
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGaN nanowires as a reusable photoredox catalyst for radical coupling of carbonyl under blacklight irradiation†
Mingxin
Liu‡
 ab,
Lida
Tan‡
a,
Roksana T.
Rashid
c,
Yunen
Cen
a,
Shaobo
Cheng
d,
Gianluigi
Botton
d,
Zetian
Mi
*bc and
Chao-Jun
Li
*a
ab,
Lida
Tan‡
a,
Roksana T.
Rashid
c,
Yunen
Cen
a,
Shaobo
Cheng
d,
Gianluigi
Botton
d,
Zetian
Mi
*bc and
Chao-Jun
Li
*a
aDepartment of Chemistry and FRQNT Centre for Green Chemistry and Catalysis, McGill University, 801 Sherbrooke Ouest, Montreal, Quebec H3A 0B8, Canada. E-mail: cj.li@mcgill.ca
bDepartment of Electrical Engineering and Computer Science, University of Michigan, 1301 Beal Ave, Ann Arbor, MI 48109, USA. E-mail: ztmi@umich.edu
cDepartment of Electrical and Computer Engineering, McGill University, 3480 University, Montreal, Quebec H3A 0E9, Canada
dDepartment of Material Science and Engineering, Canadian Centre for Electron Microscopy, McMaster University, 1280 Main Street West, Hamilton, ON L8S 4M1, Canada
First published on 1st July 2020
Abstract
Employing photo-energy to drive the desired chemical transformation has been a long pursued subject. The development of homogeneous photoredox catalysts in radical coupling reactions has been truly phenomenal, however, with apparent disadvantages such as the difficulty in separating the catalyst and the frequent requirement of scarce noble metals. We therefore envisioned the use of a hyper-stable III–V photosensitizing semiconductor with a tunable Fermi level and energy band as a readily isolable and recyclable heterogeneous photoredox catalyst for radical coupling reactions. Using the carbonyl coupling reaction as a proof-of-concept, herein, we report a photo-pinacol coupling reaction catalyzed by GaN nanowires under ambient light at room temperature with methanol as a solvent and sacrificial reagent. By simply tuning the dopant, the GaN nanowire shows significantly enhanced electronic properties. The catalyst showed excellent stability, reusability and functional tolerance. All reactions could be accomplished with a single piece of nanowire on Si-wafer.
1. Introduction
The generation of new carbon–carbon bonds in an atom-economical manner is a continuous pursuit in synthetic organic chemistry, enabling the production of many crucial pharmaceutical compounds and bio-active molecules.1 Since their discovery in the late 20th century,2 the use of photoredox catalysts has quickly realized huge potential in organic synthesis to afford facile coupling reactions enabled by photo-energy. Many excellent examples have been seen in the literature, including couplings of carbonyl-α-radicals3–5 and ketyl radicals (e.g., the Pinacol Coupling Reaction, PCR, Fig. 1A).6–9 However, despite many successful applications of photoredox catalysts in radical coupling reactions, with some exceptions,10 the scarce and expensive ruthenium (Ru) and iridium (Ir)-based homogeneous catalysts are predominantly used, which are difficult to isolate and recycle at the end of the reactions.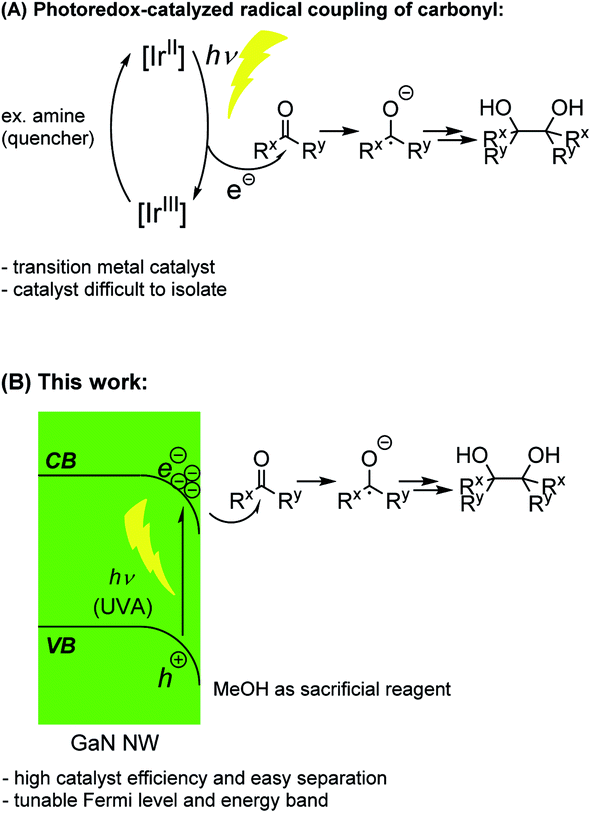 | ||
| Fig. 1 Previous radical coupling of carbonyl using a photoredox catalyst and this work. | ||
Over the past few years, we have been dedicated to the engineering of group III-nitride semiconductor nano-structures and their applications as photocatalysts. Compared to common semiconductor catalysts11 such as CdS, TiO2 and polymeric carbon nitride (PCN), the gallium nitride (GaN) semiconductor12 shows extraordinary stability13 and more than six orders of magnitude of enhancement in voltage carrying ability.14,15 In addition, the surface of the GaN manufactured in our lab is mostly non-polar,13 which does not undergo Fermi-level pinning16 and allows facile modifications of its band gap. Furthermore, the epitaxially grown GaN allows the incorporation of Si wafer, giving a dual-junction structure17 which greatly enhances catalytic reaction efficiency. We have successfully designed a GaN nanowire (NW) catalyst for water-splitting,18–23 nitrogen fixation,24,25 methane conversion,26,27 and artificial photosynthesis.28,29 In those reactions, the capability of tuning the near-surface Fermi level and energy band structure of the semiconductor is of particular importance. By carefully repositioning the conduction band (CB) energy level and the valence band (VB) energy level of GaN NWs with doping, the desired direction of the electron flow can be established upon light irradiation on the GaN NWs. The photo excited electron (e−) can then be efficiently donated from the semiconductor to the reactant (H2O, N2, or CO2, respectively) and give the desired product (H2, NH3, or CO/hydrocarbon, respectively), while the photo-excited hole (h+) can be readily neutralized by many common sacrificial reagents. Inspired by these studies, we contemplate the possibility of using those unique characters of GaN NWs to design a more practical and highly recyclable photoredox catalyst for radical coupling reactions under light (Fig. 1B). As a proof-of-concept, herein, we report GaN-catalyzed highly efficient PCRs under ambient temperature and light using methanol as both the solvent and the reagent, with no other reaction additive.
2. Results and discussion
Molecular Beam Epitaxy (MBE) is a fast and reliable method of synthesizing nano-level semiconductor units for electronic engineering. In this study, we used plasma assisted MBE (PA-MBE) under nitrogen-rich conditions to grow GaN NWs on a two-inch Si(111) wafer, resulting in a forest-like GaN NW array aligned vertically to the wafer, as confirmed by Scanning Electron Microscopy (SEM, Fig. 2 left). High-resolution Transmission Electron Microscopy (TEM, Fig. 2 right) revealed the lattice of the gallium atoms which forms the nanowire. Since GaN is a wide band semiconductor with a band gap of 3.4 eV (see Table S1 in the ESI†),13 it absorbs photons of a certain wavelength to excite e− from its VB to its CB. As confirmed by our previous research, the desired wavelength is 365 nm, or shorter, which can be provided by a commonly used black light bulb (see Fig. S33 in the ESI†). | ||
| Fig. 2 SEM (left) and TEM (right) identification of the freshly synthesized GaN NW. | ||
The surface energy band bending significantly influences the electronic properties of semiconductors.20 Upon light irradiation, the excited e− in the CB of the semiconductor tends to migrate to the potential energy well, while the h+ on the VB tends to migrate to a high potential energy (high ground). These surface band properties inhibit carrier recombination and give rise to unique reactivities. By doping the NW with a tetravalent element (e.g., silicon, germanium, etc.) as the n-type dopant, the surface energy band of GaN bends upward to give n-GaN NWs. Upon light irradiation,20 the excited e− in the CB tends to migrate to the internal region of the NW instead of the surface, while the h+ in the VB tends to migrate to the surface instead of the internal region of the NW. Such doping prohibits e− donation to the reactant and enhances the neutralization of h+ by the sacrificial reagent (Fig. 3A left). On the other hand, by using a divalent element (e.g., magnesium) as the p-type dopant, the surface energy band of the corresponding NW bends downwards (p-GaN NW). The photoexcited e− for the p-GaN NWs will therefore tend to aggregate on the NW surface, making the donation of e− to the reactant easier, while the h+ tends to migrate to the internal region of the NWs and inhibits the consumption of the sacrificial reagent (Fig. 3A right). To characterize these electronic properties, electron paramagnetic resonance (EPR) spectra were recorded for the as-synthesized intrinsic- (i-), n-, and p-GaN NWs after they were irradiated under 365 nm light for 1 hour (Fig. 3B).30 Since both Ga3+ and N3− do not possess unpaired e−, the ground state of GaN is diamagnetic (see Fig. S32 in the ESI†). Therefore, the observed EPR signal should exclusively come from photoexcited electrons (EPR spectra of i-, n-, and p-GaN NWs in the dark were silent). It can be observed from the EPR spectra that both n- and p-GaN NWs show stronger EPR signals than i-GaN NWs. This is because the photoexcited e− and h+ are more prone towards recombination in i-GaN NWs due to less significant band bending. As explained above, band bending prevents carrier recombination by driving e− and h+ to the opposite direction. At the same time, the n-GaN NWs showed a broader EPR signal compared to p-GaN NWs. The results clearly show that the photoexcited e− in n-GaN NWs tends to delocalize, suggesting the migration of e− from the NW surface to the internal region.
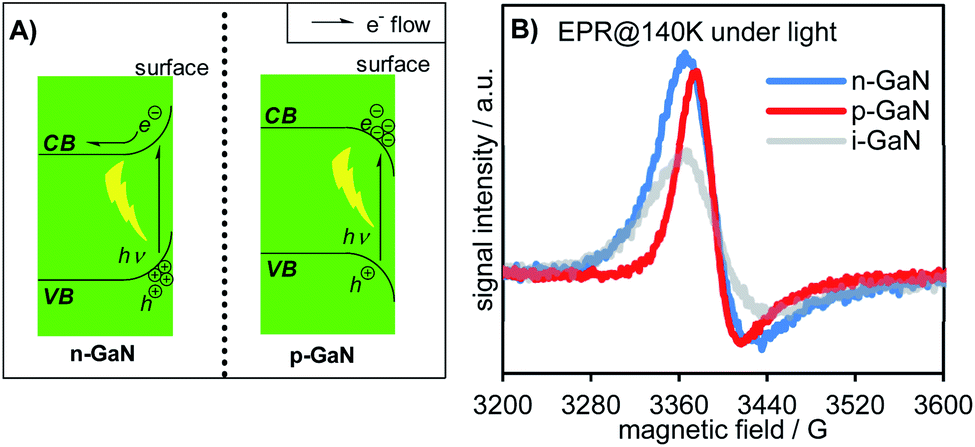 | ||
| Fig. 3 (A) Surface energy band bending of the GaN NW. (B) EPR identification of band bending. | ||
As both the e− donation to the reactant and h+ oxidation of the sacrificial reagent promote the overall reaction, it is important to find the optimum catalyst and to determine the rate-limiting step. Under the irradiation of an 8 W black light bulb (giving 1 W light irradiation at 365 nm), in 0.5 mmol methanol (MeOH) as a sacrificial reagent (oxidized by h+) and solvent, we examined i-GaN, n-GaN (Si4+ doped), and p-GaN (Mg2+ doped) as catalysts to carry out the PCR of 0.02 mmol acetophenone (1a) towards 2,3-diphenyl-2,3-butanediol (2a) as a standard reaction (Table 1, see also Fig. S31 in the ESI†). While all the GaN NWs showed negligible diastereoselectivity in the PCR product, the catalytic activity decreases gradually from p-GaN NW (Table 1 entry 3) to i-GaN NW (Table 1 entry 1) to n-GaN NW (Table 1 entry 2), affording 2a in quantitative, 93% and 81% yield, respectively. The results suggest that the e− donation from the semiconductor serves as the rate-limiting step for 1a conversion to 2a. Nevertheless, we further investigated the influence of the sacrificial reagent such as hydrogen (H2) (Table 1 entry 4), methane (CH4) (Table 1 entry 5), and water (H2O) (Table 1 entry 6) on the reaction catalyzed by p-GaN NW, none of which afforded the corresponding product 2a. Other popular sacrificial reagents such as triethylamine (Et3N) (Table 1 entry 7), ethanol (EtOH) (Table 1 entry 8), and trimethanolamine (Table 1 entry 9) also gave the desired PCR products in high yields. Control experiments were also conducted and showed that the absence of either the GaN NW (Table 1 entry 10) or light (Table 1 entry 11) inhibited the reaction completely. Using Si(111) wafer alone as the catalyst did not give any pinacol coupling product (Table 1 entry 12), and a nearly quantitative yield of 97% was obtained when p-GaN NW stripped from Si(111) was used as the catalyst (Table 1 entry 13).
|
|
||||
|---|---|---|---|---|
| Entry | GaN NW | Sacrificial reagent | Yield/% |
Rac-![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :meso- :meso- |
| a Reaction was done by injecting all reactants into a sealed quartz vessel under vacuum. NMR yields were determined by using mesitylene as the internal standard. | ||||
| 1 | i-GaN | MeOH | 93 | 1:1 |
| 2 | n-GaN | MeOH | 81 | 1:1 |
| 3 | p-GaN | MeOH | >99 | 1:1 |
| 4 | p-GaN | H2 | No reaction | N/A |
| 5 | p-GaN | CH4 | No reaction | N/A |
| 6 | p-GaN | Water | No reaction | N/A |
| 7 | p-GaN | Et3N | >99 | 1:1 |
| 8 | p-GaN | EtOH | >99 | 1:1 |
| 9 | p-GaN | Triethanolamine | 92 | 1:1 |
| 10 | p-GaN | MeOH (no GaN) | No reaction | N/A |
| 11 | p-GaN | MeOH (in dark) | No reaction | N/A |
| 12 | Si(111) | MeOH | No reaction | N/A |
| 13 | p-GaN (no Si) | MeOH | 97 | 1:1 |
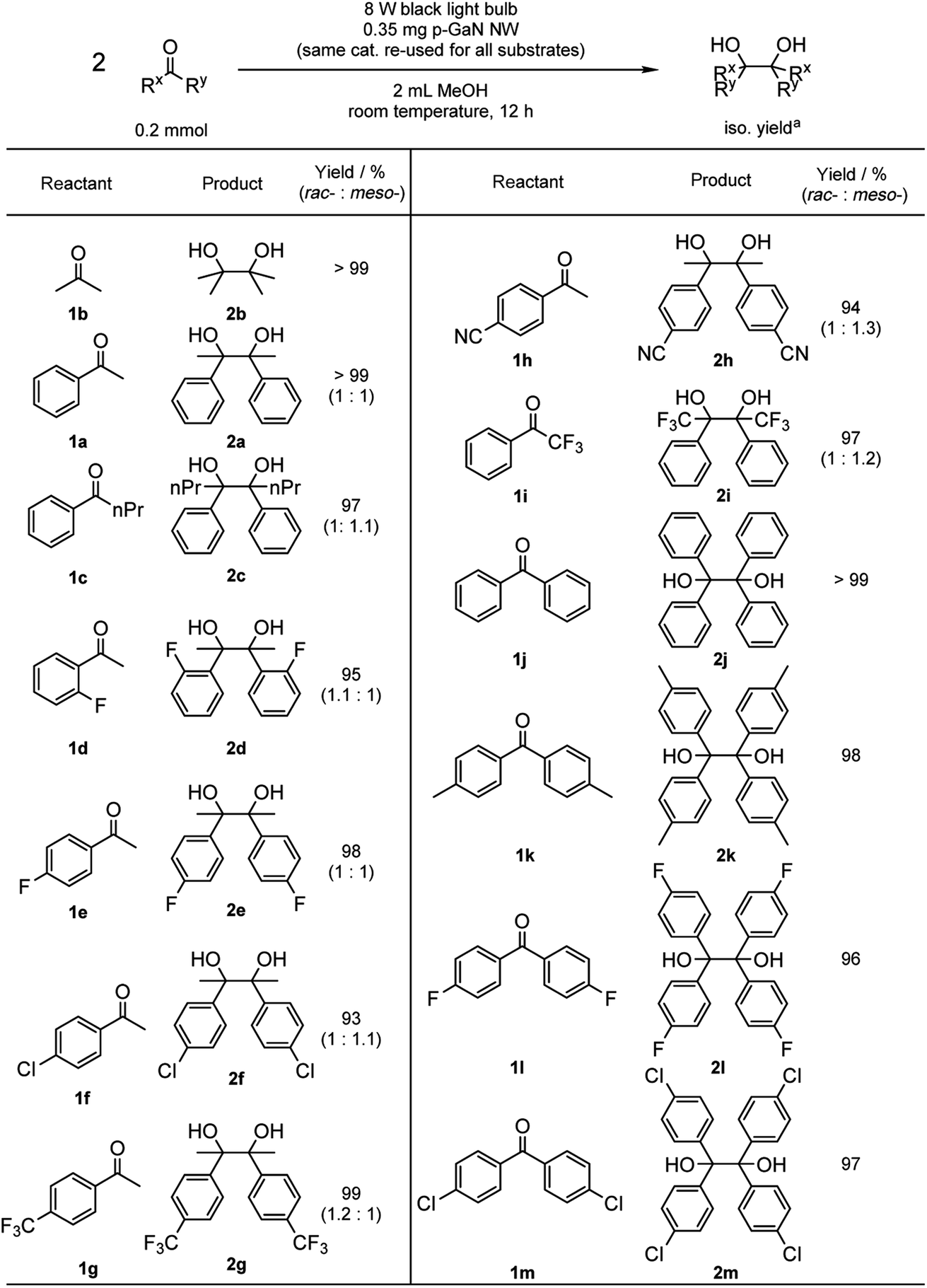
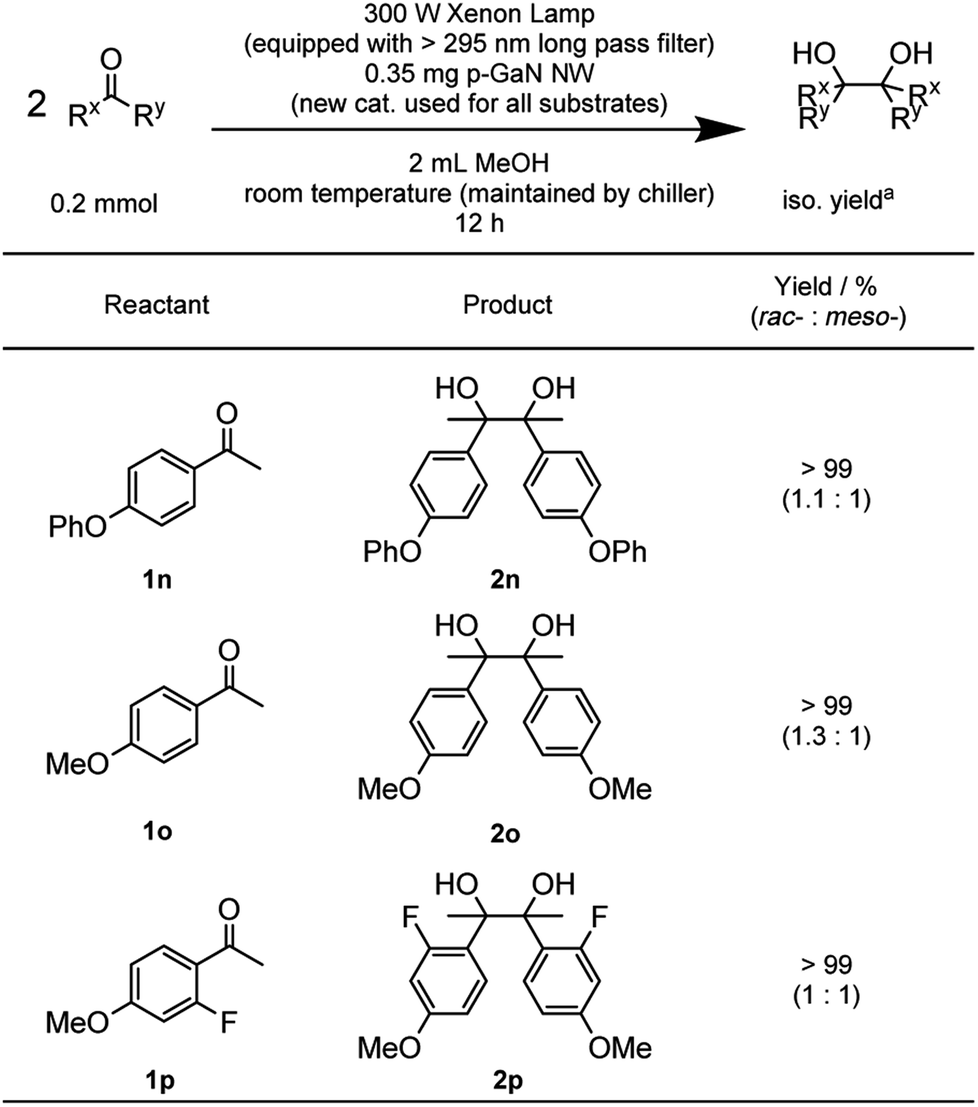
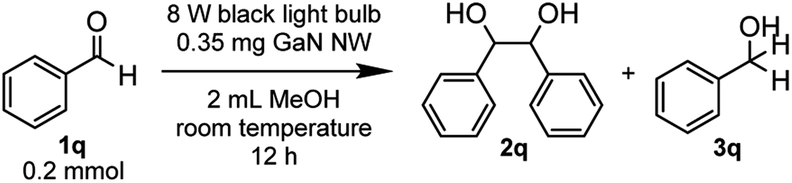
With the optimized reaction conditions in hand, we proceeded to examine the substrate scope of the photochemical PCR using 0.35 mg p-GaN NW and 0.2 mmol starting material in 2 mL MeOH. To test the stability and reusable limit of the catalyst, we recycled and reused this same catalyst over-and-over for all substrates (Table 2). We started with the simplest ketone, acetone (1b), which gave a quantitative yield of pinacol (2b). Likewise, acetophenone (1b) and butyrophenone (1c) also reacted efficiently, giving quantitative and 97% yields, respectively. Fluorinated substrates 2-fluoroacetophenone (1d) and 4-fluoroacetophenone (1e) gave 95% and 98% yields of their corresponding 2,3-bis(o-fluorophenyl)-2,3-butanediol (2d) and 2,3-bis(p-fluorophenyl)-2,3-butanediol (2e). The chloro analog 4-chloroacetophenone (1f) gave a 93% yield of 2,3-bis(p-chlorophenyl)-2,3-butanediol (2f). The e−-deficient 4-trifluoromethylacetophenone (1g) gave a 99% yield of 2,3-bis(p-trifluoromethylphenyl)-2,3-butanediol (2g). For other e−-deficient substrates, 4-cyanoacetophenone (1h) gave a 94% yield of 2,3-bis(p-cyanophenyl)-2,3-butanediol (2h) and α,α,α-trifluoroacetophenone (1i) gave a 97% yield of 1,1,1,4,4,4-hexafluoro-2,3-diphenyl-2,3-butanediol (2i). Aryl–aryl ketones were then examined. Benzophenone (1j) gave a quantitative yield of the corresponding benzopinacol (2j). The substituted benzophenone, 4,4′-dimethylbenzophenone (1k), 4,4′-difluorobenzophenone (1l), and 4,4′-dichlorobenzophenone (1m) gave 98%, 96% and 97% yields of their corresponding tetramethylbenzopinacol (2k), tetrafluorobenzopinacol (2l), and tetrachlorobenzopinacol (2m), respectively. It should be noted that even after 13 consecutive reactions of substrates with various functionalities, the same p-GaN NW showed no observable decrease in catalytic reactivity. However, certain substrates, such as 4-phenoxyacetophenone (1n), 4-methoxyacetophenone (1o), and 2-fluoro-4-methoxyacetophenone (1p), gave complete starting material recovery even after freshly synthesized catalysts were used under the given conditions. When a much stronger light source (a 300 W full-arc xenon lamp equipped with a >295 nm long pass light filter) was used instead of the black light bulb, those substrates gave quantitative yields of their corresponding PCR products (2n–2p, Table 3). These results suggest that e− transfers from the semiconductor to those substrates are more difficult, possibly due to their electron richness. We also examined the PCR of aldehyde substrates such as benzaldehyde (1q) under the black light bulb using the p-GaN NW catalyst (Table 4). Despite full conversion of the starting material and the desired PCR product of hydrobenzoin (2q) being obtained (see Fig. S30 in the ESI†), significant amounts of benzyl alcohol (3q) were observed as a side-product. After examining GaN NWs doped with other dopants, we found that the selectivity for 2q increased from p-GaN and i-GaN to n-GaN. According to the e− counting, the substrate needs to absorb two e− from the semiconductor to give the hydrogenation product, while only one e− is needed to initiate the PCR. The enhanced e− donating ability of p-GaN NW compared to n-GaN NW might cause over-donation of e− to the substrate, promoting the hydrogenation side-reaction while lowering the PCR selectivity.
| a Reaction was done by injecting all reactants into a sealed quartz vessel under vacuum. |
|---|
|
| a Reaction was done by injecting all reactants into a sealed quartz vessel under vacuum. |
|---|
|
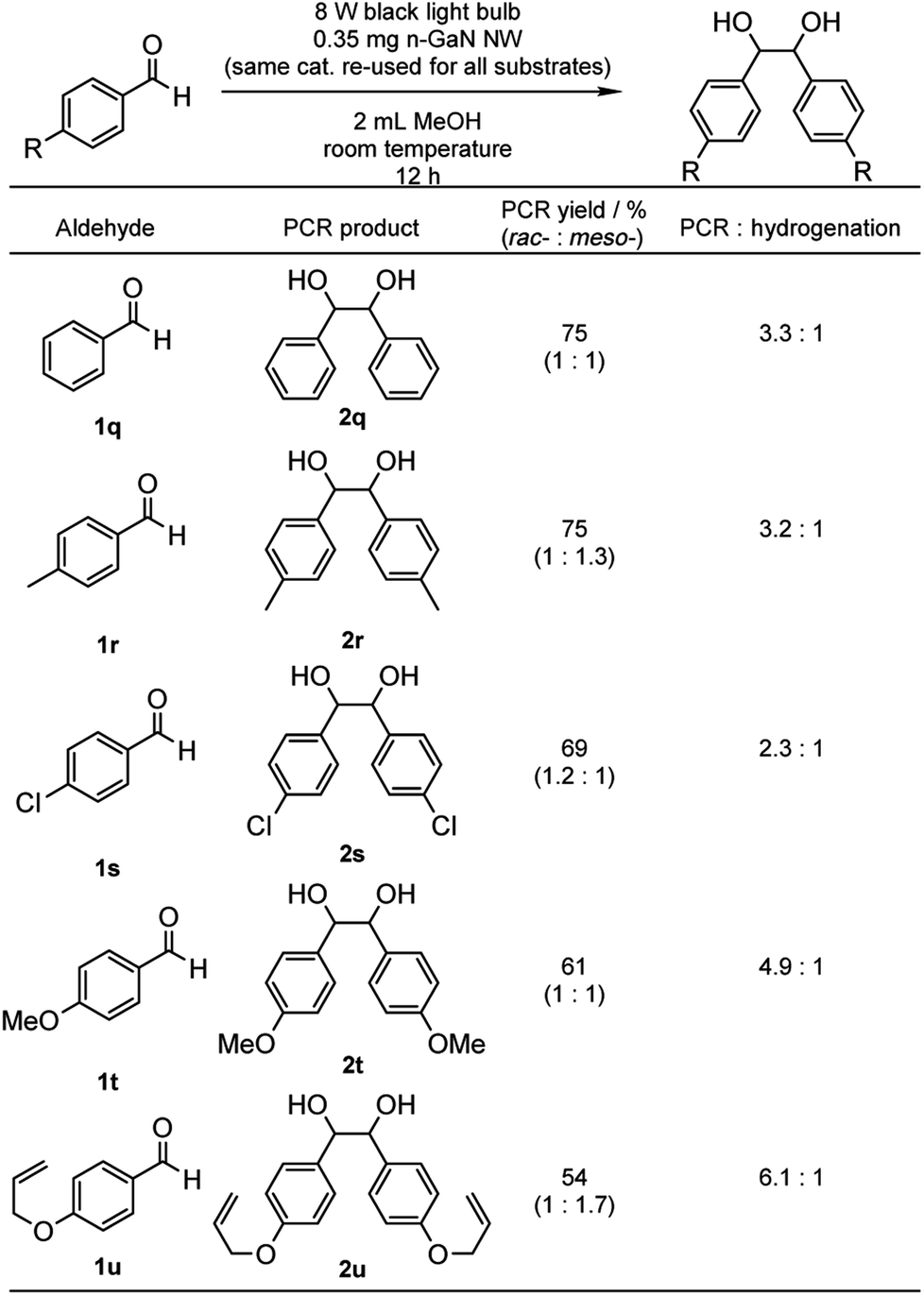
The substrate scope for aldehydes was also investigated using the catalyst recycled from Table 2 (Table 5). Although 1q and p-tolualdehyde (1r) gave the same PCR yields (2q and 1,2-bis(p-methylphenyl)-1,2-ethanediol (2r), respectively) with similar PCR/hydrogenation selectivities, the PCR/hydrogenation selectivity decreased to 2.3:1 with more e−-deficient 4-chlorobenzaldehyde (1s), which gave 69% PCR yield of 1,2-bis(4-chlorophenyl)-1,2-ethanediol (2s). On the other hand, e−-rich aldehydes, such as p-anisaldehyde (1t) and 4-allyloxybenzaldehyde (1u), led to increased PCR/hydrogenation selectivities (4.9:1 and 6.1:1, respectively) while gave 61% and 54% PCR yields of 1,2-bis(p-methoxylphenyl)-1,2-ethanediol (2t) and 1,2-bis(4-allyloxyphenyl)-1,2-ethanediol (2u), respectively.
| a Reaction was done by injecting all reactants into a sealed quartz vessel under vacuum. |
|---|
|
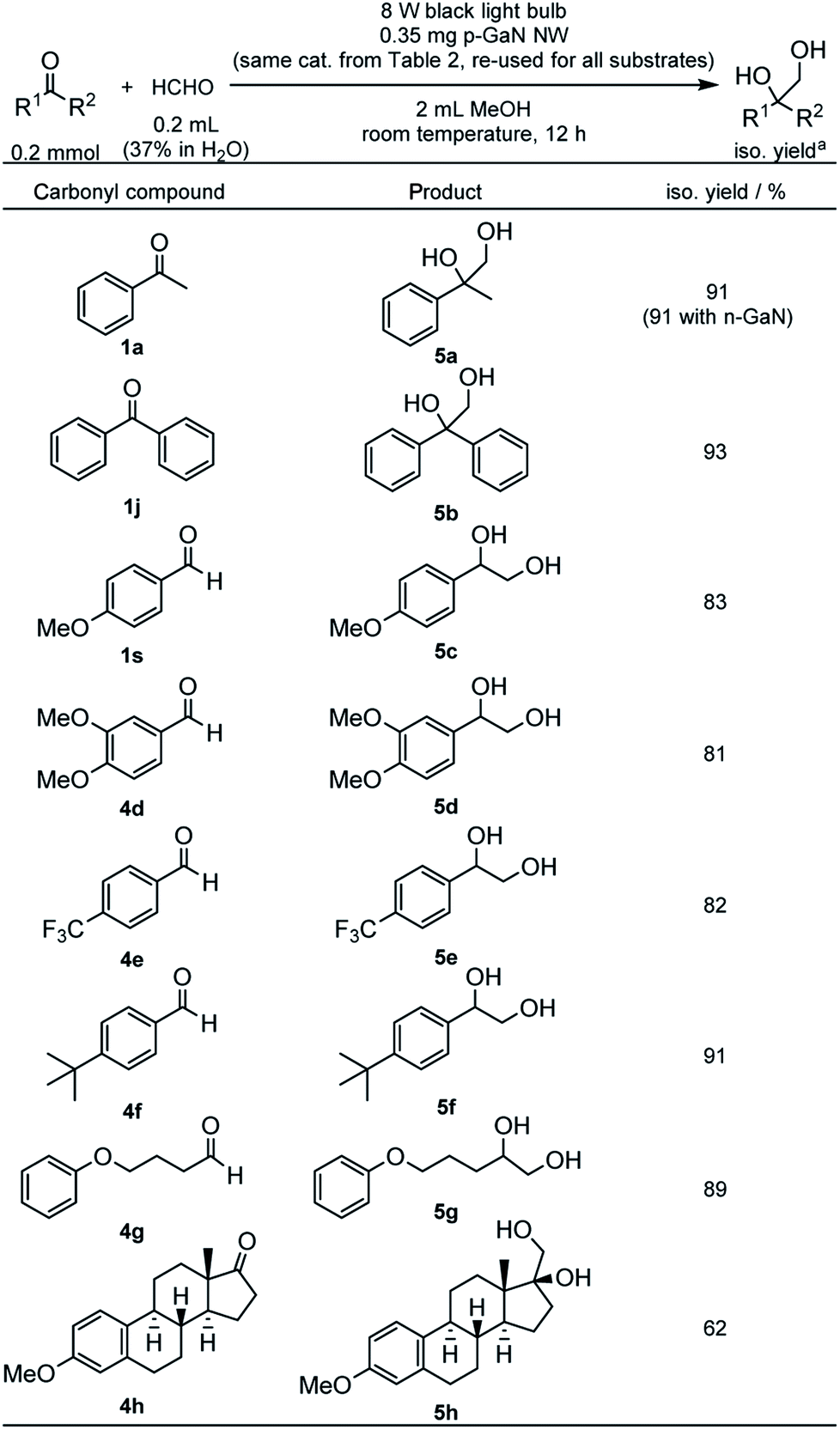
We also examined the cross-PCR of various carbonyl compounds with formaldehyde under the optimized conditions (Table 6). The reaction between 1a and formaldehyde gave an excellent 91% yield of 2-phenyl-1,2-propanediol (5a) with either n-GaN NW or p-GaN NW. Therefore, the p-GaN NW used previously in Table 2 was reused and recycled again as the catalyst. Under the same conditions, 1j also gave an excellent 93% yield of 1,1-diphenyl-1,2-ethanediol (5b). e−-rich aldehyde 1s gave a slightly reduced yield of 83% for the corresponding 1-(4-methoxyphenyl)-1,2-ethanediol (5c). An even more e−-rich aldehyde, 3,4-dimethoxybenzaldehyde (4d), gave 81% yield of 1-(3,4-dimethoxyphenyl)-1,2-ethanediol (5d). e−-deficient aldehyde, such as 4-trifluoromethylbenzaldehyde (4e), also gave 82% yield of 1-(4-trifluoromethylphenyl)-1,2-ethanediol (5e). 4-tert-Butylbenzaldehyde (4f) gave 91% yield of 1-(4-tert-butylphenyl)-1,2-ethanediol (5f). Aliphatic aldehydes such as 4-phenoxybutanal (4g) also reacted efficiently, giving 89% yield of 5-phenoxy-1,2-pentanediol (5g). Inspired by the observed functional tolerance, a natural product, 3-methoxyestrone (4h), was also examined using the recycled catalyst, which has been recycled 21 times in total, giving an astonishing 62% yield of the corresponding 17-hydroxy-3-methoxyestra-1,3,5(10)-triene-17-methanol (5h). The catalyst still shows no signs of degradation after the recycling (see Fig. S34 in the ESI†).
| a Reaction was done by injecting all reactants into a sealed quartz vessel under vacuum. Racemic mixtures were obtained for all isolated products except 5 h. |
|---|
|
Finally, we examined the optimized PCR catalyzed with p-GaN NW by adding a stoichiometric amount of TEMPO as a radical trap (Scheme 1). The TEMPO adduct was identified using mass spectroscopy (MS) while no PCR product was detected, which supports our design of a radical coupling process.
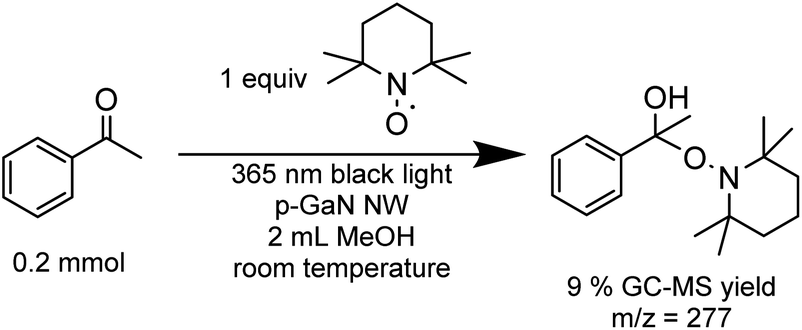 | ||
| Scheme 1 Radical-trap experiment using TEMPO. The yield was determined using GC-MS with 1,3,5-mesitylene as an internal standard. | ||
3. Conclusions
In summary, we have shown the use of epitaxially grown GaN NWs as a highly efficient, robust and readily reusable photoredox catalyst for a photo-PCR under ambient light and room temperature. Various ketones afford the corresponding products smoothly in excellent yields. Aldehydes are also effective albeit giving some over-reduction side products. By using excess formaldehyde, cross-PCRs are also effective with both aromatic and aliphatic aldehydes, as well as in complex natural products. The catalytic properties of GaN can be tuned readily with different dopants. Further investigations of GaN NWs as organic chemistry catalysts are already underway in our lab.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. L. and C. J. L. are grateful to the Canada Research Chair (Tier 1) foundation, Natural Science and Engineering Research Council of Canada, Fonds de recherche du Quebec – Nature et Technologies, Canada Foundation for Innovation (CFI), and McGill University for financial support. M. L. and Z. M. are grateful to Emissions Reduction Alberta for funding.Notes and references
- D. H. R. Barton and S. I. Parekh in Half a Century of Free Radical Chemistry, ed. L. Lincee, Cambridge University Press, Cambridge, 1993 Search PubMed.
- T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed.
- D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed.
- D. A. Nagib, M. E. Scott and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 10875–10877 CrossRef CAS PubMed.
- J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, J. Am. Chem. Soc., 2009, 131, 8756–8757 CrossRef CAS PubMed.
- L. J. Rono, H. G. Yayla, D. Y. Wang, M. F. Armstrong and R. R. Knowle, J. Am. Chem. Soc., 2013, 135, 17735–17738 CrossRef CAS PubMed.
- R. Wang, M. Ma, X. Gong, X. Fan and P. J. Walsh, Org. Lett., 2019, 21, 27–31 CrossRef PubMed.
- M. Nakajima, E. Fava, S. Loescher, Z. Jiang and M. Rueping, Angew. Chem., Int. Ed., 2015, 30, 8828–8832 CrossRef.
- A. Caron, É. Morin and S. K. Collins, ACS Catal., 2019, 9, 9458–9464 CrossRef CAS.
- R. C. McAtee, E. J. McClain and C. R. J. Stephenson, Trends Chem., 2019, 1, 111–125 CrossRef.
- H. Kisch, Angew. Chem., Int. Ed., 2013, 52, 812–847 CrossRef CAS PubMed.
- I. Akasaki, J. Cryst. Growth, 2002, 237–239, 905–911 CrossRef CAS.
- M. G. Kibria, R. Qiao, W. Yang, I. Boukahil, X. Kong, F. A. Chowdhury, M. L. Trudeau, W. Ji, H. Guo, F. J. Himpsel, L. Vayssieres and Z. Mi, Adv. Mater., 2016, 28, 8388–8397 CrossRef CAS PubMed.
- S. J. Pearton, in GaN and elated Materials, CRC Press, Boca Raton, 1997 Search PubMed.
- K. Hoshino, Chem.–Eur. J., 2001, 7, 2727–2731 CrossRef CAS.
- H. P. T. Nguyen, M. Djavid and Z. Mi, ECS Trans., 2013, 53, 93–100 CrossRef.
- Q. Cheng, W. Fan, Y. He, P. Ma, S. Vanka, S. Fan and Z. Mi, Adv. Mater., 2017, 29, 1700312 CrossRef PubMed.
- D. Wang, A. Pierre, M. G. Kibria, K. Cui, X. Han, K. H. Bevan, H. Guo, S. Paradis, A.-R. Hakima and Z. Mi, Nano Lett., 2011, 11, 2353–2357 CrossRef CAS PubMed.
- B. AlOtaibi, H. P. T. Nguyen, S. Zhao, M. G. Kibria, S. Fan and Z. Mi, Nano Lett., 2013, 13, 4356–4361 CrossRef CAS PubMed.
- M. G. Kibria, S. Zhao, F. A. Chowdhury, Q. Wang, H. P. T. Nguyen, M. L. Trudeau, H. Guo and Z. Mi, Nat. Commun., 2014, 5, 3825 CrossRef CAS PubMed.
- M. G. Kibria, F. A. Chowdhury, S. Zhao, B. AlOtaibi, M. L. Trudeau, H. Guo and Z. Mi, Nat. Commun., 2015, 6, 6797 CrossRef CAS PubMed.
- F. A. Chowdhury, M. L. Trudeau, H. Guo and Z. Mi, Nat. Commun., 2018, 9, 1707 CrossRef PubMed.
- B. Zhou, X. Kong, S. Vanka, S. Chu, P. Ghamari, Y. Wang, N. Pant, I. Shih, H. Guo and Z. Mi, Nat. Commun., 2018, 9, 3856 CrossRef PubMed.
- L. Li, Y. Wang, S. Vanka, X. Mu, Z. Mi and C.-J. Li, Angew. Chem., Int. Ed., 2017, 129, 8827–8831 CrossRef.
- M. Liu, Y. Wang, X. Kong, L. Tan, L. Li, S. Cheng, G. Botton, H. Guo, Z. Mi and C.-J. Li, iScience, 2019, 17, 208–216 CrossRef CAS PubMed.
- L. Li, S. Fan, X. Mu, Z. Mi and C.-J. Li, J. Am. Chem. Soc., 2014, 136, 7793–7796 CrossRef CAS PubMed.
- L. Li, X. Mu, W. Liu, X. Kong, S. Fan, Z. Mi and C.-J. Li, Angew. Chem., Int. Ed., 2014, 53, 14106–14109 CrossRef CAS PubMed.
- Y. Wang, S. Fan, B. AlOtaibi, Y. Wang, L. Li and Z. Mi, Chem.–Eur. J., 2016, 22, 8809–8813 CrossRef CAS PubMed.
- S. Chu, P. Ou, P. Ghamari, S. Vanka, B. Zhou, I. Shih, J. Song and Z. Mi, J. Am. Chem. Soc., 2018, 140, 7869–7877 CrossRef CAS PubMed.
- M. Foussekis, Band Bending in GaN, Master dissertation, Virginia Commonwealth University, Richmond, Virginia, 2009.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials and methods, Fig. S1–S34, and Table S1. See DOI: 10.1039/d0sc02718a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |