
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBenzannulation of isobenzopyryliums with electron-rich alkynes: a modular access to β-functionalized naphthalenes†
An
Wu
 ab,
Hui
Qian
ab,
Wanxiang
Zhao
ab and
Jianwei
Sun
*ab
ab,
Hui
Qian
ab,
Wanxiang
Zhao
ab and
Jianwei
Sun
*ab
aDepartment of Chemistry, The Hong Kong University of Science and Technology (HKUST), Clear Water Bay, Kowloon, Hong Kong SAR, China. E-mail: sunjw@ust.hk
bHKUST-Shenzhen Research Institute, No. 9 Yuexing 1st Rd, South Area, Hi-tech Park, Nanshan, Shenzhen 518057, China
First published on 6th July 2020
Abstract
Described here is a modular strategy for the rapid synthesis of β-functionalized electron-rich naphthalenes, a family of valuable molecules lacking general access previously. Our approach employs an intermolecular benzannulation of in situ generated isobenzopyrylium ions with various electron-rich alkynes, which were not well utilized for this type of reaction before. These reactions not only feature a broad scope, complete regioselectivity, and mild conditions, but also exhibit unusual product divergence depending on the substrate substitution pattern. This divergence allows further expansion of the product diversity. Control experiments provided preliminary insights into the reaction mechanism.
Functionalized naphthalenes are important structural motifs widely present in bioactive natural products, pharmaceuticals and functional materials.1,2 They also serve as valuable intermediates in organic synthesis.3 Among them, naphthalenes bearing an electron-donating group (EDG) at the β-position (e.g., β-naphthols, β-naphthylamines, and β-naphthyl thioethers) are particularly versatile (Fig. 1).2,3 For example, BINOL, which is derived from β-naphthol, is an extensively utilized synthetic precursor toward a wide range of privileged chiral ligands and catalysts.3 While various approaches have been developed for the synthesis of naphthalenes, efficient and selective de novo strategies toward these β-functionalized ones still remain in high demand. Moreover, to the best of our knowledge, a general and modular approach for rapid access to all these electron-rich β-functionalized naphthalenes remains unknown.4
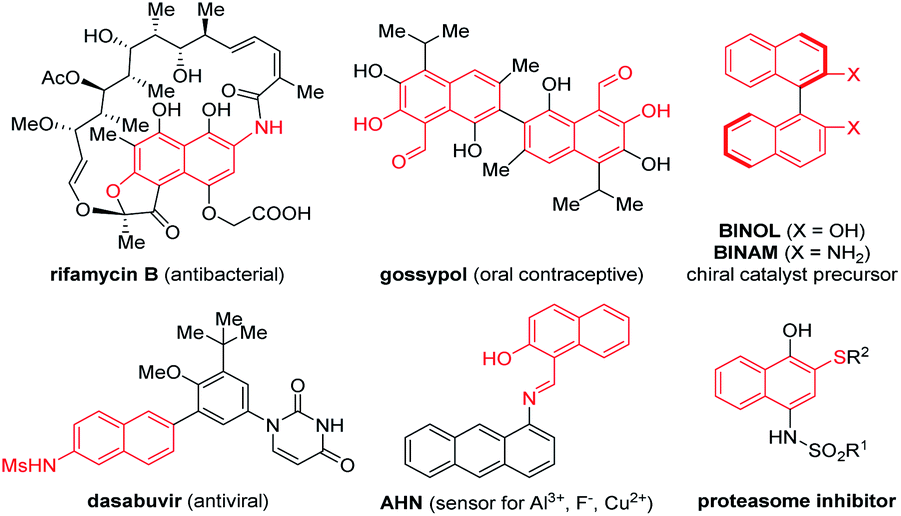 | ||
| Fig. 1 Useful β-naphthol, β-naphthylamine, and β-naphthyl thioether units. | ||
Isobenzopyrylium ions are readily accessible and versatile intermediates in organic synthesis (Scheme 1).5 They are known to participate in cycloaddition reactions with diverse carbon–carbon double bonds or triple bonds for the synthesis of naphthalenes.5,6 While extensive progress has been achieved in this topic, challenges still remain to be addressed. For example, the majority of these benzannulations with alkynes have to be executed at high temperature, except those intramolecular cases or with stoichiometric activators. Moreover, electron-rich alkynes have not been well explored as reaction partners for the benzannulations with isobenzopyrylium ions. Nevertheless, such reactions would provide expedient access to the valuable β-naphthol and β-napthylamine derivatives with diverse substitution patterns (Scheme 1). In this context, here we report our effort in achieving a general and modular strategy toward these electron-rich naphthalenes with high efficiency and regioselectivity under mild conditions. We also observed interesting selectivity divergence controlled by the substituent of the isobenzopyrylium substrates, giving rise to different types of naphthalene products (e.g., 2-hydroxy-1-naphthaldehydes, Scheme 1).
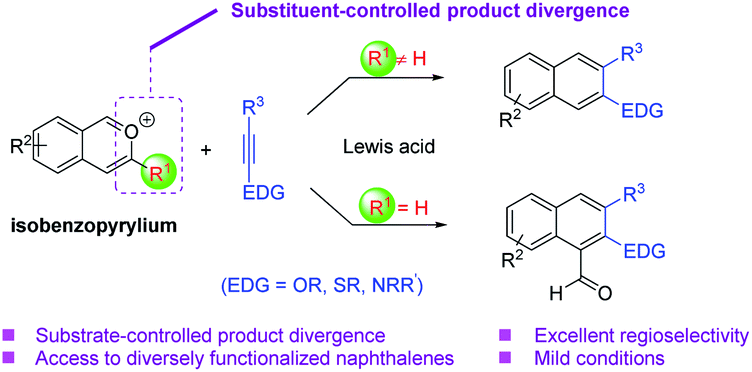 | ||
| Scheme 1 Benzannulation of isobenzopyryliums with electron-rich alkynes. | ||
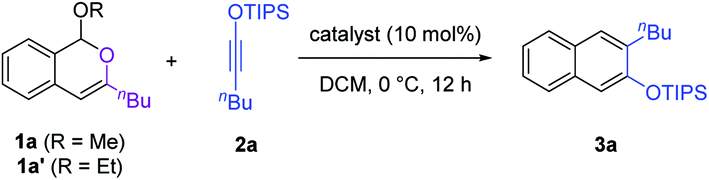
We began our study with isochromene 1a as the isobenzopyrylium precursor. Siloxy alkyne 2a was first employed as the model electron-rich alkyne in view of the extraordinary versatility of this type of alkyne in various cyclization reactions, including benzannulation.7–9 Various Brønsted and Lewis acids were evaluated as catalysts for this reaction. Unfortunately, most of them did not show obvious catalytic activity toward the formation of a naphthalene product (Table 1). These reactions either had little conversion or resulted in a mixture of undesired products. Nevertheless, further screening led to the identification of AgOTf and BF3·OEt2 as capable catalysts (entries 5–6), with the latter being superior, leading to the formation of naphthalene 3a as the major product (75% yield, entry 6). Increasing the catalyst loading to 20 mol% further improved the product yield to 85% (with an isolated yield of 81%, entry 7). Further increasing the catalyst loading proved to be not beneficial (entry 8). Notably, the use of substrate 1a′ bearing an ethoxy leaving group also provided an equally good result. Other solvents, such as toluene and MeCN, were also evaluated, but all gave lower product yields. Finally, it is worth noting that the mild conditions used here are in sharp contrast to the typical high temperature required for the previously known catalytic intermolecular benzannulations involving isobenzopyryliums and alkynes.5d
|
|
||
|---|---|---|
| Entry | Catalyst | Yielda (%) |
| a Reaction scale: 1a (0.05 mmol), 2a (0.06 mmol), catalyst (10 mol%), and DCM (0.5 mL). Yield is based on the analysis of the 1H NMR spectrum of the crude product using CH2Br2 as the internal standard. b Unreacted substrates account for the major remainder of the mass balance. c An unidentifiable mixture accounts for the major remainder of the mass balance. d Yield in parentheses is isolated yield. | ||
| 1 | MeSO3H | <5b |
| 2 | HNTf2 | <5c |
| 3 | Sc(OTf)3 | <5c |
| 4 | TiCl4 | <5b |
| 5 | AgOTf | 30c |
| 6 | BF3·OEt2 | 75 |
| 7 | BF 3 ·OEt 2 (20 mol%) | 85 (81) |
| 8 | BF3·OEt2 (1.0 equiv.) | 83 |
| 9 | BF3·OEt2 (20 mol%), with 1a′ | (75)d |
With the optimized conditions, the generality of this process was examined (Scheme 2). A range of isochromenes 1 and siloxy alkynes 2 participated in this benzannulation to form the desired silylated β-naphthols 3 with moderate to good efficiency. Notably, these products were all generated as a single isomer. This is particularly noteworthy when compared with the cases using phthalazines, which gave a mixture of regioisomers if the phthalazine was unsymmetrically substituted.8g The excellent regioselectivity observed in our reaction is likely attributed to the significant polarization of both cycloaddition partners. While alkyl-substituted siloxy alkynes reacted efficiently, unfortunately aryl-substituted ones led to low yield.
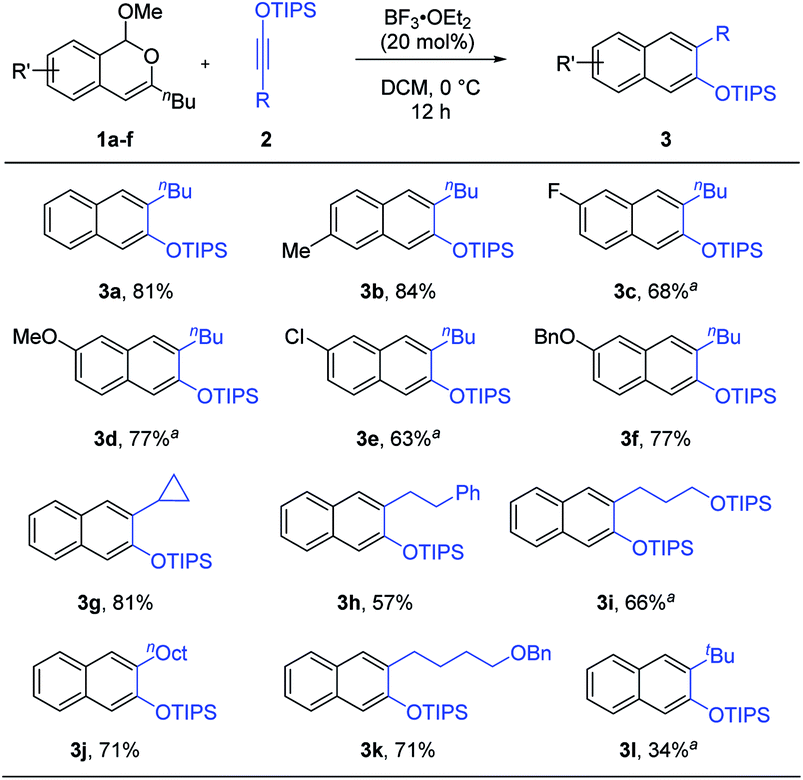 | ||
| Scheme 2 Reaction scope. Reaction scale: 1 (0.5 mmol), 2 (0.6 mmol), DCM (5 mL), 12 h, and isolated yield. a Run for 24 h. | ||
During the above scope study, we found that the reaction of 3-unsubstituted isochromene 1g and siloxy alkyne 2a did not form the desired product 3a. Instead, 2-hydroxy-1-naphthaldehyde 4a was formed as the major product (64% yield, eqn (1)). Careful analysis of this product structure indicated that the original leaving part in 1a (in the case of 3a) was not completely cleaved from the molecule. Instead, only the C–O bond cleavage took place, which led to a dangling aldehyde group. Another important observation is that the TIPS group was lost in the naphthalene product. It was proposed that the methoxide leaving group in 1a might help remove this silyl group and assist the above C–O bond cleavage. In contrast, in the formation of 3a, this methoxide group was a part of the whole leaving unit during rearomatization and thus unavailable for desilylation (vide infra).
In fact, such 2-hydroxy-1-naphthaldehyde products, resembling the highly versatile salicylaldehyde family, are indeed a useful substructure of bioactive molecules, such as gossypol (Fig. 1).2b They can also serve as key intermediates toward functional materials, such as molecular sensors (e.g., AHN, Fig. 1).2e In view of these important applications, considerable efforts were next devoted to further improving the reaction efficiency (see the ESI for details†). Finally, we found that the reaction yield of 4a could be improved to 85% with 2.0 equivalents of BF3·OEt2 and one equivalent of 2,4,6-collidine as the additive. We believe that the role of collidine is to help reversibly stabilize the isobenzopyrylium intermediate and prevent its decomposition during the reaction progress.9b
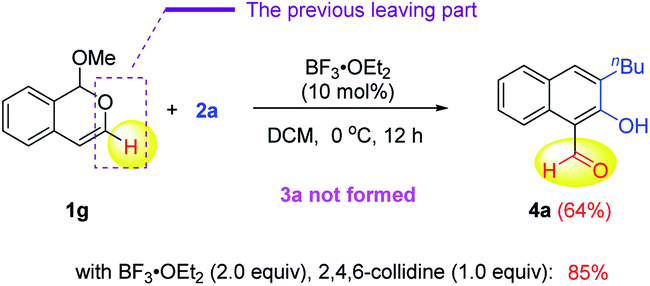 | (1) |
The above protocol for the synthesis of 2-hydroxy-1-naphthaldehydes is general for a range of 3-substituted isobenzopyryliums (Scheme 3). The reaction efficiency was not affected by electron-donating or electron-withdrawing groups. Various siloxy alkynes were also suitable reaction partners, including aryl- and alkyl-substituted ones. Again, these products were all formed as a single regioisomer. While the majority of these reactions were highly chemoselective toward the formation of aldehydes 4, it is worth noting that tBu- and TBS-substituted alkynes resulted in a mixture of 3 and 4, with the former being major. This is likely due to the substantial steric hindrance in close proximity to the silyl group, whose cleavage was obstructed and thus the driving force for the subsequent C–O bond cleavage was weakened, thereby altering the pathway to preferentially form 3 (vide infra). Finally, the structures of 4a and 4b were unambiguously confirmed by X-ray crystallography.
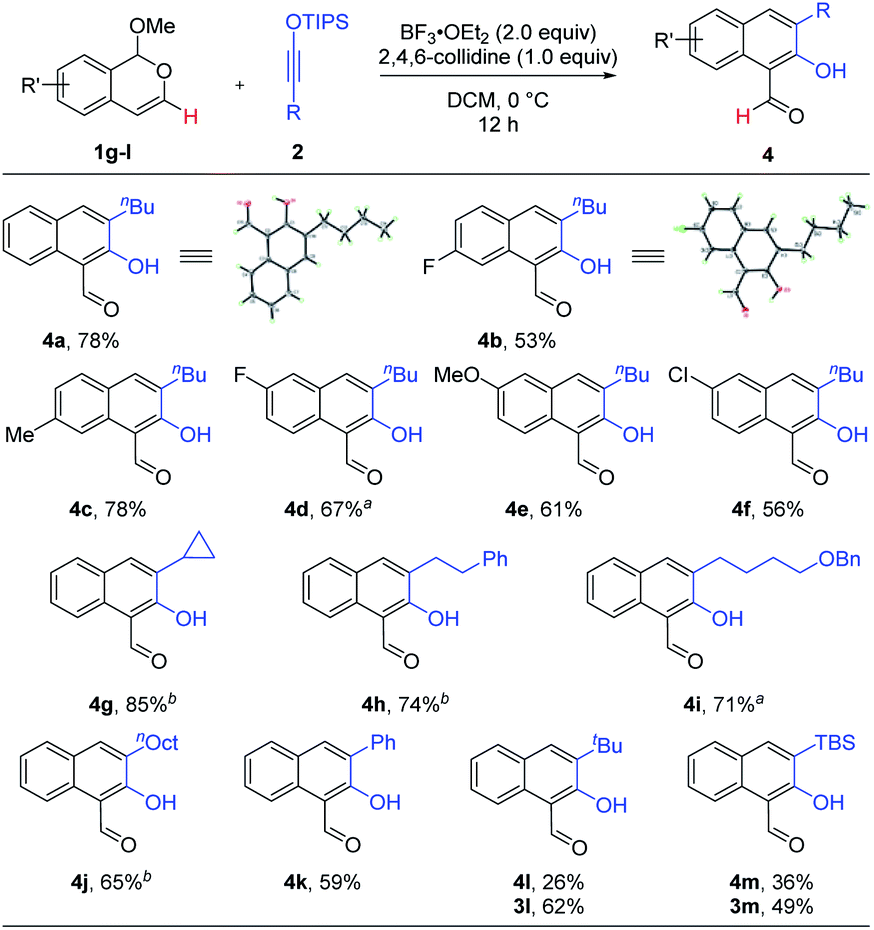 | ||
| Scheme 3 Scope for the synthesis of 2-hydroxy-1-naphthaldehydes 4. Reaction scale: 1 (0.5 mmol), 2 (0.6 mmol), DCM (5 mL), 12 h, and isolated yield. a Run for 16 h. b Run for 19 h. | ||
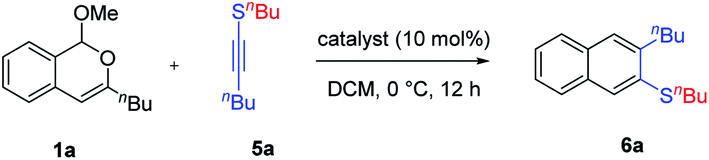
The divergent reaction patterns observed with siloxy alkynes prompted us to explore other electron-rich alkynes. Thioalkyne 5a was next employed for the reaction with 1a. Indeed, direct extension with BF3·OEt2 as the catalyst successfully resulted in regioselective formation of β-naphthyl thioethers 6a in about 70% yield (Table 2, entries 1 and 2). Further screening of other catalysts was performed to further improve the reaction efficiency (see the ESI for details†). While Brønsted acids led to a significant drop in the yield, we were pleased to find that the Lewis acid AgNTf2 exhibited excellent catalytic activity, furnishing 6a in 89% yield (entry 6).
|
|
||
|---|---|---|
| Entry | Catalyst | Yieldb (%) |
| a Reaction scale: 1a (0.05 mmol), 5a (0.06 mmol), catalyst (10 mol%), and DCM (0.5 mL). The yields are based on NMR analysis of the crude product using CH2Br2 as the internal standard. b Isolated yield. | ||
| 1 | BF3·OEt2 | 70 |
| 2 | BF3·OEt2 (20 mol%) | 72 (68)b |
| 3 | HNTf2 | 20 |
| 4 | MeSO3H | 7 |
| 5 | AgOTf | 80 |
| 6 | AgNTf 2 | 89 (83) |
With the above conditions, we carried out a brief examination of the reaction scope (Scheme 4). Interestingly, a similar selectivity divergence was also observed with thioalkynes. Indeed, under identical conditions, the 3-butyl-substituted and 3-unsubstituted isobenzopyryliums all reacted efficiently, with the former leading to only 2-naphthyl thioethers 6, but the latter preferentially to 2-sulfenyl-1-naphthaldehydes 7. It is worth noting that only one regioisomer was observed in all these products.
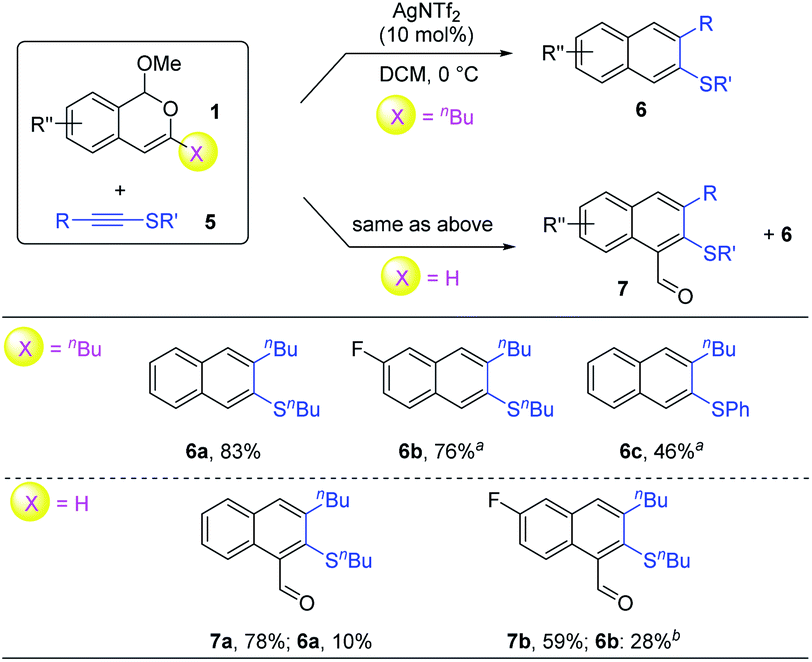 | ||
| Scheme 4 Scope for the synthesis of 2-naphthyl thioethers. Reaction scale: 1 (0.3 mmol), 5 (0.36 mmol), 12 h, and isolated yield. a Run for 37 h. b Run for 19 h. The unreacted alkyne accounted for the major mass balance. | ||
Finally, we were curious about the reactivity of ynamides in such benzannulations.10 To our delight, in the presence of 20 mol% of BF3·OEt2, the reaction between isochromene 1a and different ynamides 8 successfully afforded the desired 2-naphthylamine products 9 with excellent regioselectivity and good efficiency (Scheme 5). Different electron-withdrawing groups on the ynamides were all compatible with this protocol. However, to our surprise, the use of 3-unsubstituted isochromene 1g did not lead to the expected naphthaldehyde 10 at all. Instead, the same product 9a was obtained as the only product, even if two equivalents of BF3·OEt2 were used together with collidine as the additive (conditions in Scheme 3). This result is in dramatic contrast to the cases of siloxy alkynes and thioalkynes.
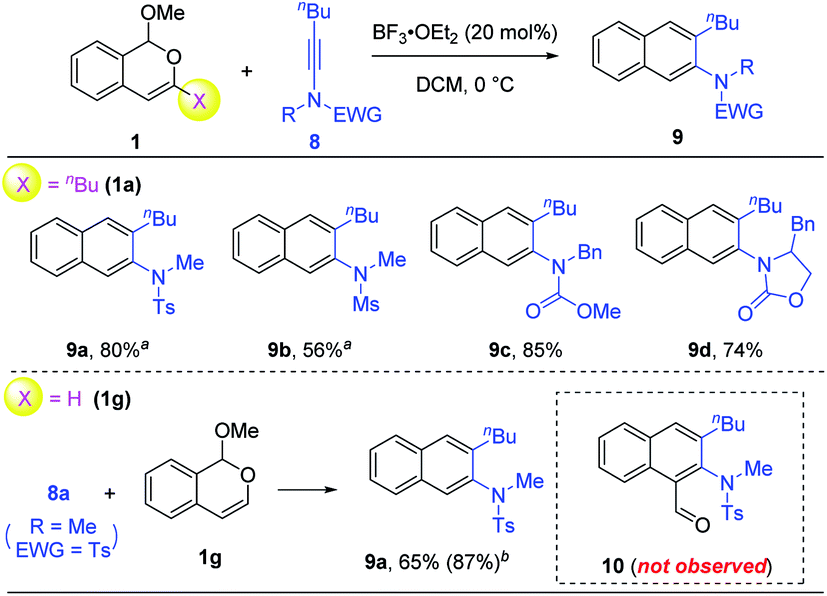 | ||
| Scheme 5 Scope of ynamides. Reaction scale: 1 (0.5 mmol), 8 (0.6 mmol), DCM (5 mL), 12 h, and isolated yield. a Run for 30 h. b Yield in parentheses is obtained with BF3·OEt2 (2.0 equiv.), 2,4,6-collidine (1.0 equiv.), 0 °C, and DCM. | ||
Next, further studies were performed to help understand the unusual selectivity divergence observed with siloxy alkynes and thioalkynes. It is obvious that the substituent at the 3-position of the isochromene substrates has a crucial impact on the product distribution. To further probe the possible steric and electronic effects, we incorporated various other substituents, such as different aryl groups and the bulky tBu group (Table 3). We found that these reactions afforded products 3a and 4′ with variable ratios. With the tBu group, almost the same product distribution as the nBu group was observed, exclusively forming 3a (Table 3, entry 2). However, aryl substitution reversed this ratio, giving product 4′ as the major product (entries 3–5). More importantly, the preference toward 4′ is more pronounced with electron-deficient aryl groups. p-Fluorophenyl substitution led to almost only 4′. These results suggested that the product distribution appeared to be more related to the electronic effect than the steric effect.

Possible mechanisms are depicted in Scheme 6 using a siloxy alkyne as an example. The reaction begins with Lewis acid activation on the acetal motif to form the isobenzopyrylium intermediate I. The subsequent cycloaddition with the alkyne triple bond forms the key bicyclic oxonium intermediate II, which has a resonance form II′. This step might also be stepwise by forming one C–C bond first via ketenium III. In either case, the regioselectivity is precisely controlled by matching the polarity of the two partners, which explains the exclusive formation of only one regioisomer in all the cases. Depending on the R substituent, oxonium II can proceed via three possible pathways. In path a (R = alkyl), the methoxide attacks the oxonium carbon to form intermediate IV, which then undergoes retro-[4 + 2] cycloaddition with concomitant rearomatization to form naphthalene 3, together with the release of ester RCO2Me. However, if the oxonium carbon is unsubstituted (R = H), this intermediate is relatively unstable due to less stabilization of the positive charge (also viewed as a secondary cation in II′, versus tertiary carbocation if R ≠ H). Thus, the silyl enol ether motif tends to push the electron toward this unstable oxonium (a “push–pull” scenario) to cleave the bridging C–O bond. This step, likely assisted by the methoxide attack on the silyl unit, leads to 1,3-dicarbonyl intermediate V. The subsequent tautomerization/aromatization leads to the observed 2-hydroxy-1-naphthaldehyde 4.6b As an exception, if the siloxy alkyne is bulky (e.g., R′ = tBu or TBS), the reaction preferentially gives product 3 (Scheme 3, 4l–m). We believe that the increased steric repulsion by bulky R′ significantly disfavors the approach of methoxide to the neighboring silyl group in IIb. Instead, the methoxide prefers to add to the oxonium carbon, which allows path a to operate and forms 3 as the major product. Another exceptional case is the use of ynamides, which exclusively give products 3 even if R = H. Although actual rationalization would require more sophisticated mechanistic study, we currently reason that the enamide unit in the IIb analogue might not be electron-rich enough to exert sufficient driving force to cleave the C–O bond. This can also be viewed from the standpoint of the limited stabilization of the resulting iminium ion next to an electron-withdrawing group if this C–O bond is cleaved. Consequently, the methoxide prefers to attack the oxonium carbon to favor path a. Finally, with 3-aryl-substituted isochromenes (R = Ar), we believe that the oxonium intermediate IIc is well stabilized by aryl resonance. Thus, the effective delocalization of the positive charge makes this oxonium carbon less electrophilic for nucleophilic attack by methoxide. Instead, the methoxide serves as a base to deprotonate the bridgehead hydrogen, which triggers rearomatization and C–O cleavage to form product 4′. The more electron-deficient aryl group makes this bridgehead hydrogen more acidic, thus further favoring this pathway, which explains the trend in entries 3–5, Table 3.
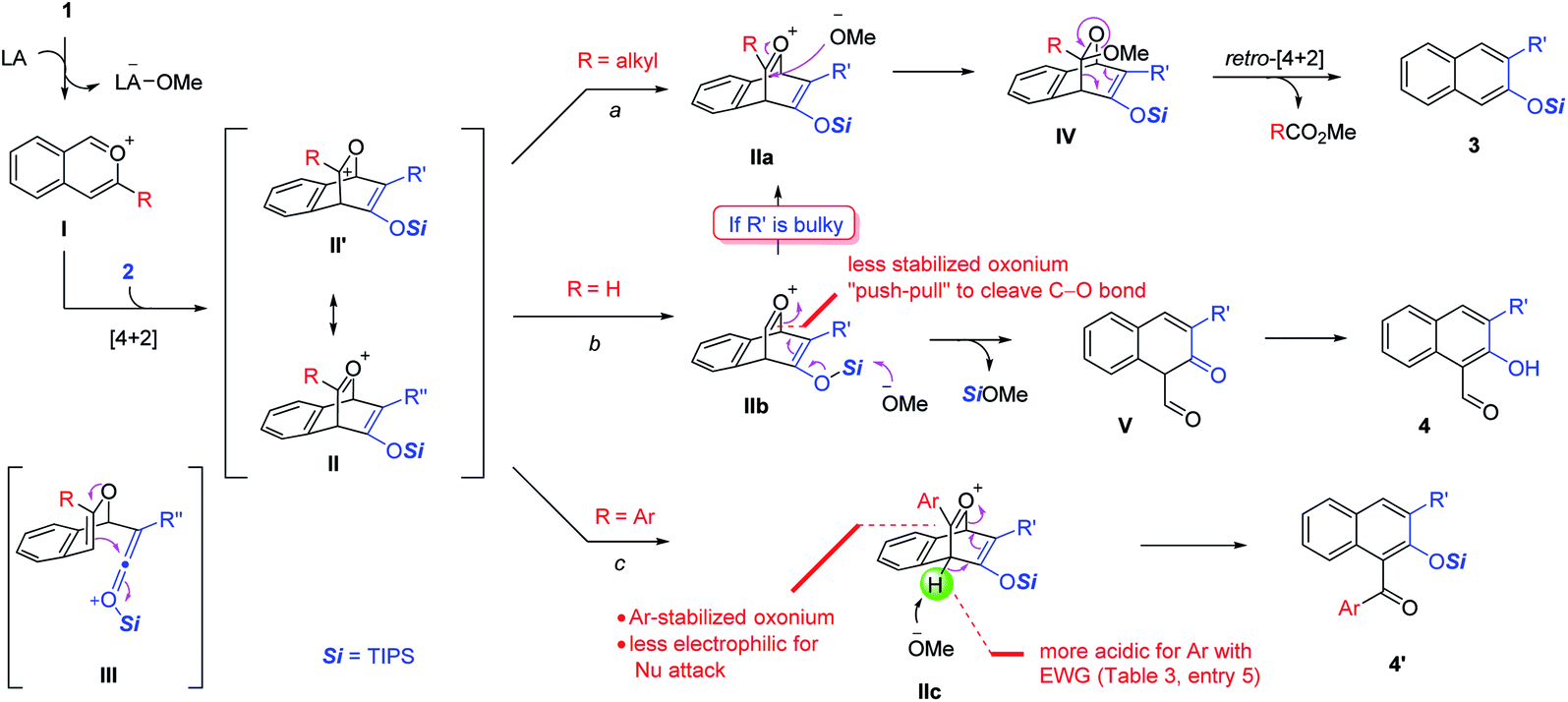 | ||
| Scheme 6 Proposed mechanism. | ||
To further substantiate the above rationale, we carried out some control experiments. First of all, to confirm the fate of the leaving part in the isochromene substrates when β-naphthols 3 were formed, we used a large homobenzyl group in isochromene 1p (eqn (2)). After its reaction with 2a, we were able to isolate ester 11, which is consistent with path a of the proposed mechanism. Next, we also suspected that products 3 and 4 might interconvert to each other depending on the reaction conditions. To probe this possibility, a mixture of 3a and methyl formate was treated with BF3·OEt2 and 2,4,6-collidine, the standard conditions toward products 4 (eqn (3)). However, product 4a was not observed at all, indicating that 3 is not an intermediate toward 4. Similarly, subjecting 4a and MeOH (or MeONa) to BF3·OEt2 did not lead to the deformylation product β-naphthol 3a′, suggesting that 4 is unlikely an intermediate in the formation of 3 (eqn (4)). These observations are consistent with the proposed mechanism, in which the product distribution is kinetically controlled by the barrier in each case and these paths are likely irreversible, but not thermodynamically controlled by product stability.
 | (2) |
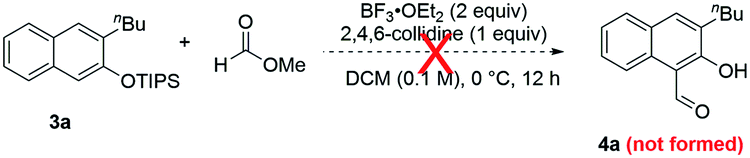 | (3) |
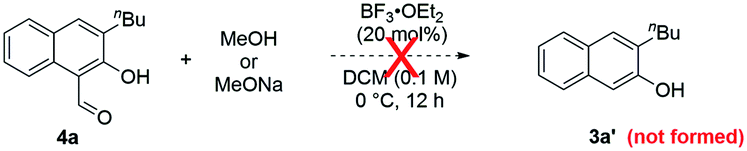 | (4) |
In summary, we have developed a modular strategy for the rapid synthesis of valuable β-functionalized electron-rich naphthalenes, specifically, β-naphthol, β-naphthylamine, and β-naphthyl thioether derivatives. It also represents the first systematic study of the benzannulations of versatile isobenzopyryliums with general electron-rich alkynes. With suitable choice of the isochromene substrates and the Lewis acid catalysts, different types of electron-rich alkynes, such as siloxy alkynes, ynamides, and thioalkynes, participated in the intermolecular cycloaddition reactions under mild conditions with high efficiency and complete regioselectivity. Moreover, depending on the substitution pattern of the isochromene substrates, unusual divergence toward different naphthalene products was observed, thus allowing further diversification of the naphthalene products. Control experiments provided preliminary insights into the intriguing mechanism. Further detailed investigations toward a better understanding are ongoing.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by the Shenzhen Science and Technology Innovation Committee (JCYJ20160229205441091 and JCYJ20170818113708560) and Hong Kong RGC (16302719).References
- (a) N. Abdissa, F. Pan, A. Gruhonjic, J. Grafenstein, P. A. Fitzpatrick, G. Landberg, K. Rissanen, A. Yenesew and M. Erdelyi, J. Nat. Prod., 2016, 79, 2181–2187 CrossRef CAS PubMed; (b) M. C. Kozlowski, B. J. Morgan and E. C. Linton, Chem. Soc. Rev., 2009, 38, 3193–3207 RSC; (c) L. Pu, Chem. Rev., 2004, 104, 1687–1716 CrossRef CAS PubMed.
- (a) K. L. Rinehart Jr, Acc. Chem. Res., 1972, 5, 57–64 CrossRef; (b) E. M. Coutinho, Contraception, 2002, 65, 259–263 CrossRef CAS PubMed; (c) Y. Ge, A. Kazi, F. Marsilio, Y. Luo, S. Jain, W. Brooks, K. G. Daniel, W. C. Guida, S. M. Sebti and H. R. Lawrence, J. Med. Chem., 2012, 55, 1978–1998 CrossRef CAS PubMed; (d) W. Kati, G. Koev, M. Irvin, J. Beyer, Y. Liu, P. Krishnan, T. Reisch, R. Mondal, R. Wagner, A. Molla, C. Maring and C. Collins, Antimicrob. Agents Chemother., 2015, 59, 1505–1511 CrossRef PubMed; (e) B. Kaur, A. gupta and N. Kaur, J. Photochem. Photobiol., A, 2020, 389, 112140 CrossRef.
- (a) S. G. Telfer and R. Kuroda, Coord. Chem. Rev., 2003, 242, 33–46 CrossRef CAS; (b) J. M. Brunel, Chem. Rev., 2007, 107, PR1–PR45 CrossRef CAS; (c) Q.-X. Guo, Z.-J. Wu, Z.-B. Luo, Q.-Z. Liu, J.-L. Ye, S.-W. Luo, L.-F. Cun and L.-Z. Gong, J. Am. Chem. Soc., 2007, 129, 13927–13938 CrossRef CAS PubMed; (d) H. Egami, K. Matumoto, T. Oguma, T. Kunisu and T. Katsuki, J. Am. Chem. Soc., 2010, 132, 13633–13635 CrossRef CAS PubMed; (e) D. C. Patel, Z. S. Breitbach, R. M. Woods, Y. Lim, A. Wang, F. W. Foss Jr and D. W. Armstrong, J. Org. Chem., 2016, 81, 1295–1299 CrossRef CAS PubMed.
- For a review: (a) C. B. de Koning, A. L. Rousseau and W. A. L. van Otterlo, Tetrahedron, 2003, 59, 7–36 CrossRef CAS; for selected examples: (b) R. B. Dateer, B. S. Shaibu and R.-S. Liu, Angew. Chem., Int. Ed., 2012, 51, 113–117 CrossRef CAS PubMed; (c) T. P. Willumstad, O. Haze, X. Y. Mak, T. Y. Lam, Y.-P. Wang and R. L. Danheiser, J. Org. Chem., 2013, 78, 11450–11469 CrossRef CAS PubMed; (d) M. Murai, K. Origuchi and K. Takai, Org. Lett., 2014, 16, 3828–3831 CrossRef CAS PubMed; (e) Z. Chen, W. Zeng, H. Jiang and L. Liu, Org. Lett., 2012, 14, 5385–5387 CrossRef CAS PubMed; (f) Y. Bai, J. Yin, Z. Liu and G. Zhu, J. Org. Chem., 2015, 80, 10226–10233 CrossRef CAS PubMed; (g) D. Wei, T.-J. Hu, C.-G. Feng and G.-Q. Lin, Chin. J. Chem., 2018, 36, 743–748 CrossRef CAS; (h) Y. Bao, Y. Yang, Z. Dai, S. Ji, Q. Zhou and F. Yang, Adv. Synth. Catal., 2019, 361, 2154–2158 CrossRef CAS.
- Reviews on isobenzopyrylium ions: (a) E.-V. Kuznetsov, I.-V. Shcherbakova and A.-T. Balaban, Adv. Heterocycl. Chem., 1990, 50, 157–254 CrossRef CAS; (b) M. Nogradi, Sci. Synth., 2003, 14, 201–273 CAS; (c) J. Santamaría and C. Valdés, Mod. Heterocycl. Chem., 2011, 3, 1631–2682 Search PubMed; (d) J.-R. Chen, X.-Q. Hu and W.-J. Xiao, Angew. Chem., Int. Ed., 2014, 53, 4038–4040 CrossRef CAS PubMed; (e) S. J. Hein, D. Lehnherr, H. Arslan, F. J. Uribe-Romo and W. R. Dithtel, Acc. Chem. Res., 2017, 50, 2776–2788 CrossRef CAS PubMed; (f) L. Chen, K. Chen and S. Zhu, Chem, 2018, 4, 1–55 CrossRef.
- Ynol ethers or ynamines/ynamides (O- or N-substituted alkynes) have not been used as reaction partners before. For selected cycloadditions of isobenzopyryliums with alkenes and alkynes to form naphthalenes (high temperature was typically used for catalytic intermolecular ones of alkynes), see: (a) N. Asao, K. Takahashi, S. Lee, T. Kasahara and Y. Yamamoto, J. Am. Chem. Soc., 2002, 124, 12650–12651 CrossRef CAS PubMed; (b) N. Asao, T. Nogami, S. Lee and Y. Yamamoto, J. Am. Chem. Soc., 2003, 125, 10921–10925 CrossRef CAS PubMed; (c) J. Barluenga, H. Vazquez-Villa, A. Ballesteros and J. M. Gonzalez, Org. Lett., 2003, 5, 4121–4123 CrossRef CAS PubMed; (d) N. Asao, T. Kasahara and Y. Yamamoto, Angew. Chem., Int. Ed., 2003, 42, 3504–3506 CrossRef CAS PubMed; (e) N. Asao, H. Aikawa and Y. Yamamoto, J. Am. Chem. Soc., 2004, 126, 7458–7459 CrossRef CAS PubMed; (f) N. Asao, K. Sato, Menggenbateer and Y. Yamamoto, J. Org. Chem., 2005, 70, 3682–3685 CrossRef CAS PubMed; (g) J. Barluenga, H. Vazquez-Villa, I. Merino, A. Ballesteros and J. M. Gonzalez, Chem.–Eur. J., 2006, 12, 5790–5805 CrossRef CAS PubMed; (h) D. Yue, N. D. Cá and R. C. Larock, J. Org. Chem., 2006, 71, 3381–3388 CrossRef CAS PubMed; (i) X. Zhao, X.-G. Zhang, R.-Y. Tang, C.-L. Deng and J.-H. Li, Eur. J. Org. Chem., 2010, 4211–4217 CrossRef CAS; (j) X.-L. Fang, R.-Y. Tang, X.-G. Zhang, P. Zhong, C.-L. Deng and J.-H. Li, J. Organomet. Chem., 2011, 696, 352–356 CrossRef CAS; (k) D. Lehnherr, J. M. Alzola, E. B. Lobkovsky and W. R. Dichtel, Chem.–Eur. J., 2015, 21, 18122–18127 CrossRef CAS PubMed; (l) C. Zhang, L. Chen, K. Chen, H. Jiang and S. Zhu, Org. Chem. Front., 2018, 5, 1028–1033 RSC; (m) R. Liang, J. Zhang, L. Chen and S. Zhu, Asian J. Org. Chem., 2018, 7, 545–549 CrossRef CAS; sporadic examples with thioalkynes or selenoalkynes are known, but all under heating conditions, see ref. 6b and j, and (n) A. C. Mantovani, D. F. Back and G. Zeni, Eur. J. Org. Chem., 2012, 4574–4579 CrossRef CAS; for an example of our effort on the study of isobenzopyryliums: (o) H. Qian, W. Zhao, Z. Wang and J. Sun, J. Am. Chem. Soc., 2015, 137, 560–563 CrossRef CAS PubMed.
- Reviews on siloxy alkynes: (a) M. Shindo, Tetrahedron, 2007, 63, 10–36 CrossRef CAS; (b) H. Qian, W. Zhao and J. Sun, Chem. Rec., 2014, 14, 1070–1085 CrossRef CAS PubMed.
- Examples of siloxy alkynes in benzannulation reactions: (a) C. J. Kowalski and G. S. Lal, J. Am. Chem. Soc., 1988, 110, 3693–3695 CrossRef CAS; (b) R. L. Danheiser, R. G. Brisbois, J. L. Kowalczyk and R. F. Miller, J. Am. Chem. Soc., 1990, 112, 3093–3100 CrossRef CAS; (c) W. F. Austin, Y. Zhang and R. L. Danheiser, Org. Lett., 2005, 7, 3905–3908 CrossRef CAS PubMed; (d) M. Movassaghi, M. D. Hill and O. K. Ahmad, J. Am. Chem. Soc., 2007, 129, 10096–10097 CrossRef CAS PubMed; (e) W. F. Austin, Y. Zhang and R. L. Danheiser, Tetrahedron, 2008, 64, 915–925 CrossRef CAS PubMed; (f) J. R. Cabrera-Pardo, D. I. Chai and S. A. Kozmin, Adv. Synth. Catal., 2013, 355, 2495–2498 CrossRef CAS PubMed; (g) Y. E. Türkmen, T. J. Montavon, S. A. Kozmin and V. H. Rawal, J. Am. Chem. Soc., 2012, 134, 9062–9065 CrossRef PubMed; (h) J. R. Cabrera-Pardo, D. I. Chai, S. Liu, M. Mrksich and S. A. Kozmin, Nat. Chem., 2013, 5, 423–427 CrossRef CAS PubMed; (i) C. S. Sumaria, Y. E. Türkmen and V. H. Rawal, Org. Lett., 2014, 16, 3236–3239 CrossRef CAS PubMed.
- Our efforts in the study of siloxy aklynes: (a) W. Zhao, Z. Wang and J. Sun, Angew. Chem., Int. Ed., 2012, 51, 6209–6213 CrossRef CAS PubMed; (b) W. Zhao, Z. Li and J. Sun, J. Am. Chem. Soc., 2013, 135, 4680–4683 CrossRef CAS PubMed; (c) W. Zhao, H. Qian, Z. Li and J. Sun, Angew. Chem., Int. Ed., 2015, 54, 10005–10008 CrossRef CAS PubMed; (d) A. Wu, Q. Feng, H. H. Y. Sung, I. D. Williams and J. Sun, Angew. Chem., Int. Ed., 2019, 58, 6776–6780 CrossRef CAS PubMed.
- Examples of ynamides/ynamines for benzannulations: (a) E. Oishi, N. Taido, K.-i. Iwamoto, A. Miyashita and T. Higashino, Chem. Pharm. Bull., 1990, 38, 3268–3272 CrossRef CAS; (b) K.-i. Iwamoto, S. Suzuki, E. Oishi, K.-i. Tanji, A. Miyashita and T. Higashino, Chem. Pharm. Bull., 1995, 43, 679–682 CrossRef CAS; (c) T. Y. Lam, Y.-P. Wang and R. L. Danheiser, J. Org. Chem., 2013, 78, 9396–9414 CrossRef CAS PubMed; (d) J. Xue, E. Gao, X.-N. Wang and J. Chang, Org. Lett., 2018, 20, 6055–6058 CrossRef CAS PubMed; (e) Y. Wang, L.-J. Song, X. Zhang and J. Sun, Angew. Chem., Int. Ed., 2016, 55, 9704–9708 CrossRef CAS PubMed . See also ref. 4.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1995830 and 1995829. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc02502j |
| This journal is © The Royal Society of Chemistry 2020 |