
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConversion between triplet pair states is controlled by molecular coupling in pentadithiophene thin films†
Natalie A.
Pace
 a,
Brandon K.
Rugg
a,
Christopher H.
Chang
a,
Obadiah G.
Reid
ab,
Karl J.
Thorley
c,
Sean
Parkin
c,
John E.
Anthony
c and
Justin C.
Johnson
*a
a,
Brandon K.
Rugg
a,
Christopher H.
Chang
a,
Obadiah G.
Reid
ab,
Karl J.
Thorley
c,
Sean
Parkin
c,
John E.
Anthony
c and
Justin C.
Johnson
*a
aNational Renewable Energy Laboratory, 15013 Denver West Parkway, Golden, Colorado 80401, USA. E-mail: Justin.Johnson@nrel.gov
bRenewable and Sustainable Energy Institute, University of Colorado Boulder, Boulder, Colorado 80309, USA
cDepartment of Chemistry, University of Kentucky, Lexington, KY 40506, USA
First published on 22nd June 2020
Abstract
In singlet fission (SF) the initially formed correlated triplet pair state, 1(TT), may evolve toward independent triplet excitons or higher spin states of the (TT) species. The latter result is often considered undesirable from a light harvesting perspective but may be attractive for quantum information sciences (QIS) applications, as the final exciton pair can be spin-entangled and magnetically active with relatively long room temperature decoherence times. In this study we use ultrafast transient absorption (TA) and time-resolved electron paramagnetic resonance (TR-EPR) spectroscopy to monitor SF and triplet pair evolution in a series of alkyl silyl-functionalized pentadithiophene (PDT) thin films designed with systematically varying pairwise and long-range molecular interactions between PDT chromophores. The lifetime of the (TT) species varies from 40 ns to 1.5 μs, the latter of which is associated with extremely weak intermolecular coupling, sharp optical spectroscopic features, and complex TR-EPR spectra that are composed of a mixture of triplet and quintet-like features. On the other hand, more tightly coupled films produce broader transient optical spectra but simpler TR-EPR spectra consistent with significant population in 5(TT)0. These distinctions are rationalized through the role of exciton diffusion and predictions of TT state mixing with low exchange coupling J versus pure spin substate population with larger J. The connection between population evolution using electronic and spin spectroscopies enables assignments that provide a more detailed picture of triplet pair evolution than previously presented and provides critical guidance for designing molecular QIS systems based on light-induced spin coherence.
Introduction
Singlet fission (SF) is most commonly studied in systems with the energy of the initially excited singlet state (S1) approximately equal to the sum of the energies of the generated triplet states (2 × T1). This criterion limits intrinsic energetic losses in photovoltaic devices, where SF has the potential to increase device efficiencies past the Shockley–Queisser limit.1 As a consequence of the focus on photovoltaic applications, larger acenes like hexacene have received significantly less attention due to highly exothermic SF (ΔE ≈ −0.5 eV), a low triplet energy, and chemical instability.2,3 However, the energetic separation between S1 and triplet pair states in larger acenes provides an opportunity for greater mechanistic clarity heretofore unrealized in smaller acenes. If higher acenes can be made sufficiently stable, they represent a promising platform for quantum information applications, where clearly defined spin coherence between multiple magnetically active states would be favoured over efficient exciton multiplication using all available potential energy.When compared to SF mainstays like tetracene and pentacene, relatively little is known about the photophysical properties of larger acenes. SF has been observed in crystalline hexacene with time constants between ∼50 fs and 500 fs.2,4 The large variation in time constants and the shortage of measurements makes it difficult to discern whether the SF rate continues to increase as acene size increases, or whether hexacene displays a turnover in SF rate due to excess exoergicity.2 A recent report on a hexacene dimer suggests the latter may be true.5 In order to effectively utilize SF systems in a quantum information context, chromophores must be efficiently prepared in a state of pure and well-defined character (e.g. as in the ms = 0 sublevel of the triplet ground state of a nitrogen vacancy in diamond).6 The simplest kinetic scheme for the SF process is shown below:
| S1 + S0 ⇄ 1(TT) ⇆ T1 + T1 | (1) |
This general picture has been confirmed through spectroscopic measurements like transient absorption (TA) on coupled chromophore systems. S1 photoinduced absorption (PIA) can be assigned based on correlation with dilute monomer solution measurements where singlets are long-lived, whereas T1 PIA can usually be accurately assigned based on comparison with TA data from triplet sensitization of isolated molecules in solution. The spectroscopic signatures of 1(TT) are less definitively discerned but are typically differentiated from T1 through near-IR PIA features that are absent from the triplet sensitization spectrum but are kinetically correlated with triplet features in the visible region.8–11 In the work of Trinh et al.,8 these near-IR features have previously been attributed to 1(TT) → Sn transitions based on energetic proximity to the S1 → Sn transition. Increased coupling between chromophores in dimers is linked to increased transition strength and peak narrowing. However, these transitions are often weak and broad in crystals, which leads to uncertainty about whether SF proceeds fully to produce two independent triplets in these systems. Thus, the standard definition of the full SF rate gleaned from eqn (1)12 is often discarded in favour of the rate of formation of 1(TT) or the disappearance of S1 in systems where 1(TT) and T1 + T1 are not easily distinguished.
Recent work by Pensack et al.13 has suggested that this picture should be further elaborated due to the observation of multiple triplet pair states in the near-IR TA spectra of TIPS pentacene nanoparticles. In that work, an initial triplet pair spectrum with bands near ∼950 nm and ∼1050 nm and broad absorption in the 1200–1400 nm region evolves into a second triplet pair spectrum with decreased PIA in the 1200–1400 nm region and an altered spectral envelope on a timescale of ∼1 ps. The first spectrum is assigned to the 1(TT) state and the second spectrum is assigned to a state where two triplets reside on non-neighbouring molecules, 1(T⋯T), with the following kinetic scheme:
| S1 + S0 ⇄ 1(TT) ⇄ 1(T⋯T) ⇆ T1 + T1 | (2) |
This assignment is based on correlation with the timescale for triplet transfer, which had been previously estimated at ∼1 ps for a single triplet hop in pentacene thin films,14 although in related systems diffusion coefficients range between ∼1 × 10−3 and ∼1 × 10−6 cm2 s−1,15,16 which correspond to timescales for a single triplet hop between ∼400 fs and ∼400 ps. Later studies found a temperature dependence for the triplet separation kinetics,9,17 which is consistent with a thermally activated process like triplet hopping, with time constants of ∼20 ps (ref. 17) and ∼250 ps (ref. 9) reported at ∼100 K across both visible and near-IR PIA features in TIPS pentacene films. The assignment of this spectral evolution to the transition from 1(TT) to 1(T⋯T) has not yet been corroborated by theoretical studies.
Apart from spatial correlation, an additional feature of the triplet pair is its spin state. Recent time-resolved electron paramagnetic resonance (TR-EPR) studies on bipentacene dimers,18 pentacene aggregates,19,20 TIPS tetracene thin films,21 and terrylene diimide dimers22 have revealed quintet states, 5(TT), on a response-limited timescale (<100 ns). The 5(TT) state holds particular promise for quantum information applications, as this state can be optically prepared in a long-lived spin-polarized configuration at room temperature.6,19,23 The flow of population from 5(TT) into states with triplet character (either 3(TT) or T1 + T1), primarily occurs on a ∼100 ns to 1 μs timescale.18,21,22 The generation dynamics of 5(TT) and 3(TT) are not well-understood, but TR-EPR measurements indicate population evolution as in Scheme 1.
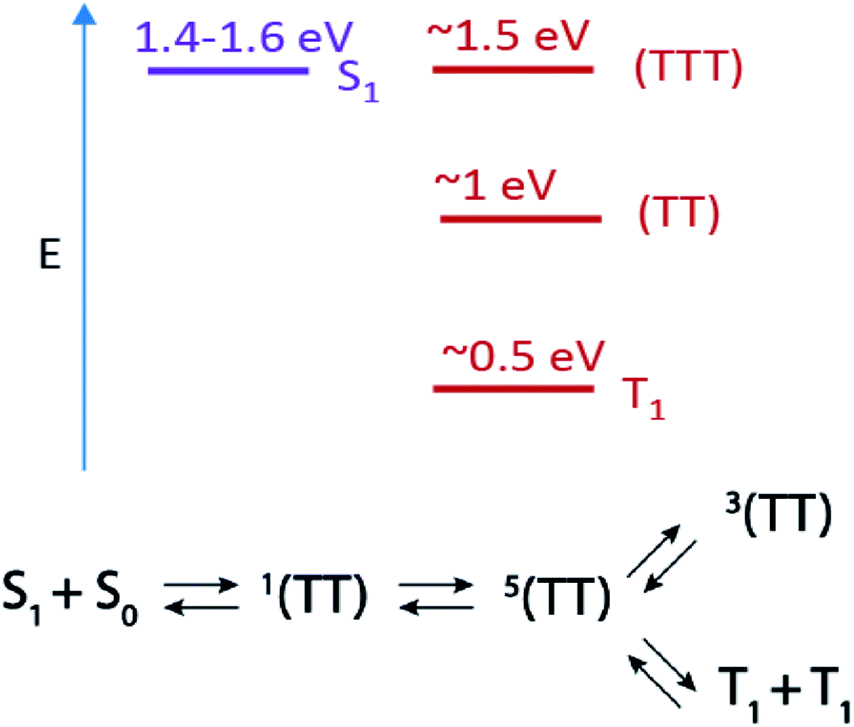 | ||
| Scheme 1 Top: Estimated energy level diagram for PDT singlet and triplet states and Bottom: scheme for singlet fission and subsequent triplet pair state evolution. | ||
In order to exploit the unique properties of the 5(TT) or 3(TT) state, their characteristics must be fully mapped using techniques with appropriate time resolution and spin selectivity, and unfortunately no single technique possesses both. Though features in TA spectra have not previously been assigned to high-spin (TT) states, the conversion between 1(TT) and 5(TT) likely occurs on an appropriate timescale (ps–ns) for this evolution to be detected. Furthermore, although their energies are likely similar (i.e., within kBT), these states are connected to different manifolds of higher lying states in PIA experiments, as we will discuss below.
In this work we use TA to identify two triplet pair spectra in a series of pentadithiophene (PDT) films with systematically varied crystal structures. The PDT derivatives used here are expected to be photophysically analogous to hexacene,9 with a large energy offset between 1(TT) and S1 (Scheme 1), enhanced stability through the substitution of thiophene rings and fluoro groups, and increased solubility through the addition of bulky trialkylsilyl ethynyl groups.24 This PDT series gives us unprecedented access to two well-defined triplet pair spectra with resolvable and tuneable dynamics. We are thus able to explore the connection between triplet pair spectra, molecular coupling, and the two aforementioned kinetic schemes, which propose either diffusion-mediated (1(TT) to 1(T⋯T)) or spin-specific (1(TT) to 5(TT) to 3(TT)) mechanisms for their evolution. We find that spatial separation of triplets alone is inconsistent with the full set of observations, although short-range hopping may play some role in the spin state conversion. We also rule out other common potential mechanisms of excited state spectral evolution in SF films, such as formation of excimers or species segregated between crystalline and amorphous regions, and we make firm connections between the unique transient optical spectra and TR-EPR spectra.
Results
Structural and steady-state properties
The molecular structure, crystal structure, and optical absorption spectra for TIPS (triisopropylsilylethynyl) PDT, TSPS (tri-2-pentylsilylethynyl) PDT, and TSBS (trisecbutylsilylethynl) PDT are shown in Fig. 1. The three molecules are named for their different core substituents for brevity, but they also differ by their peripheral substituents, with triisopropylsilyl groups attached to the thiophene rings in TIPS PDT and fluoro groups attached to the thiophene rings in TSPS PDT and TSBS PDT. Synthesis details can be found in the ESI.† | ||
| Fig. 1 (a) TIPS PDT, (b) TSPS PDT, and (c) TSBS PDT normalized solution and film absorbance spectra, with molecular structures inset. Right, crystal structures (viewed along the a-axis for TIPS PDT, b-axis for TSPS PDT, and c-axis for TSBS PDT). Trialkylsilyl substituents are omitted from crystal structures for clarity. Crystal structure measurements are between the centroids of nearest neighbours. | ||
The combination of core substitution and bulky peripheral substitution in TIPS PDT produces a crystal structure not typically encountered for acenes in which pairs of TIPS PDT molecules are significantly offset from each other (Fig. 1a right, ∼8.5 Å between nearest acene cores) as well as tilted and rotated with respect to other pairs. This limits orbital overlap between nearest neighbours as well as long-range molecular interactions. TSPS PDT lacks bulky alkyl silyl groups on the thiophene rings, which allows for a crystal structure in which dimer pairs are relatively close together, and orbital overlap is expected to be large (Fig. 1b right, ∼3.8 Å between acene cores). However, the large TSPS group separates dimer pairs, such that long-range molecular coupling is limited (Fig. 1b right, ∼11.3 Å). TSBS PDT has both small peripheral substituents and a somewhat smaller alkyl silyl group attached to the acene core, which produces a brickwork crystal structure where both short-range and long-range molecular coupling is strong (Fig. 1c right, 3.7–3.9 Å between all nearest neighbours). Additional views of the unit cell (Fig. S1†) and crystallographic files can be found in the SI. This series thus represents a gradual increase in molecular coupling strength, from weak coupling between dimer pairs in TIPS PDT to strong coupling between dimer pairs in TSPS PDT to strong coupling between all neighbours in TSBS PDT.
The differences in crystal structure in the series produce clear distinctions in the optical spectra. The TIPS PDT film absorption spectrum retains the main vibronic progression of the TIPS PDT solution-phase absorption spectrum, with only minor blue-shifting (∼3–4 nm) of the peaks at ∼640 nm, ∼705 nm, and ∼785 nm. There is some redistribution of oscillator strength, which may be partially due to film texturing. Overall, this spectrum indicates a small amount of excitonic coupling consistent with the absence of short-range interactions deduced from the TIPS PDT crystal structure. The TSPS PDT film absorption spectrum has significant redistribution of oscillator strength compared to the TSPS PDT solution-phase spectrum, with the most prominent new peaks at ∼700 nm and ∼780 nm, indicating strong excitonic coupling.25 The TSBS PDT film absorption spectrum shows similar signs of strong excitonic coupling, including increased spectral broadening relative to the TSPS PDT film absorption spectrum. All films show weak emission, with the TIPS PDT fluorescence being the strongest and exhibiting a small Stokes shift and vibronic structure in the spectrum (Fig. S2†).
The crystallinity of the films can be varied through annealing, Fig. S3 and S4.† TSPS and TSBS PDT films tended toward high crystallinity immediately upon casting, whereas TIPS PDT films required some solvent vapor annealing to reach high crystallinity. Studies as a function of crystallinity revealed some changes in spectral characteristics (i.e., broadening and weakening of triplet pair features in as-cast films) but no clear modifications in excited state kinetics, thus we present data on solvent vapor annealed TIPS PDT films.
Transient absorption spectroscopy
We use TA spectroscopy to monitor SF dynamics in the PDT compounds in films and solution. In solution, all three PDT compounds have photoinduced absorption (PIA) features at ∼565 nm, ∼980 nm, ∼1270 nm, and ∼1570 nm (Fig. S5†). All PIA features are present at early times and decay concurrently on a nanosecond timescale, which is consistent with the dynamics of a singlet state that decays primarily by fluorescence.We begin by analysing the kinetics of the ostensibly more strongly coupled films of TSPS and TSBS PDT, as their steady-state optical behaviour bears resemblance to the well-studied TIPS pentacene. The TSPS PDT film displays fast excited state evolution consistent with SF, with an instrument response-limited decay of singlet PIA at ∼1570 nm and a concomitant rise of PIA at ∼550 nm, ∼595 nm, ∼895 nm, ∼1020 nm, ∼1150 nm, and broad amplitude in the ∼1300–1600 nm region (Fig. 2, Fig. S7†). The peaks at ∼550 nm and ∼595 nm correspond to peaks in the sensitized triplet spectrum for TSPS PDT, with some red-shifting and broadening (Fig. 2). The energetic offset and broadening between the two spectra has previously been observed in other strongly-coupled acene thin films and underscores the strong coupling that perturbs even fairly localized triplet states.26,27 There are no triplet features in the sensitized triplet spectrum in the near-IR, which affirms the assignment of observed long-lived bands to the (TT) state in the directly excited film.
 | ||
| Fig. 2 TA spectra for TSPS PDT film. Note distinct ΔOD scales used for visible and near-IR regions. 700 nm excitation. | ||
The TSPS PDT (TT) spectrum further evolves on a ∼10 ps timescale, where the broad amplitude in the ∼1300–1600 nm region as well as the peak at ∼895 nm decay as the peaks at ∼1020 nm and ∼1150 nm rise (Fig. 2, 1 ps to 50 ps). Spectral broadening and substantial overlap prevent further deconvolution of the two (TT) spectra. However, the peaks at ∼1020 nm and ∼1150 nm are clearly distinct at longer time scales and are assigned as the long-lived triplet pair, (TT)B, whereas peaks at 895 nm and 1300–1600 nm comprise the initially formed triplet pair, (TT)A. The (TT)B species lifetime (Fig. S6,† 42 ± 2 ns), is similar to that observed in TIPS-pentacene and related derivatives that possess strong pairwise molecular interactions leading to triplet pair decay in channels such as (TT) → T1 or (TT) → S0.8 Both (TT)A and (TT)B TSPS PDT spectra resemble those observed by Pensack et al. in TIPS pentacene nanoparticles,13 though the conversion between the two (TT) spectra is slower in TSPS films by an order of magnitude.
The fluence dependence for TSPS PDT films reveals a slight hastening of the decay of (TT)B at higher fluences, Fig. S8.† A rough estimate of the diffusion coefficient is approximately ∼10−4 cm2 s−1, corresponding to about 6 ps per hop between the farthest separated TSPS molecules in the unit cell. The relatively small change in decay constant with fluence leaves this value with considerable uncertainty.
Temperature dependent studies reveal essentially no change in the spectroscopic features in transient absorption (data not shown). The kinetics of the rise of the (TT)A feature, Fig. S10,† remains within the instrument response at all temperatures; however, the emergence of the assigned (TT)B feature near 1020 nm is resolvable and reveals a slight slowing at 77 K (7 ps) vs. 298 K (6 ps), suggesting at most a weakly activated transition.
The TSBS PDT film exhibits similar dynamics to TSPS PDT, with instrument response-limited singlet PIA decay in the 1400–1600 nm region (Fig. 3 and S11†) and simultaneous rise of triplet PIA at ∼550 nm and ∼595 nm (Fig. 4a). Once again, there are PIA features in the near-IR at ∼895 nm, ∼1020 nm, ∼1170 nm, and in the broad 1300–1600 nm region that do not correspond to any peaks in the sensitized triplet spectrum but are kinetically correlated with the triplet PIA features at ∼550 nm and ∼595 nm. As in the TSPS PDT film, the (TT)A spectrum is distinguishable from the (TT)B spectrum only by increased PIA amplitude at ∼895 nm and in the ∼1300–1600 nm region. The evolution between (TT)A and (TT)B species in TSBS PDT films approaches the conversion observed by Pensack et al.13 both spectrally and temporally (∼3 ps for TSBS PDT compared to ∼1 ps for TIPS pentacene nanoparticles, Fig. S11†). The relatively strong molecular coupling in TSBS PDT films is also accompanied by a short (TT)B lifetime (56 ± 10 ns, Fig. S6†).
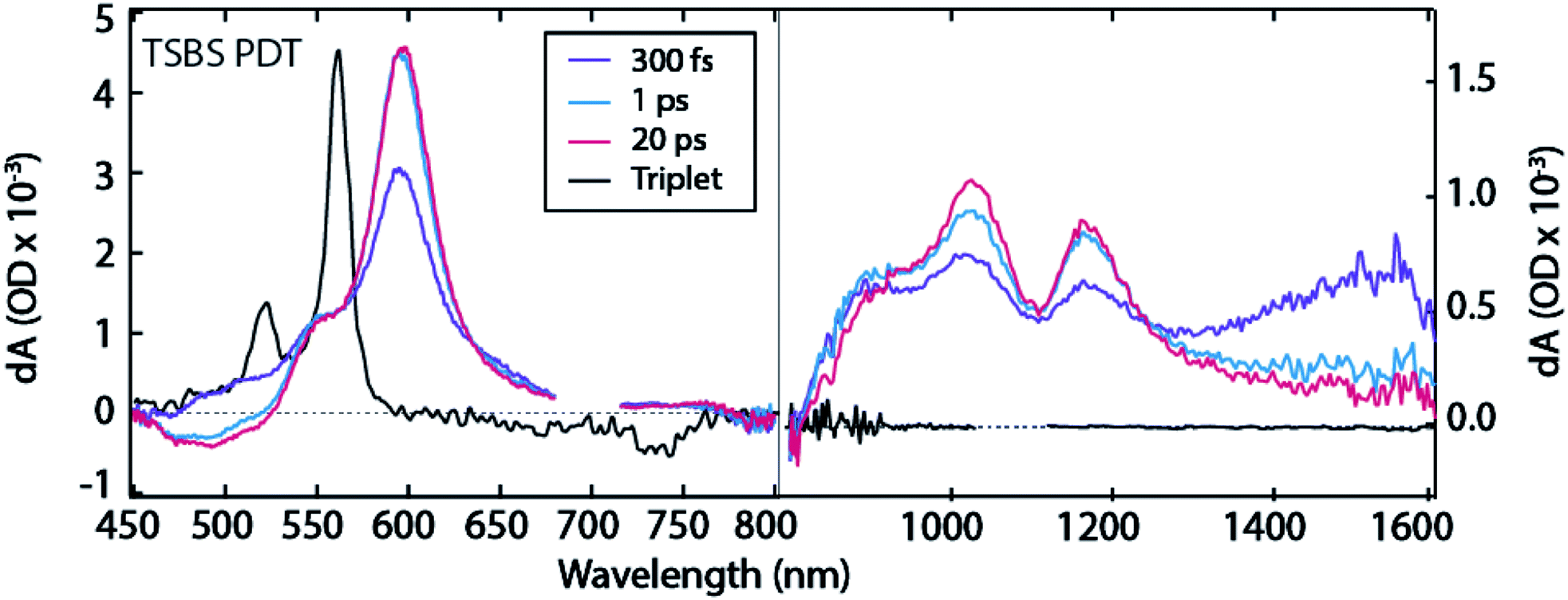 | ||
| Fig. 3 TA spectra for TSBS PDT film. Note distinct ΔOD scales used for visible and near-IR regions. 700 nm excitation. | ||
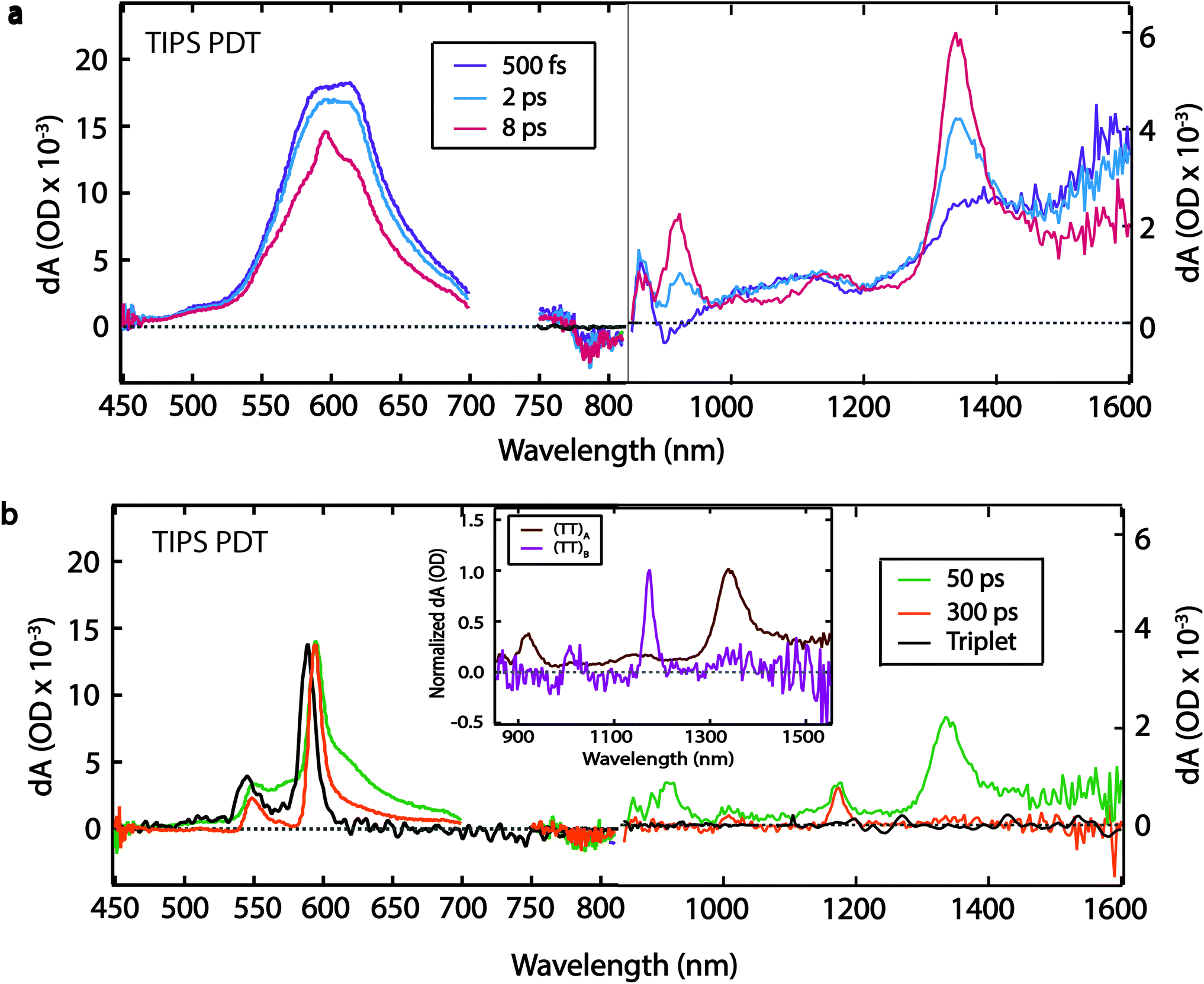 | ||
| Fig. 4 TA spectra for TIPS PDT film. (a) Initial evolution to 8 ps and (b) later time evolution, including triplet spectrum from solution sensitization for comparison. Inset shows normalized (TT)A (8 ps) and (TT)B spectra (300 ps). Note distinct ΔOD scales used for visible and near-IR (>800 nm) regions. 725 nm excitation. | ||
Fluence dependence for the TSBS derivative (Fig. S12–S14†) is the strongest of the series, and analysis using a bimolecular decay constant is straightforward, Fig. S13.† The diffusion coefficient is ∼10−4 cm2 s−1, corresponding to a triplet energy transfer time of around 2 ps. This value matches the time of conversion between (TT)A and (TT)B and agrees with similar measurements on TIPS-pentacene28 and pentacene.29
Temperature dependence of transient absorption indicates no substantive change in spectral features but a considerable slowing in the (TT)A to (TT)B conversion, from 1.5 ps at 298 K to roughly 6 ps at 77 K (Fig. S15†). Thermal activation is similar to although somewhat weaker than the trend found in Lee et al. with unsubstituted pentacene.17
The TIPS PDT compound exhibits a similar TA spectrum in thin film and solution at early times (500 fs), with a PIA feature at ∼1570 nm, and corresponding red-shifted PIA features at ∼605 nm, ∼1100 nm, and ∼1370 nm (Fig. 4a). We attribute these initial features to the singlet state based on the similarities between the solid-state and solution-phase spectra. The singlet decay is accompanied by a concurrent rise in narrow peaks at ∼550 nm, ∼595 nm, ∼865 nm, ∼915 nm, ∼1020 nm, ∼1140 nm, and ∼1340 nm. The peaks at ∼550 nm and ∼595 nm are excellent matches for the PIA features produced by triplet sensitization of TIPS PDT by anthracene in solution (Fig. 4b, black trace). The ∼4 ps rise of these visible triplet features is definitively extracted from the global fit (vide infra) but is difficult to visually discern from the raw spectra due to overlapping singlet PIA decay and spectral narrowing. The near-IR peaks have no corollary in the sensitized triplet spectrum (Fig. 4a, 8 ps spectrum), and thus we assign them to formation of (TT)A.
The (TT)A spectrum evolves into a second, distinct spectrum, (TT)B, on a 50 ps timescale with sharp, well-separated peaks at ∼895 nm, ∼1020 nm, ∼1170 nm, and a broader peak at ∼1330 nm (Fig. 4b). The (TT)B spectrum is also assigned based on its kinetic correlation with the rise of triplet PIA features at ∼550 nm and ∼595 nm, where the concurrent emergence of narrowed visible triplet features is evident. Unlike with TSPS and TSBS, there is no analogue in films of previously studied pentacene derivatives that displays stark and unambiguous near-IR evolution from (TT)A to a distinct set of peaks associated with (TT)B, which are unencumbered by spectral overlap that exists in the visible portion of the spectrum. The prominent peak in the (TT)A spectrum (∼1340 nm) is ∼2.7 times as broad as the most prominent peak in the (TT)B spectrum (∼1170 nm) (Fig. 4b, inset). The (TT)B species has a long lifetime of 1.5 ± 0.2 μs (Fig. S6†). This decay time was found to be independent of the pump pulse fluence, suggesting unimolecular decay (Fig. S16†).
We find the evolution between (TT)A and (TT)B for TIPS PDT to be temperature-independent (Table S1†). Spectral narrowing in low-temperature spectra reveals that the transition at ∼1330 nm in the (TT)B spectrum is distinct from the transition at ∼1340 nm in the (TT)A spectrum (Fig. S18†). Both (TT) spectra contain pairs of transitions separated by typical vibrational energies for hexacene (∼1300 cm−1, ∼1700 cm−1), suggesting significant vibronic coupling (Table S2†).
Global fitting analysis was applied to the TA data sets to determine rate constants and define an appropriate kinetic scheme. After singular value decomposition (SVD), parallel and sequential population evolution involving exponential components were used to extract evolution associated spectra, EAS, which are shown in Fig. S19† for TIPS PDT. Comparing sequential and parallel decay models leads to a preference for the sequential model, which faithfully reproduces the raw kinetics of intermediate rise and decay (Fig. S19a†). The EAS (Fig. S19b†) match the raw spectra at different times provided in Fig. 2–4. The success of this approach leads to a kinetic scheme summarized in Fig. 5a (with time constants in the table below and described in more detail in Fig. S20†) that tracks population of all species vs. time. Global target analysis using this scheme was applied to the data for each film up to time delays of 5 ns and provides rate constants for each transition and produces species associated spectra (SAS) (Fig. S21–S23†). The results confirm the improved fit consistency for the sequential vs. parallel model.
 | ||
| Fig. 5 (a) General kinetic scheme used in target analysis and associated time constants determined for all three PDT derivatives, where GS is the ground state. t3 was determined using separate single wavelength fit to long time TA signals. The asterisk for TSBS t2 indicates a secondary decay component of 280 ps was included in the fit. (b) Comparison between (TT)B spectra in each film as derived from global target analysis. | ||
The scheme was successful at fitting the data without reversibility for each film, although it was discovered that the decay pathway from (TT)A to the ground state (red arrow in Fig. 5a) is only needed to account for reduction in the ground state bleach that follows the (TT)A population profile in TIPS PDT, but found to be negligible for TSPS PDT and TSBS PDT. Similar decay from 1(TT) directly to the ground state has been previously inferred in crystals of long acenes and competes with formation of the (TT)B species, thus reducing its yield in TIPS PDT.30 Without an accurate measure of the triplet yield and due to potential overlapping photoinduced absorptions with the ground state bleach, the rate constant for this pathway is uncertain.
As molecular coupling increases between pairs of molecules in TSBS PDT films compared to TSPS PDT and especially TIPS PDT films, the time constants for formation and decay of (TT)A and (TT)B decrease. This is consistent with dynamics assigned to (TT) states in dimers, where both formation and decay time constants decrease with increased coupling strength.8 Furthermore, the evolution between (TT)A and (TT)B becomes more subtle with increasing electronic coupling, and although the peak positions of the final (TT)B state are similar among the films, the linewidths decrease dramatically for TIPS PDT (Fig. 5b). A secondary decay of (TT)A of roughly 280 ps was found to be necessary for TSBS PDT films, although the SAS is identical to that of the much faster few ps decay. This type of biexponential decay of a single species is likely due to sample inhomogeneity that offers different decay pathways (e.g., via trapping or annihilation) for triplet pairs born in distinct film regions.30
Time-resolved electron paramagnetic resonance spectroscopy
Although TA spectroscopy of the PDT films strongly suggests triplet pairs are formed within a few ps after photoexcitation and evolve further in 10s of ps, the technique does not directly measure the associated spin state. In order to investigate the spin evolution of the long-lived (TT)B species observed in TA, TR-EPR spectra were collected after pulsed photoexcitation of PDT crystalline powders.Observation of transient EPR signals precludes the possibility of a pure, diamagnetic 1(TT) state with S = 0 at long times. For distinguishing triplet pair states of higher spin, e.g.3(TT) and 5(TT), it is necessary to have information about the magnetic dipole–dipole interaction between the electron spins within an individual triplet. This interaction is parameterized in the form of D and E, which represent its axial (z) and transversal (x/y) components, respectively, relative to a set of axes defined by the molecular frame. Values of D = 1148 MHz, and E = 0 MHz (Fig. S25†) were obtained by fitting the spectrum of 3*PDT formed through heavy-atom induced intersystem crossing, observed in a sample of TIPS PDT in a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of iodobutane and toluene. Substitution to the TSPS derivative had no discernible effect on the derived D and E values (data not shown).
:1 mixture of iodobutane and toluene. Substitution to the TSPS derivative had no discernible effect on the derived D and E values (data not shown).
TR-EPR spectra obtained after photoexcitation of a crystalline powder sample of TSPS PDT, shown in Fig. 6a, were fitted to a I → II → GS model. The 15 ± 7 ns lifetime derived from global fitting (Fig. 6b) is reasonably associated with (TT)B from TA considering its weak fluence dependence observed in the nsTA experiment that would accelerate the decay under TR-EPR fluences (approximately 2–3 orders of magnitude higher than TA, for which the decay is 42 ns). EAS II is small and relatively featureless, but nonetheless a second kinetic component was necessary to produce a satisfactory fit. This is likely caused by slightly different rates of relaxation from different spin states, which would cause the signal represented in EAS I (resulting from a mixture of different spin sublevel populations and molecular orientations) to decay at different rates at various field points. Interpretation of the TSPS PDT data is made with the assumption that singlet fission primarily takes place within the mostly strongly exchanged pair of molecules and that this is reflected in the TR-EPR spectra. The large magnitude of the splitting between n(TT) manifolds, justified below with calculations of J, indicates that the spin manifolds are well-separated and that EPR transitions take place between states with well-defined spin quantum numbers. The molecular axes of the two chromophores within candidate dimer pairs are also aligned, which simplifies analysis. We note that the most prominent peaks in EAS I are split by D/3 and appear respectively as emissive and absorptive features. This observation is consistent with transitions from ms = 0 to ms = ±1 within the 5(TT) spin manifold, occurring in dimer pairs for which the applied magnetic field B0 is perpendicular to the z-axis associated with the molecular frame that defines D and E.18,21 The broad shoulders and width of the signal imply that population of the 5(TT) ms = 0 state is not entirely selective—there also appears to be transitions involving either the 5(TT) ms = ±2 states, the 3(TT) manifold, or a combination thereof. Pathways toward population of various quintet and triplet sublevels are possible due to both the availability of triplet pair sites with varying J and the different orientations of the dimer pairs with respect to the magnetic field in the powder sample.
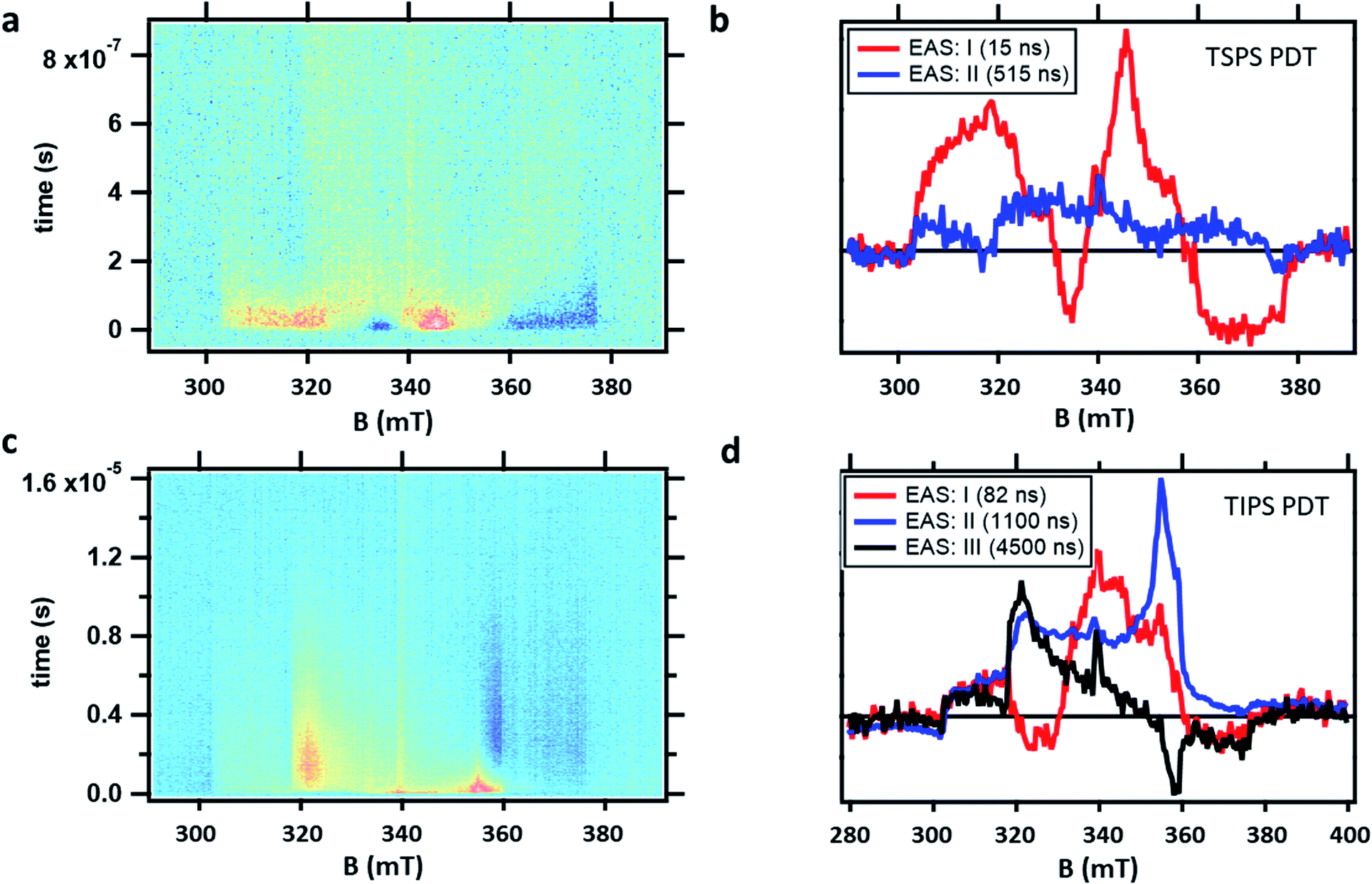 | ||
| Fig. 6 Raw TR-EPR data obtained at X-band (9.5 GHz) at room temperature after photoexcitation and corresponding EAS extracted from global fits for (a and b) TSPS PDT powder and (c and d) TIPS powder. Fit component lifetimes are shown in the legend. | ||
In the case of the PDT derivatives under discussion, for which E is negligible, there is no discernible difference in energy level splitting between the dimer orientations x‖B0 and y‖B0. With this in mind, an energy level plot of the triplet pair spin states was calculated for x‖B0 to be representative of z⊥B0, which is statistically the most represented orientation within a powder sample. Comparison of this plot (Fig. 7) to EAS I highlights the 5(TT) character of the spectrum, since, as mentioned previously, the two spin-allowed transitions from the 5(TT) ms = 0 state are predicted to occur at the same field position and with the same polarization as the two central peaks. While it is tempting to assign the outer shoulders of the spectrum to transitions from the 3(TT) ms = 0 state to the ms = ±1 states because of their respective absorptive and emissive character, we avoid doing so in light of the complex relationship between energy level splitting and molecular orientation relative to B0. This is further complicated by recent models which suggest additional orientation dependence in the way that spin sublevels are populated by singlet fission,31 as well as the presence of two different dimers within the TSPS PDT powder samples (vide infra).
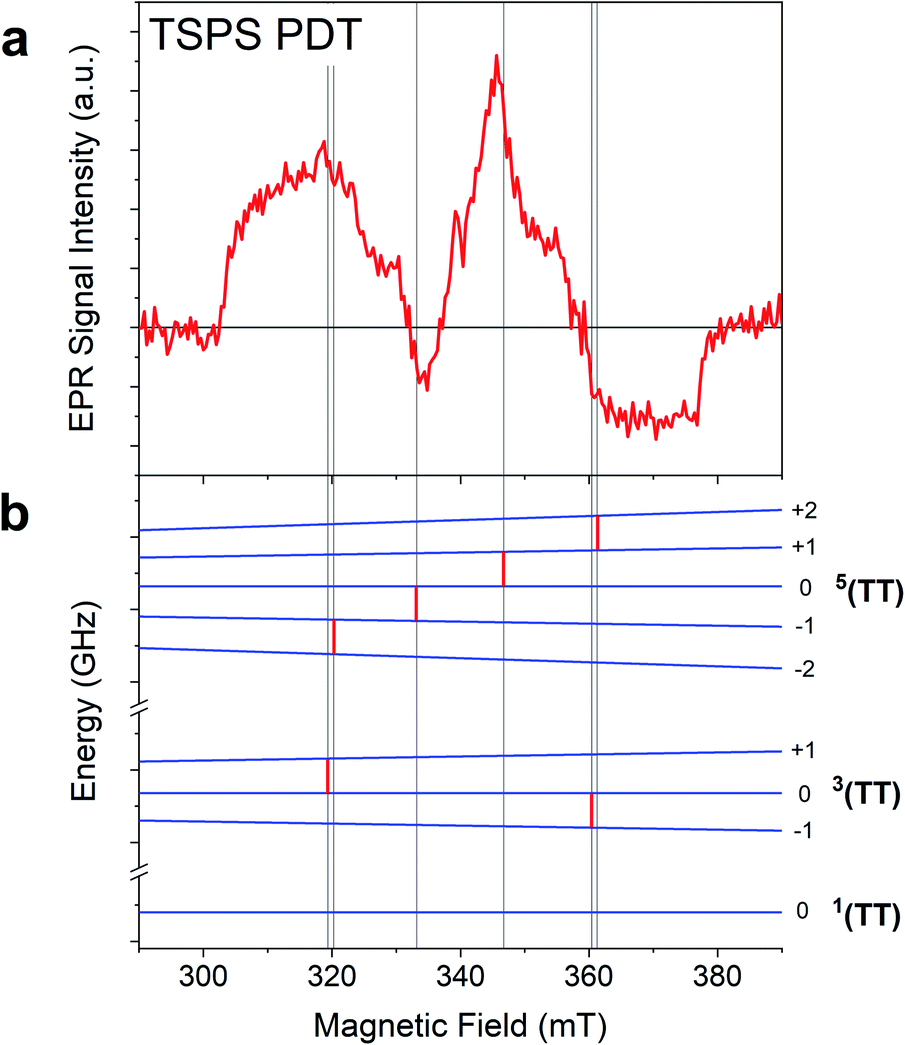 | ||
| Fig. 7 Comparison of (a) EAS I and (b) the energy level plot calculated for the TSPS A dimer in the z⊥B0 orientation. Red lines placed on the energy level plot indicate field positions at which the energy level splitting between neighbouring spin states is equal to the experimental microwave frequency of 9.536 GHz. These lines are extended for easier comparison to EAS I. | ||
TSBS PDT powder produced no detectable signal in the TR-EPR experiment, which is unsurprising given the strong fluence dependence due to triplet–triplet annihilation that would reduce the lifetime to less than 1 ns for TSBS PDT (Fig. S15†), which is significantly shorter than the >20 ns response time of the resonator used for the TR-EPR experiments.
The TR-EPR spectra for TIPS PDT powder (Fig. 6c) were fitted with three kinetic components, indicating more complex spin evolution than observed in the corresponding TA spectra. The first spectral component I of TIPS PDT (Fig. 6d) shares some features with the longer-lived EAS II. However, it is distinguished by two peaks (one emissive, and one absorptive) that are closer to centre field than the peaks observed in EAS II and III, as well as in the monomer triplet spectrum in Fig. S25.† As noted during discussion of the TSPS PDT data, pairs of peaks split by D/3 have been considered evidence for the formation of an S = 2 quintet species with preferential population of the ms = 0 state.18,21 The pair of peaks observed in EAS I do not perfectly match this quintet pattern—they are broad and not split symmetrically about centre field. However, neither can they be assigned to transitions associated with free triplets or occurring within the 3(TT) manifold when considering the D and E parameters extracted from the TIPS PDT monomer triplet spectrum.
In EAS II, which has a lifetime of approximately 1.1 μs, there are two peaks close to where transitions within the triplet manifold would be expected, i.e. they appear in similar locations as the prominent peaks observed in the monomer triplet spectrum, as well as the peaks observed in EAS III, which strongly resembles a triplet formed by ISC. However, EAS II is unusual in that it appears predominately absorptive. Triplets formed by ISC, as well as triplets formed from SF in which the quintet ms = 0 state is preferentially populated, inevitably have polarizations which lead to nearly equal mixtures of absorptive and emissive features. The features of EAS II are roughly consistent with preferential population of the triplet ms = −1 state, with some population of ms = 0, which would lead to the two absorptive transitions ms = −1 → 0 and 0 → +1. In the context of SF, a similar observation was made of an all-emissive triplet signal in the work of Chen et al.,22 in which the unusual polarization was caused by mixing of the 5(TT) and 3(TT) manifolds induced by the magnitude of the magnetic field at X-band. This explanation was verified by the disappearance of the emissive triplet species in higher field Q-band EPR measurements, as in the stronger magnetic field, the relevant spin sublevels no longer mixed. In contrast, we observe nearly identical spectra at both X- and Q-band (Fig. S26†). Therefore, the observed polarization must be due to the specifics of the SF process in the TIPS PDT dimers under investigation. The TA kinetics suggest a strong correlation between (TT)B and EAS I and II from TR-EPR, but no species persists long enough in TA to be associated with EAS III. The presence of the magnetic field or the different sensitivities to spin vs. optical transitions may contribute to this discrepancy.
Exchange energy calculations
Because the exchange energy between the two constituent triplet excitons in a triplet pair largely defines the eigenstates and population flow between them, an estimate of this value is important. Calculations of the inter-triplet exchange energy were performed for molecules in the unit cell of each type of PDT crystal (Fig. 8). Exchange coupling magnitudes were found in a range from almost 0 cm−1 to 87 cm−1. A clear pattern was observed requiring both facial interaction between members of the dimer, and direct contact of atomic p orbitals of the two π systems (Fig. S27†). This observation is in line with expectations around the nature of spin-exchange interaction32 and provides guidelines for crystalline design to control the magnitude of triplet–triplet exchange. Notably, both plausible dimers from the TIPS PDT structure show very small exchange couplings, which demonstrates that TIPS PDT falls outside the diabatic regime in which J is greater than D, which is about 0.037 cm−1. In contrast, TSPS PDT has a large coupling for one dimer (∼87 cm−1) and a small coupling in the other (≪1 cm−1). TSBS shows one intermediate coupling between all relevant dimer pairs (∼15 cm−1). We note that these trends map directly onto our qualitative considerations for degree of short- vs. long-range order for the three crystals described above. The two possible TSPS unit cell dimers present an interesting situation in which J is large for one pair, thus predicting formation of pure triplet pair spin states, but very small for a second pair, which could harbour mixed states and potentially independent triplets after a single hop. We note that TSPS A and TSBS pairs have very similar intermolecular disposition but produce a six-fold difference in calculated J (Fig. S27†).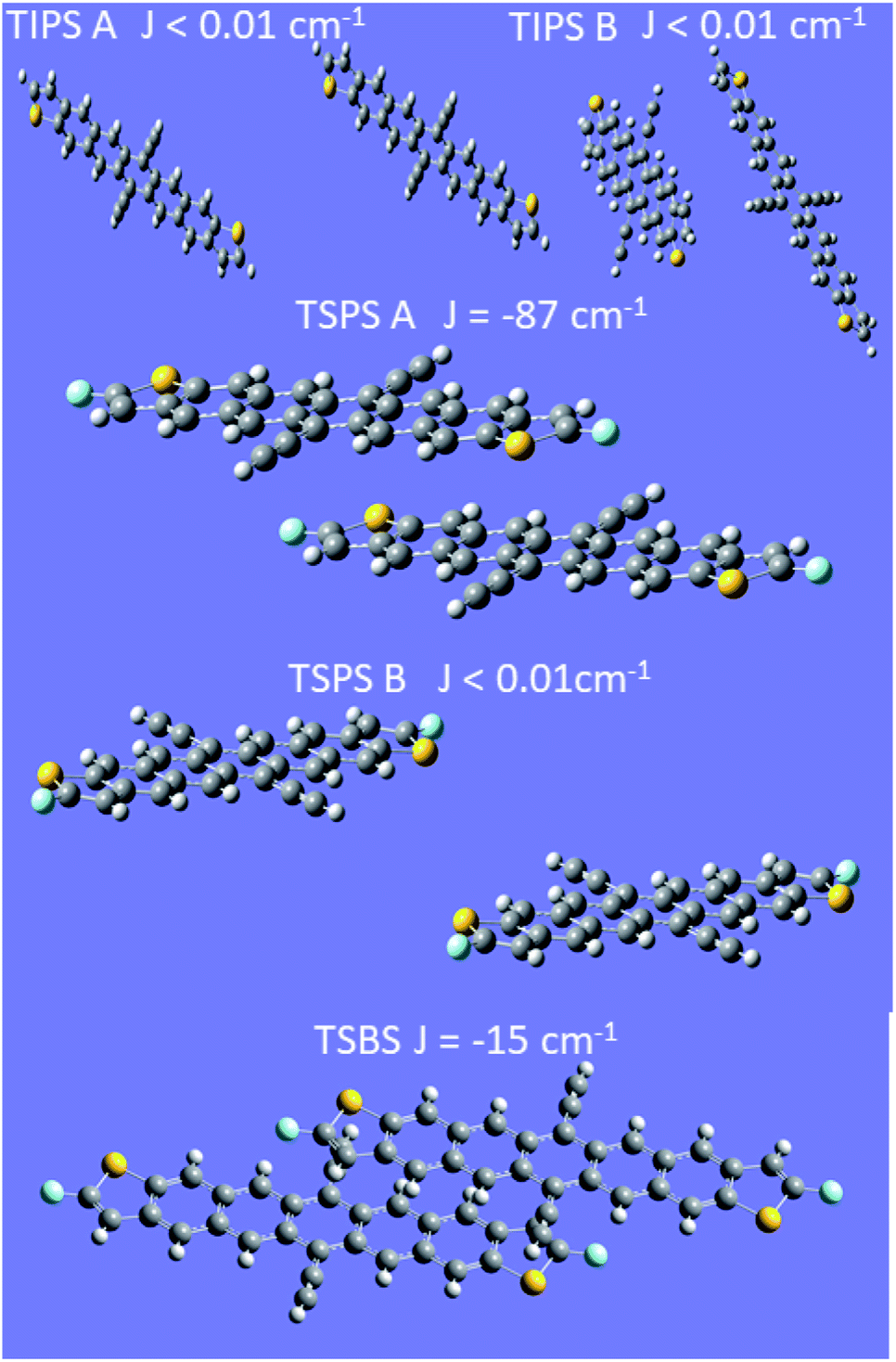 | ||
| Fig. 8 Calculations of exchange coupling between dimer pairs in crystals. In all cases the energy ordering of the states is antiferromagnetic. | ||
Discussion
Although the behaviour of TSBS, and to some extent TSPS PDT, is at first glance consistent with other crystalline SF systems like TIPS-pentacene that exhibit significant exciton delocalization, consideration of the entire series including TIPS PDT impels us to consider a different perspective, outlined in Fig. 9. Broadened and red-shifted triplet absorption features in TSBS and TSPS PDT films signify moderately perturbed excited states due to intermolecular coupling, and ps-scale evolution in the broad NIR spectra, correlated with estimated triplet diffusion times, implies a hopping-induced transition from tightly to more weakly bound triplet pairs. Biexciton binding of the triplets may modulate the barriers to triplet diffusion, but it does not apparently alter the expected trend, which is fastest motion in TSBS PDT and slowest in TIPS PDT. In the environment provided by a TSBS crystal (long-range, 3D brickwork coupling), triplet pairs transition quickly from the strong to weak exchange coupling regime with large inter-triplet distance engendered through hopping, and persistent population in diabatic triplet pair states detectable by TR-EPR (e.g., 3(TT) or 5(TT)) is unlikely to be observed. The situation in TSPS is somewhat modified (i.e., strong pairwise but limited long-range coupling), and despite similar transient optical spectral evolution to TSBS PDT, quintet features are observed from ∼15 ns in TR-EPR spectra. As the long-lived (TT)B apparently possesses significant 5(TT) character, the transient spectral evolution cannot be assigned to triplet pair separation per se, because this separation would destroy 5(TT) signatures the way it apparently does for singlet fission systems with fast inherent triplet diffusion (e.g., pentacene and tetracene crystals). Furthermore, the lack of apparent triplet hopping in TIPS PDT, such that the decay is unimolecular, puts any possible diffusion time several orders magnitude longer than the transition time, τ2, between (TT)A and (TT)B, negating the notion that triplet migration could mediate the spectral evolution observed in transient absorption experiments.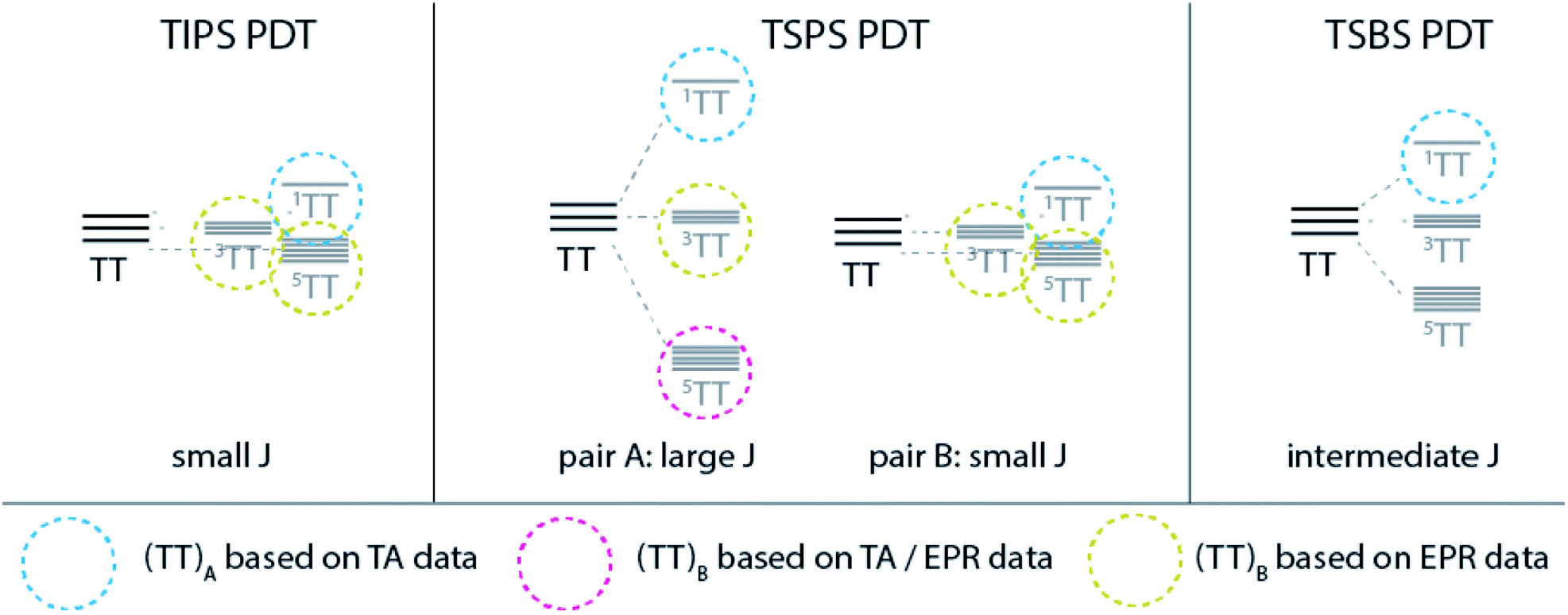 | ||
| Fig. 9 Schematic description of triplet pair manifolds for each PDT film and identified species based on TA and EPR results. | ||
Why is this strong contrast in transient optical and EPR behaviour observable in the PDT series and not in pentacene derivatives? We conjecture that a combination of atypical crystal structure and a large amount of driving force for SF have made the transition between (TT) manifolds clearly optically distinguishable for the first time. Whereas most prior observations of (TT) spectra involved significant mixing with the S1 state,33–35 the large exothermicity of SF in PDT more clearly separates the associated S1vs. TT transitions and reduces this mixing, which is more likely with the closer S1/TT resonance in pentacene derivatives.8,13,36 This prior work has suggested that transitions from 1(TT) to Sn states, energetic companions to the S1–Sn transitions, dominate the near-IR portion of the pentacene spectrum, but for PDT the roughly 0.5 eV energy separation between (TT) and S1 removes any expected correlation between (TT) ESA and the S1–Sn spectra. Furthermore, as shown by TR-EPR, the (TT) spin state at times beyond a few 10s of ps is not likely 1(TT), and although the 5(TT) and 3(TT) energies are very similar to 1(TT), transitions from the non-singlet states will not occur to Sn due to optical selection rules. The expected transitions are thus altered, and for example, if 3(TT) lies near T2 or another Tn state, then mixing may facilitate a 3(TT) to Tn transition. Accordingly, although no monomer quintet state is nearby in energy, 5(TT) to 5(TT)* is the relevant expected transition, which may be entirely distinct from those in the singlet or triplet manifolds. Thus, stark spectral evolution, less reasonable for a 1(TT) to 1(T⋯T) transition, is expected for a transfer between spin manifolds. Calculated predictions of these transitions, made more cumbersome by the molecular size and multiple spin state manifolds, are currently being pursued. Other possible explanations for significant spectral evolution on a ps timescale, such as excimer formation or transport through domains of amorphous aggregation, are ruled out by the lack of exciton diffusion or appropriate intermolecular geometries in TIPS PDT samples. Furthermore, the lack of strong temperature dependence of observed rates renders these scenarios unlikely.
The previously observed unusual behaviour of polycrystalline films of TIPS-tetracene vs. the parent tetracene is a worthy comparison of distinct intermolecular coupling regimes that are similar to the PDT series, despite the different energetics from PDT. TIPS-tetracene exhibits weak electronic coupling as judged by optical spectra and diffusion properties,10 yet it has been shown to form population almost exclusively in 5(TT)0 in <100 ns by TR-EPR.21 Furthermore, sharp bands in the NIR appear at early times, similar to what is observed for TIPS PDT, although these bands do not show significant change on a ps or ns timescale and thus reveal nothing about (TT) evolution. In contrast, tetracene has broad and unstructured NIR features37 and no reported triplet pair signatures by TR-EPR, which is behaviour more similar to TSBS PDT. As stated earlier, the primary novel observation for TIPS PDT—clear distinction in optical spectra as the triplet pair evolves—may originate in S1/(TT) energy level offsets, despite otherwise similar regimes of intermolecular coupling to other acenes.
Returning to the remarkable evolution measured in TA, the most distinct and resolvable (TT)A and (TT)B spectra are observed in TIPS PDT films, where molecular coupling is the weakest, and the features evolve on the slowest timescale. Vibrational satellites are found that are rarely observed for other intermolecular species such as excimers or charge-transfer states.38,39 Although both triplet pair spectra possess unusually narrow and intense peaks, the (TT)A species has broader features relative to the (TT)B species, which may indicate increased delocalization often characteristic of singlet states (i.e., 1(TT)). Changes in (TT)B peak ratios and line widths among the three compounds might be related to changes in excitonic coupling,13 where the narrower peaks in both the near-IR and visible regions for TIPS PDT are consistent with a more localized (TT) state than is expected in the more strongly coupled pairs and networks found in films of the TSPS and TSBS derivatives. Furthermore, the relatively short lifetime of (TT)B in the case of TSPS PDT implies its spin character is not purely quintet (spin forbidden return to the ground state) but rather possesses singlet character that facilitates faster internal conversion to S0. The somewhat subtle evolution in TA spectral features between (TT)A and (TT)B for both TSPS and TSBS PDT further supports shared spin character in each species, as either inherent mixing between singlet and quintet states (expected as exchange coupling weakens21) or fast and reversible transitions between them are plausible and would produce these observations. The longer lifetime and starker spectral evolution found in the triplet pair of TIPS PDT might then imply 3(TT) character, which is unmixed with singlet and quintet states. We note that despite some narrowing and shifting, triplet bands in the visible largely reflect overall triplet population and not the spin character or boundedness of the triplet pair.
Although TA results demonstrate evolution away from 1(TT) toward other triplet pair states on a ps time scale (most completely for TIPS PDT), definitive assignment of spectral transitions relies upon TR-EPR. EPR spectra clearly reveal formation of states with quintet and triplet character that can be connected with long-lived states from TA, but the exact distribution and evolution of population between spin states is not simple, particularly for TIPS PDT. Existing models for SF focus on situations in which J ≫ D, for which the 1(TT), 3(TT), and 5(TT) manifolds are energetically well-separated, and EPR transitions only occur within the sublevels of a particular spin manifold. The TIPS PDT data are unique in that they represent SF occurring within dimers that are much more weakly coupled than those studied previously. Typically, weakly exchanged triplet exciton pairs have been observed after thermally activated hopping from the strongly exchanged dimer sites at which they formed. This means that the exciton pairs have already undergone significant spin evolution before reaching the weakly exchanged regime, and the associated TR-EPR signal tends to resemble that of free triplets with preferential population of the triplet ms = 0 state. This is not what we observe, and we hypothesize that with the small J value and non-parallel magnetic axes between neighbouring chromophores, fast transitions occur among the weakly exchange coupled eigenstates, which include mixed singlet and quintet states, as well as pure triplet states.31,40 Nevertheless, the current data imply that (TT)B in TIPS PDT is associated with a paramagnetic species with evidence of triplet and quintet character. Further analysis of the spectrum and its evolution may require the development of a detailed model that explores the possibility of mixing between spin manifolds in the weak exchange coupling regime.
In terms of designing interactions that encourage 5(TT)0 production and its subsequent protection from dephasing/de-excitation (besides geometrical factors discussed below), it seems that a balance must be struck that enlarges J without simultaneously facilitating triplet hopping. TSPS PDT appears to lie in between the extremes of TSBS and TIPS derivatives and comes closest to striking this balance. According to theory, the alignment of the molecular axes in all dimer pairs in the TSPS PDT crystal is advantageous for funnelling population to 5(TT)0vs. other spin states and sublevels.31 However, the “powder” nature of the sample likely reduces this state selectivity. Despite no TR-EPR signal, we hypothesize that TSBS PDT, with its similar unit cell properties, may undergo similar initial triplet pair evolution as TSPS PDT, but that resulting quintet and triplet signatures are lost well within the TR-EPR response time because of triplet motion and subsequent annihilation that is facilitated by the long-range intermolecular coupling. The fact that the same spectral transitions, though broadened, are observed in TSPS PDT as in TIPS PDT, but that hopping and spectral evolution times are vastly different, further implies a unique change in triplet pair spin state as potentially responsible.
Conclusions
In summary, we assign component spectra with unusually sharp and intense transitions in PDT films to two distinct correlated triplet pair states. Since the evolution between the two spectra is 103 times faster than diffusion in the TIPS PDT system, it is possible that the conversion represents a change in (TT) spin state and that the sharp spectral transitions are of the type 3(TT) → Tn or 5(TT) → 5(TT)* rather than previously assigned 1(T⋯T) → Sn. TR-EPR spectra confirm the presence of higher spin species such as 5(TT) forming at the earliest detection times (<15 ns). This newfound correlation opens a window into the previously envisioned “black box” of triplet pair spin evolution on a ps and ns timescale. The strong 5(TT)0 character of the EPR spectrum of TSPS PDT can be rationalized by its large exchange coupling and parallel molecular orientation in the unit cell. Evidence that other spin states are also populated can be explained by spin evolution that is dependent on the dimer orientation relative to the applied magnetic field and hopping within the unit cell that modulates exchange coupling. This behaviour contrasts with TIPS PDT, which exhibits complex EPR dynamics suggesting mixed spin states dictated by weak coupling and non-parallel orientation of molecular pairs. The elaboration of dynamics and spectral features with the PDT series has improved our understanding of the design rules for promoting a specific fate of the triplet pair, which has important implications for applications involving optically driven quantum entanglement.Experimental methods
Film fabrication
TSBS PDT was synthesized according to a previously reported procedure.1 The syntheses of TIPS PDT and TSPS PDT are described in the ESI.† All solutions for thin film fabrication were made at a concentration of 7.5 mg mL−1 in chlorobenzene. TIPS PDT, TSPS, and TSBS PDT film were either spin-cast or drop-cast from solution using a volume of 100 μL on quartz substrates. The TIPS PDT film was further solvent-annealed with dichloromethane in order to induce crystallization.Steady-state absorption
Absorption spectra were acquired using a Varian Cary-6000i with integrating sphere.X-ray diffraction
A Rigaku DMAX 2500 diffractometer with Cu Kα radiation was used to measure intensity as a function of 2Θ angle (3–12°).Transient absorption
Samples were excited with ∼150 fs pulses (1 kHz repetition rate) by a Coherent Ti:sapphire laser. A TOPAS optical parametric amplifier was used to select excitation wavelengths of 700 or 725 nm. In order to generate probe light in the visible and near-IR on a ∼5 ns timescale, a small amount of 800 nm light was used to pump sapphire crystals of different thicknesses, which was then detected by photodiode arrays (HELIOS, Ultrafast Systems). Time-dependent spectra were produced by mechanically stepping the time delay, t, of the probe beam from t = −1 ps to 5 ns. An electro-optical system was used to extend the time window to the μs regime (EOS, Ultrafast Systems).Time-resolved EPR
TR-EPR experiments were performed using a Bruker Elexsys E-580 X/Q band spectrometer. Measurements at X-band (∼9.5 GHz) used an ER 4118X-MS5 resonator, while Q-band (∼33.7 GHz) measurements were performed with an EN 5107D2 resonator. Samples were prepared by placing glass capillaries coated with small amounts of the appropriate crystalline powder into quartz EPR tubes, which were then flame-sealed under vacuum. Spectra were collected after photoexcitation at 700 nm with 7 ns, ∼2.5 mJ pulses from an Opotek Radiant 355 LD laser system under constant irradiation with microwave power of 2.4 mW at X-band, and 0.2 mW at Q-band.The monomer triplet spectrum of TIPS PDT was fit using the pepper function in EasySpin,41 a MATLAB package designed for the analysis of EPR spectra. The spin energy level diagram associated with TSPS PDT was calculated using the levels function of EasySpin.
Calculations
Experimental crystal structures were manipulated with the Gaussview version 6 program to create a block of crystal, then whole molecules extracted by filtering out bonded assemblies with atom counts less than the maximal found. From the resulting molecular array, plausible symmetry-unique dimeric structures were selected for further analysis. Alkyl side groups and acetylenic bridges that have not been found to participate in the π conjugation of the PDT system were deleted. Both the TIPS PDT and TSPS PDT structures had two alternative dimers each, whereas the TSBS PDT crystal only showed one (see Fig. 8). Broken-symmetry exchange couplings were calculated with ORCA 4.2.1 (ref. 42 and 43) on the Eagle high-performance computing cluster. Spin-unrestricted DFT calculations with the PBE0 hybrid functional and TZVPP basis were carried out using the resolution-of-the-identity approximation44 with the def2/JK auxiliary fitting basis4. This resulted in 2388 contracted basis functions and 5516 contracted auxiliary basis functions. The broken symmetry automation feature of the code was used with an input spin-quintet wave function, two electrons on “site A” and two on “site B” (each corresponding to one molecular component of the dimer) in order to calculate exchange coupling between the two molecular triplets in the dimer.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was authored in part by the National Renewable Energy Laboratory, operated by Alliance for Sustainable Energy, LLC, for the U.S. Department of Energy (DOE) under Contract No. DE-AC36-08GO28308. Funding provided by the U.S. Department of Energy, Office of Basic Energy Sciences (ERW7404) for spectroscopy, film fabrication and characterization at NREL and for pentadithiophene synthesis at the University of Kentucky. Single-crystal structure determination was supported by NSF-MRI (CHE-1625732). K. J. T., S. P, and J. E. A. thank the National Science Foundation (DMREF 1627428) for support of development of synthetic routes for pentadithiophene derivatives. We thank Kori Smyser and Joel Eaves for useful discussions. The views expressed in the article do not necessarily represent the views of the DOE or the U.S. Government. The U.S. Government retains and the publisher, by accepting the article for publication, acknowledges that the U.S. Government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this work, or allow others to do so, for U.S. Government purposes.References
- M. C. Hanna and A. J. Nozik, J. Appl. Phys., 2006, 100, 074510 CrossRef
.
- E. Busby, T. C. Berkelbach, B. Kumar, A. Chernikov, Y. Zhong, H. Hlaing, X. Y. Zhu, T. F. Heinz, M. S. Hybertsen, M. Y. Sfeir, D. R. Reichman, C. Nuckolls and O. Yaffe, J. Am. Chem. Soc., 2014, 136, 10654–10660 CrossRef CAS PubMed
- J. Lee, M. J. Bruzek, N. J. Thompson, M. Y. Sfeir, J. E. Anthony and M. A. Baldo, Adv. Mater., 2013, 25, 1445–1448 CrossRef CAS PubMed
- N. Monahan and X.-Y. Zhu, Annu. Rev. Phys. Chem., 2015, 66, 601–618 CrossRef CAS PubMed
- S. N. Sanders, E. Kumarasamy, K. J. Fallon, M. Y. Sfeir and L. M. Campos, Chem. Sci., 2020, 11, 1079–1084 RSC
- L. Childress, M. V. Gurudev Dutt, J. M. Taylor, A. S. Zibrov, F. Jelezko, J. Wrachtrup, P. R. Hemmer and M. D. Lukin, Science, 2006, 314, 281–285 CrossRef CAS PubMed
- M. Smith and J. Michl, Chem. Rev., 2010, 110, 6891 CrossRef CAS PubMed
- M. T. Trinh, A. Pinkard, A. B. Pun, S. N. Sanders, E. Kumarasamy, M. Y. Sfeir, L. M. Campos, X. Roy and X.-Y. Zhu, Sci. Adv., 2017, 3, e1700241 CrossRef PubMed
- C. K. Yong, A. J. Musser, S. L. Bayliss, S. Lukman, H. Tamura, O. Bubnova, R. K. Hallani, A. Meneau, R. Resel, M. Maruyama, S. Hotta, L. M. Herz, D. Beljonne, J. E. Anthony, J. Clark and H. Sirringhaus, Nat. Commun., 2017, 8, 15953 CrossRef CAS PubMed
- H. L. Stern, A. Cheminal, S. R. Yost, K. Broch, S. L. Bayliss, K. Chen, M. Tabachnyk, K. Thorley, N. Greenham, J. M. Hodgkiss, J. Anthony, M. Head-Gordon, A. J. Musser, A. Rao and R. H. Friend, Nat. Chem., 2017, 9, 1205–1212 CrossRef CAS PubMed
- S. Khan and S. Mazumdar, J. Phys. Chem. Lett., 2017, 8, 5943–5948 CrossRef CAS PubMed
- K. Miyata, F. S. Conrad-Burton, F. L. Geyer and X. Y. Zhu, Chem. Rev., 2019, 119, 4261–4292 CrossRef CAS PubMed
- R. D. Pensack, E. E. Ostroumov, A. J. Tilley, S. Mazza, C. Grieco, K. J. Thorley, J. B. Asbury, D. S. Seferos, J. E. Anthony and G. D. Scholes, J. Phys. Chem. Lett., 2016, 7, 2370–2375 CrossRef CAS PubMed
- A. R. Srimath Kandada, A. Petrozza and G. Lanzani, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 075310 CrossRef
- C. Grieco, G. S. Doucette, R. D. Pensack, M. M. Payne, A. Rimshaw, G. D. Scholes, J. E. Anthony and J. B. Asbury, J. Am. Chem. Soc., 2016, 138, 16069–16080 CrossRef CAS PubMed
- T. Zhu, Y. Wan, Z. Guo, J. Johnson and L. Huang, Adv. Mater., 2016, 28, 7539–7547 CrossRef CAS PubMed
- T. S. Lee, Y. L. Lin, H. Kim, B. P. Rand and G. D. Scholes, Can. J. Chem., 2019, 97, 465–473 CrossRef CAS
- M. J. Y. Tayebjee, S. N. Sanders, E. Kumarasamy, L. M. Campos, M. Y. Sfeir and D. R. McCamey, Nat. Phys., 2017, 13, 182–188 Search PubMed
- D. Lubert-Perquel, E. Salvadori, M. Dyson, P. N. Stavrinou, R. Montis, H. Nagashima, Y. Kobori, S. Heutz and C. W. M. Kay, Nat. Commun., 2018, 9, 4222 Search PubMed
- H. Nagashima, S. Kawaoka, S. Akimoto, T. Tachikawa, Y. Matsui, H. Ikeda and Y. Kobori, J. Phys. Chem. Lett., 2018, 9, 5855–5861 Search PubMed
- L. R. Weiss, S. L. Bayliss, F. Kraffert, K. J. Thorley, J. E. Anthony, R. Bittl, R. H. Friend, A. Rao, N. C. Greenham and J. Behrends, Nat. Phys., 2017, 13, 176–181 Search PubMed
- M. Chen, M. D. Krzyaniak, J. N. Nelson, Y. J. Bae, S. M. Harvey, R. D. Schaller, R. M. Young and M. R. Wasielewski, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 8178–8183 CrossRef CAS PubMed
- M. Anderlini, P. J. Lee, B. L. Brown, J. Sebby-Strabley, W. D. Phillips and J. V. Porto, Nature, 2007, 448, 452–456 CrossRef CAS PubMed
- K. P. Goetz, Z. Li, J. W. Ward, C. Bougher, J. Rivnay, J. Smith, B. R. Conrad, S. R. Parkin, T. D. Anthopoulos, A. Salleo, J. E. Anthony and O. D. Jurchescu, Adv. Mater., 2011, 23, 3698–3703 CrossRef CAS PubMed
- N. J. Hestand and F. C. Spano, Chem. Rev., 2018, 118, 7069–7163 CrossRef CAS PubMed
- C. Ramanan, A. L. Smeigh, J. E. Anthony, T. J. Marks and M. R. Wasielewski, J. Am. Chem. Soc., 2012, 134, 386–397 CrossRef CAS PubMed
- N. A. Pace, D. H. Arias, D. B. Granger, S. Christensen, J. E. Anthony and J. C. Johnson, Chem. Sci., 2018, 9, 3004–3013 RSC
- C. Grieco, G. S. Doucette, J. M. Munro, E. R. Kennehan, Y. Lee, A. Rimshaw, M. M. Payne, N. Wonderling, J. E. Anthony, I. Dabo, E. D. Gomez and J. B. Asbury, Adv. Funct. Mater., 2017, 27, 1703929 CrossRef
- M. Tabachnyk, B. Ehrler, S. Bayliss, R. H. Friend and N. C. Greenham, Appl. Phys. Lett., 2013, 103, 153302 CrossRef
- R. D. Pensack, A. J. Tilley, C. Grieco, G. E. Purdum, E. E. Ostroumov, D. B. Granger, D. G. Oblinsky, J. C. Dean, G. S. Doucette, J. B. Asbury, Y.-L. Loo, D. S. Seferos, J. E. Anthony and G. D. Scholes, Chem. Sci., 2018, 9, 6240–6259 RSC
- K. Smyser and J. D. Eaves, 2020, arXiv:2004.00103.
-
A. Bencini and D. Gatteschi, EPR of Exchange Coupled Systems, Spring-Nature, New York, 1990 Search PubMed
- N. V. Korovina, S. Das, Z. Nett, X. Feng, J. Joy, R. Haiges, A. I. Krylov, S. E. Bradforth and M. E. Thompson, J. Am. Chem. Soc., 2016, 138, 617–627 CrossRef CAS PubMed
- A. T. Gilligan, E. G. Miller, T. Sammakia and N. H. Damrauer, J. Am. Chem. Soc., 2019, 141, 5961–5971 CrossRef CAS PubMed
- S. N. Sanders, E. Kumarasamy, A. B. Pun, M. T. Trinh, B. Choi, J. Xia, E. J. Taffet, J. Z. Low, J. R. Miller, X. Roy, X. Y. Zhu, M. L. Steigerwald, M. Y. Sfeir and L. M. Campos, J. Am. Chem. Soc., 2015, 137, 8965–8972 CrossRef CAS PubMed
- S. Khan and S. Mazumdar, 2019, arXiv.org > cond-mat > arXiv:1904.08539.
- D. H. Arias, J. L. Ryerson, J. D. Cook, N. H. Damrauer and J. C. Johnson, Chem. Sci., 2016, 7, 1185–1191 RSC
- R. Katoh, E. Katoh, N. Nakashima, M. Yuuki and M. Kotani, J. Phys. Chem. A, 1997, 101, 7725–7728 CrossRef CAS
- K. E. Brown, W. A. Salamant, L. E. Shoer, R. M. Young and M. R. Wasielewski, J. Phys. Chem. Lett., 2014, 5, 2588–2593 CrossRef CAS PubMed
- R. C. Johnson and R. E. Merrifield, Phys. Rev. B: Solid State, 1970, 1, 896–902 CrossRef
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed
- S. Kossmann and F. Neese, Chem. Phys. Lett., 2009, 481, 240–243 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Crystal structure information, additional transient spectroscopic data, global analysis, film X-ray diffraction, temperature and power dependence, raw TR-EPR spectra, calculation details, synthesis details. CCDC 2000560 and 2000561. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc02497j |
| This journal is © The Royal Society of Chemistry 2020 |