawane, F. Reilmann, S. Schulz, D. Schuchmann and K. Kohse-Höinghaus, J. Am. Soc. Mass Spectrom., 2008, 19, 1336–1342 CrossRef CAS PubMed.
- M. Johnson, A. Bodi, L. Schulz and T. Gerber, Nucl. Instrum. Methods Phys. Res., Sect. A, 2009, 610, 597–603 CrossRef CAS.
- (a) E. Welz, J. Böhnke, R. D. Dewhurst, H. Braunschweig and B. Engels, J. Am. Chem. Soc., 2018, 140, 12580–12591 CrossRef CAS PubMed; (b) J. Böhnke, T. Dellermann, M. A. Celik, I. Krummenacher, R. D. Dewhurst, S. Demeshko, W. C. Ewing, K. Hammond, M. Heß, E. Bill, E. Welz, M. I. S. Röhr, R. Mitrić, B. Engels, F. Meyer and H. Braunschweig, Nat. Commun., 2018, 9, 1197 CrossRef PubMed.
- V. Settels, W. Liu, J. Pflaum, R. F. Fink and B. Engels, J. Comput. Chem., 2012, 33, 1544–1553 CrossRef CAS PubMed.
- (a) J.-D. Chai and M. Head-Gordon, J. Chem. Phys., 2008, 128, 084106 CrossRef PubMed; (b) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- (a) C. Angeli, R. Cimiraglia, S. Evangelisti, T. Leininger and J.-P. Malrieu, J. Chem. Phys., 2001, 114, 10252–10264 CrossRef CAS; (b) C. Angeli, R. Cimiraglia and J.-P. Malrieu, Chem. Phys. Lett., 2001, 350, 297–305 CrossRef CAS; (c) C. Angeli, R. Cimiraglia and J.-P. Malrieu, J. Chem. Phys., 2002, 117, 9138–9153 CrossRef CAS.
- D. A. Pantazis and F. Neese, Theor. Chem. Acc., 2012, 131, 1292 Search PubMed.
- It should be noted that methyl nitrene (N–CH3) and methyl phosphinidene (P–CH3) also have triplet ground states. But the structures with methylene (rather than methyl) functional groups, i.e. HNCH2 and HPCH2, are energetically favored over N–CH3 and P–CH3. See ref. 27–32 and A. G. Kutateladze, Computational Methods in Photochemistry, CSC Press Taylor and Francis Group, 2019.
- Bismuth-containing radical species were not detected under these conditions. It has been reported that even persistent dialkyl bismuth radicals may not be detectable by EPR spectroscopy, which has been ascribed to fast relaxation as a result of spin orbit coupling: S. Ishida, F. Hirakawa, K. Furukawa, K. Yoza and T. Iwamoto, Angew. Chem., Int. Ed., 2014, 53, 11172–11176 CrossRef CAS PubMed.
- Trapping of alkyl radical species in reactions of bismuth compounds has been reported: J. Ramler, I. Krummenacher and C. Lichtenberg, Angew. Chem., Int. Ed., 2019, 58, 12924–12929 CrossRef CAS PubMed.
- (a) F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS; (b) F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1991253. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc02410d |
| This journal is © The Royal Society of Chemistry 2020 |