
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRuthenium-catalyzed hydrogenation of CO2 as a route to methyl esters for use as biofuels or fine chemicals†
Zheng
Wang
 abc,
Ziwei
Zhao
a,
Yong
Li
a,
Yanxia
Zhong
d,
Qiuyue
Zhang
b,
Qingbin
Liu
*a,
Gregory A.
Solan
*be,
Yanping
Ma
b and
Wen-Hua
Sun
*b
abc,
Ziwei
Zhao
a,
Yong
Li
a,
Yanxia
Zhong
d,
Qiuyue
Zhang
b,
Qingbin
Liu
*a,
Gregory A.
Solan
*be,
Yanping
Ma
b and
Wen-Hua
Sun
*b
aHebei Key Laboratory of Organic Functional Molecules, College of Chemistry and Material Science, Hebei Normal University, Shijiazhuang 050024, China. E-mail: liuqingb@sina.com
bKey Laboratory of Engineering Plastics and Beijing National Laboratory for Molecular Science, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: gas8@leicester.ac.uk; whsun@iccas.ac.cn; Fax: +86-10-62618239; Tel: +86-10-62557955
cCollege of Science, Agricultural University of Hebei, Baoding 071001, China
dDepartment of Nursing Shijiazhuang Medical College, Shijiazhuang 050000, China
eDepartment of Chemistry, University of Leicester, University Road, Leicester LE1 7RH, UK
First published on 5th June 2020
Abstract
A novel robust diphosphine–ruthenium(II) complex has been developed that can efficiently catalyze both the hydrogenation of CO2 to methanol and its in situ condensation with carboxylic acids to form methyl esters; a TON of up to 3260 is achievable for the CO2 to methanol step. Both aromatic and aliphatic carboxylic acids can be transformed to their corresponding methyl esters with high conversion and selectivity (17 aliphatic and 18 aromatic examples). On the basis of a series of experiments, a mechanism has been proposed to account for the various steps involved in the catalytic pathway. More importantly, this approach provides a promising route for using CO2 as a C1 source for the production of biofuels, fine chemicals and methanol.
Introduction
The steadily rising levels of carbon dioxide in the atmosphere over the last hundred years or so have made a major contribution to the earth's greenhouse effect. These higher concentrations can be attributed, in large measure, to increasing worldwide energy consumption that is generated through power plants that make use of fossil resources.1 However, in order to maintain the carbon dioxide balance in the atmosphere and to reduce the dependency on a limited fossil resource, alternative ways for the sustainable production of fuels and chemicals represent a major global challenge. A strategy that has been gaining attention is the use of captured CO2 as a feedstock for the synthesis of biofuels and chemicals.2 In recent decades, chemists have explored and developed more than one hundred laboratory processes for using CO2 as an alternative carbon source in fields that interface the chemical and energy sectors (Scheme 1).1b Such processes include the direct metal-catalyzed hydrogenation of CO2 to formic acid,3 formides,4 methanol5 and ethanol.6 In addition, there has been the use of CO2 as a C1 building block in hydroxymethylation,7N-methylation of amines/amides,8N-methylation of imines,9 methylation of C–H bonds (sp3)10 as well as the carboxylation of C–H (sp2)11 and N–H bonds.12 Elsewhere, the non-reductive incorporation of a CO2 molecule into organic products, such as cyclic carbonates and polycarbonates/polyethercarbonates has been demonstrated.1a,1b,13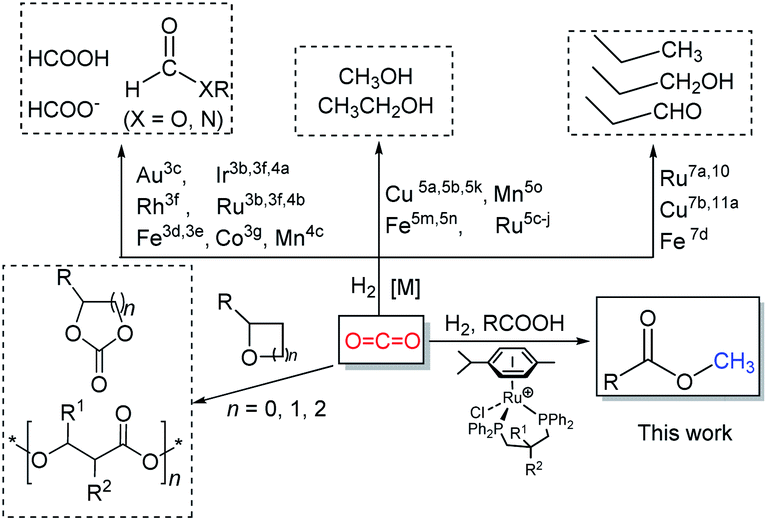 | ||
| Scheme 1 Capturing CO2 as a carbon source for the production of fine chemicals and biofuels. | ||
From an industrial standpoint, relatively few processes employ CO2 as a starting material for the manufacture of organic products.2,13e Nevertheless, those processes that do operate allow access to a number of high demand materials including urea, methanol, salicylic acid, organic carbonates and polycarbonates.1a,1b,13 Staggeringly however, this use of CO2 as a feedstock for chemicals only accounts for about 0.36% of global CO2 emissions.14 Elsewhere, it has been stated that only by completely using biomass energy (and without fossil energy), can the concentration of carbon dioxide reach equilibrium in the atmosphere.1a Moreover, it could be argued that one of the best ways to reduce CO2 emissions would be to synthesize biofuels that could be recycled.
As part of an ongoing program, we have been interested in developing methods of using CO2 as a feedstock to form carboxylic acid methyl esters as their aliphatic examples could then be used as biofuels while their aromatic methyl esters as fine chemicals (Scheme 2). In particular, a cascade strategy involving metal-catalyzed hydrogenation of CO2 to methanol and then in situ condensation of methanol with a carboxylic acid has been envisioned. Of course, the carboxylic acids represent attractive family of reactants as they can in principle be obtained from biomass. Furthermore, the catalytic hydrogenation of methyl esters has been shown by us15 and others,5c–5e,16 to give alkyl/aryl alcohols and methanol which can, in their own right, serve as important fuels and synthetic building blocks.15,16 Alternatively, methanol can be produced by the hydrolysis of a methyl ester and the corresponding carboxylic acid by-product recycled. Overall, this sequence of reactions could present an elegant and green sustainable route for the production of fuels and chemicals.
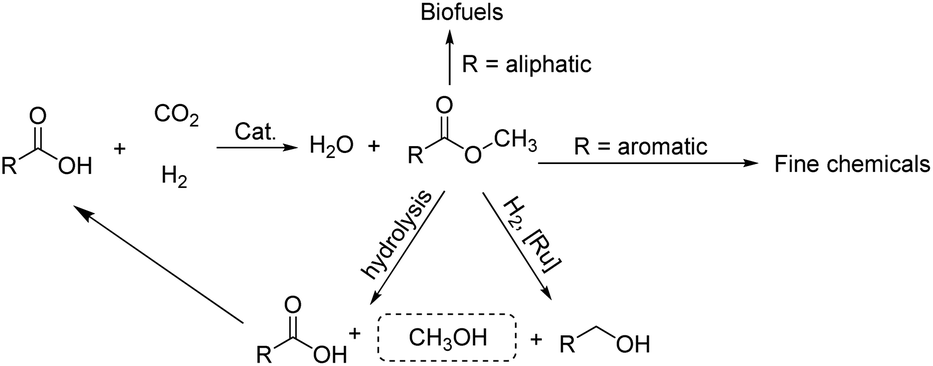 | ||
| Scheme 2 Potential strategy involving CO2 utilization to form industrially useful methyl esters and their amenability to recycling and use in other applications. | ||
To realize our goal, we herein disclose a new family of well-defined diphosphine–ruthenium(II) cationic complexes that can serve as versatile (pre)catalysts for the conversion of CO2 to a raft of different types of methyl ester (Scheme 1). By performing the reactions in the presence of a carboxylic acid, we show that this acid plays two key roles; (i) as a reactant in the conversion of the methanol intermediate to the target methyl ester and (ii) as a promoter in the CO2 hydrogenation step. Unlike the ruthenium-triphos catalysts previously reported by Leitner and others for the hydrogenation of CO2,5e,5j the current (pre)catalysts are based on an organometallic η6-arene-ruthenium core that incorporates a chelating diphosphine of the type, CR1R2(CH2PPh2)2 [R1 = CH2PPh2, R2 = Me; R1 = CH2P(O)Ph2, R2 = Me; R1 = CH2P(O)Ph2, R2 = Et; R1 = R2 = H].
Results and discussion
Synthesis and characterization of Ru1–Ru4
Cationic [RuCl(κP,κP-triphos)(η6-p-cymene)][Cl] (Ru1) was prepared in excellent yield by the reaction of [RuCl2(η6-p-cymene)]2 with 1,1,1-tris(diphenylphosphinomethyl)ethane (triphos) in a mixture of CH2Cl2 and EtOH at 50 °C (Scheme 3, see ESI†); related ruthenium(II) complexes and reaction conditions have been reported elsewhere.17 The 31P NMR spectrum of Ru1 exhibited two singlet peaks at δ 26.02 and δ 24.81, corresponding to the inequivalent phosphine donors, as well as one single peak at δ −29.77 for the pendant phosphine (see ESI, Fig. S3†).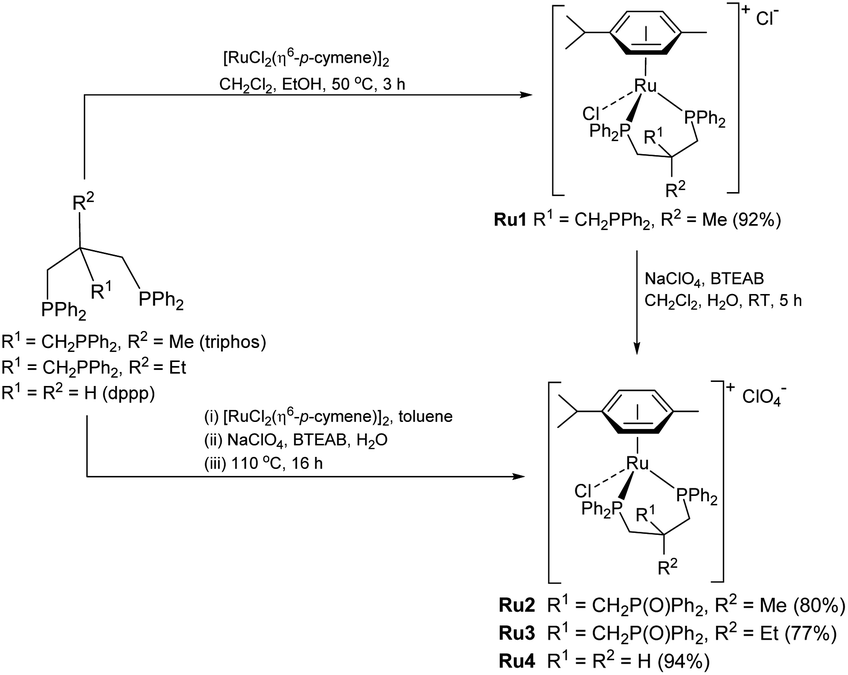 | ||
| Scheme 3 Synthetic route to Ru1–Ru4. | ||
Counteranion exchange and oxidation of the pendant phosphine arm in Ru1 and could be readily achieved by its reaction with sodium perchlorate in a mixture of dichloromethane and water at room temperature to give [RuCl(κP,κP-triphos(O))(η6-p-cymene)][ClO4] (Ru2) (Scheme 3). Alternatively, Ru2 could be obtained by a one-pot reaction involving successive treatment of triphos with [RuCl2(η6-p-cymene)]2 and sodium perchlorate in a water/toluene mixture; benzyl-triethylammonium bromide (BTEAB) was used as a phase transfer catalyst in both routes. The 31P NMR spectrum of Ru2 in DMSO-d6 exhibited two mutually coupled doublets at δ 26.18 and δ 25.05, which were assigned to the signals for the inequivalent phosphine donors; a single peak at δ 28.32 was attributed to the non-coordinated phosphine oxide (see ESI, Fig. S9†).
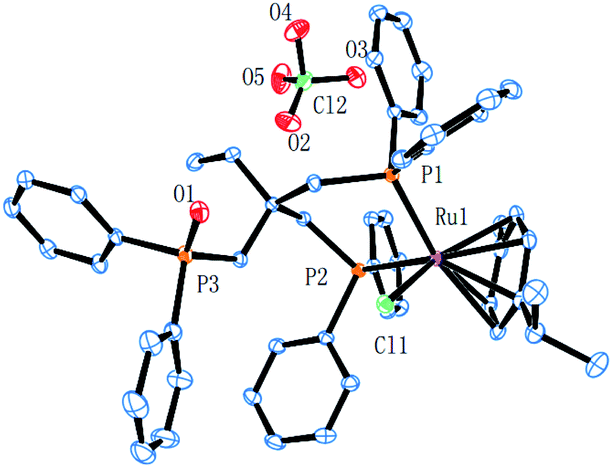
Complex cations [RuCl{κP,κP-(CH2PPh2)2CR1R2}(η6-p-cymene)] [ClO4] [Ru3 R1 = CH2P(O)Ph2, R2 = Et; Ru4 R1 = R2 = H] could be obtained from CEt(CH2PPh2)3 or CH2(CH2PPh2)2 (dppp), respectively, in high yield by using a one-pot route based on that employed for the synthesis of Ru2. As with Ru2, Ru3 showed two doublets in the 31P NMR spectrum for the inequivalent coordinated phosphines and a singlet for the pendant phosphine oxide group, while Ru4 showed two distinct singlets for the bidentate dppp ligand. Besides 31P NMR spectroscopy, all four ruthenium complexes were characterized by 1H, 13C NMR spectroscopy, elemental analysis and by ESI-mass spectrometry (Table S3, see ESI†). To confirm their structural identity, Ru2 and Ru3 were the subject of single crystal X-ray diffraction studies (Fig. 1 and 2); selected bond distances and angles are given in the figure captions. Both structures consist of a cationic ruthenium(II) unit and a non-coordinating perchlorate anion. The cationic unit adopts a three-legged piano stool geometry comprising a η6-p-cymene, a monodentate chloride and a bidentate CR2(CH2P(O)Ph2)(CH2PPh2)2 (R2 = Me Ru2, Et Ru3) ligand which binds through the diphenylphosphine phosphorus atoms while the oxidized phosphine arm remains uncoordinated; the bond parameters around ruthenium are not exceptional and indeed similar to related half-sandwich structures. To try and explain the origin of the phosphine oxide units in Ru2 and Ru3, the four different ruthenium complexes were separately evaluated as catalysts for the oxidation of PPh3 in a toluene–water mixture at 100 °C over 24 hours (Table S2, ESI†). Inspection of the results revealed conversions to triphenylphosphine oxide of 24% for [RuCl2(η6-p-cymene)]2, 94% for Ru1, 43% for Ru2 and 97% for [[RuCl2(η6-p-cymene)]2 + triphos]. Evidently, the uncoordinated phosphine oxide present in Ru2 and Ru3 derives from a ruthenium-mediated oxidation of the corresponding –CH2PPh2 unit.
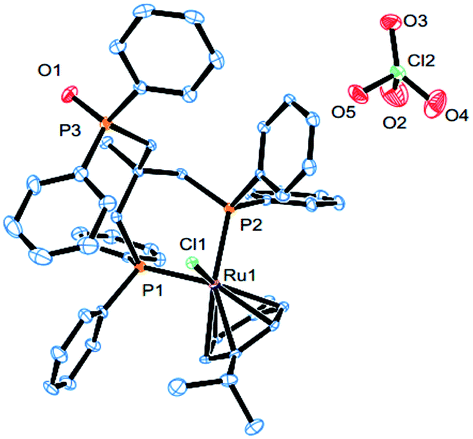 | ||
| Fig. 1 ORTEP representation of Ru2. Thermal ellipsoids are shown at the 30% probability level; hydrogen atoms have been omitted for clarity. Selected bond distances (Å): Ru1–Cl1 = 2.4086(11), Ru1–P2 = 2.3276(12), Ru1–P1 = 2.3286(12), Ru1–C47 = 2.221(4), Ru1–C46 = 2.269(4), Ru1–C45 = 2.322(4), Ru1–C43 = 2.254(4), Ru1–C44 = 2.260(4), Ru1–C48 = 2.295(5), P3–O1 = 1.480(3); Selected angles (deg (°)): P1–Ru1–Cl1 = 85.07(3), P2–Ru1–Cl1 = 87.77(4), P2–Ru1–P1 = 86.85(2). | ||
 | ||
| Fig. 2 ORTEP representation of Ru3. Thermal ellipsoids are shown at the 30% probability level; hydrogen atoms have been omitted for clarity. Selected bond distances (Å): Ru1–Cl1 = 2.4249(7), Ru1–P1 = 2.3333(7), Ru1–P2 = 2.3193(6), Ru1–C43 = 2.322(2), Ru1–C44 = 2.285(2), Ru1–C45 = 2.224(2), Ru1–C46 = 2.298(2), Ru1–C47 = 2.241(2), Ru1–C48 = 2.248(2), P3–O1 = 1.4893(15); Selected angles (deg (°)): P1–Ru1–Cl1 = 82.55(3), P2–Ru1–Cl1 = 90.39(3), P2–Ru1–P1 = 86.76(2). | ||
Catalytic hydrogenation of CO2 to give carboxylic acid methyl esters
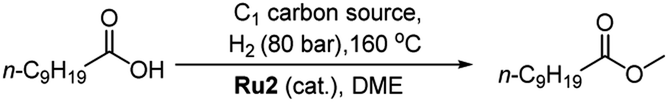
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :68 ratio) and the substrate to catalyst molar ratio (S:C) at 100:1, a 28% conversion was observed after 20 hours of which 25% constituted methyl decanoate (2) and 3% n-decanol (3) (entry 1, Table 1). By slightly increasing the hydrogen pressure in the gaseous mixture to 12:70, the conversion decreased to 14% with a larger proportion now being 3 (entry 2, Table 1). Under the same conditions but with the volume of solvent reduced to 10 mL, to increase the substrate concentration, a 91% conversion was achieved with 83% being 2 (entry 3, Table 1). To our delight, by reverting to the pressure ratio of CO2:H2 = 12:68 and maintaining the higher concentration of substrate, a high conversion and selectivity (conv. = 99%, selectivity = 100%) were realized (entry 4, Table 1).
:68 ratio) and the substrate to catalyst molar ratio (S:C) at 100:1, a 28% conversion was observed after 20 hours of which 25% constituted methyl decanoate (2) and 3% n-decanol (3) (entry 1, Table 1). By slightly increasing the hydrogen pressure in the gaseous mixture to 12:70, the conversion decreased to 14% with a larger proportion now being 3 (entry 2, Table 1). Under the same conditions but with the volume of solvent reduced to 10 mL, to increase the substrate concentration, a 91% conversion was achieved with 83% being 2 (entry 3, Table 1). To our delight, by reverting to the pressure ratio of CO2:H2 = 12:68 and maintaining the higher concentration of substrate, a high conversion and selectivity (conv. = 99%, selectivity = 100%) were realized (entry 4, Table 1).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | S:C |
[Ru] | Solvent | CO2:H2 (bar) |
Conv.b (%) | 2/3/4b (% conv. to each) |
|
a Conditions: decanoic acid (1.0 mmol), [Ru] (1.0–10.0 μmol), solvent (10 mL), PH2 = 68–70 bar (at RT), PCO2 = 0–12 bar (at RT), temp. = 160 °C, time = 20 h, S:C = the molar substrate to catalyst ratio, DME is 1,2-dimethoxyethane.
b The conversion, with reference to decanoic acid (1), was determined by GC (using mesitylene as the internal standard) and by GC-MS.
c 30 mL of DME in place of 10 mL.
d DME (2.5 mL), the ester products are methyl decanoate (95%) and ethyl decanoate (5%).
e In the absence of CO2.
f In the absence of decanoic acid (1), no CH3OH was observed.
|
||||||
| 1c | 100:1 |
Ru2 | DME | 12:68 |
28 | 25/3/0 |
| 2c | 100:1 |
Ru2 | DME | 12:70 |
14 | 12/2/0 |
| 3 | 100:1 |
Ru2 | DME | 12:70 |
91 | 83/8/0 |
| 4 | 100:1 |
Ru2 | DME | 12:68 |
99 | 99/0/0 |
| 5 | 100:1 |
Ru2 | 1,2-Diethoxyethane | 12:68 |
76 | 70/6/0 |
| 6 | 100:1 |
Ru2 | Diglyme | 12:68 |
36 | 30/6/0 |
| 7 | 100:1 |
Ru2 | Triglyme | 12/68 | 65 | 60/5/0 |
| 8 | 200:1 |
Ru2 | DME | 12:68 |
96 | 94/2/0 |
| 9 | 500:1 |
Ru2 | DME | 12:68 |
100 | 100/0/0 |
| 10 | 1000:1 |
Ru2 | DME | 12:68 |
42 | 40/2/0 |
| 11d | 100:1 |
Ru2 | DME | 12:68 |
100 | 100/0/0 |
| 12 | 100:1 |
[RuCl2(p-cymene)]2 | DME | 12:68 |
3 | 0/2/1 |
| 13 | 100:1 |
Ru1 | DME | 12:68 |
76 | 76/0/0 |
| 14 | 100:1 |
Ru3 | DME | 12:68 |
67 | 67/0/0 |
| 15 | 100:1 |
Ru4 | DME | 12:68 |
56 | 56/0/0 |
| 16e | 100:1 |
Ru2 | DME | 00:80 |
n.d. | n.d. |
| 17f | 100:1 |
Ru2 | DME | 12:68 |
n.d. | n.d. |
With a view to establishing the most compatible reaction medium for the hydrogenation using Ru2, four related solvents were screened, namely, DME, 1,2-diethoxyethane, diglyme and triglyme (entries 4–7, Table 1). On examination of the data, DME was the standout performer in terms of the conversion (99%) and selectivity for 2 (100%). When 1,2-diethoxyethane was employed as solvent, a lower conversion (76%) and selectivity (92%) was observed while with the longer chain solvents, diglyme and triglyme, the conversions observed were markedly less at 36% and 65%, respectively.
In order to determine the optimal S:C ratio, four different combinations, 100:1, 200:1, 500:1 and 1000:1, were screened using Ru2 (entries 4 and 8–10, Table 1). It was found that with ratios between 100:1 and 500:1, high conversions (96–100%) could be achieved. By contrast with the ratio at 1000:1, the CO2 hydrogenation was incomplete with only 42% conversion achievable (entry 10, Table 1). With the volume of DME decreased to 2.5 mL and a 100:1 ratio re-deployed, 100% conversion was obtained, but the ester products consisted of 2 (95%) and ethyl decanoate (5%) (entry 11, Table 1, see ESI†). By contrast, when [RuCl2(p-cymene)]2 was employed as catalyst, no methyl decanoate was obtained instead minor amounts of 3 and decyl decanoate (4) were detected (entry 12, Table 1). In addition, when the catalysis was conducted using Ru2 but in the absence of decanoic acid, no CH3OH was observed (entry 17, Table 1), which suggests that the decanoic acid acts as a promoter.
Using the best overall set of conditions established for Ru2 [T = 160 °C, CO2/H2 = 12:68 (80 bar in total), S:C = 100:1, 20 h, DME (10 mL)], the remaining ruthenium complexes Ru1, Ru3 and Ru4 were also evaluated. Indeed, all of these complexes were active and selective catalysts for methyl decanoate with their conversions, when compared to Ru2, falling in the order: Ru2 > Ru1 > Ru3 > Ru4 (entries 4, 13–15, Table 1). As a control, no conversion to 2 was achieved when the hydrogenation was performed in the absence of CO2 under the same conditions (entry 16, Table 1).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | S:C |
t (h) | Conv.b (%) | Ester/alcoholb (% conv. to each) |
|
a Reaction conditions: substrate (1.0 mmol), Ru2 (2.0–10.0 μmol), DME (10 mL), PH2 = 68 bar (at RT), PCO2 = 12 bar (at RT), temp. = 160 °C, S:C = the molar substrate to catalyst ratio.
b The conversion with reference to the carboxylic acid was determined by GC (using mesitylene as the internal standard) and by GC-MS.
c In the absence of solvent; 54% of methyl hexanoate and 12% of hexyl hexanoate were produced.
d In the presence of decanoic acid (0.1 mmol) as additive.
e 5% of the product is 6-methoxy-6-oxohexanoic acid.
f The product is methyl stearate.
g The product is methyl benzenepropanoate.
|
|||||
| 1 |
|
100:1 |
20 | 16 | 10/6 |
| 2 |
|
100:1 |
20 | 10 | 6/4 |
| 3 |
|
500:1 |
20 | 100 | 100/0 |
| 4c |
|
500:1 |
20 | 99 | 66(54/12)/33 |
| 5 |
|
500:1 |
20 | 100 | 100/0 |
| 6 |
|
500:1 |
20 | 99 | 99/0 |
| 7 |
|
500:1 |
20 | 100 | 100/0 |
| 8 |
|
500:1 |
20 | 96 | 78/18 |
| 9d |
|
500:1 |
20 | 99 | 99/0 |
| 10d |
|
500:1 |
20 | 99 | 99/0 |
| 11d |
|
500:1 |
20 | 99 | 99/0 |
| 12d |
|
100:1 |
20 | 99 | 99/0 |
| 13d |
|
100:1 |
20 | 99 | 99/0 |
| 14e |
|
500:1 |
24 | 99 | 94/5 |
| 15 |
|
500:1 |
24 | 99 | 99/0 |
| 16 |
|
500:1 |
24 | 99 | 99/0 |
| 17d,f |
|
500:1 |
20 | 98 | 98/0 |
| 18g |
|
500:1 |
20 | 99 | 99/0 |
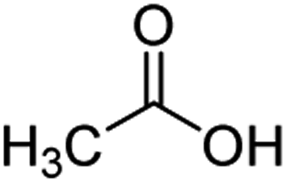
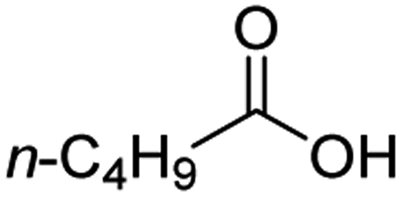
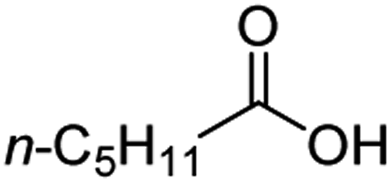
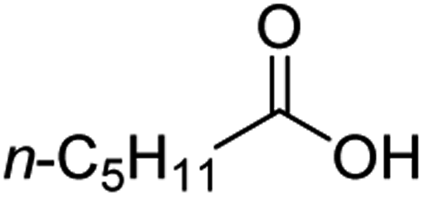
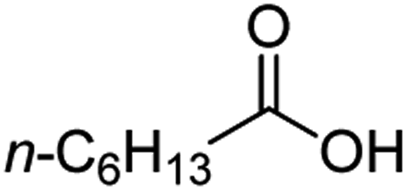
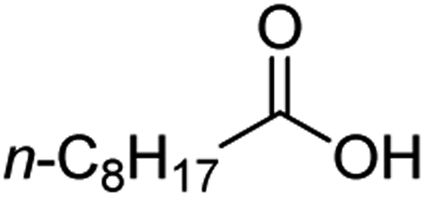
For aliphatic carboxylic acids containing carbon chain lengths of C ≤ 5, low conversions and low selectivity were evident after a 20 hour run time. For example, only 10% of methyl acetate was observed when using acetic acid and 6% of methyl pentanoate when using pentanoic acid (entries 1 and 2, Table 2). By contrast, the longer chain lengths (6 ≤ C ≤ 10) showed excellent conversions and high selectivity for the corresponding methyl ester (entries 3–7, Table 2). In the case of hexanoic acid, it was shown that in the absence of solvent the selectivity dramatically reduced [100% (with DME) to 54% (without), entries 3 and 4, Table 2] producing 54% methyl hexanoate, 12% of hexyl hexanoate and 33% 1-hexanol (entry 4, Table 2).
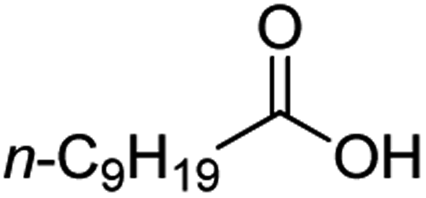
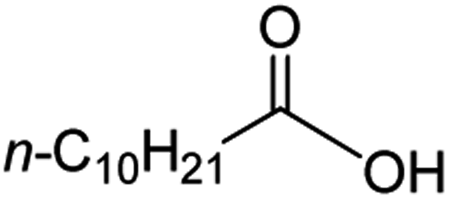
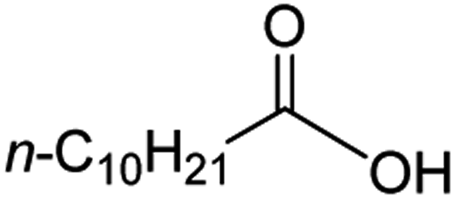
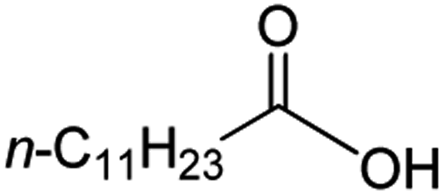
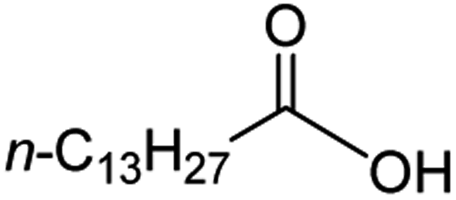
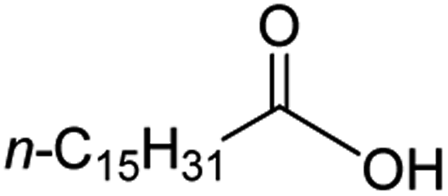
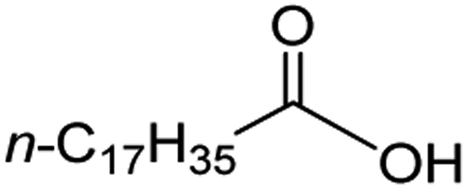
Intriguingly, decanoic acid (1) can also be used as additive for adjusting the activity and selectivity of the ruthenium catalyst. For example, using undecanoic acid as the substrate, the selectivity for the methyl ester increased from 81% to 100% and the conversion from 96% to 99% when 10 mol% decanoic acid was introduced (entries 8 vs. 9, Table 2). The longer carbon chain aliphatic acids (12 ≤ C ≤ 19) also gave excellent results in the presence of decanoic acid as an additive (entries 10–13, Table 2). Significantly, many of the resulting aliphatic esters are in the range of biodiesels.18
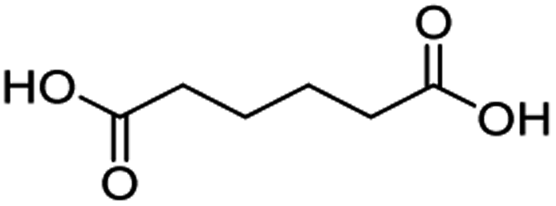
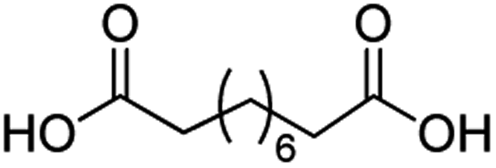
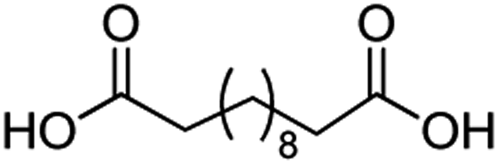
For dicarboxylic acids incorporating aliphatic linkers of a range of chain lengths, all underwent 99% conversion with 95–100% selectivity. However, it was noted that longer reaction times (up to 24 hours) were needed to complete the transformation when compared to those containing only one carboxyl group (entries 14–16, Table 2). It was also apparent that the shorter carbon chain dicarboxylic acid, adipic acid, was at the lower end of the selectivity (95%) range with 94% dimethyl adipate and 5% of 6-methoxy-6-oxohexanoic acid produced (entry 14, Table 2).
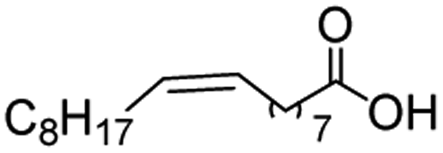
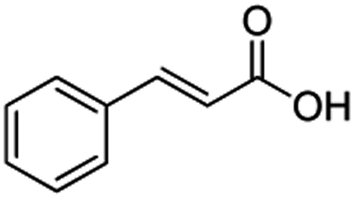
In the case of aliphatic carboxylic acids incorporating some unsaturation, methyl ester formation was also achievable but at the expense of double bond (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) hydrogenation. For example, 99% of methyl benzenepropanoate was obtained when using cinnamic acid while 98% of methyl octadecanoate were observed from octadec-9-enoic acid (entries 17 and 18, Table 2).
C) hydrogenation. For example, 99% of methyl benzenepropanoate was obtained when using cinnamic acid while 98% of methyl octadecanoate were observed from octadec-9-enoic acid (entries 17 and 18, Table 2).
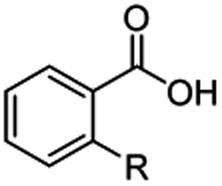
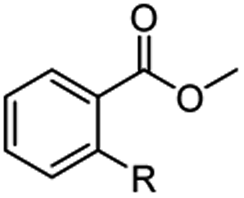
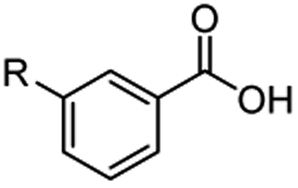
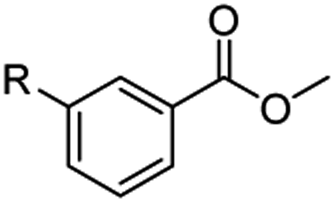
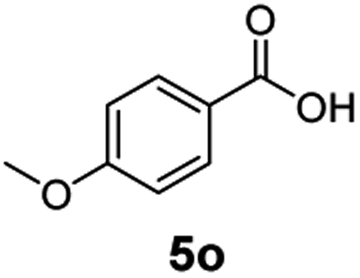
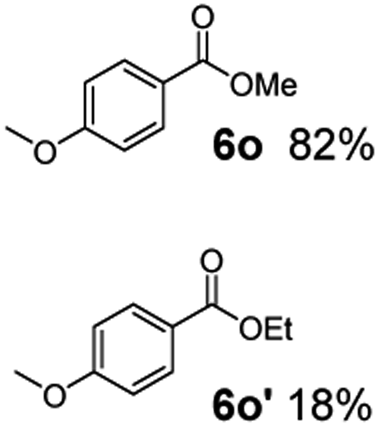
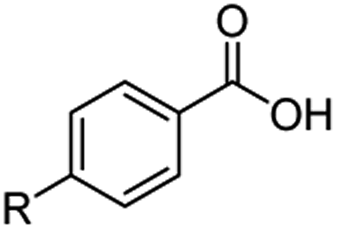
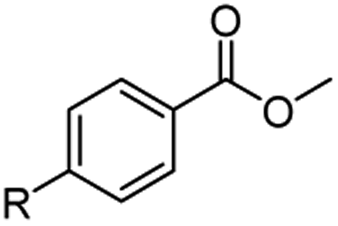
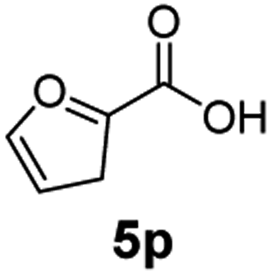
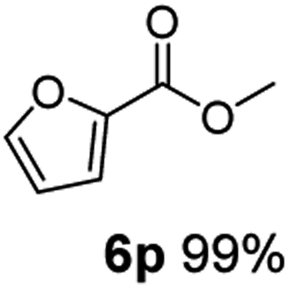
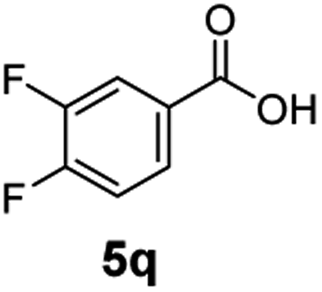
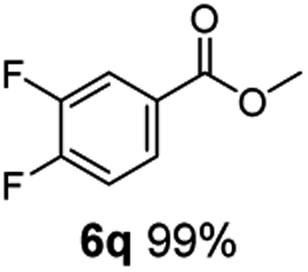
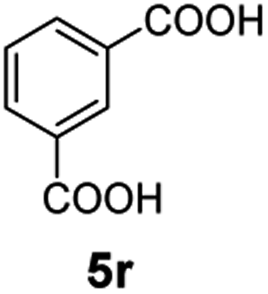
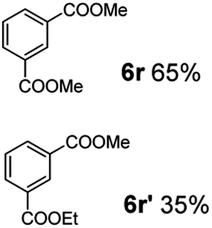
The hydrogenation capacity of Ru2 is not limited to aliphatic carboxylic acids. Indeed, a wide variety of aromatic examples differing in their ortho-, meta- and para-substitution patterns, also showed excellent conversions and selectivity for methyl esters (6a–6r) (Table 3). For example, benzoic acid (5a) underwent 93% conversion to methyl benzoate (6a) with a S:C ratio of 100:1 that only reduced to 69% (99% selectivity) when the ratio was increased to 500:1. Interestingly, benzoic acids (5b–5e, 5f–5h, 5j–5l) incorporating electron withdrawing groups such as halides (F, Cl, Br) or CF3 at the ortho- (5b–5e), meta- (5f–5h) or para-positions (5j–5l) showed little variation in conversion (73–99%) and selectivity (almost 100%) for the methyl ester. However, dehalogenation was an outcome of all benzoic acids containing either Cl or Br substituents resulting in methyl benzoate as the sole methyl ester product in these cases (see substrates 5c, 5d, 5g, 5h, 5k, 5l); this finding can likely be attributed to the high temperature and the reactivity of Ru2 towards the Caryl–X bonds present in these particular substrates. In comparison, the electron-rich benzoic acids with methyl or methoxy substituents (5n and 5o) at the para-positions gave 100% conversions to their methyl esters (6n and 6o). Interestingly, p-methoxybenzoic acid (5o) produced two kinds of esters, methyl 4-methoxybenzoate (6o, 82%) and ethyl 4-methoxybenzoate (6o′, 18%). The presence of 6o′ would suggest that the hydrogenation of CO2 produced a small amount of the higher alcohol ethanol which then underwent condensation with p-methoxybenzoic acid (5o) to give 6o′; the mechanism of ethanol production from CO2 is assumed to be similar to that reported by Han.6 Furan-2-carboxylic acid (5p) also resulted in 99% conversion and 100% selectivity (6p). Notably, the difluoro-substitution on the aryl ring did not affect the reaction with 3,4-difluorobenzoic acid (5q) also producing 99% of methyl 3,4-difluorobenzoate (6q). In addition, the dicarboxyl-substituted isophthalic acid (5r), though reaching 100% conversion, displayed a slight difference in reactivity when compared to benzoic acid by producing 65% of the dimethylester (6r) and 35% of the methyl ethyl ester (6r′). Once again the hydrogenation of CO2 to form a small amount of ethanol seems likely to account for the formation of 6r′.
|
|
|||
|---|---|---|---|
| Substrate | Productb (conv.%) | Substrate | Productb (conv.%) |
|
a Conditions: substrate (1.0 mmol), Ru2 (10.0 μmol), S:C = 100:1, DME (10 mL), PH2 = 68 bar (at RT), PCO2 = 12 bar (at RT), temp. = 160 °C, time = 20 h.
b The conversion, with reference to the carboxylic acid, was determined by GC (using mesitylene as the internal standard) and by GC-MS.
c
Ru2 (2.0 μmol), S:C = 500:1.
d A dehalogenation reaction occurs with the product being methyl benzoate (6a).
|
|||
|
|
|
|
| 5a R = H | 6a R = H, 93%, 6a 69%c | 5f R = F | 6f R = F, 99% |
| 5b R = F | 6b R = F, 99% | 5g R = Cld | 6a 99% |
| 5c R = Cld | 6a 99% | 5h R = Brd | 6a 99% |
| 5d R = Brd | 6a 99% | 5i R = CF3 | 6i R = CF3, 99% |
| 5e R = CF3 | 6e R = CF3,99% |
|
|
|
|
||
| 5j R = F | 6j R = F, 78% | ||
| 5k R = Cld | 6a 99% |
|
|
| 5l R = Brd | 6a 99% | ||
| 5m R = CF3 | 6m R = CF3, 73% | ||
| 5n R = Me | 6n R = Me, 100% | ||
|
|
|
|
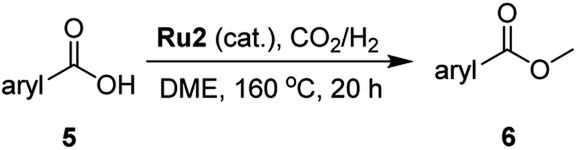
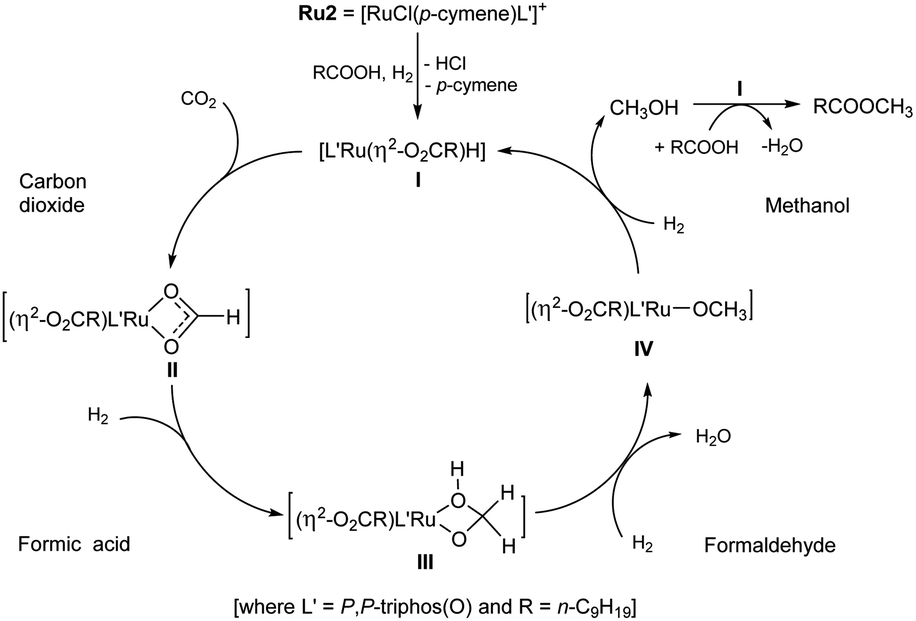 | ||
| Scheme 4 Proposed mechanism for the Ru2 catalyzed hydrogenation of CO2 to methyl decanoate in the present of decanoic acid via Ru(II) intermediates derived from formic acid, formaldehyde and methanol. | ||
In attempt to shed some light on this proposed mechanism, the following set of experiments were conducted. Firstly, 1H NMR spectroscopy was used to detect for methanol in the catalytic system. Using the optimal operating conditions established in entry 4 (Table 1), free methanol was indeed detected by 1H NMR spectroscopy after 20 hours (Fig. S16, see ESI†). Furthermore, when the volume of DME was decreased from 10 mL to 2.5 mL a small amount of ethanol was also observed. This finding can explain the formation of methyl decanoate (95%) and ethyl decanoate (5%) that was observed in entry 11 (Table 1).6 On the other hand, there was no methanol observed when the hydrogenation was conducted in the absence of CO2 (entry 16, Table 1, see ESI†). More importantly, in the absence of decanoic acid, no CH3OH was observed (entry 17, Table 1), which would imply decanoic acid acts as a promoter.
Secondly, for each ruthenium catalyst (Ru1–Ru4), the amount of methanol produced during the hydrogenation of CO2 when in the presence of decanoic acid (1 mmol), was quantitatively determined: conditions, 10 mL of DME at 160 °C with PCO2 = 12 bar (at RT) and PH2 = 68 bar (at RT) (Table 4, ESI 6.2†). To our surprise, the turnover number (TON) of Ru2 for the direct transformation of CO2 to methanol was up to 3262 which noticeably exceeds that previously reported by Leitner et al. (TON = 442, 2-MTHF as solvent, at 140 °C with PCO2 = 20 bar (at RT) and PH2 = 60 bar (at RT)).5e,5j Although the origin of this increase remains uncertain, it does support the assertion that decanoic acid present in the catalytic system can act as a promoter in the conversion of CO2 to methanol. It was also found that the amount of methanol produced using Ru2 and Ru4 proved consistent with the percentage conversions to the methyl decanoate. On the other hand, that observed for catalysts Ru1 and Ru3 showed some discrepancy (entries 1 and 3, Table 4), which may be due to the structural differences of the ligands leading to the different activities for catalytic esterification.
| Entry | [Ru] | Conv.b (%) | Conv.c (mg mL−1) | MeOHd (n mmol) | TONMeOHe |
|---|---|---|---|---|---|
|
a Conditions: decanoic acid (1.0 mmol), [Ru] (10.0 μmol), DME (10 mL), PCO2 = 12 bar (at RT), PH2 = 68 bar (at RT), time = 20 hours, temp. = 160 °C, S:C = 100:1.
b The conversion with reference to decanoic acid was determined by GC (using mesitylene as the internal standard) and by GC-MS.
c The conversion to methanol was determined by GC (using a standard concentration curve of methanol as the external standard (see ESI 6.2)).
d The total volume of the reaction solution was 10 mL.
e TON is the ratio of the moles of methanol (n) to the moles of catalyst (10 μmol).
|
|||||
| 1 | Ru1 | 76 | 90.03 | 28.12 | 2812 |
| 2 | Ru2 | 99 | 104.4 | 32.62 | 3262 |
| 3 | Ru3 | 67 | 99.31 | 30.94 | 3094 |
| 4 | Ru4 | 56 | 62.36 | 19.49 | 1949 |
Finally, we set about attempting to confirm that the catalytic mechanism proceeded via three steps involving intermediates derived from formic acid, formaldehyde and methanol.3h,5j,5k,19 Hence, we used MeOH, HCHO (37% wt aqueous solution) and HCOOH independently as the C1 sources in the absence of CO2 under the same conditions. In all cases methyl decanoate was smoothly formed. However, the different carbon sources required different run times to complete the transformation (Table 5, ESI 6.1†). For example, the use of one or fifty equivalents of methanol needed less time (2–4 hours) to be converted to methyl decanoate than that using formaldehyde and formic acid. Indeed, using formic acid took close to 20 hours to allow a comparable conversion. Although uncertain at this stage, it would seem likely that the rate-determining step in the catalytic cycle is located in the sequence of steps leading from HCOOH to CH3OH.3h,5j,5k,19 As a final point, only 5% conversion to methyl decanoate was observed in the absence of Ru2 (entry 4, Table 5).
|
|
|||||
|---|---|---|---|---|---|
| Entry | C1 carbon source | S:C |
t (h) | Conv.b (%) | Ester/alcoholb (yield%) |
|
a Reaction conditions: decanoic acid (1 mmol), Ru2 (10 μmol), DME (10 mL), PH2 = 80 bar (at RT), temp. = 160 °C, S:C = the molar substrate to catalyst ratio.
b The conversion with reference to the decanoic acid was determined by GC (using mesitylene as the internal standard) and by GC-MS.
c 37% wt aqueous solution.
|
|||||
| 1 | MeOH (50 eq.) | 100:1 |
2 | 99 | 99/0 |
| 2 | MeOH (1 eq.) | 100:1 |
4 | 99 | 99/0 |
| 3 | HCHOc (1 eq.) | 100:1 |
10 | 99 | 99/0 |
| 4 | HCO2H (1 eq.) | 100:1 |
20 | 99 | 99/0 |
| 5 | MeOH (1 eq.) | 100:0 |
4 | 5 | 5/0 |
Conclusions
We have developed a novel route to prepare a wide range of methyl esters from CO2via a ruthenium catalyzed hydrogenation to methanol and in situ condensation with a carboxylic acid. By evaluating four structurally related diphosphine-ruthenium (pre)catalysts, complex [RuCl(κP,κP-triphos(O))(η6-p-cymene)][ClO4] (Ru2) has shown the greatest efficiency for the transformation. Indeed using Ru2, both aromatic and aliphatic carboxylic acids can be converted with high selectively to the corresponding carboxylic acid methyl ester. Furthermore, we have also probed the mechanism through a series of experiments, which has shown that the pathway involves the direct hydrogenation of CO2 to form consecutively formic acid, formaldehyde and methanol which then finally undergoes condensation with carboxylic acids to the form the corresponding methyl ester. Moreover, this work also presents an original and effective ruthenium catalyzed process for producing methanol from CO2 with the TON for the CO2 to methanol step up to 3262; the role of the carboxylic acid as a promoter in this reaction has been demonstrated. More importantly, this work provides an attractive route for the utilization of CO2 as a carbon source for the production of biofuels and fine chemicals. We therefore hope that this report will inspire the development of more efficient metal-catalyzed hydrogenation reactions of CO2 and ultimately reduce the demand for fossil resources.Experimental section
Catalytic study
Under an atmosphere of argon, a stainless steel 100 mL autoclave, equipped with a magnetic stir bar, was charged with Ru1–Ru4 (2.5–10 μmol) and the solvent to be used (2.5–5 mL). A solution of the carboxylic acid (1 mmol) in the solvent (5–25 mL) was then added via a syringe. The autoclave was purged by three cycles of pressurization/venting with CO2 (5–10 bar), and then pressurized with the desired mixture of CO2 and H2. The autoclave was heated to the desired temperature and the contents stirred. After the pre-determined reaction time, the autoclave was cooled to room temperature and the pressure slowly released. The reaction mixture was filtered through a plug of silica gel and then analyzed by GC and GC-MS.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the support from the National Natural Science Foundation of China (21871275), the Nature Science Foundation of Hebei Province (B2019205149) and Talent Introduction Foundation of Agricultural University of Hebei (YJ201931). GAS thanks the Chinese Academy of Sciences for a President's International Fellowship for visiting scientists.Notes and references
-
(a) M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed
; (b) J. Klankermayer, S. Wesselbaum, K. Beydoun and W. Leitner, Angew. Chem., Int. Ed., 2016, 55, 7296–7343 CrossRef CAS PubMed
-
(a) P. Markewitz, W. Kuckshinrichs, W. Leitner, J. Linssen, P. Zapp, R. Bongartz, A. Schreiber and T. E. Müller, Energy Environ. Sci., 2012, 5, 7281–7305 RSC
- Select recent examples for forming formic acid:
(a) P. G. Jessop, T. Ikariya and R. Noyori, Nature, 1994, 368, 231–233 CrossRef CAS
- Select recent examples for forming formides:
(a) Q.-Y. Bi, J.-D. Lin, Y.-M. Liu, S.-H. Xie, H.-Y. He and Y. Cao, Chem. Commun., 2014, 50, 9138–9140 RSC
- Select recent examples for methanol:
(a) M. Behrens, F. Studt, I. Kasatkin, S. Kuhl, M. Havecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B.-L. Kniep, M. Tovar, R. W. Fischer, J. K. Nørskov and R. Schlçgl, Science, 2012, 336, 893–897 CrossRef CAS PubMed
- Q. L. Qian, M. Cui, Z. H. He, C. Y. Wu, Q. G. Zhu, Z. F. Zhang, J. Ma, G. Y. Yang, J. J. Zhang and B. X. Han, Chem. Sci., 2015, 6, 5685–5689 RSC
-
(a) Q. Liu, L. Wu, I. Fleischer, D. Selent, R. Franke, R. Jackstell and M. Beller, Chem–Eur. J., 2014, 20, 6888–6894 CrossRef CAS PubMed
-
(a) J. Klankermayer and W. Leitner, Science, 2015, 350, 629–630 CrossRef CAS PubMed
- K. Beydoun, G. Ghattas, K. Thenert, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2014, 53, 11010–11014 CrossRef CAS PubMed
- Y. Li, T. Yan, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 10476–10480 CrossRef CAS PubMed
-
(a) L. Zhang, J. Cheng, T. Ohishi and Z. Hou, Angew. Chem., Int. Ed., 2010, 49, 8670–8673 CrossRef CAS PubMed
- I. I. F. Boogaerts, G. C. Fortman, M. R. L. Furst, C. S. J. Cazin and S. P. Nolan, Angew. Chem., Int. Ed., 2010, 49, 8674–8677 CrossRef CAS PubMed
- Select recent examples for cyclic carbonates and polycarbonates:
(a) A. M. Chapman, C. Keyworth, M. R. Kember, A. J. J. Lennox and C. K. Williams, ACS Catal., 2015, 5, 1581–1588 CrossRef CAS
- A. Otto, T. Grube, S. Schiebahn and D. Stolten, Energy Environ. Sci., 2015, 8, 3283–3297 RSC
- Z. Wang, X.-Y. Chen, B. Liu, Q. Liu, G. A. Solan, X.-Z. Yang and W.-H. Sun, Catal. Sci. Technol., 2017, 7, 1297–1304 RSC
- Select recent examples for hydrogenation of esters
(a) T. Zell and D. Milstein, Acc. Chem. Res., 2015, 48, 1979–1994 CrossRef CAS PubMed
-
(a) K. Mashima, K. Kusano, N. Sate, Y. Matsumura, K. Nozaki, H. Kumobayashi, N. Sayo, Y. Hori, T. Ishizaki, S. Akutagawa and H. Takaya, J. Org. Chem., 1994, 59, 3064–3076 CrossRef CAS
-
(a)
A. Pandey, C. Larroche, S. C. Ricke, C.-G. Dussap and E. Gnansounou, Biofuels alternative feedstocks and conversion processes, Elsevier Inc, 2011 Search PubMed
-
(a) M. Nielsen, E. Alberico, W. Baumann, H. J. Drexler, H. Junge, S. Gladiali and M. Beller, Nature, 2013, 495, 85–89 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, spectra (NMR, GC-MS, LC-MS), Fig. S1–S19, Tables S1–S6. CCDC 1961855 (Ru2) and 1961856 (Ru3). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc02942d |
| This journal is © The Royal Society of Chemistry 2020 |