
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEncoded, click-reactive DNA-binding domains for programmable capture of specific chromatin segments†
Anna
Witte
a,
Álvaro
Muñoz-López
a,
Malte
Metz
b,
Michal R.
Schweiger
cd,
Petra
Janning
b and
Daniel
Summerer
 *a
*a
aFaculty of Chemistry and Chemical Biology, TU Dortmund University, Otto-Hahn Str. 4a, 44227 Dortmund, Germany
bMax-Planck Institute for Molecular Physiology, Otto-Hahn Str. 4a, 44227 Dortmund, Germany
cInstitute for Translational Epigenetics, Medical Faculty, University of Cologne, Weyertal 115b, 50931 Köln, Germany
dCenter for Molecular Medicine Cologne, Robert-Koch-Str. 21, 50931 Cologne, Germany. E-mail: Daniel.summerer@tu-dortmund.de
First published on 20th October 2020
Abstract
Enrichment of chromatin segments from specific genomic loci of living cells is an important goal in chromatin biology, since it enables establishing local molecular compositions as the basis of locus function. A central enrichment strategy relies on the expression of DNA-binding domains that selectively interact with a local target sequence followed by fixation and isolation of the associated chromatin segment. The efficiency and selectivity of this approach critically depend on the employed enrichment tag and the strategy used for its introduction into the DNA-binding domain or close-by proteins. We here report chromatin enrichment by expressing programmable transcription-activator-like effectors (TALEs) bearing single strained alkynes or alkenes introduced via genetic code expansion. This enables in situ biotinylation at a defined TALE site via strain-promoted inverse electron demand Diels Alder cycloadditions for single-step, high affinity enrichment. By targeting human pericentromeric SATIII repeats, the origin of nuclear stress bodies, we demonstrate enrichment of SATIII DNA and SATIII-associated proteins, and identify factors enriched during heat stress.
Identifying chromatin proteins associated with specific genomic loci is an important prerequisite for establishing locus functions. Studies in this direction are typically based on chromatin immuno-precipitation (ChIP) of target proteins, and correlations with transcriptional activity or other genome activities.1 An emerging alternative approach relies on the selective, tag-based enrichment of chromatin from specific genomic loci for mass spectrometry analyses of the bound proteins, since this allows hypothesis-free analyses, and is independent of the availability of suited antibodies.2,3 A key strategy to select genomic loci is based on the expression of programmable DNA binding domains (DBDs) such as CRISPR-dCas9 or TALE constructs that recognize a given target sequence and thereby introduce an enrichment tag into the bound chromatin segment. The low abundance of target proteins typically associated with a particular chromatin locus of interest thereby results in significant sensitivity challenges.
A critical component of expressed DBDs is thus the employed tag and the affinity and selectivity of the interaction used for capturing the target chromatin segments. Moreover, the strategy used for introducing the tag into the DBD itself or into other proteins in proximity critically affects the selectivity and thus the successful enrichment and identification of locus-bound chromatin proteins. Besides the use of standard epitope tags for immunoprecipitation that are fused to the employed DBD, strategies to introduce biotin moieties into the target chromatin segment have become increasingly important, given the superior affinity and selectivity of streptavidin-based enrichment.2 This has so far been achieved by enzymatic approaches, such as the use of BirA biotin ligase that is either co-expressed as a wild-type enzyme to biotinylate target peptides contained in the DBD,4 or fused to the DBD itself as a mutant with nonspecific activity (BirA*) to biotinylate other proteins.5 Alternatively, biotinylation has been reported with fusions of the DBD to APEX2, an ascorbate peroxidase that can generate highly reactive biotin–phenoxyl radicals for nonspecific tagging of other proteins.6–8 These concepts differ in the extent of off-target biotinylation and thus overall selectivity due to differential inherent needs for overexpressing the biotinylating enzyme, to diffusion of reactive species, or to biotinylation of proteins that are in spatial proximity without being associated with the target locus.
We aimed to introduce a complementary chemical–biological biotinylation approach that substantially differs in concept and thus in off-target biotinylation behavior. We envisaged to develop encoded programmable DNA binding domains that bear a single strained alkene or alkyne moiety for rapid and bioorthogonal click-biotinylation at defined sites for subsequent streptavidin-bead enrichment of target loci (Fig. 1a). We chose to install alkenes/alkynes in the form of noncanonical amino acid (ncAA) via genetic code expansion.9–11 A range of lysine-based ncAA bearing strained alkene and alkyne side chains are available that can be incorporated in response to the amber codon (TAG) with high efficiency and fidelity12via co-expression of orthogonal pyrrolysyl-tRNA-synthetase (PylRS)/tRNAPyl pairs.13 These react with tetrazines in strain-promoted inverse electron demand Diels–Alder cycloadditions (SPIEDAC) with high bioorthogonality and rapid kinetics.12 For engineering, we employed transcription-activator-like effector (TALE) proteins that feature a central repeat domain (CRD) recognizing DNA in a one-repeat versus one-nucleobase-pairing mode via a variable di-residue in each repeat.14,15 This offers a high degree of programmability and selective targeting of genomic loci in vivo.16 Incorporation of suited ncAA via amber suppression should allow the expression and target-binding of click-functionalized TALEs in mammalian cells, followed by chromatin fixation, click-biotinylation with biotin–tetrazine conjugates, and single step, high-affinity enrichment with streptavidin beads for downstream analysis (Fig. 1a and b).
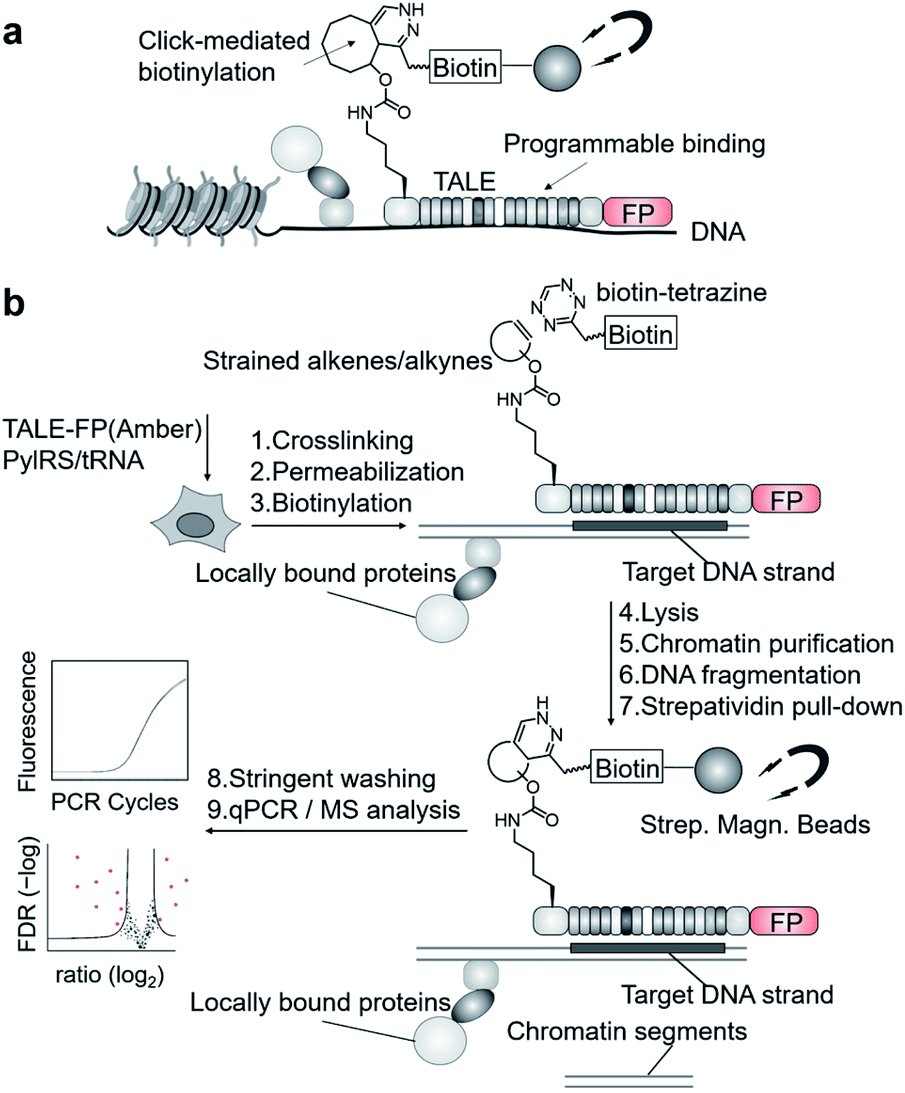 | ||
| Fig. 1 Enrichment of user-defined chromatin segments by encoded, click-reactive DNA binding domains. (a) Concept. (b) Workflow. FP: fluorescent protein. | ||
We targeted repetitive pericentromeric satellite III DNA (SATIII) for enrichment. SATIII is the origin of nuclear stress bodies (nSB),17,18 a class of membrane-less organelle19,20 that is activated by recruitment of the transcription factor heat shock factor 1 (HSF1) upon stress conditions such as heat. This leads to transcription of long noncoding SATIII RNA that seem to act as architectural RNA in stress body formation. Besides the poorly understood mechanisms of nSB formation, the relatively high genomic abundance of repeats makes SATIII a realistic initial target for the development of a single step enrichment methodology.21 We designed the construct “SATIII-TALE_wt”, bearing an N-terminal nuclear localization sequence, the TALE N-terminal region (NTR), the programmable CRD and a C-terminal mCherry domain (Fig. 2a). SATIII-TALE_wt targets the CpG-free SATIII sequence TGATTCCATTCCATTCCATT,22 allowing DNA-binding without potential interference by CpG cytosine 5-methylation.23 We initially confirmed selective binding of SATIII-TALE_wt to its target sequence in HEK293T cells by co-imaging with HSF1 after heat shock (Fig. S2†), and by activation of a luciferase reporter model (Fig. S3†).
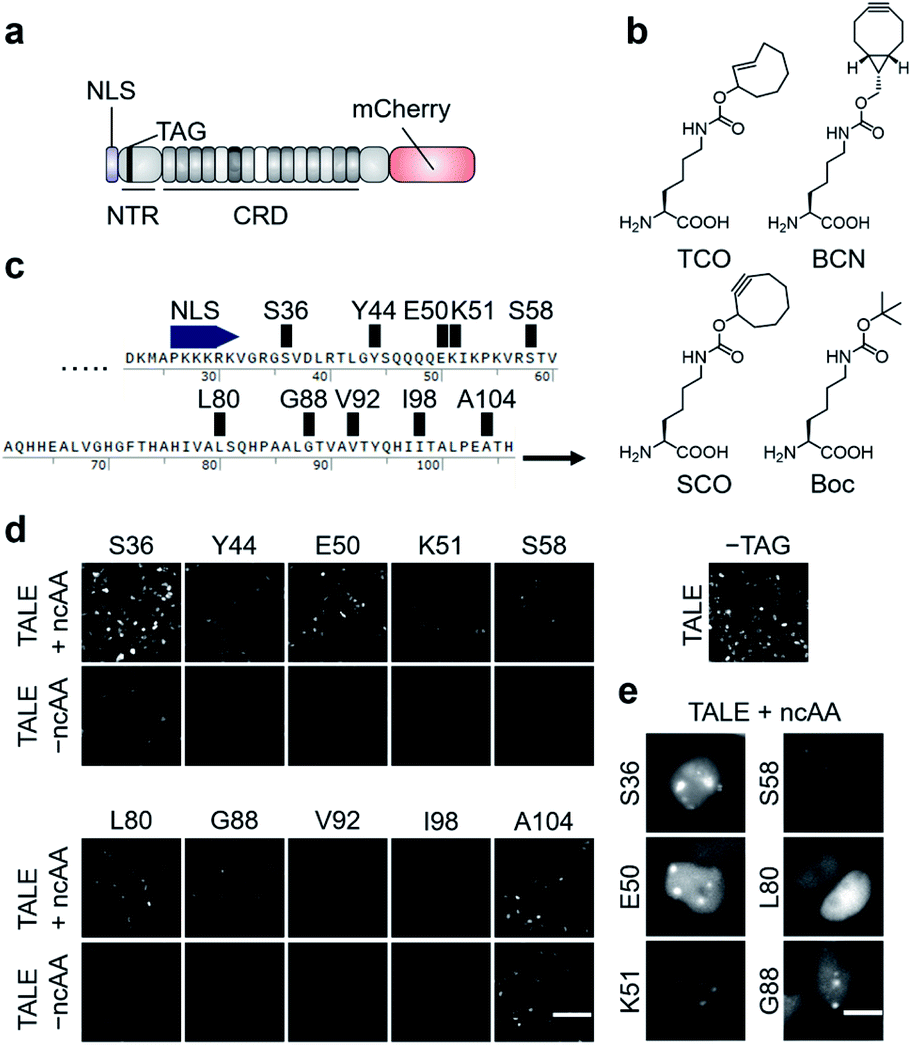 | ||
| Fig. 2 Expression of click-reactive TALE proteins in HEK293T cells. (a) Overview of the employed TALE-mCherry fusion constructs used in this study. (b) Alkene- and alkyne-bearing ncAAs used in this study (Boc = Nε-Boc-L-lysine serving as a non-reactive control ncAA). (c) Amber codon positions within the TALE NTR studied for the efficiency and fidelity of ncAA incorporation. (d) Fluorescence images of HEK293T cells transfected with PylRS_AF and indicated pSATIII-TALE amber mutants grown in the presence or absence of ncAA TCO. The image on the right is from cells transfected with non-amber pSATIII-TALE_wt only (scale bar: 100 μm). (e) Cell images of indicated pSATIII-TALE amber mutants showing ability or inability to bind SATIII loci (scale bars: 10 μm). See Fig. S1† for Hoechst co-stain. | ||
We selected three alkene- or alkyne-bearing lysine ncAAs for incorporation, bearing either a trans-cyclooctene (TCO24,25), a [6.1.0]-bicyclononyne (BCN25,26), or a cyclooctyne (SCO27) moiety attached to the lysine Nε-atom via a carbamate linker (Fig. 2b). These ncAAs are substrates for tRNAPyl aminoacylation by the polyspecific Methanosarcina mazei PylRS mutant Y306A, Y384F (PylRS_AF, bearing an additional nuclear export sequence28) for incorporation in response to the amber codon in mammalian cells, and can be employed for SPIEDAC reactions with a variety of tetrazines. Though arylazide ncAAs have successfully been used for enrichment and MS analysis of overexpressed proteins,29 the higher reaction rates of tetrazines should better match the particular sensitivity challenges associated with the enrichment of highly dilute chromatin segments from repetitive or even single loci.2 Since the efficiency and fidelity of ncAA-incorporation are critical for sensitive and selective enrichment, we initially evaluated a total of ten different amino acid positions in the TALE NTR (Fig. 2c).
We chose positions to be surface-exposed, and to not be involved in DNA-binding or stabilization of important secondary structures. We co-transfected HEK293T cells with vectors pPylRS_AF encoding the (PylRS)/tRNAPyl pair as well as pSATIII-TALE vectors encoding the respective TALE amber mutant, and grew the cells for 24 h in the presence or absence of 0.25 mM ncAA TCO. We imaged the cells, and recorded the fluorescence signals of the C-terminal mCherry domain to judge the efficiency and fidelity of suppression with TCO at each amber codon. We observed a high degree of context-dependence,30 with certain mutants exhibiting low efficiency (Fig. 2d, see V92, I98), or low fidelity of TCO incorporation (indicated by fluorescence in the absence of ncAA, see A104). Moreover, even for positions with good incorporation efficiency and fidelity, the ncAA was able to cause a loss of SATIII DNA binding, visible by the absence of the characteristic foci (i.e. position L80, Fig. 2e).
We selected the SATIII-TALE_S36, _E50 and _S58 amber mutants for further analyses, since they exhibited correct binding and high incorporation fidelity, with SATIII-TALE_S36 amber even showing expression levels comparable to those of the non-amber SATIII-TALE_wt (Fig. 2d). We employed these mutants in SPIEDAC reactions with fluorescein (FAM)-bearing H-tetrazine conjugates (FAM-tetrazine) in fixed cells, since this allows simple visual assessment of correct SATIII binding after the treatment, but also of SPIEDAC reactivity and selectivity directly in the relevant, cross-linked chromatin context. We transfected HEK293T cells and grew them in the presence of TCO as before, fixed them with formaldehyde, and reacted them with FAM-tetrazine. Imaging of mCherry revealed the SATIII-typical genomic localization of all three TALEs (Fig. 3a). However, only SATIII-TALE_S36TCO showed a co-localized FAM signal, indicating that the TCO side chain was accessible for the tetrazine only at this incorporation site (Fig. 3a). We repeated the experiment with this TALE in cells grown either in the presence of TCO or the control ncAA Boc (Fig. 2b). Reactions with a biotin–tetrazine conjugate followed by staining with streptavidin-Cy7 and imaging revealed a Cy7 signal co-localized to mCherry only for the TCO cells (Fig. 3b). This shows that biotin can be introduced site-selectively at the trans-cyclooctene moiety of SATIII-TALE_S36TCO by SPIEDAC, and that the biotin is available for later streptavidin binding.
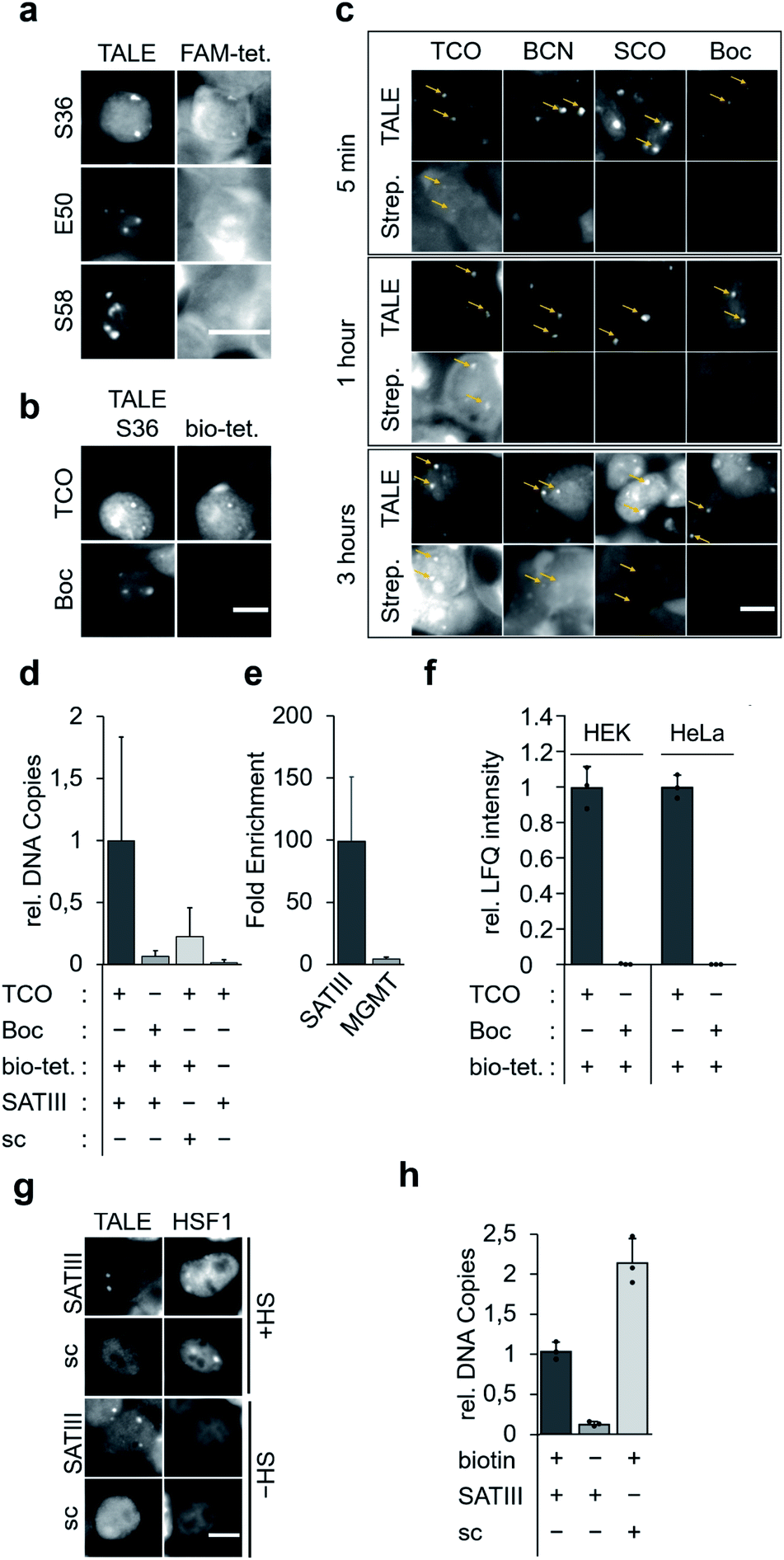 | ||
| Fig. 3 Click reactions of TALEs in fixed cellular chromatin and enrichment of target loci. (a) Reaction of FAM-tetrazine (FAM-tet.) conjugates with TCO-bearing SATIII-TALEs with indicated amber positions in formaldehyde-fixed cells (scale bar: 10 μm). (b) Reactions of biotin–tetrazine (bio-tet.) with SATIII-TALE_S36TCO or SATIII-TALE_S36Boc in formaldehyde-fixed cells followed by staining with streptavidin_Cy7 conjugates and imaging indicate chemoselectivity of click-conjugation (scale bar: 10 μm). (c) Comparative kinetics of reactions between bio-tet. and SATIII-TALE_S36TCO, BCN, SCO and Boc (scale bar: 10 μm). (d) Click-mediated enrichment of SATIII DNA with SATIII-TALE_S36TCO, SATIII-TALE_S36Boc or scSATIII-TALES36TCO (sc) in the presence or absence of bio-tet. measured by qPCR. Error bars are from N = 12 (six biological replicates, two qPCR duplicates). (e) SATIII TALE specifically enriches SATIII DNA. Fold enrichment of each indicated target relative to −bio-tet. cells. Error bars are from N = 12 (six biological replicates, each two technical qPCR duplicates). (f) Click-mediated enrichment with SATIII-TALE_S36TCO or SATIII-TALE_S36Boc in HEK293T or HeLa cells (error bars from N = 3 biological triplicates). (g) Colocalization study of SATIII-TALE_BirA* (wt) or scTALE_BirA* (sc) and HSF1-mCherry. HEK293T cells were cotransfected with SATIII-TALE_BirA* and HSF1-mCherry in HEK293T cells, grown either at 37 °C or under heat shock conditions (HS, 1 h 44 °C) and imaged after immunostaining for the HA tag contained in the TALE-BirA* constructs. (h) Enrichment of SATIII DNA with SATIII-TALE_BirA* (SATIII) or scTALE_BirA* (sc) in the presence or absence of biotin in the cell culture. Error bars from N = 3 biological triplicates. | ||
To evaluate the three alkene- and alkyne-ncAA TCO, SCO and BCN for their biotinylation efficiencies in cross-linked chromatin, we performed time-resolved measurements. We stained cells grown in the presence of each ncAA or the control ncAA Boc with Cy7-streptavidin after incubation with biotin–tetrazine for 5 min, 1 hour, or 3 hours. We observed a weak Cy7 signal for TCO already after 5 min, whereas signals for BCN (and faintly for SCO), were visible only after 3 hours (Fig. 3c; no signal was observed for the Boc negative control). In model reactions using ncAA-bearing GFP protein, SCO reacts comparatively slowly in SPIEDAC with H-tetrazines (0.67 × 103 M−1 s−1) in vitro.31 In contrast, under the same experimental conditions, both BCN and TCO have been reported to react much faster, and with similar rate constants (1.6 × 104 M−1 s−1 for BCN and 1.3 × 104 M−1 s−1 for TCO). The difference we observe for TCO and BCN compared to the in vitro model system could arise for various reasons, including differential ncAA accessibility in fixed chromatin, or different off-target reactivity with thioles.31,32
We next asked, if SATIII loci could be selectively enriched by streptavidin bead capture with sheared chromatin from fixed cells expressing SATIII-TALE_S36TCO. We cultured, fixed, and reacted cells with biotin–tetrazine, purified the nuclei, and sheared them with ultrasound. We performed a capture/purification step with streptavidin magnetic beads, reversed the formaldehyde crosslinks with heat, and purified the released DNA. qPCR analyses showed a clear enrichment of SATIII DNA for cells expressing SATIII-TALE_S36TCO and reacted with biotin–tetrazine compared to identically treated cells expressing TALE_S36Boc (Fig. 3d). Similarly, cells expressing SATIII-TALE_S36TCO and not reacted with biotin–tetrazine also showed low enrichment (Fig. 3d). This indicates that enrichment of SATIII DNA depends on a successful click reaction between the installed TCO and biotin–tetrazine. We performed identical experiments with cells expressing a TALE_S36TCO with the same repeat composition, but randomized repeat sequence (scrambled TALE, sc) that does not bind to the SATIII target sequence in our in vivo luciferase reporter model (Fig. S3†). We again observed a low enrichment of SATIII DNA, indicating that the enrichment depends on the selective interaction of the SATIII TALE with its target sequence. Moreover, a potential background of endogenous, amber-terminated proteins bearing TCO via off-target amber suppression seems to not strongly contribute to the enrichment of the SATIII target (this background should be identical for SATIII-TALE_S36TCO and scTALE_S36TCO). Finally, we analysed DNA captured from cells expressing SATIII-TALE_S36TCO and reacted or not reacted with biotin–tetrazine for enrichment of an off-target locus in the MGMT gene by qPCR. However we observed a ∼100-fold enrichment of the target SATIII locus for biotin–tetrazine-reacted as compared to non-reacted cells, we observed only a ∼4-fold enrichment of the MGMT locus (Fig. 3e). This again indicates enrichment via a selective interaction of the TALE with the SATIII target sequence. It should be noted that SATIII repeats are heterogenous in sequence and our qPCR based on two (30 and 22 nt) primers is thus expected to amplify only a very small subfraction of SATIII DNA, whereas the single 20 nt sequence bound by our TALE is expected to occur much more frequently. Hence, our qPCR data are expected to strongly underestimate the actual enrichment by the TALE and should not directly be extrapolated to proteomics analyses of enriched DNA, since this will measure all TALE-enriched chromatin segments.
To compare the enrichment selectivity of our approach with an alternative, established biotinylation approach for targeted chromatin enrichment, we designed fusion constructs consisting of the SATIII-TALE or the scTALE and a BirA* domain. In imaging studies with HEK293T cells, both TALEs exhibited expected localization under heat shock (HS) and normal (-HS) conditions, i.e. colocalization with HSF1 of the SATIII-TALE under HS conditions, and diffuse nuclear localization of the scTALE under both conditions (Fig. 3g). Enrichment experiments conducted under identical conditions to those used to generate the results shown in Fig. 3d showed successful enrichment of SATIII DNA with the SATIII-TALE-BirA* that depended on the presence of biotin in the growth medium (Fig. 3h). However, we observed an even higher enrichment of SATIII DNA for the scTALE-BirA*, indicating that this enrichment was not dependent on the selective binding of the TALE to the SATIII target sequence (Fig. 3h). Though the two approaches are not fully comparable because of their different biotinylation concepts, the data show a higher enrichment selectivity of our approach for this DNA target.
We next subjected enriched chromatin samples to on-bead tryptic digests and nano-high performance liquid chromatography mass spectrometry (nanoHPLC-MS/MS), and analysed the amount of enriched TALE by label-free quantification (LFQ). We detected TALE-specific peptides in chromatin of SATIII-TALE_S36TCO expressing cells only if they had been reacted with biotin–tetrazine (Fig. 3e). Similarly, no TALE peptides were detectable for HEK293T or HeLa cells expressing TALE_S36Boc (Fig. 3e, similar data were observed for cells grown under heat-shock conditions, Fig. S4†).
To discover proteins that are recruited to SATIII DNA during nuclear stress body formation, we next performed proteomics studies with cells grown under heat shock and normal conditions. To take advantage of our two-step biotinylation approach compared to potential direct incorporation of a biotin ncAA,33 we blocked endogenous biotin sites before click-biotinylation to reduce background enrichment. Specifically, we followed the protocol as before, but before the addition of biotin–tetrazine, we incubated the cells with avidin, added an excess of free biotin to block remaining binding sites at the avidin, and washed the cells. HEK293T and HeLa cells expressing SATIII-TALE_S36TCO showed normal behaviour after HS, i.e. co-localization of endogenous, immuno-stained HSF1 and TALE at SATIII-typical foci (Fig. 4a). We first compared HEK293T and HeLa cells transfected with pSATIII-TALE_S36TCO or an empty vector (EV) negative control driving the expression of a non-TALE, mCherry-containing truncation product under nHS conditions. In both cases, the TALE was significantly enriched compared to the negative control, whereas mCherry was not (Fig. 4b). We further found enrichment of proteins known to be associated with centromeric and/or pericentromeric regions, including satellite DNA (XRCC5 for HEK293T cells; NAP1L4, YWHAH for HeLa cells, Fig. 4b).34–37 We next compared cells expressing SATIII-TALE_S36TCO under HS and nHS conditions. To our surprise, we did not observe enrichment of HSF1 under HS conditions, despite the co-localization with TALE-marked foci we observed in the immunostainings. To evaluate, if this co-localization corresponds to an actual interaction of HSF1 with SATIII DNA that can be stabilized by fixation as prerequisite for enrichment and MS-detection, we conducted a chromatin-immunoprecipitation (ChIP) with an anti-HSF1 antibody after fixation, and analysed the enrichment by qPCR. HSF1 was strongly enriched under HS conditions, suggesting that the particular detection limit of HSF1 in nanoHPLC-MS/MS analyses is higher than the amount of protein after enrichment (Fig. 4c). Apart from the particular case of HSF1, we found heat-shock-associated enrichment of other proteins. This included the nuclear ubiquitin-activating enzyme UBA1 that is involved in stress-induced DNA damage repair38 and was enriched in both cell types (Fig. 4d). Similarly, the purine biosynthesis gene ATIC was enriched that is known to associate with a number of heat shock proteins as part of the purinosome.39 We also found for heat-stressed HeLa cells enrichment of LGALS3 that has been proposed in stabilizing DNA-interactions of CREB,40 a transcription factor associated with SATIII DNA18 (Fig. 4d). Finally, though not enriched for TCO HS versus TCO nHS cells, we found the nucleotide-exchange factor HSPH1 to be enriched in TCO HS versus EV HS cells (Fig. S5†). HSPH1 is known to be heat-induced and to control substrate turnover of the chaperones HSPA1A/B by ADP release, and its expression is strictly dependent on HSF1.41 These combined results suggest protein enrichment by our click-mediated biotinylation and single step, high affinity capture that is sufficient for MS-identification of proteins that are recruited to our target locus by heat-stress.
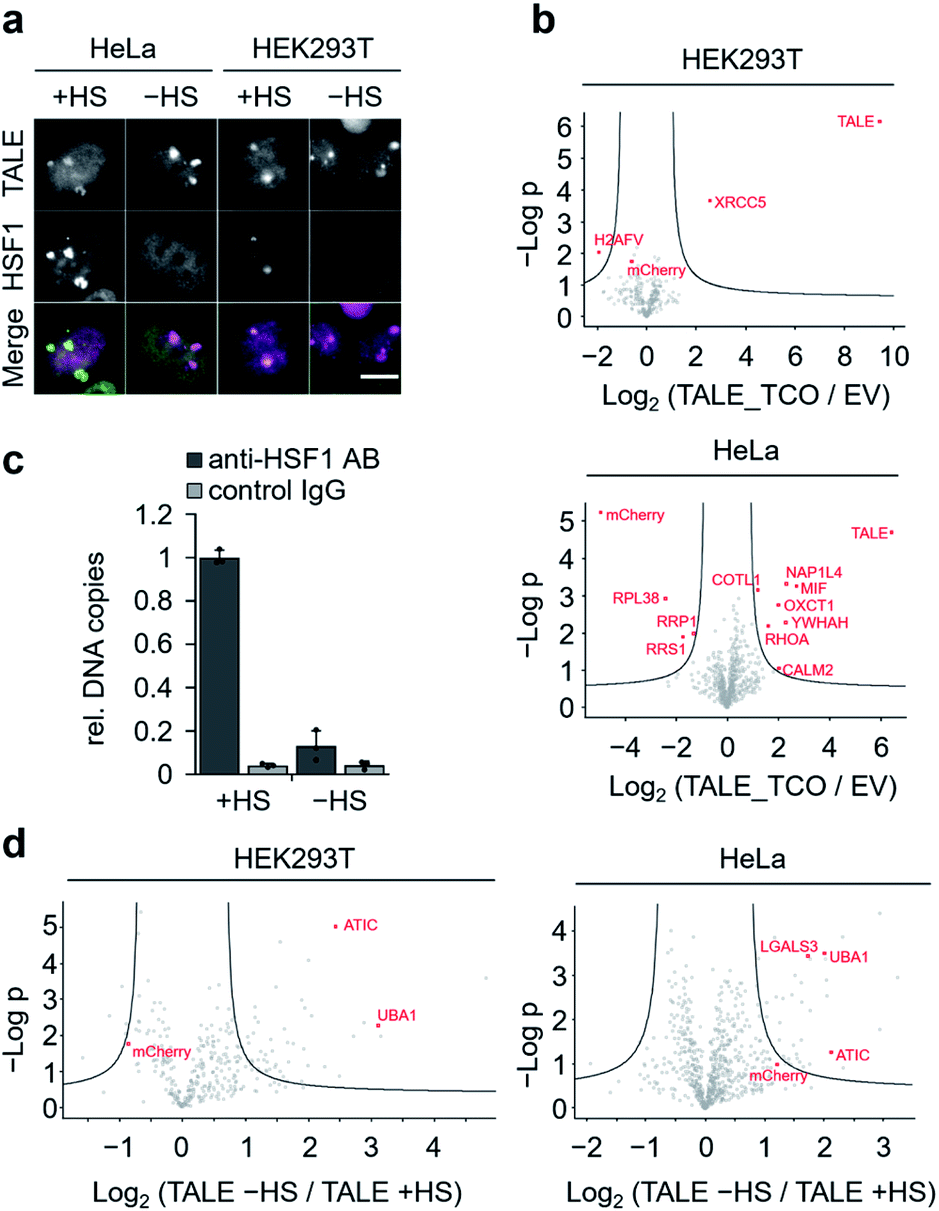 | ||
| Fig. 4 Proteomic analysis of SATIII loci under heat-shock and non-heat-shock conditions. (a) Images of cells expressing SATIII-TALE_S36TOC after subjecting to HS or nHS conditions and immunostaining for endogenous HSF1 (scale bar: 10 μm). (b) Volcano plots depicting the enrichment and significance (FDR: 0.05) of the identified proteins for SATIII-TALE_S36TCO in non-heat shocked HEK293T and HeLa cells (biological triplicates). (c) HSF1 qPCR enrichment upon heat shock. Error bars from biological triplicates. (d) Volcano plots depicting the enrichment and significance (FDR: 0.05) of the identified proteins for SATIII-TALE_S36TCO upon heat shock in HEK293T and HeLa cells (biological triplicates). | ||
Conclusions
In conclusion, we report a novel chemical–biological strategy for the click-biotinylation of user-defined genomic loci by incorporation of strained alkene amino acids into encoded, programmable DNA binding domains in live cells. We define optimal ncAA types and incorporation positions for correct target binding and in situ biotinylation, and provide first proof of principle and benchmark experiments for chromatin enrichment combined with qPCR analysis and MS-based identification of chromatin-associated proteins. We are aware of the high sensitivity and selectivity challenges generally associated with the enrichment of multiple or even single user-defined chromatin segments.2,3 Moreover, each biotinylation concept will likely result in complementary information. For example, while BirA*-based approaches have high potential to mark and identify proteins with accessible surfaces, a direct crosslinking of a DBD to DNA may more likely identify proteins that may be buried in larger complexes, but that directly interact with the DNA. For our approach, we here demonstrate selective enrichment of SATIII loci on the basis of DNA, and establish protocols for downstream proteomic analyses, enabling the identification of SATIII-associated proteins during normal and heat-shock conditions. Nevertheless, a full judgement of the performance of this and other strategies will require larger-scale comparative studies including genome-wide analyses of enriched DNA as well as of proteomes, and potentially transcriptomes. These will reveal the true potential and inherent differences of each approach to identify specific classes of chromatin factors, ranging from proteins to potentially associated RNAs and proximal DNA elements.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (project number 418983006 to D. S.; KFO-RP2 and SFB-1399-B03 to M. R. S), the ERC (CoG EPICODE No. 723863 to D. S.), and the Volkswagen Stiftung (Lichtenberg Professorship to M. R. S.).References
- B. E. Bernstein, E. Birney, I. Dunham, E. D. Green, C. Gunter and M. Snyder, Nature, 2012, 489, 57–74 CrossRef.
- M. Gauchier, G. van Mierlo, M. Vermeulen and J. Dejardin, Nat. Methods, 2020, 17, 380–389 CrossRef CAS.
- M. Vermeulen and J. Dejardin, Nat. Rev. Mol. Cell Biol., 2020, 21, 249–250 CrossRef CAS.
- X. Liu, Y. Zhang, Y. Chen, M. Li, F. Zhou, K. Li, H. Cao, M. Ni, Y. Liu, Z. Gu, K. E. Dickerson, S. Xie, G. C. Hon, Z. Xuan, M. Q. Zhang, Z. Shao and J. Xu, Cell, 2017, 170(5), 1028–1043 CrossRef CAS.
- E. Schmidtmann, T. Anton, P. Rombaut, F. Herzog and H. Leonhardt, Nucleus, 2016, 7, 476–484 CrossRef CAS.
- W. Qiu, Z. Xu, M. Zhang, D. Zhang, H. Fan, T. Li, Q. Wang, P. Liu, Z. Zhu, D. Du, M. Tan, B. Wen and Y. Liu, Nucleic Acids Res., 2019, 47, e52 CrossRef CAS.
- X. D. Gao, L. C. Tu, A. Mir, T. Rodriguez, Y. Ding, J. Leszyk, J. Dekker, S. A. Shaffer, L. J. Zhu, S. A. Wolfe and E. J. Sontheimer, Nat. Methods, 2018, 15, 433–436 CrossRef CAS.
- S. A. Myers, J. Wright, R. Peckner, B. T. Kalish, F. Zhang and S. A. Carr, Nat. Methods, 2018, 15, 437–439 CrossRef CAS.
- H. Xiao and P. G. Schultz, Cold Spring Harbor Perspect. Biol., 2016, 8, a023945 CrossRef.
- J. W. Chin, Nature, 2017, 550, 53–60 CrossRef CAS.
- L. Wang, Acc. Chem. Res., 2017, 50, 2767–2775 CrossRef CAS.
- K. Lang and J. W. Chin, Chem. Rev., 2014, 114, 4764–4806 CrossRef CAS.
- S. K. Blight, R. C. Larue, A. Mahapatra, D. G. Longstaff, E. Chang, G. Zhao, P. T. Kang, K. B. Church-Church, M. K. Chan and J. A. Krzycki, Nature, 2004, 431, 333 CrossRef CAS.
- D. Deng, C. Yan, X. Pan, M. Mahfouz, J. Wang, J. K. Zhu, Y. Shi and N. Yan, Science, 2012, 335, 720–723 CrossRef CAS.
- A. N. S. Mak, P. Bradley, R. A. Cernadas, A. J. Bogdanove and B. L. Stoddard, Science, 2012, 335, 716–719 CrossRef CAS.
- J. Boch and U. Bonas, Annu. Rev. Phytopathol., 2010, 48, 419–436 CrossRef CAS.
- R. Valgardsdottir, I. Chiodi, M. Giordano, A. Rossi, S. Bazzini, C. Ghigna, S. Riva and G. Biamonti, Nucleic Acids Res., 2008, 36, 423–434 CrossRef CAS.
- G. Biamonti and C. Vourc'h, Cold Spring Harbor Perspect. Biol., 2010, 2, a000695 Search PubMed.
- N. I. Enukashvily, R. Donev, I. S. R. Waisertreiger and O. I. Podgornaya, Cytogenet. Genome Res., 2007, 118, 42–54 CrossRef CAS.
- Y. Saito, Y. Kanai, M. Sakamoto, H. Saito, H. Ishii and S. Hirohashi, Hepatology, 2001, 33, 561–568 CrossRef CAS.
- M. Gauchier, G. van Mierlo, M. Vermeulen and J. Dejardin, Nat. Methods, 2020, 17(4), 380–389 CrossRef CAS.
- A. Munoz-Lopez, B. Buchmuller, J. Wolffgramm, A. Jung, M. Hussong, J. Kanne, M. R. Schweiger and D. Summerer, Angew. Chem., Int. Ed. Engl., 2020, 59(23), 8927–8931 CrossRef CAS.
- G. Kubik, M. J. Schmidt, J. E. Penner and D. Summerer, Angew. Chem., Int. Ed. Engl., 2014, 53, 6002–6006 CrossRef CAS.
- T. Plass, S. Milles, C. Koehler, J. Szymanski, R. Mueller, M. Wiessler, C. Schultz and E. A. Lemke, Angew. Chem., Int. Ed. Engl., 2012, 51, 4166–4170 CrossRef CAS.
- K. Lang, L. Davis, S. Wallace, M. Mahesh, D. J. Cox, M. L. Blackman, J. M. Fox and J. W. Chin, J. Am. Chem. Soc., 2012, 134, 10317–10320 CrossRef CAS.
- A. Borrmann, S. Milles, T. Plass, J. Dommerholt, J. M. Verkade, M. Wiessler, C. Schultz, J. C. van Hest, F. L. van Delft and E. A. Lemke, Chembiochem, 2012, 13, 2094–2099 CrossRef CAS.
- T. Plass, S. Milles, C. Koehler, C. Schultz and E. A. Lemke, Angew. Chem., Int. Ed. Engl., 2011, 50, 3878 CrossRef CAS.
- I. Nikic, G. Estrada Girona, J. H. Kang, G. Paci, S. Mikhaleva, C. Koehler, N. V. Shymanska, C. Ventura Santos, D. Spitz and E. A. Lemke, Angew. Chem., Int. Ed. Engl., 2016, 55, 16172–16176 CrossRef CAS.
- A. H. Smits, A. Borrmann, M. Roosjen, J. C. van Hest and M. Vermeulen, ACS Chem. Biol., 2016, 11, 3245–3250 CrossRef CAS.
- M. Pott, M. J. Schmidt and D. Summerer, ACS Chem. Biol., 2014, 9, 2815–2822 CrossRef CAS.
- I. Nikic, T. Plass, O. Schraidt, J. Szymanski, J. A. Briggs, C. Schultz and E. A. Lemke, Angew. Chem., Int. Ed. Engl., 2014, 53, 2245–2249 CrossRef CAS.
- R. van Geel, G. J. Pruijn, F. L. van Delft and W. C. Boelens, Bioconjugate Chem., 2012, 23, 392–398 CrossRef CAS.
- A. Hohl, R. Karan, A. Akal, D. Renn, X. Liu, S. Ghorpade, M. Groll, M. Rueping and J. Eppinger, Sci. Rep., 2019, 9, 11971 CrossRef.
- V. Barra and D. Fachinetti, Nat. Commun., 2018, 9, 4340 CrossRef CAS.
- K. Tsouroula, A. Furst, M. Rogier, V. Heyer, A. Maglott-Roth, A. Ferrand, B. Reina-San-Martin and E. Soutoglou, Mol. Cell, 2016, 63, 293–305 CrossRef CAS.
- H. Tachiwana, Y. Miya, N. Shono, J. Ohzeki, A. Osakabe, K. Otake, V. Larionov, W. C. Earnshaw, H. Kimura, H. Masumoto and H. Kurumizaka, Nucleic Acids Res., 2013, 41, 2869–2880 CrossRef CAS.
- D. Goutte-Gattat, M. Shuaib, K. Ouararhni, T. Gautier, D. A. Skoufias, A. Hamiche and S. Dimitrov, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8579–8584 CrossRef CAS.
- P. Moudry, C. Lukas, L. Macurek, H. Hanzlikova, Z. Hodny, J. Lukas and J. Bartek, Cell Cycle, 2012, 11, 1573–1582 CrossRef CAS.
- J. B. French, H. Zhao, S. O. An, S. Niessen, Y. J. Deng, B. F. Cravatt and S. J. Benkovic, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2528–2533 CrossRef CAS.
- H. M. Lin, R. G. Pestell, A. Raz and H. R. Kim, Oncogene, 2002, 21, 8001–8010 CrossRef CAS.
- A. Vihervaara, C. Sergelius, J. Vasara, M. A. Blom, A. N. Elsing, P. Roos-Mattjus and L. Sistonen, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E3388–E3397 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc02707c |
| This journal is © The Royal Society of Chemistry 2020 |