
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organophotoredox/palladium dual catalytic decarboxylative Csp3–Csp3 coupling of carboxylic acids and π-electrophiles†‡
Kaitie C.
Cartwright
and
Jon A.
Tunge
 *
*
Department of Chemistry, The University of Kansas, 1567 Irving Rd., Lawrence, KS 66045, USA. E-mail: tunge@ku.edu
First published on 16th July 2020
Abstract
A dual catalytic decarboxylative allylation and benzylation method for the construction of new C(sp3)–C(sp3) bonds between readily available carboxylic acids and functionally diverse carbonate electrophiles has been developed. The new process is mild, operationally simple, and has greatly improved upon the efficiency and generality of previous methodology. In addition, new insights into the reaction mechanism have been realized and provide further understanding of the harnessed reactivity.
Introduction
Photoredox catalysis has emerged as a powerful strategy in the functionalization of feed-stock carboxylic acids.1 Using the carboxylic acid as a traceless activating group has allowed for the site-specific generation of radical species under mild conditions, while producing carbon dioxide as the only stoichiometric byproduct. Moreover, as compared to traditional anionic decarboxylation, radical decarboxylation often provides a facile and more generally applicable pathway for activation.2One reaction that has benefited from the employment of photoredox-promoted decarboxylation is decarboxylative allylation (DcA). This reaction is a subset of the Tsuji–Trost allylation that distinguishes itself by the use of carboxylic acids as latent carbanions, bypassing the need for pre-formed organometallics or strong alkaline conditions.3 Despite being an attractive and powerful transformation, anionic decarboxylation is limited to carboxylic acid substrates which provide sufficient stabilization of the resulting carbanion following the decarboxylation event. A pKa of less than 25 is often required for facile decarboxylation, and substrates with pKa values between 25 and 30 demand elevated temperatures.3a,4
Recently, our lab has developed methodology that overcomes this limitation in pKa through the development of a photoredox/Pd dual catalytic coupling strategy (Schemes 1A and B).5,6 In this system, an oxidative radical decarboxylation is facilitated by an iridium photosensitizer, which allows for a facile radical decarboxylation. Inverting the electronic demand for the decarboxylation event allows access to carboxylic acid nucleophiles that fall outside the pKa requirement for thermal decarboxylation. Once the carbon radical species is generated, combination with an electrophilic Pd-π-allyl intermediate provides a new C(sp3)–C(sp3) bond.7
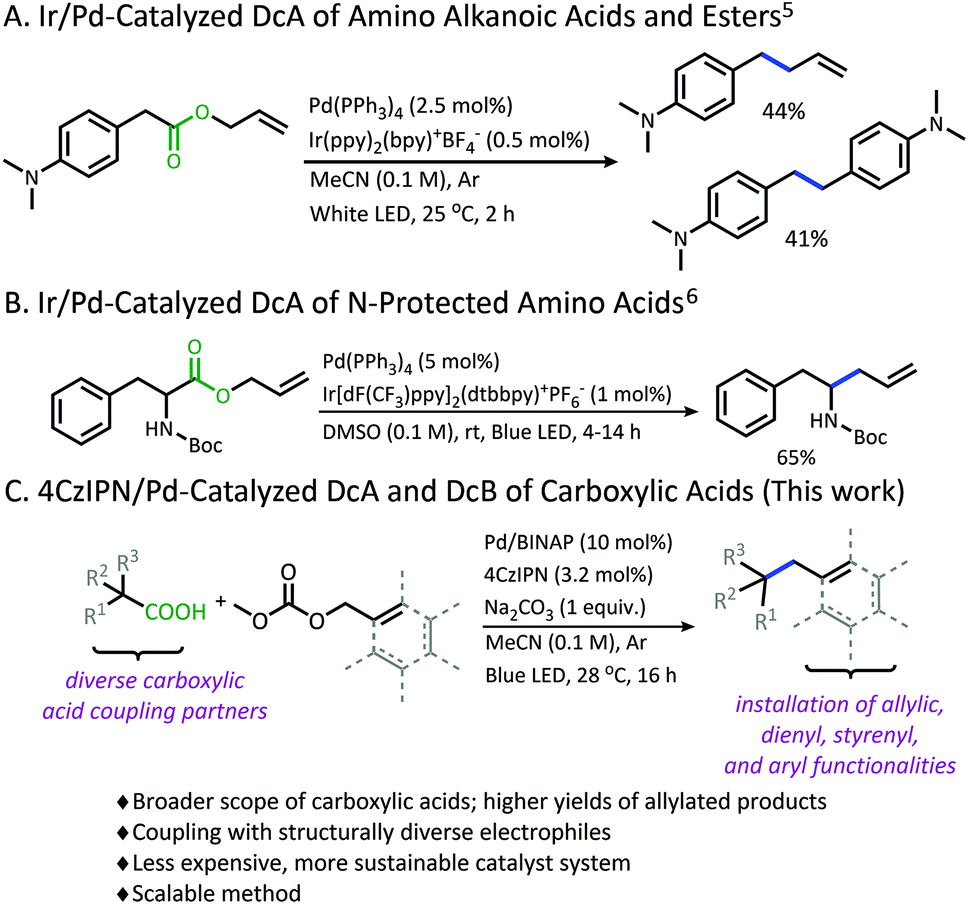 | ||
| Scheme 1 Tunge dual catalytic decarboxylative couplings. | ||
Although a great advancement in DcA, this chemistry remains underdeveloped. Specifically, the reaction was only demonstrated on activated carboxylic acids that contain α-amino5,6 and/or α-benzylic5 functionalities which provide significant stabilization to the radical intermediate. Within these substrate classes, the α-amino precursors tend to provide higher yields than benzylic radical precursors due to an undesired homocoupling observed between benzylic radicals (Scheme 1A).5,6 Allylation of carboxylic acids without these radical-stabilizing functionalities has remained elusive. Additionally, the desire to utilize this reactivity with more complex electrophilic coupling partners has not been realized. In fact, the original method was only operable with a narrow range of 2-substituted allyl electrophiles, while the more challenging 3-substituted allyl electrophiles were not effectively coupled.5,6,8 Finally, the methods reported thus far utilized iridium-based photosensitizers which are ultimately expensive and unsustainable.9
Herein, a new organophotoredox/palladium dual catalytic process is described that has greatly improved the utility of this technology through (1) achieving higher yields and greater generality in carboxylic acid nucleophiles, and (2) allowing access to a variety of structurally diverse allylic electrophiles for the installation of various alkenyl, styrenyl, dienyl, and aryl functionalities (Scheme 1C). In addition, evaluating this methodology has provided new insights into the advantages and limitations of radical DcA, as well as the dominant catalytic pathway.
Results and discussion
With the goal of developing a broadly applicable catalyst system for decarboxylative allylation of alkanes, a complete re-evaluation of the catalyst system was undertaken. In 2016, Zhang reported a set of carbazole-based donor–acceptor fluorophores that are inexpensive and easily accessible organic photoredox catalysts.10 Further, this group of catalysts possesses a range of redox potentials resembling that of the iridium photosensitizers originally employed (Fig. 1, [Ir(ppy)2(bpy)]+ BF4−: P*/P− = +0.95 V & P/P− = −1.05 V; [Ir[dF(CF3)ppy]2(dtbbpy)]+ PF6−: P*/P− = +1.21 V & P/P− = −1.37 V). Employing the carbazole-based photosensitizers (Fig. 1) in the photoredox-facilitated DcA revealed the 4CzIPN catalyst to be the most compatible with the transformation (Table S1†).10a,11 The only other catalyst to provide notable conversion in the DcA was 4CzPN, but produced less allylated product than 4CzIPN (Table S1†).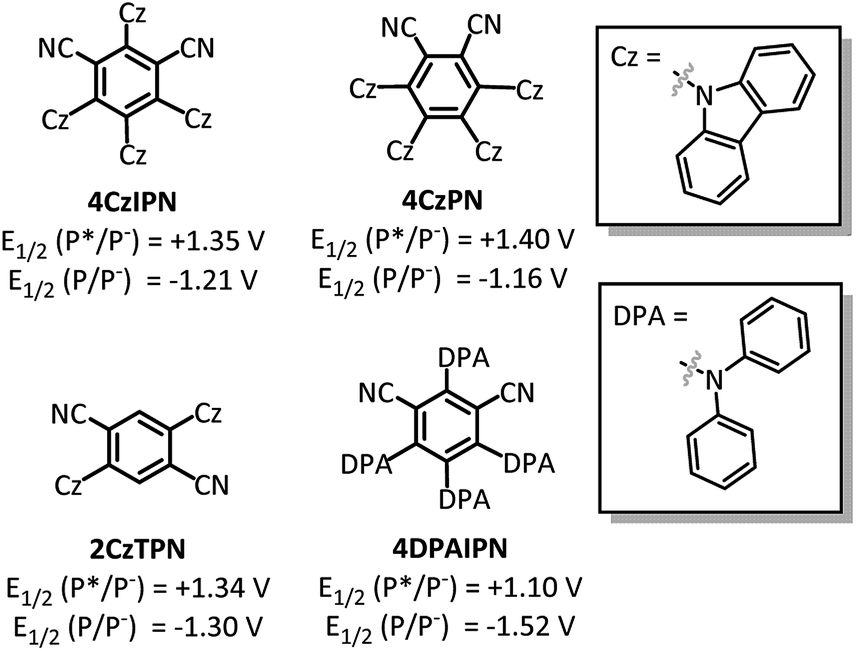 | ||
| Fig. 1 Dicyanobenzene carbazole-based fluorophores. | ||
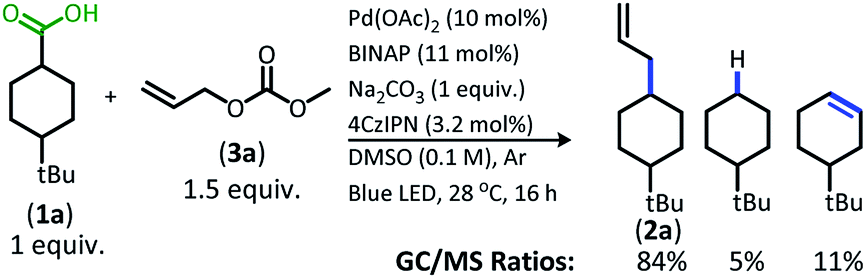
The change from Ir to 4CzIPN was first adopted into reaction conditions that were found to be successful in our previous dual catalytic DcA (Scheme 2).5,6,12 This reaction produced the allylated product, but also produced an alkane and alkene side product. Additionally, this reaction did not proceed to high conversion.
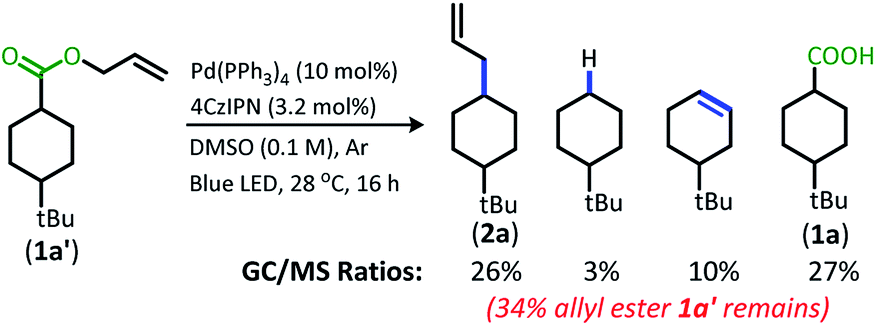 | ||
| Scheme 2 Previous DcA conditions with 4CzIPN photosensitizer (GC/MS ratios list represent area percent out of 100% of all products and starting materials present in the final reaction mixture). | ||
In order to maximize production of the allylated product, the influence of changing the Pd ligands was investigated (Table 1, see Table S2† for full ligand screening and Table S3† for Pd pre-catalyst evaluation). The change in ligand proved to have a significant influence on the success of the reaction. Change to the ligand resulted in either (1) low conversion, (2) high conversion, but little decarboxylation, (3) the alkene/alkane side products were formed in large quantity, or (4) the allylated product was the major product.
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entrya | Ligand | (2a) | Alkane | Alkene | (1a′) | (1a) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
a All reactions were performed on 0.2 mmol scale and product ratios were determined by GC/MS (GC/MS ratios list represent area percent out of 100% of all products and starting materials present in the final reaction mixture).
b
2a formed as a 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 trans:cis mixture. :40 trans:cis mixture.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1b | BINAP | 82 | 9 | 9 | 0 | 0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | (R)-C3-TUNEPHOS | 78 | 8 | 14 | 0 | 0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | (R)-SEGPHOS | 76 | 9 | 15 | 0 | 0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | (S)–SYNPHOS | 75 | 8 | 17 | 0 | 0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | (R)-Tol-BINAP | 73 | 8 | 19 | 0 | 0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | DTMB SegPhos | 70 | 8 | 22 | 0 | 0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
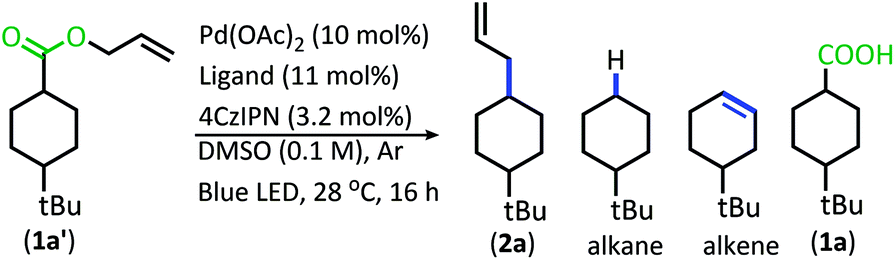
The ligands that allowed the reaction to proceed with full conversion, complete decarboxylation, and produced the allylated product as the major product consisted of bidentate phosphine ligands with similar molecular scaffolds (Table 1, entries 1–6). Of these catalysts, the BINAP and SegPhos ligands provided a higher percentage of allylated product 2a (Table 1, entries 1 & 3) as compared to their more sterically demanding counterparts, Tol-BINAP and DTBM-SegPhos (Table 1, entries 5 & 6). Gratifyingly, BINAP proved to be the superior ligand and is also one of the least expensive and most readily accessible of the ligand set (Table 1, entry 1).
To improve upon the reaction economy and operational simplicity, an intermolecular process that directly utilizes a carboxylic acid for reaction with various allylic carbonates was devised (Table S5†). To our delight, the intermolecular reaction proceeded well. Allyl methyl carbonate was identified as the ideal allylic electrophile and sodium carbonate proved to be the ideal base (Scheme 3). Lastly, the reaction was found to produce comparable results in acetonitrile (73% yield 2a, 60:40 d.r.), which was preferred over the use of dimethyl sulfoxide due to the more straightforward isolation of the allylated product.13
 | ||
| Scheme 3 Intermolecular DcA of 1a (GC/MS ratios list represent area percent out of 100% of all products and starting materials present in the final reaction mixture). | ||
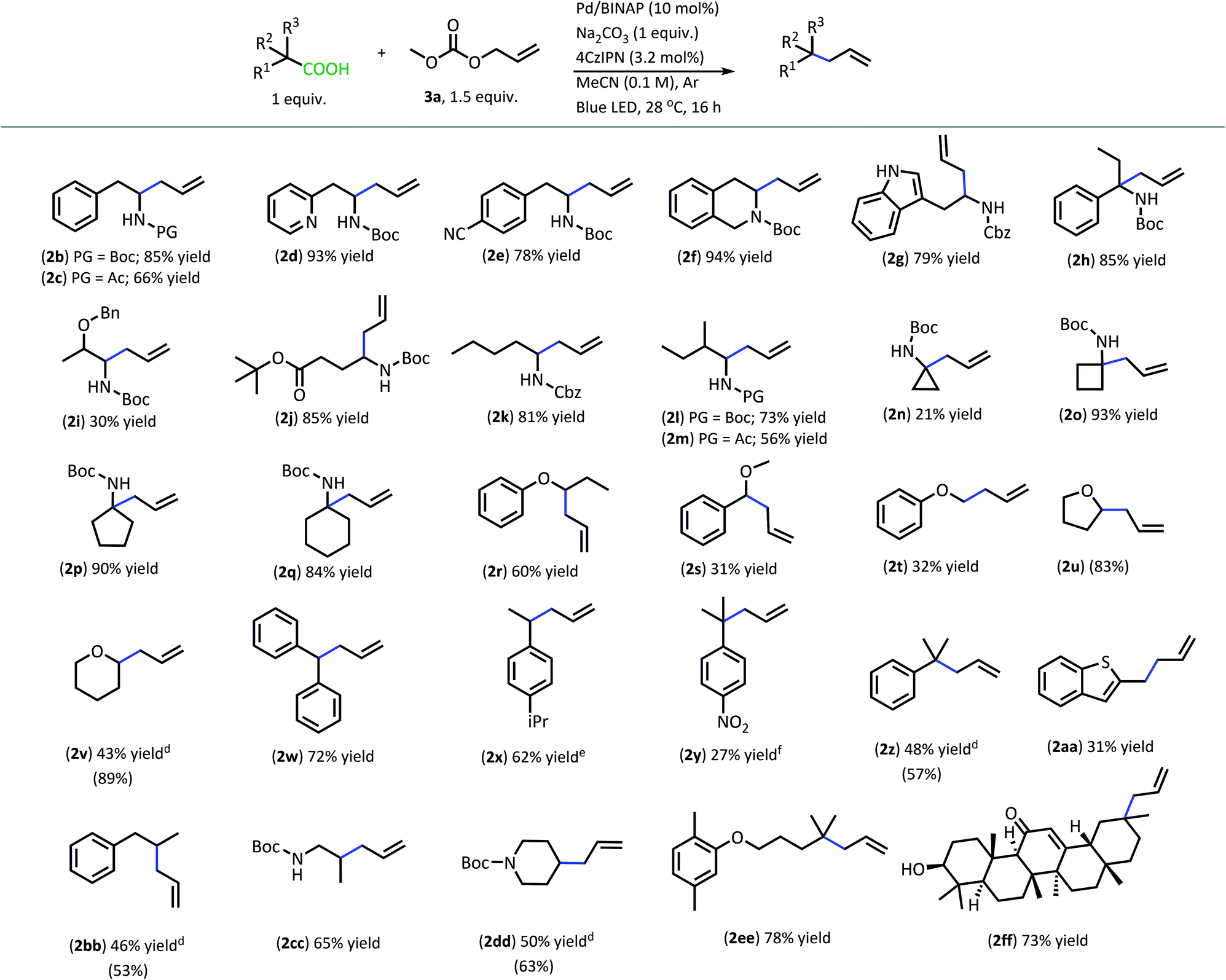
With superior decarboxylative allylation conditions established, attention was turned to the allylation of a variety of carboxylic acids (Table 2). Initially, the allylation was performed with N-protected amino acids (2b–q). No alkene or alkane side products were observed with these substrates. Gratifyingly, the amino acid substrates that underwent allylation with our first-generation method were able to be allylated in similar to increased yields under our new conditions.
| a Reactions were performed on 0.2 mmol scale. b Isolated yields are shown. c (Yields) were determined by q1H NMR with pyridine as the internal standard for products that were found to be volatile. d Yields are an average of two reactions. e 95% pure, see ESI for details. f Product was isolated with alkane side product and mass adjusted; product previously used in thermal DcA.17 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Interestingly, under these conditions, carbamate-protected amino acids produced higher yields than their acyl-protected counterparts, in contrast to first-generation conditions that resulted in comparable yields for both groups (2b–c, 2l–m).6 The N-Boc- and N-Cbz-protected amino acid substrates generally performed well with a variety of different side chain functionalities. Substrates containing N-heterocycle side chains proceeded smoothly, allowing for high yields of the allylated alkaloids 2d and 2g (93% & 79%, respectively).14 Increased steric demand around the carboxylic acid did not interfere with the efficiency of DcA (2h & 2l). Cyclic scaffolds also generally underwent DcA well (2n–q). Interestingly, the cyclopropane amino acid substrate 2n was allylated in low yield (21%) but larger ring sizes such as a cyclobutane (2o), cyclopentane (2p), and cyclohexane (2q) were allylated in high yields.15 The DcA of 1b was successfully run in batch on the 1 mmol scale, which provided 2b in 93% yield (see ESI† for details).
In addition to the successful decarboxylative allylation of amino acids, other carboxylic acids that produce more reactive radical intermediates were allylated with this method (2r–2ff). Substrates that produced monosubstituted and unsubstituted α-oxy radicals were generally allylated in moderate yields (2r–v). The alkene and alkane side products do arise in the DcA of these substrates. Benzylic carboxylic acid substrates that provide 3°, 2°, and 1° benzylic radicals upon decarboxylation could be allylated in moderate to good yields (2w–2aa). Of these, the substrates 1w and 1x that produce 2° benzylic radicals provided yields exceeding 60% (2w & 2x). This is notable since the diphenylmethane nucleophile has previously been employed in anionic allylation under thermal conditions, but the yield of allylated product is much less (10% yield) than what can be obtained in the 4CzIPN/Pd DcA conditions.16 The homodimeric products frequently observed under the Ir/Pd conditions with benzylic carboxylic acids were not observed with the disubstituted benzylic acids allylated under the new conditions. Instead, alkene and/or alkane side products were observed. Conversely, benzylic acid substrate (1aa) was able to be allylated in 31% yield (2aa) but did produce the homocoupled product in 48% yield.
The dual catalytic DcA was also applicable to carboxylic acids that possess weak radical-stabilizing functionalities (2a, 2bb–2ff). The highest yields of allylated products, 70–80%, were observed with quaternary carboxylic acids such as gemfibrozil (1ee) and 18-β-glycyrrhetinic acid (1ff). The tertiary carboxylic acids of this group such as β-amino acid (1cc) and γ-amino acid (1dd), were allylated in moderate yields (65% 2cc & 50% 2dd). Unfortunately, unsubstituted acids that do not possess functionalities to provide moderate stabilization of the radical intermediate underwent poor conversion and only produced trace amounts of allylated product. To demonstrate the difference in reactivity, a disubstituted acid (1w) was allylated selectively in the presence of an unsubstituted acid under standard reaction conditions (Scheme 4).18 Substrate 1gg remained intact during this reaction.19
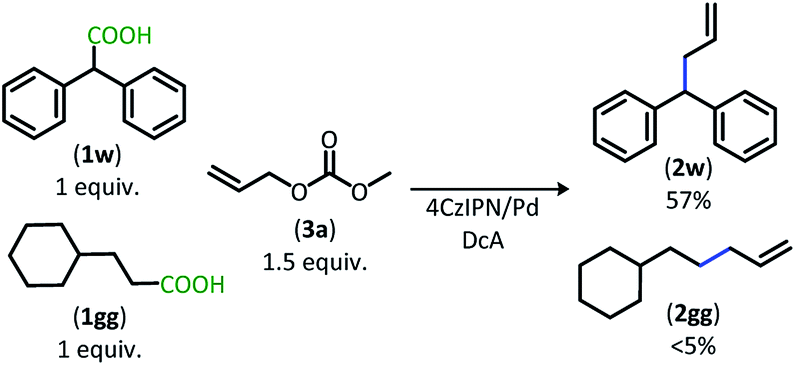 | ||
| Scheme 4 Preferential DcA of disubstituted carboxylic acid. | ||
While substrates that contain strong radical-stabilizing groups such as the α-amino acids only produced the allylated product, more reactive radicals resulted in the formation of the alkene and alkane side products as was observed in the optimization of DcA of 1a’ and 1a. Potentially, these products form as a result of off-cycle pathways such as disproportionation or β-hydride elimination.20
In addition to the alkane- and alkene-forming side reactions, a recent report by König brought to our attention the possibility of competitive radical addition to the 4CzIPN photocatalyst.21 Sure enough, the “Allyl-4CzIPN” was isolated along with intact 4CzIPN after the catalytic DcA of 1w (Scheme 5A).22 When the “Allyl-4CzIPN” was utilized as the photocatalyst in the DcA of 1b, a greatly diminished yield results (Scheme 5B). Thus, the side reaction with 4CzIPN ultimately degrades the photocatalyst activity.
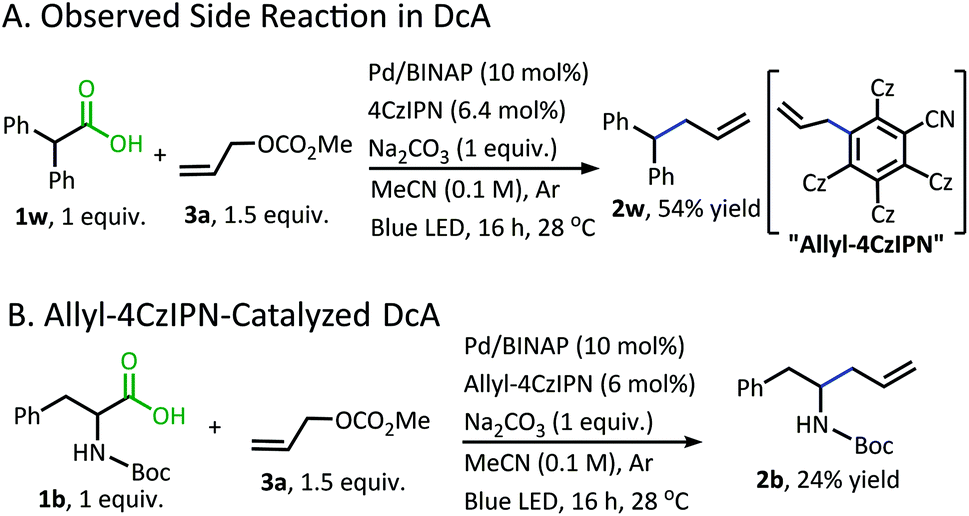 | ||
| Scheme 5 Allylation of 4CzIPN. | ||
Apart from the improved accessibility of carboxylic acid nucleophiles, another compelling aspect of this methodology is its efficient coupling of carboxylic acids with more functionally diverse π-electrophiles (Table 3). Allylic carbonates with substituents in the 2-position proceeded well, with the electron-withdrawing phenyl substituent producing higher yield (4c, 82%) compared with 2-methyl allyl carbonate (4b, 64%). Having Cl in the 2-position unfortunately resulted in a poor yield (4d, 14%) and formation of a plethora of side products. This may be due to poor chemoselectivity between allyl carbonate and vinyl halide electrophiles.
1- and 3-substituted allylic carbonates having a variety of carbon chains also operate well in the radical DcA (4e–4h). These carbonates functioned comparably regardless of the geometric configuration of the alkene or the structure of the allylic carbonate (branched/linear). They also produced the E-configured olefin and the linear product despite the variation in the carbonate starting material. Cyclic allylic carbonates also provided allylated products, however the yields were about half what is realized with their acyclic counterparts (4i–4j).
Utilizing branched acyclic allylic carbonates was quite advantageous (e.g.3e′vs.3e).23 For one, they can be synthesized from aldehydes, providing a plethora of inexpensive options for the electrophilic coupling partner.24 Also, utilizing branched carbonates provided cleaner reactions than their 3-substituted allyl carbonate counterparts in some cases. For instance, using carbonates derived from cinnamyl alcohols produced a complex mixture of products under these reaction conditions and provided poor yields of allylated product. In contrast, switching to the branched carbonates allowed for the formation of styrenyl products in good yields (4k–4o, 70–95% yield). Potentially, this reaction benefits from a more facile oxidative addition that is achieved with the branched carbonates as opposed to the cinnamyl carbonates. The conjugated olefin products are produced as a mixture of geometric isomers which is likely the result of a background photoisomerization.24 Here, the styrenyl products were obtained in higher yields and with greater E-selectivity when the aryl moiety contained electron-withdrawing functionalities (4m & 4o, 86% & 81% yield, respectively, >90:10 E:Z) as opposed to electron-donating functionalities (4l & 4n, 79% & 72% yield, respectively, ∼70:30 E:Z). The branched carbonates also proved highly useful for the installation of other conjugated functionalities such as dienes (4p & 4q, 72% & 45% yield, respectively), but these too provided a mix of geometric isomers.
Apart from DcA, decarboxylative benzylation (DcB) with Pd-π-benzyl electrophiles would be an attractive application of dual catalytic couplings.25 Through the use of benzylic carbonates, toxic benzylic halides that are traditionally employed in cross-coupling could be avoided.26 However, Pd-catalyzed DcB is challenging since aromatic moieties receive additional stability from conjugation, making the barrier for oxidative addition higher.27 Therefore, benzylic carbonates with extended conjugation are generally utilized because their lower resonance energies lead to lower activation barriers.27,28
Benzylic carbonates with extended conjugation underwent high conversion under our mild reaction conditions. The highest yielding benzylations are those that install naphthalene (4r, 71% yield) and phenanthrene tethers (4t, 78% yield). The benzylic carbonates react to full conversion to produce the cross-coupled product and the mass-balance is typically made up of the benzyl dimer.
The regiochemistry of the alkylcarbonate group does influence the reaction yield. With the naphthalene system, the higher yield is obtained when the naphthalene is substituted in the 2-position (3r) vs. the 1-position (3s) (4r & 4s isolated in 71% & 35% yield, respectively). The reaction with carbonate (3s) resulted in 46% yield of naphthalene dimer. Similarly, the 9-anthracenyl carbonate (3u) provides the cross-coupled product (4u) in just 21% yield due to favorable formation of the homocoupled bianthracenyl dimer.
Heteroaromatic systems also underwent successful cross-coupling, but with lower yields.14 Compared to the aromatic hydrocarbon systems, the heteroaromatic benzylic carbonates produced a variety of oligomer side products. Nevertheless, these carbonates have been utilized to provide the cross-coupled product between 1f and furyl carbonates as well as with pyrrolyl carbonate, with yields between 20–40% (4v–x). Like the naphthalene system, the regiochemistry of the carbonate leaving group influences the yield of the reaction. With the furyl carbonate, the 2-alkyl carbonate (3v) provided higher yields than the analogous 3-substitution (3w). An N-Boc pyrrole group was also able to be installed through this coupling, albeit with a low isolated yield that is reminiscent of what was observed with the related furyl carbonates (4x).
After exploration of scope and reactivity, mechanistic probes were utilized to shed light on the dominant catalytic cycle. First, it is important to note that the reaction does not proceed (<5% yield) without the Pd, photocatalyst, or light, indicating that this process relies on a light-promoted dual catalytic system (Table S6†).
When the radical trap, TEMPO, is incorporated into the reaction, the allylated product is not produced (Scheme 6). Instead, conversion is interrupted, and low yields of TEMPO trapped products result. This result supports the formation of radicals along the reaction pathway.
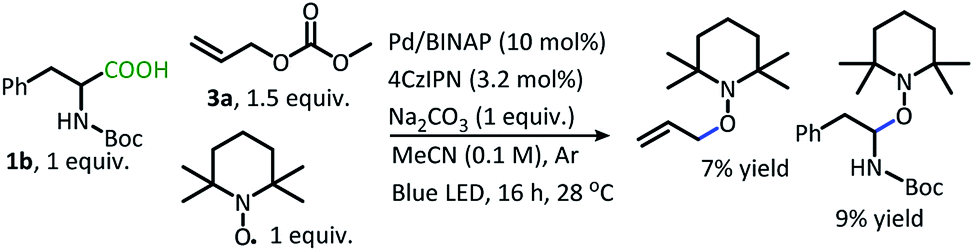 | ||
| Scheme 6 DcA in the presence of TEMPO. | ||
A Stern–Volmer analysis was completed to assess the photocatalytic processes taking place. The potential fluorescence emission quenching of 4CzIPN with diphenylcarboxylate tetrabutylammonium salt, as well as with a variety of Pd species, was investigated (see ESI† for experimental details). Quenching of 4CzIPN with diphenylcarboxylate tetrabutylammonium salt was observed, which was not surprising since 4CzIPN (+1.35 V)10 is sufficiently reactive to oxidize the carboxylate (+1.0 to +1.3 V).18 However, the observed 4CzIPN quenching with the carboxylate salt was significantly less efficient (Fig. S2†) than the observed quenching with relevant Pd species investigated.
The first Pd species investigated was the Pd(OAc)2/BINAP pre-catalyst. When these reactions were assembled, an initial stirring of the Pd(OAc)2 pre-catalyst and BINAP was performed to allow for reduction of the Pd(II) to Pd(0).29 However, this process was not anticipated to fully reduce the Pd(II) species and thus higher oxidation states of Pd are expected to be present at the onset of the reaction.29 The mixture of Pd(OAc)2/BINAP was found to be an efficient quencher of 4CzIPN (∼10-fold more than carboxylate, Fig. S3†). Similar Pd species are reported to have reduction potentials between −1.1 and −1.3 V, which places the potentials for the Pd catalyst reduction close to the excited state oxidation potential of 4CzIPN (−1.04 V).10,30–32 If such a reduction initiates the cycle, the resulting 4CzIPN radical cation (+1.52 V)10 should easily oxidize carboxylates (+1.0 to +1.3 V)17 in the reaction, thus turning over the catalyst.
Alternatively, it is possible that the dominant catalytic pathway proceeds via oxidative addition of Pd(0) and allyl carbonate to provide the cationic π-allyl-Pd. This species is anticipated to be highly relevant to the mechanistic pathway, thus its ability to quench 4CzIPN was also considered. After an evaluation of reported redox potentials, the cationic π-allyl-Pd is not expected to be reduced by 4CzIPN as the potential reported for this species is −1.35 V, which is not a favorable match for oxidative quenching of 4CzIPN (−1.04 V).10,33 In agreement with this redox potential mismatch, a π-allyl-PdCl dimer/BINAP mixture did not quench 4CzIPN (Fig. S4†). However, π-allyl-Pd(OAc)/BINAP was found to be an efficient quencher of 4CzIPN (∼10-fold more efficient than carboxylate, Fig. S4†). The high quenching ability of the BINAP complex of π-allyl-Pd(OAc) as opposed to π-allyl-PdCl points to a carboxylate oxidation pathway reminiscent of other metal-carboxylate species.34 Furthermore, the higher quenching constant for the π-allyl-PdOAc complex, as compared to tetrabutylammonium carboxylate, indicates the key role of the Pd complex in facilitating quenching.
After the Pd-facilitated quenching and decarboxylation events, there are several possibilities for the C–C bond formation. First, the possibility of a radical-polar cross-over event must be considered.21,35 But, due to the large negative reduction potentials of most alkyl radicals utilized herein (<−1.3 V),36 this pathway is not favorable and thus determined to be irrelevant in the dominant catalytic cycle. An alternate route is the radical addition to a π-allyl-Pd intermediate. This addition can occur at the allyl ligand resulting in the new C–C bond or through coordination to the Pd centre followed by inner-sphere reductive elimination to form the new C–C bond.6,37 In either event, the cross-coupled product would be liberated along with a Pd(I) species. This species is expected to have a reduction potential around −1.26 V (ref. 31) which would be well-matched with the 4CzIPN radical anion (−1.21 V).10 The decreased efficiency seen with 4CzPN may be due to the less negative oxidation potential (−1.16 V)10 that is not as well-matched for Pd(I) reduction as that of 4CzIPN, further supporting the relevancy of a Pd(I) reduction to the catalytic cycle (Table S1†).
Taken together, we believe the dominant catalytic pathway to proceed via a reductive quenching pathway in which the excited 4CzIPN is quenched by a π-allyl-Pd carboxylate species (Scheme 7).38 Single electron transfer from carboxylate to Pd then facilitates decarboxylation and the formation of a carbon radical. Rebound of the radical with the π-allyl-Pd results in the formation of the allylated product and a Pd(I). The Pd(I) can be reduced by one electron via the 4CzIPN radical anion to complete both catalytic cycles.39
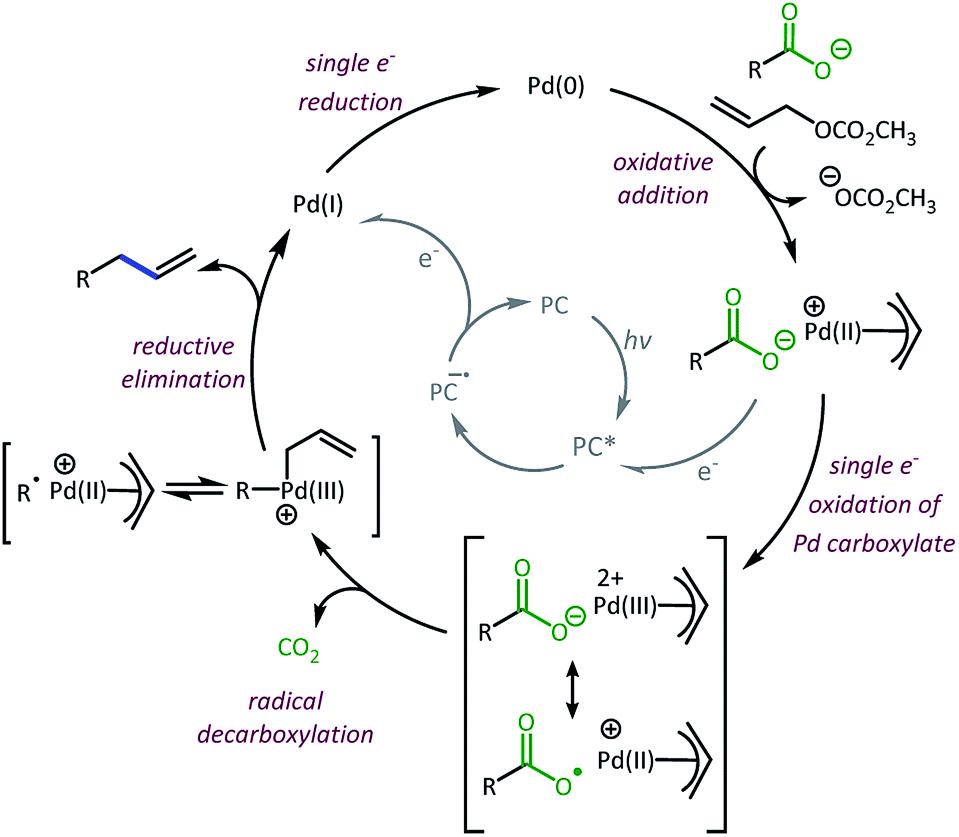 | ||
| Scheme 7 Proposed dominant catalytic pathway.38,39 | ||
Conclusions
A catalytic, mild, operationally simple, and minimal waste-producing decarboxylative method for the site-specific installation of allylic, dienyl, styrenyl, and benzylic functionalities on carboxylic acids has been realized. This method has drastically improved upon our previously reported methodology through the change to a more sustainable and less expensive organophotocatalyst which also led to higher yields across a much broader substrate scope. In addition, the cross-coupling methodology described herein provides a general way to incorporate diverse electrophiles as well as utilize carboxylic acid nucleophiles that do not undergo decarboxylation under thermal control. Thus, the methodology herein may provide a simple and general approach towards building molecular complexity from easily accessible and inexpensive starting materials that would be otherwise difficult to achieve.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation (CHE-1800147) and the Kansas Bioscience Authority Rising Star program. Support for the NMR instrumentation was provided by NSF Academic Research Infrastructure Grant No. 9512331, NIH Shared Instrumentation Grant No. S10RR024664, and NSF Major Research Instrumentation Grant No. 0320648.Notes and references
- Reviews: (a) H. Huang, K. Jia and Y. Chen, ACS Catal., 2016, 6, 4983–4988 CrossRef CAS; (b) J. Xuan, Z.-G. Zhang and W.-J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 15632–15641 CrossRef CAS PubMed; (c) R. A. Angnes, Z. Li, C. R. D. Correia and G. B. Hammond, Org. Biomol. Chem., 2015, 13, 9152–9167 RSC. Select examples: (d) Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillian, Science, 2014, 345, 436–440 CrossRef PubMed; (e) Y. Miyake, K. Nakajima and Y. Nishibayashi, Chem. Commun., 2013, 49, 7854–7856 RSC; (f) J. Schwarz and B. König, Green Chem., 2018, 20, 323361 RSC.
- Select seminal reports and reviews on the radical decarboxylation strategy: (a) R. G. Johnson and R. K. Ingham, Chem. Rev., 1956, 56, 219–269 CrossRef CAS; (b) F. Minisci, R. Galli, M. Cecere, V. Malatesta and T. Caronna, Tetrahedron Lett., 1968, 5609–5612 CrossRef CAS; (c) B. C. L. Weedon, Q. Rev., Chem. Soc., 1952, 6, 380–398 RSC; (d) W. B. Smith and H. G. Glide, J. Am. Chem. Soc., 1959, 5325–5329 CrossRef CAS; (e) Y. A. Serguchev and I. P. Beletskaya, Russ. Chem. Rev., 1980, 49, 2257–2285 CAS; (f) W. W. Cleland, Acc. Chem. Res., 1999, 32, 862–868 CrossRef CAS; (g) D. H. R. Barton and E. P. Serebryakov, Proc. Chem. Soc., 1962, 309 Search PubMed; (h) K. Okada, K. Okamoto and M. Oda, J. Am. Chem. Soc., 1988, 110, 8736–8738 CrossRef CAS; (i) R. A. Sheldon and J. K. Kochi, Org. React., 2011, 19, 279–421 Search PubMed; (j) B. R. Brown and M. A. D. L. Phil, Q. Rev., Chem. Soc., 1951, 5, 131–146 RSC.
- Review on decarboxylative allylation and benzylation: (a) J. D. Weaver, A. Recio, A. J. Grenning and J. A. Tunge, Chem. Rev., 2011, 111, 1846–1913 CrossRef CAS PubMed. Tsuji–Trost reaction: (b) J. Tsuji, H. Takahashi and M. Morikawa, Tetrahedron Lett., 1965, 6, 4387 CrossRef; (c) B. M. Trost and C. R. Self, J. Org. Chem., 1984, 49, 468 CrossRef CAS; (d) B. M. Trost and E. Keinan, Tetrahedron Lett., 1980, 21, 2591 CrossRef CAS. Recent review on ionic decarboxylative cross-coupling reactions of C(sp3) acids: P. J. Moon and R. J. Lundgren, ACS Catal., 2020, 10, 1742–1753 CrossRef CAS.
- Review on pKa limitation in DcA: (a) J. A. Tunge, Isr. J. Chem., 2020, 60, 1–10 CrossRef. Select representative examples of various nucleophiles in thermal decarboxylative allylation: (b) J. Tsuji, Tetrahedron Lett., 1983, 24, 1793 CrossRef CAS; (c) T. Saegusa, J. Am. Chem. Soc., 1980, 102, 6381 CrossRef; (d) A. J. Grenning and J. A. Tunge, Org. Lett., 2010, 12, 740 CrossRef CAS PubMed; (e) A. Recio and J. A. Tunge, Org. Lett., 2009, 11, 5630 CrossRef CAS; (f) J. D. Weaver and J. A. Tunge, Org. Lett., 2008, 10, 4657 CrossRef CAS.
- S. B. Lang, K. O'Nele and J. A. Tunge, J. Am. Chem. Soc., 2014, 136, 13606–13609 CrossRef CAS PubMed.
- S. B. Lang, K. O'Nele and J. A. Tunge, Chem.–Eur. J., 2015, 51, 18589–18593 CrossRef.
- Other method employing radical addition to Pd-π-allyl: (a) H.-H. Zhang, J.-J. Zhao and S. Yu, J. Am. Chem. Soc., 2018, 140, 16914–16919 CrossRef CAS PubMed. Method utilizing Ni/Ir in single electron Tsuji–Trost reaction: (b) J. K. Matsui, A. Gutiérrez-Bonet, M. Rotella, R. Alam, O. Gutierrez and G. A. Molander, Angew. Chem., Int. Ed., 2018, 57, 15847–15851 CrossRef CAS PubMed.
- Y. Duan, M. Zhang, R. Ruzi, Z. Wu and C. Zhu, Org. Chem. Front., 2017, 4, 525–528 RSC.
- (a) K. Teegardin, J. I. Day, J. Chan and J. Weaver, Org. Process Res. Dev., 2016, 20, 1156–1163 CrossRef CAS; (b) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS.
- (a) J. Luo and J. Zhang, ACS Catal., 2016, 6, 873–877 CrossRef CAS; (b) E. Speckmeier, T. G. Fisher and K. Zeitler, J. Am. Chem. Soc., 2018, 140, 15353–15365 CrossRef CAS.
- 4CzIPN was also found to be a successful alternative to Ir by the Zhang group in a decarboxylative cross-coupling: ref. 9a: Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillian, Science, 2014, 345, 437–440 CrossRef CAS PubMed.
- Ir[dF(CF3)ppy]2(dtbbpy)PF6 can be purchased from Sigma for $834 per gram. 4CzIPN cost can be synthesized from carbazole and tetrafluoroisophthalonitrile for $4.01 per gram.7a.
-
2a q1H NMR yield 73%, ∼60:40 d.r. under optimal intermolecular reaction conditions in acetonitrile (0.1 M).
- A. R. Katritzky, Chem. Rev., 2004, 104, 2125–2126 CrossRef CAS.
- The ring size influence was speculated to be due to a less favorable radical decarboxylation that results from the formation of a carbon radical in an orbital with increased s-character: E. V. Anslyn and D. A. Dougherty, Modern Physical Organic Chemistry, University Science Books, 1st edn, 2006, p. 100 Search PubMed.
- S.-C. Sha, J. Zhang, P. J. Carroll and P. J. Walsh, J. Am. Chem. Soc., 2013, 135, 17602–17609 CrossRef CAS.
- S. R. Waetzig and J. A. Tunge, J. Am. Chem. Soc., 2007, 129, 14860–14861 CrossRef CAS.
- Carboxylate oxidation potentials: (a) J. D. Griffin, M. A. Zeller and D. A. Nicewicz, J. Am. Chem. Soc., 2015, 137, 11340–11348 CrossRef CAS; (b) L. Capaldo, L. Buzzetti, D. Merli, M. Fagnoni and D. Ravelli, J. Org. Chem., 2016, 81, 7102–7109 CrossRef CAS.
- Standard DcA reaction with 1gg provided 15% yield of 2gg. Irradiating the reaction 4x as long (65 hours) increased the yield of 2gg to 20%. Irradiation for 170 hours resulted in 27% yield of 2gg. Note, yields of 2gg were determined by q1H NMR with pyridine internal standard from crude reaction mixture.
- S. W. Benson, J. Phys. Chem., 1985, 89(20), 4366–4369 CrossRef CAS.
- K. Donabauer, M. Maity, A. L. Berger, G. S. Huff, S. Crespi and B. König, Chem. Sci., 2019, 10, 5162–5166 RSC.
- Regiochemistry of 4CzIPN allylation not known, see ESI† Section 8 for spectral data.
- Allylic carbonates could be made from commercially available allylic alcohols and various allylic alcohols could be easily accessed via reduction of α,β-unsaturated aldehydes or from vinyl magnesium bromide Grignard additions to aldehydes (see ESI† for details).
- J. Lu, B. Pattengale, Q. Liu, S. Yang, W. Shi, S. Li, J. Huang and J. Zhang, J. Am. Chem. Soc., 2018, 140(42), 13719–13725 CrossRef CAS PubMed.
- B. Liegault, J.-L. Renaud and C. Bruneau, Chem. Soc. Rev., 2008, 37, 290–299 RSC.
- S. Marchini, L. Passerini, M. D. Hoglund, A. Pinno and M. Nendza, Environ. Toxicol. Chem., 1999, 18, 2759 CAS.
- (a) Y. Hayashi, M. Nishizawa and T. Sakan, Tetrahedron Lett., 1977, 33, 2509 CrossRef CAS; (b) J. Y. Legros, M. Toffano and J.-C. Fiaud, Tetrahedron, 1995, 51, 3235 CrossRef CAS; (c) J.-Y. Legros, G. Primault, M. Toffano, M. A. Rivière and J.-C. Fiaud, Org. Lett., 2000, 2, 433 CrossRef CAS; (d) J.-Y. Legros, A. Boutros, J.-C. Fiaud and M. Toffano, J. Mol. Catal. A: Chem., 2003, 196, 21 CrossRef CAS.
- Select examples of thermal benzylations with Pd-π-benzyl intermediates: (a) R. R. Torregrosa, Y. Ariyarathna, K. Chattopadhyay and J. A. Tunge, J. Am. Chem. Soc., 2010, 132, 9280 CrossRef CAS PubMed; (b) W. H. Fields and J. J. Chruma, Org. Lett., 2010, 12, 316 CrossRef CAS; (c) M. F. Grünberg and L. J. Gooßen, Chem.–Eur. J., 2013, 19, 7334 CrossRef; (d) S. N. Mendis and J. A. Tunge, Chem. Commun., 2016, 7695 RSC; (e) S. N. Mendis and J. A. Tunge, Org. Lett., 2015, 17, 5164 CrossRef CAS PubMed; (f) M.-H. Yang, J. R. Hunt, N. Sharifi and R. A. Altman, Angew. Chem., Int. Ed., 2016, 55, 9080 CrossRef CAS; (g) A. Recio III, J. D. Heinzman and J. A. Tunge, Chem. Commun., 2012, 48, 142–144 RSC; (h) R. Shang, Z. Huang, X. Xiao, X. Lu, Y. Fu and L. Liu, Adv. Synth. Catal., 2012, 354, 2465 CAS; (i) T. D. Montgomery, Y. Zhu, N. Kagawa and V. H. Rawal, Org. Lett., 2013, 15, 1140 CrossRef CAS; (j) R. Kuwano and H. Kusano, Org. Lett., 2008, 10, 1979 CrossRef CAS; (k) T. Maji, K. Ramakumar and J. A. Tunge, Chem. Commun., 2014, 14045 RSC; (l) R. R. P. Torregrosa, S. N. Mendis, A. Davies and J. A. Tunge, Synthesis, 2018, 50, 3205–3216 CrossRef CAS; (m) F. Azambuja, M.-H. Yang, T. Feoktistova, M. Selvaraju, A. C. Brueckner, M. A. Grove, S. Koley, P. H.-Y. Cheong and R. A. Altman, Nat. Chem., 2020, 12, 489–496 CrossRef.
- (a) F. Ozawa, A. Kubo and T. Hayashi, Chem. Lett., 1992, 2177–2180 CrossRef CAS; (b) Z. Csákai, R. Skoda-FÖldes and L. Kollá, Inorg. Chim. Acta, 1999, 286, 93–97 CrossRef.
- See ESI Scheme S1† for a possible 4CzIPN-facilitated Pd reduction mechanism.
- (a) C. Amatore, A. Jutand, F. Khalil and M. F. Nielsent, J. Am. Chem. Soc., 1992, 114, 7076–7085 CrossRef CAS; (b) C. Amatore, E. Carré, A. Jutand and M. A. M'Barki', Organometallics, 1995, 14, 1818–1826 CrossRef CAS.
- Pd(0) can also be competitively oxidized under the reaction conditions (+0.8 V): C. Amatore, C. Cammoun and A. Jutand, Adv. Synth. Catal., 2007, 349, 292–296 CrossRef CAS.
- P. Zhang, W. Zhang, T. Zhang, Z. Wang and W. Zhou, J. Chem. Soc., Chem. Commun., 1991, 491–492 RSC.
- Metal carboxylate oxidative decarboxylation select examples: With Ag: (a) P. Lu, C. Sanchez, J. Cornella and I. Larrosa, Org. Lett., 2009, 11, 5710–5713 CrossRef CAS; (b) L. J. Goossen, C. Lunder, N. Rodríguez, P. P. Lange and A. Fromm, Chem. Commun., 2009, 7173–7175 RSC; (c) J. Cornella, C. Sanchez, D. Banawa and I. Larrosa, Chem. Commun., 2009, 7176–7178 RSC; (d) L. J. Goossen, N. Rodríguez, C. Linder, P. P. Lange and A. Fromm, ChemCatChem, 2010, 2, 430–442 CrossRef CAS; (e) L. Xue, W. Su and Z. Lin, Dalton Trans., 2011, 40, 11926–11936 RSC; (f) S. Seo, M. Slater and M. F. Greaney, Org. Lett., 2012, 14, 2650–2653 CrossRef CAS; (g) S. Seo, J. B. Taylor and M. F. Greaney, Chem. Commun., 2012, 48, 8270–8272 RSC; (h) J. Kan, S. Huang, J. Lin, M. Zhang and W. Su, Angew. Chem., Int. Ed., 2015, 54, 2199–2203 CrossRef CAS PubMed. With Pb: (i) J. K. Kochi, J. Am. Chem. Soc., 1965, 87, 3609 CrossRef CAS; (j) J. K. Kochi, R. A. Sheldon and S. S. Lande, Tetrahedron, 1969, 25, 1197 CrossRef CAS. With Ce: (k) R. A. Sheldon and J. K. Kochi, J. Am. Chem. Soc., 1968, 90, 6688 CrossRef CAS; (l) E. A. I. Heiba and R. M. Dessau, J. Am. Chem. Soc., 1971, 995–999 CrossRef CAS.
- Select recent examples of decarboxylation in radical-polar cross-over to a carbanion: (a) S. Ni, N. M. Padial, C. Kingston, J. C. Vantourout, D. C. Schmitt, J. T. Edwards, M. M. Kruszyk, R. R. Merchant, P. K. Mykhailiuk, B. B. Sanchez, S. Yang, M. A. Perry, G. M. Gallego, J. J. Mousseau, M. R. Collins, R. J. Cherney, P. S. Lebed, J. S. Chen, T. Qin and P. S. Baran, J. Am. Chem. Soc., 2019, 141, 6726–6739 CrossRef CAS; (b) C. Shu, R. S. Mega, B. J. Andreassen, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2018, 57, 15430–15434 CrossRef CAS.
- Alkyl radical redox potentials (all are reported vs. SCE): (a) D. D. M. Wayner, D. J. McPhee and D. Griller, J. Am. Chem. Soc., 1988, 110, 132–137 CrossRef CAS; (b) B. Jaun, J. Schwarz and R. Breslow, J. Am. Chem. Soc., 1980, 102, 5741–5748 CrossRef CAS; (c) L. Álvarez-Griera, I. Gallardo and G. Guirdo, Electrochim. Acta, 2009, 54, 5098–5108 CrossRef.
- B. M. Trost and D. L. Van Vranken, Chem. Rev., 1996, 96(1), 395–422 CrossRef CAS.
- The Pd-π-allyl carboxylate is depicted as an ion pair, but the possibility of a covalently bound Pd carboxylate species has not been ruled out. For discussions on Pd addition to allylic esters, see: (a) C. Amatore, A. Jutand, G. Meyer and L. Mottier, Chem.–Eur. J., 1999, 5, 466–473 CrossRef CAS; (b) T. Cantat, E. Génin, C. Giroud, G. Meyer and A. Jutand, J. Organomet. Chem., 2003, 687, 365–376 CrossRef CAS; (c) C. Amatore, A. A. Bahsoun, A. Jutand, L. Mensah, G. Meyer and L. Ricard, Organometallics, 2005, 24, 1569–1577 CrossRef CAS.
- See the ESI† Section 5 for alternative mechanisms.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc02609c |
| ‡ All potentials listed are listed as V vs. SCE and were obtained in MeCN. |
| This journal is © The Royal Society of Chemistry 2020 |